Introduction
ARD1 was originally described as
N-acetyltransferase in Saccharomyces cerevisiae,
where it is required for regulation of the cell cycle, mating, and
sporulation (1). Subsequently,
mammalian ARD1 was identified and is known to acetylate lysine
residues of several proteins, including hypoxia-inducible factor-1α
(HIF-1α), β-catenin, myosin light chain kinase, the androgen
receptor, and the tubulin complex (2–5).
Several ARD1 variants produced from alternative splicing of mRNA
have been identified in mouse and human cells (6,7).
Thus far, three mouse (mARD1198, mARD1225,
mARD1235) and two human (hARD1131, hARD1235)
ARD1 variants were reported. Among these, mARD1225,
mARD1235, and hARD1235 have been most
extensively studied and characterized.
mARD1225 was first identified in mouse
and found to negatively regulate angiogenesis. mARD1225
acetylates HIF-1α protein leading to its degradation via the
ubiquitin-proteasome pathway (2).
However, it was reported subsequently that other homologs of ARD1
(mARD1235 and hARD1235) could not alter
HIF-1α stability, suggesting different roles of ARD1 variants in
the regulation of HIF-1α (8–10).
In contrast to the tumor suppression effects of
mARD1225, hARD1235 is mainly known to
contribute to tumorigenesis by enhancing cell proliferation. In
many studies, the downregulation of hARD1235 reduces
cellular growth and induces cell cycle arrest (3,11,12).
Furthermore, increased expression of hARD1235 is
frequently observed in various human cancers, including breast,
lung, and colorectal cancers (13–16).
Thus, hARD1235 is recognized as a critical oncogenic
protein in cancer progression.
In a previous study, we reported that
hARD1235 has auto-acetylation activity that is required
for the stimulation of cancer growth by hARD1235
(11). Based on this information,
the present study was designed to compare the regulatory mechanisms
of ARD1 variants and to investigate how they selectively regulate
distinct functions of ARD1 variants that are involved in
tumorigenesis. The results demonstrate that ARD1 variants have
conserved autoacetylation activity that stimulates their catalytic
activities. However, depending on the physiological conditions,
this autoacetylation differentially regulates the biological
functions of ARD1 variants in angiogenesis and cell proliferation
in an isoform-specific manner.
Materials and methods
Reagents and antibodies
Anti-HIF-1α antibody was purchased from BD
Pharmingen. Anti-Myc and green fluorescent protein (GFP) antibodies
were purchased from Santa Cruz Biotechnology. Anti-acetyl-lysine
antibody was