Introduction
Glioblastoma (GBM) is the most frequent and
malignant brain tumor in the human central nervous system (CNS).
Despite research progress on its molecular mechanisms and optimal
treatments, including aggressive surgery, chemotherapy,
radiotherapy and immunotherapy, the prognosis for patients with GBM
is still relatively poor (1,2). The
current model of glioma stem cells (GSCs) has significantly
contributed to a better understanding of the cellular origin and
failure of current conventional treatments (3,4).
GSCs, which are characterized by their ability to self-renew,
multi-potentiality and tumorigenicity, are thought to contribute to
the tumor propagation, recurrence and resistance to chemotherapy
and radiotherapy, and they represent a critical therapeutic target
(5–9).
Neural stem cell/neural progenitor cell (NSC/NPC)
transplantation was initially applied in the study of Parkinson’s
disease (10,11) and subsequently in spinal cord
injury (12), stroke and multiple
sclerosis (13,14). Until 2000, several research groups
demonstrated that NSCs exhibited a unique migratory capacity of
efficiently crossing the blood-brain barrier to target brain tumors
located distantly from the original transplant site throughout the
experimental brain (15–17). Subsequent studies have proposed
that NSCs may possess some natural abilities to suppress tumor
growth and to induce tumor cell apoptosis. For example, Staflin and
colleagues injected rat embryonic neural progenitor cell lines into
the nucleus caudatus of Fisher rats combined with glioma cells and
found that injected NPCs could inhibit glioma outgrowth in
vivo, resulting in an extension in the life span of glioma
inoculated rats (18). Consistent
with this finding, Glass et al showed that endogenous NPCs
in the adult brain exhibited a strong tropism for glioblastomas
(19). The addition of neural
precursors into glioblastoma grafts promoted tumor cell death and
prolonged survival in vivo. Furthermore, the NSC inherent
tropism towards brain tumors resulted in the pursuit of applying
NSCs as a promising therapeutic tool and/or vehicle for tracking
and suppressing malignant glioma cells (20–25).
However, limitations exist when applying a small number of NSCs to
restrain the overwhelming number of bulk tumor cells. In the
present study, we aimed at exploring the preferential tropism of
NSCs to GSCs and the effect of migrated NSCs on GSC stemness
phenotypes.
Materials and methods
Cell culture
Human neural stem cells (NSCs; ReNcell CX
immortalized cells, SCC007, Millipore, MA, USA) were cultured as an
adherent monolayer in a laminin-coated flask in NSC maintenance
medium (Millipore) supplemented with epidermal growth factor (EGF;
PeproTech Inc., NJ, USA) and basic fibroblast growth factor (bFGF;
PeproTech) at 20 ng/ml, respectively, and were used within ten
passages as recommended. Human primary glioma stem cells (GSCs)
were derived from tumor resection. Tumor tissue was enzymatically
dissociated and cultured as floating neurospheres in serum-free
supplemented medium [DMEM/F-12 medium containing 20% BIT serum-free
supplement (Stemcell Technologies Inc., Vancouver, Canada), EGF and
bFGF at 20 ng/ml, respectively] (26,27).
Glioma U251 stem cells (U251-SC) were also cultured as non-adherent
neurospheres in serum-free supplemented medium as previously
described, which were derived from the human glioma cell line U251.
GSCs of more than six passages were used for further experiments.
To induce GSC differentiation, we used the conventional
serum-containing medium (10% FBS in DMEM) for seven days. For
hypoxic incubation, the cells were grown at 1% oxygen for 24 h in a
hypoxic work-station. The Human human fibroblast cell line HFL1
(ATCC® CCL-153™) was used as control cells.
Immunofluorescence staining
The primary antibodies used in this study included
rabbit anti-human CD133 (1:500, Abcam, MA, USA) and mouse
anti-human Nestin (1:500, Abcam) and incubated for 16 h at 4°C,
followed by detection with the corresponding fluorescent secondary
antibodies. Nuclei were stained with DAPI, and the slides were
detected using fluorescence microscopy.
CD133 flow cytometry analysis
Up to 106 cells were resuspended in the
recommended buffer (containing PBS pH 7.2, 0.5% BSA, and 2 mM EDTA)
and incubated for 10 min at 4°C with CD133/1 (AC133) antibody
(1:11; Miltenyi Biotec, Bergisch Gladbach, Germany). Mouse IgG1
(1:11; Miltenyi Biotec) was used as isotype control antibody. CD133
detection and analysis were performed on BD FACS Aria.
Western blot analysis
The primary antibodies used included rabbit
anti-human CD133 (1:500, Abcam), mouse anti-human Nestin (1:500,
Abcam), rabbit anti-human GFAP (1:500, Abcam) and mouse anti-human
GAPDH (1:1000, Boster, Wuhan, China).
Conditioned medium (CM)
GSC and U251-SC conditioned medium were harvested as
previously described (27). For
hypoxic cells CM, GSCs and their differentiated cells were shifted
to DMEM/F-12 medium without any supplement at 1% oxygen for 24 h.
The CM was harvested with viable cell counting and stored at −80°C
for further use. To obtain the growth factor medium, VEGF, EGF and
bFGF were added into DMEM/F12 at a concentration of 20, 10, 5 and 0
ng/ml immediately before use.
Cell migration transwell assay
The in vitro migration of NSCs to GSCs or
differentiated cells was detected using Transwell assay. Each well
of 24-well cell culture plates was separated into two chambers by
an insert membrane of 8-μm pores. Briefly, 600 μl of CM was placed
into each lower chamber and DMEM/F12 without supplement was used as
a basal migration control. NSCs (1×104 cells in 100 μl
of DMEM/F12) were then seeded into the upper chamber. After 6 h of
incubation at 37°C, NSCs in the chambers were fixed with 95%
ethanol. Non-migrating cells on the upper side of the insert
membrane were wiped off. Migrating cells on the bottom of the
membrane were stained using 0.5% crystal violet and quantified
under a microscope.
Mixed culture of NSCs and GSCs
For mixed culture, NSCs and GSCs were labeled using
the fluorescent tracer Dio and Dil (Molecular Probes, Inc., Eugene,
OR, USA), respectively, as recommended. Single cell suspensions of
NSCs, GSCs or U251-SCs were prepared at a density of
1×106/ml in serum-free culture medium and 5 μl of the
cell-labeling solution (Dio for NSC, Dil for GSC and U251-SC) was
added per ml of cell suspension. After incubation for 20 min at
37°C and a rinse in warm (37°C) medium, 2×105 NSCs were
seeded in per laminin-coated 6-well plates as an adherent monolayer
and 1×105 GSCs were seeded in per uncoated plates as
suspended neurospheres. On the second day, the GSC neurospheres
were mixed and cultured with NSCs. The migration of NSCs and
proliferation of GSC neurospheres were detected using fluorescent
microscopy.
Co-culture of NSCs and GSCs
For co-culture of NSCs and GSCs, the transwell assay
was used. Each well of 6-well cell culture plates was separated
into two chambers by an insert membrane of 0.4-μm pores. Briefly,
2×105 NSCs (or HFL1 cells as control) were cultured as
an adherent monolayer in the laminin-coated lower chamber and
1×105 GSCs were cultured as neurospheres in the upper
chamber. NSC maintenance medium supplemented with EGF and bFGF at 2
ng/ml, respectively, was used and added every three days for one
week. The expression of cell markers and the self-renewal ability
of GSCs co-cultured with NSCs (or control cells) were subsequently
determined.
Secondary neurosphere formation
assay
For secondary neurosphere formation assay, primary
GSC neurospheres co-cultured with NSCs (or control cells) after a
week were harvested and dissociated mechanically using a mechanical
cellular filter. Single cell suspension washed with PBS was
confirmed microscopically and suspended at 5,000 cells/ml in
serum-free supplemented medium. Using the limiting dilution assay,
1,000, 500, 200, 100, 50, 20 and 10 cells in 200-μl suspensions
were plated into 12 wells of each row in a 96-well microplate.
Fifty μl of serum-free supplemented medium was added to each well
every third day for two weeks. Neurospheres (non-adherent, tight
and spherical masses >75 μm in diameter) per well were
quantified using an ocular micrometer. To confirm the gradient
dilution results described above, we performed a more stringent
clonal assay by plating single cells into a 96-well plate, i.e.,
one viable cell per well, and at the end of two weeks, the wells
containing the clonal spheres derived from a single cell were
calculated.
Intracranial cell migration and
tumorigenesis assay
Female BALB/c nude mice, six to eight weeks of age,
were housed under specific pathogen-free conditions. All animal
experimental protocols were approved by the Institutional Animal
Care and Use Committee, Huazhong University of Science and
Technology. Briefly, NSCs and GSCs were labeled using the
fluorescent tracer Dio (green fluorescent) and Dil (red
fluorescent), respectively, as previously described. For the group
of unilateral intracranial xenografts, GSCs (1×105 cells
in 5 μl PBS) were stereotactically implanted into the right basal
ganglia of the nude mouse brain (AP +1.0 mm, ML +2.0 mm and DV −3.0
mm from bregma and dura) using a 10-μl Hamilton syringe at a speed
of 1 μl/min and the NSCs (2×105 cells in 5 μl PBS) were
then implanted 2.0 mm to the right of the injection site (which was
AP +1.0 mm, ML +4.0 mm and DV −3.0 mm from bregma and dura). For
the bilateral intracranial xenograft group, GSCs were implanted at
the same site, and the NSCs were symmetrically injected into the
contralateral side of the mouse brain, 2.0 mm left from bregma.
Injection of HFL1 cells instead of NSCs served as the control. To
detect the migration of NSCs, mouse brain samples were collected at
one and two weeks and were snap-frozen for subsequent examination.
To determine tumorigenicity, the mice were maintained until weight
loss of >10% occurred or when neurological signs appeared.
Statistical analyses
Statistics were performed using SPSS 17.0 software.
Comparisons among the groups were performed with analysis of
variance (ANOVA) or Student’s t-test. Significance was established
at p<0.05.
Results
Compared with differentiated cells, GSCs
induced enhanced NSC tropism and secreted more chemotactic
factors
To determinate the migratory capacity of NSCs to
GSCs and their differentiated cells, we first established human
GSCs from primary glioblastoma and U251 glioma stem cells (U251-SC)
from the U251 glioma cell line. GSCs were cultured as floating
neurospheres in an uncoated flask and serum-free supplemented
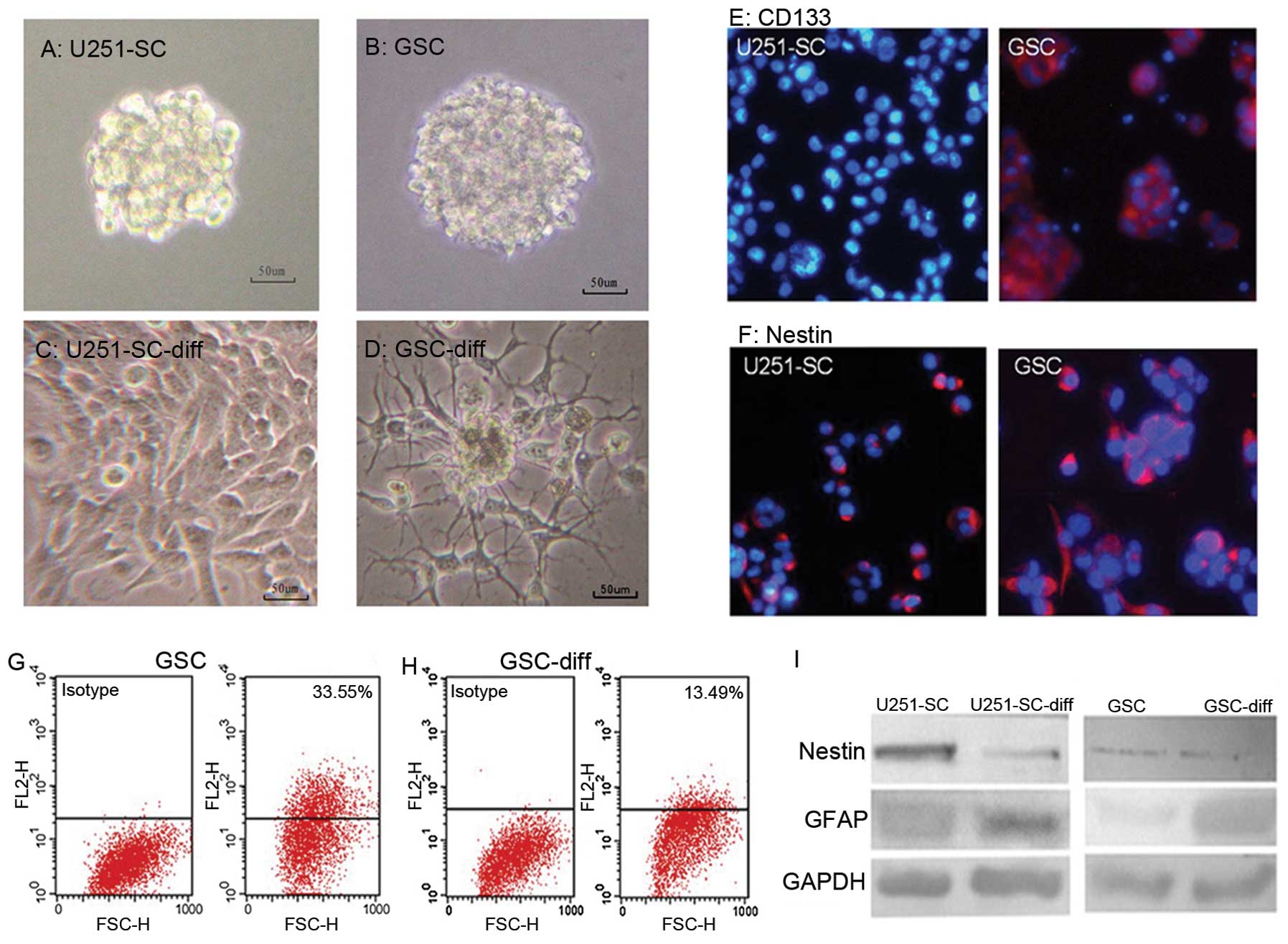
medium (Fig. 1A and B). The GSCs
and U251-SC were derived from serum-containing medium in seven
days. Upon differentiation in serum-containing medium, the GSC
neurospheres quickly attached to the culture flask and branched
out, instead of forming a suspension (Fig. 1C and D). Cell markers (CD133,
Nestin and GFAP) of GSCs and their differentiated cells showed
contrasting effects. A marked decrease in Nestin and/or CD133
expression in GSCs and a notable increase in GFAP expression in
differentiated cells were revealed (Fig. 1E–I).
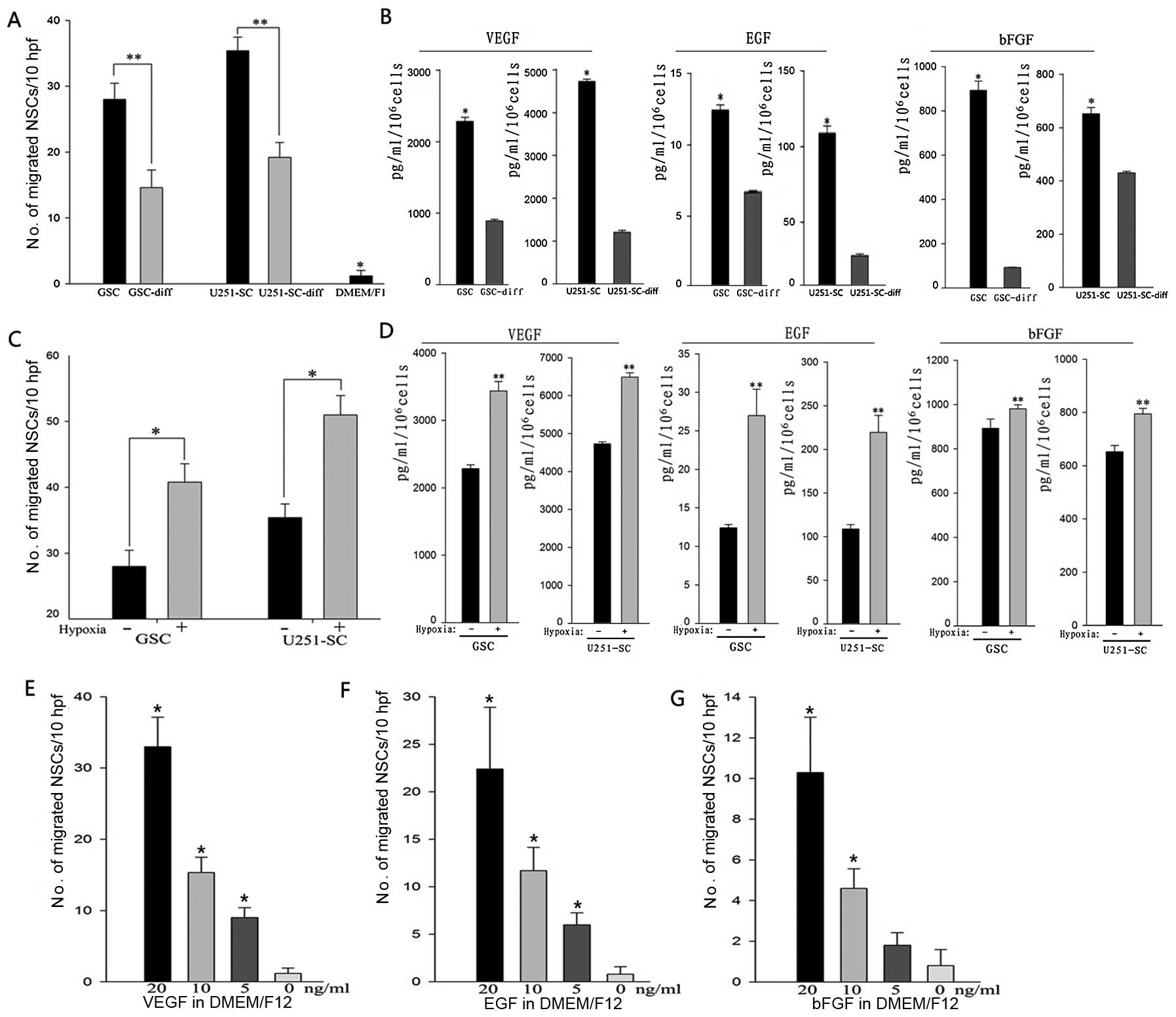
To detect NSC migration in vitro, conditioned
medium of GSCs and their differentiated cells were harvested. An
in vitro migration assay was used to test the migratory
capacity of NSCs to GSCs and their differentiated cells. These
results showed that the conditioned medium of GSCs and their
differentiated cells caused significantly more NSCs to migrate from
the top well, through the porous membrane to the lower surface when
compared to unconditioned medium (*P<0.001).
Interestingly, NSCs exhibited robust mobility toward conditioned
medium of GSCs, compared to that of their differentiated cells,
with an increase of ~2-fold in primary GSCs and U251-SCs (Fig. 2A; **P<0.05). Next, we
detected the levels of chemotactic factors (VEGF, EGF and bFGF) in
the conditioned medium. Notably, an incremental secretion of
chemotactic factors was detected in the conditioned medium of GSCs
compared to their differentiated cells (Fig. 2B; *P<0.05).
Hypoxia promoted NSC tropism to GSCs by
upregulating the expression of chemotactic factors, which played
critical roles in NSC migration
Hypoxia has been identified to play a critical role
in promoting tropism and mobilization of multiple stem cells,
including NSCs. To assess the effects of hypoxia on NSC migration
toward GSCs, we preconditioned GSCs to DMEM/F-12 medium without any
supplement at 1% oxygen for 24 h and then harvested the hypoxic
preconditioned medium for in vitro migration assay.
Increasing NSC migration rates of ~1.5 times were found in primary
GSCs and U251-SC (Fig. 2C;
*P<0.05).
Since hypoxia promoted NSC tropism to GSCs, we
further detected the effects of hypoxia on the expression of
chemotactic factors. Quantified GSCs were preconditioned in the
DMEM/F-12 medium without any supplement at 1% oxygen for 24 h and
the hypoxic preconditioned medium were then harvested for ELISA
analysis. These results showed that primary GSCs and U251-SC under
hypoxia secreted much greater amounts of VEGF, EGF and bFGF
compared to cells cultured in normoxia (Fig. 2D; **p<0.05).
To confirm the role of chemokines in NSC migration,
we investigated the effects of grow factor concentration gradient
medium on NSC tropism. VEGF, EGF and bFGF were added into DMEM/F12
at a concentration gradient of 20, 10, 5 and 0 ng/ml for in
vitro migration assay. A concentration-dependent migration of
NSCs was elicited by VEGF, EGF and bFGF, which suggested that
chemotactic factors induced by hypoxia played critical roles in NSC
migration (Fig. 2E–G).
In vitro migration of NSCs to GSCs
displays cytostatic effect
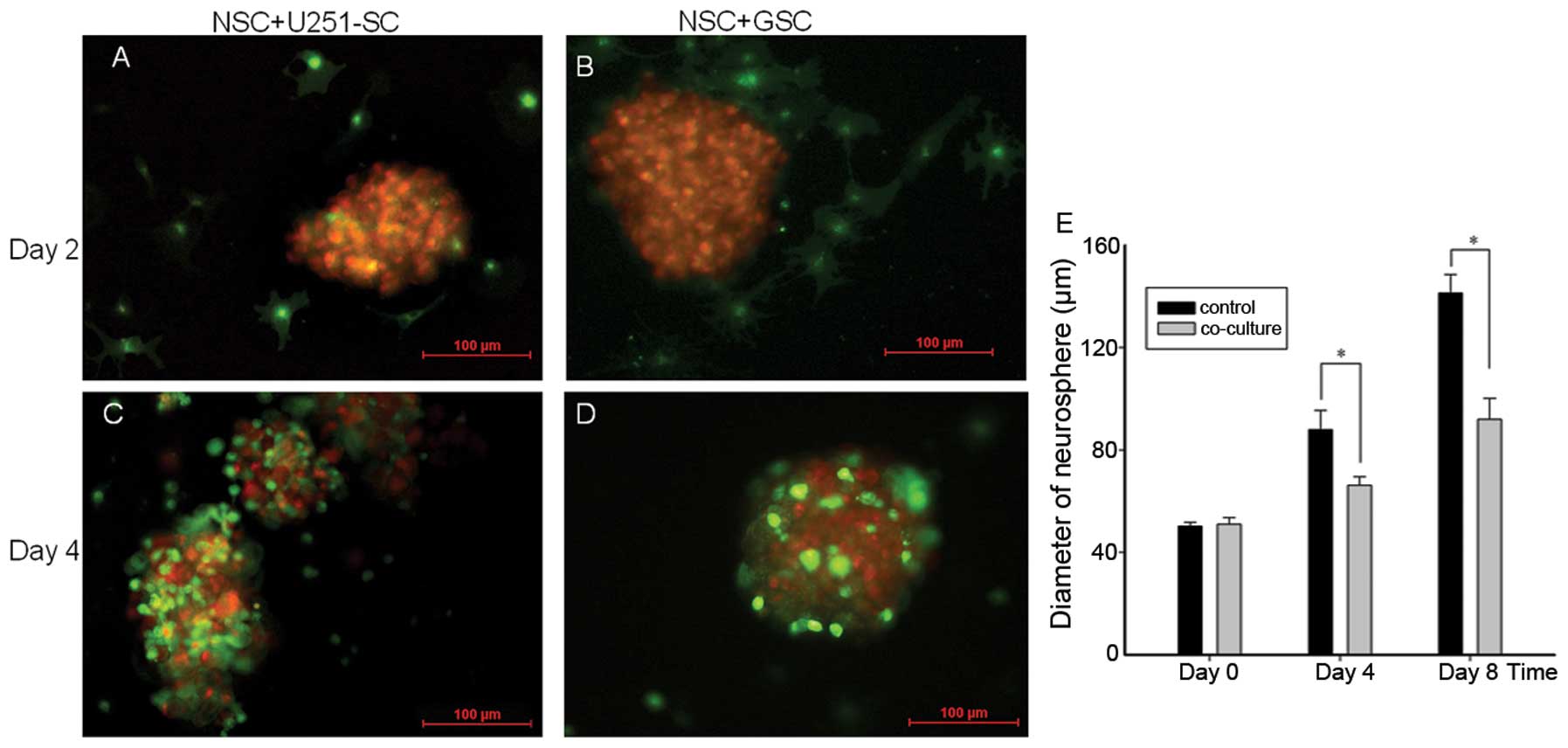
To explore the effects of in vitro NSC
migration on GSC growth, we labeled NSCs and GSCs using the
fluorescent tracer Dio (green) and Dil (red), respectively, and we
co-cultured labeled NSCs and GSC neurospheres on the second day. On
the second day after the co-culture, NSCs were detected to migrate
toward GSC neurospheres (Fig. 3A and
B). On the fourth day after the co-culture, numerous NSCs had
migrated to the GSC neurospheres and surrounded the entire
neurospheres (Fig. 3C and D).
To assess the effects of NSC migration on GSC
neurosphere growth, the diameters of GSC neurospheres were measured
and analyzed using a computerized fluorescence microscope after
co-culture. Compared with the control, co-cultures with NSCs caused
an inhibition of GSC neurosphere growth, with a remarkable decrease
in the neurosphere diameters (Fig.
3E; P<0.05). Stem-like phenotypes and self renewal
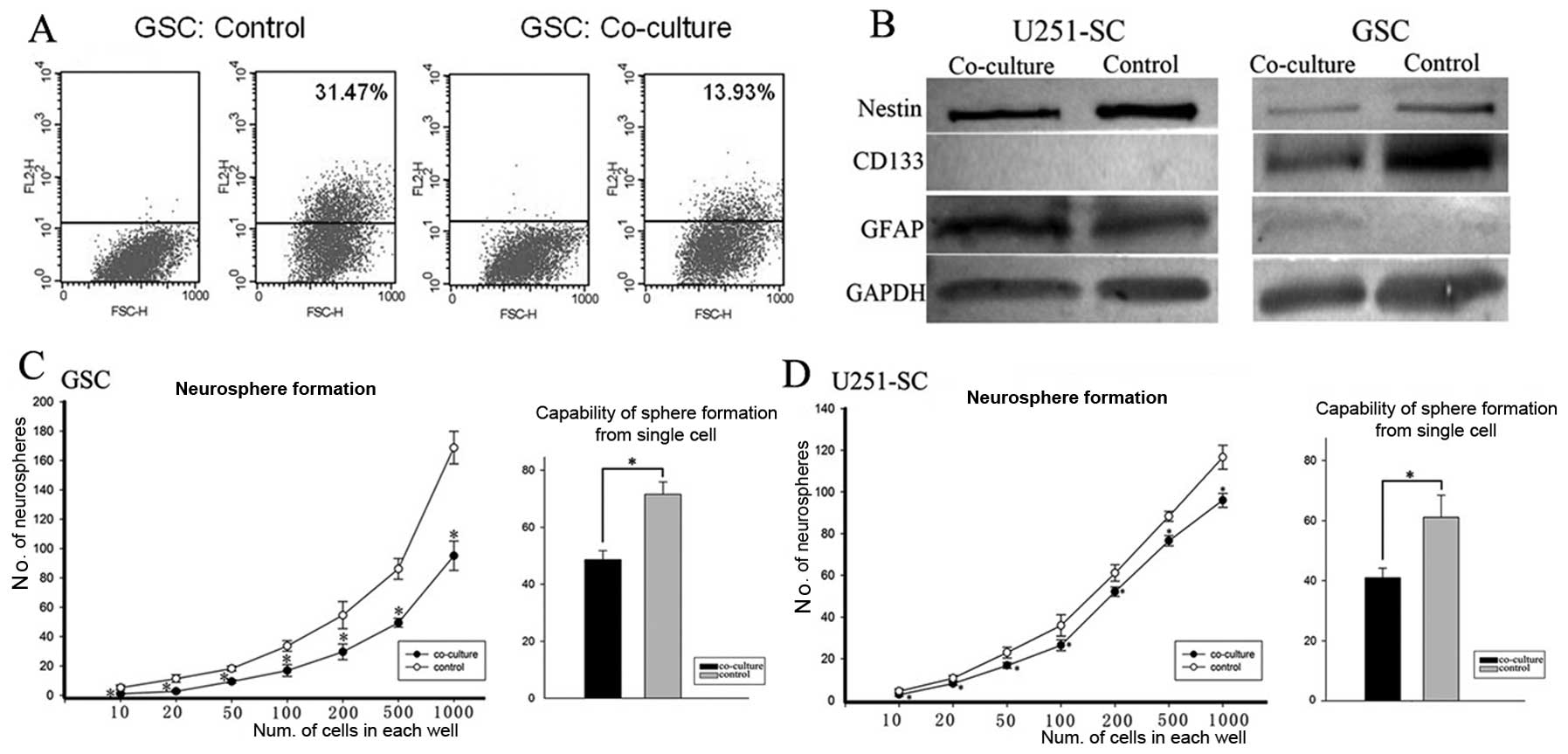
capability of GSCs were reduced after co-culture with NSCs. For the
purpose of detecting the effects of NSC migration on GSC stemness,
we co-cultured GSCs with NSCs. After one-week of co-culture,
CD133-positive cells were analyzed using the flow cytometry assay.
Compared with the control (co-culture with HFL1), there was a
notable decrease in the rate of CD133-positive GSC cells after
co-culture with NSCs, which indicated a decrease in the GSC
subpopulation (Fig. 4A). Further
detection using western blot analyses confirmed this finding. The
expression of CD133 and Nestin in GSCs were decreased, while GFAP
expression was increased in the co-culture (Fig. 4B). The data indicated that GSCs
were induced to differentiate in co-cultures with NSCs.
The ability to self-renew is one of the most
important stem cell phenotypes. The capability to neurosphere
formation in vitro has been applied to identify self-renewal
of GSCs and is considered a significant and independent predictor
of the clinical outcome of glioma patients (28). As manifested by the limiting
dilution clone assay and further supported by the more stringent
single-cell sphere formation assay, GSCs after co-culture showed
decreased self-renewal potentiality (Fig. 4C and D; P<0.05).
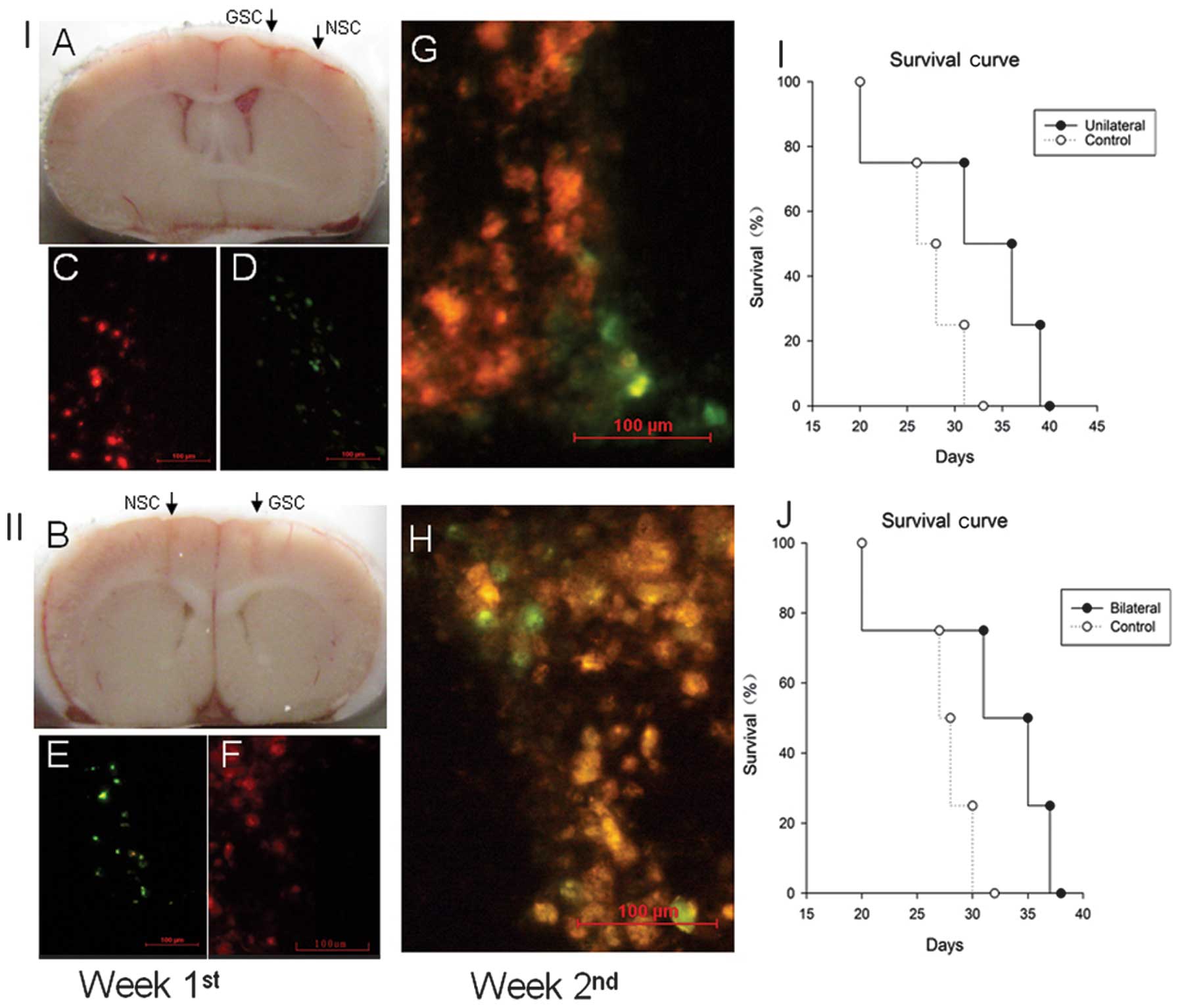
In vivo migration of NSCs to GSCs
improves survival
To detect the in vivo migration of NSCs to
GSCs, we labeled NSCs with Dio (green fluorescent) and GSCs with
Dil (red fluorescent), respectively. Next, GSCs and NSCs were
implanted stereotactically either unilaterally (Fig. 5A) or bilaterally (Fig. 5B) in the mouse brain. At the end of
the first week after the xenograft, there was no migrating NSCs
detected nearby the implanted region of GSCs (Fig. 5C and F). After two weeks, the
migrated NSCs around the GSCs were detected in both the unilateral
and bilateral grafted mouse brains (Fig. 5G and H).
To determine the effect of NSC migration on GSC
tumorigenicity, tumor-burdened mice that developed weight loss
>10% or showed the presence of neurological signs were recorded.
There were prolonged survivals in both the unilateral and bilateral
grafted mice, compared to the control group (Fig. 5I and J), suggesting that the in
vivo migration of NSCs to GSCs improved survival.
Discussion
Failure of current therapies for malignant gliomas
suggests that effective treatment may be dependent on the
development of new therapeutic strategies, which can eradicate the
arch criminal origin and/or the residual reservoirs of tumor cells
left behind after conventional treatments. Increasing studies on
GSCs have provided support for a new paradigm in tumor biology and
therapeutic targeting. It has been identified that NSCs exhibit an
inherent tropism to target malignant gliomas and inhibit tumor cell
growth (15–19). In the present study, we evaluated
the specific tropism of NSC to GSCs and its potential therapeutic
significance.
There have been numerous studies demonstrating that
stem cell migration is largely driven by various chemotactic
cytokines (29–31). Tumor upregulated expression of
chemotactic cytokines and the microvasculature contain relevant
guidance signals for NSC tropism toward malignant brain tumors
(32,33). We have previously demonstrated that
the secretion of VEGF and bFGF by GSCs was dramatically higher than
that of differentiated tumor cells (27). Furthermore, considering that EGF,
in addition to bFGF, is one of the key growth factors, which
suppresses differentiation and enables the in vitro
expansion of highly pure populations of stem cells (34–36),
we further compared the distinct secretion of EGF by GSCs and their
differentiated cells, which showed a similarly high secretion by
GSCs. A concentration-dependent NSC migration elicited by these
factors confirmed their critical roles in NSC tropism. These
findings strongly suggested that GSCs might possess enhanced
tropism for NSC compared with their differentiated
counterparts.
Furthermore, we found that this growth
factor-regulating NSC migration was dramatically upregulated by
hypoxia, which was consistent with previous reports that hypoxia
plays an important role in regulating NSC migration toward gliomas
(29,37,38).
Importantly, given the intratumoral hypoxic gradient in the tumor
microenvironment, which may drive heterogeneous GSC distribution
(39), NSCs could preferentially
target the GSC compartment in the tumor mass. Indeed, NSCs
exhibited a robust mobility toward GSCs in vitro in a
migration model compared to their differentiated cells.
Importantly, our results showed that the preferential migration of
NSCs to GSCs exhibited an antitumor effect. In vitro, a
mixed culture of NSCs with GSCs resulted in a direct inhibition of
GSC neurosphere growth. Such antitumor effects have been previously
reported for both endogenous neural precursor cells (19) and exogenously added NSCs from
newborn mice (16,18). Interestingly, our results
demonstrated that co-culture with NSCs induced GSC differentiation,
attenuated stem cell marker expression and reduced their
self-renewal capability. Importantly, in vivo, the
orthotopic xenografted NSCs prolonged the survival of the mice
bearing xenografted GSCs. Consistently, it has been demonstrated
that neural precursor cells may suppress the tumorigenicity of GSCs
by releasing bone morphogenetic protein-7 (40). Taken together, NSCs exhibited
preferential tropism to GSCs and reduced their stemness
phenotypes.
Intracranially or intravenously injected genetically
engineered NSCs have been applied to eradicate the invasive tumor
microsatellite and/or remnant tumor cells prior to their
development into recurrent gliomas (17,41–43).
Under the scenario of the GSCs model, our results introduce the
potential strategy of applying exogenous NSCs to specifically
target and reduce the most invasive and therapy-resistant tumor
stem cell compartment, which can eventually result in tumor
relapse. Thus, it would be interesting to further explore when and
how the endogenous NSCs undergo this process and what could be the
ultimate fate of the migrated NSCs when encountering their aberrant
counterparts.
Acknowledgements
This study was supported by grants of the Natural
Science Foundation of China (NSFC: 81272423, 81101620 and
81402058).
References
|
1
|
Wen PY and Kesari S: Malignant gliomas in
adults. N Engl J Med. 359:492–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fuller GN and Scheithauer BW: The 2007
Revised World Health Organization (WHO) Classification of Tumours
of the Central Nervous System: newly codified entities. Brain
Pathol. 17:304–307. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Galli R, Binda E, Orfanelli U, et al:
Isolation and characterization of tumorigenic, stem-like neural
precursors from human glioblastoma. Cancer Res. 64:7011–7021. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Singh SK, Clarke ID, Terasaki M, et al:
Identification of a cancer stem cell in human brain tumors. Cancer
Res. 63:5821–5828. 2003.PubMed/NCBI
|
|
5
|
Bao S, Wu Q, McLendon RE, et al: Glioma
stem cells promote radioresistance by preferential activation of
the DNA damage response. Nature. 444:756–760. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sakariassen PO, Immervoll H and Chekenya
M: Cancer stem cells as mediators of treatment resistance in brain
tumors: status and controversies. Neoplasia. 9:882–892. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Eyler CE and Rich JN: Survival of the
fittest: cancer stem cells in therapeutic resistance and
angiogenesis. J Clin Oncol. 26:2839–2845. 2008. View Article : Google Scholar
|
|
8
|
Clarke MF, Dick JE, Dirks PB, et al:
Cancer stem cells - perspectives on current status and future
directions: AACR Workshop on cancer stem cells. Cancer Res.
66:9339–9344. 2006. View Article : Google Scholar
|
|
9
|
Liu Q, Nguyen DH, Dong Q, et al: Molecular
properties of CD133+ glioblastoma stem cells derived
from treatment-refractory recurrent brain tumors. J Neurooncol.
94:1–19. 2009.
|
|
10
|
Spencer DD, Robbins RJ, Naftolin F, et al:
Unilateral transplantation of human fetal mesencephalic tissue into
the caudate nucleus of patients with Parkinson’s disease. N Engl J
Med. 327:1541–1548. 1992.
|
|
11
|
Freed CR, Breeze RE, Rosenberg NL, et al:
Survival of implanted fetal dopamine cells and neurologic
improvement 12 to 46 months after transplantation for Parkinson’s
disease. N Engl J Med. 327:1549–1555. 1992.PubMed/NCBI
|
|
12
|
Imitola J, Raddassi K, Park KI, et al:
Directed migration of neural stem cells to sites of CNS injury by
the stromal cell-derived factor 1alpha/CXC chemokine receptor 4
pathway. Proc Natl Acad Sci USA. 101:18117–18122. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Armstrong RJ and Svendsen CN: Neural stem
cells: from cell biology to cell replacement. Cell Transplant.
9:139–152. 2000.PubMed/NCBI
|
|
14
|
Carbajal KS, Schaumburg C, Strieter R,
Kane J and Lane TE: Migration of engrafted neural stem cells is
mediated by CXCL12 signaling through CXCR4 in a viral model of
multiple sclerosis. Proc Natl Acad Sci USA. 107:11068–11073. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Aboody KS, Brown A, Rainov NG, et al:
Neural stem cells display extensive tropism for pathology in adult
brain: evidence from intracranial gliomas. Proc Natl Acad Sci USA.
97:12846–12851. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Benedetti S, Pirola B, Pollo B, et al:
Gene therapy of experimental brain tumors using neural progenitor
cells. Nat Med. 6:447–450. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Herrlinger U, Woiciechowski C,
Sena-Esteves M, et al: Neural precursor cells for delivery of
replication-conditional HSV-1 vectors to intracerebral gliomas. Mol
Ther. 1:347–357. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Staflin K, Honeth G, Kalliomaki S,
Kjellman C, Edvardsen K and Lindvall M: Neural progenitor cell
lines inhibit rat tumor growth in vivo. Cancer Res. 64:5347–5354.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Glass R, Synowitz M, Kronenberg G, et al:
Glioblastoma-induced attraction of endogenous neural precursor
cells is associated with improved survival. J Neurosci.
25:2637–2646. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yip S, Sabetrasekh R, Sidman RL and Snyder
EY: Neural stem cells as novel cancer therapeutic vehicles. Eur J
Cancer. 42:1298–1308. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim SU: Neural stem cell-based gene
therapy for brain tumors. Stem Cell Rev. 7:130–140. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Spencer D: Fighting brain tumors while
protecting the brain: the stem cell story. Neurology. 76:e69–e70.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yu JJ, Sun X, Yuan X, Lee JW, Snyder EY
and Yu JS: Immunomodulatory neural stem cells for brain tumour
therapy. Expert Opin Biol Ther. 6:1255–1262. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ahmed AU, Alexiades NG and Lesniak MS: The
use of neural stem cells in cancer gene therapy: predicting the
path to the clinic. Curr Opin Mol Ther. 12:546–552. 2010.PubMed/NCBI
|
|
25
|
Aboody KS, Najbauer J and Danks MK: Stem
and progenitor cell-mediated tumor selective gene therapy. Gene
Ther. 15:739–752. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang SJ, Ye F, Xie RF, et al: Comparative
study on the stem cell phenotypes of C6 cells under different
culture conditions. Chin Med J. 124:3118–3126. 2011.PubMed/NCBI
|
|
27
|
Campos B, Wan F, Farhadi M, et al:
Differentiation therapy exerts antitumor effects on stem-like
glioma cells. Clin Cancer Res. 16:2715–2728. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Laks DR, Masterman-Smith M, Visnyei K, et
al: Neurosphere formation is an independent predictor of clinical
outcome in malignant glioma. Stem Cells. 27:980–987. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang S, Luo X, Wan F and Lei T: The roles
of hypoxia-inducible factors in regulating neural stem cells
migration to glioma stem cells and determinating their fates.
Neurochem Res. 37:2659–2666. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Busletta C, Novo E, Valfre DBL, et al:
Dissection of the biphasic nature of hypoxia-induced motogenic
action in bone marrow-derived human mesenchymal stem cells. Stem
Cells. 29:952–963. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kendall SE, Najbauer J, Johnston HF, et
al: Neural stem cell targeting of glioma is dependent on
phosphoinositide 3-kinase signaling. Stem Cells. 26:1575–1586.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Schmidt NO, Przylecki W, Yang W, et al:
Brain tumor tropism of transplanted human neural stem cells is
induced by vascular endothelial growth factor. Neoplasia.
7:623–629. 2005. View Article : Google Scholar
|
|
33
|
Mercapide J, Rappa G, Anzanello F, King J,
Fodstad O and Lorico A: Primary gene-engineered neural
stem/progenitor cells demonstrate tumor-selective migration and
antitumor effects in glioma. Int J Cancer. 126:1206–1215. 2010.
|
|
34
|
Pollard SM, Conti L, Sun Y, Goffredo D and
Smith A: Adherent neural stem (NS) cells from fetal and adult
forebrain. Cereb Cortex. 16(Suppl 1): i112–i120. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sun Y, Pollard S, Conti L, et al:
Long-term tripotent differentiation capacity of human neural stem
(NS) cells in adherent culture. Mol Cell Neurosci. 38:245–258.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pollard SM, Yoshikawa K, Clarke ID, et al:
Glioma stem cell lines expanded in adherent culture have
tumor-specific phenotypes and are suitable for chemical and genetic
screens. Cell Stem Cell. 4:568–580. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Xu Q, Wang S, Jiang X, et al:
Hypoxia-induced astrocytes promote the migration of neural
progenitor cells via vascular endothelial factor, stem cell factor,
stromal-derived factor-1alpha and monocyte chemoattractant
protein-1 upregulation in vitro. Clin Exp Pharmacol Physiol.
34:624–631. 2007. View Article : Google Scholar
|
|
38
|
Zhao D, Najbauer J, Garcia E, et al:
Neural stem cell tropism to glioma: critical role of tumor hypoxia.
Mol Cancer Res. 6:1819–1829. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Pistollato F, Abbadi S, Rampazzo E, et al:
Intratumoral hypoxic gradient drives stem cells distribution and
MGMT expression in glioblastoma. Stem Cells. 28:851–862.
2010.PubMed/NCBI
|
|
40
|
Chirasani SR, Sternjak A, Wend P, et al:
Bone morphogenetic protein-7 release from endogenous neural
precursor cells suppresses the tumourigenicity of stem-like
glioblastoma cells. Brain. 133:1961–1972. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kim SK, Kim SU, Park IH, et al: Human
neural stem cells target experimental intracranial medulloblastoma
and deliver a therapeutic gene leading to tumor regression. Clin
Cancer Res. 12:5550–5556. 2006. View Article : Google Scholar
|
|
42
|
van Eekelen M, Sasportas LS, Kasmieh R, et
al: Human stem cells expressing novel TSP-1 variant have
anti-angiogenic effect on brain tumors. Oncogene. 29:3185–3195.
2010.PubMed/NCBI
|
|
43
|
Ehtesham M, Kabos P, Gutierrez MA, et al:
Induction of glioblastoma apoptosis using neural stem cell-mediated
delivery of tumor necrosis factor-related apoptosis-inducing
ligand. Cancer Res. 62:7170–7174. 2002.
|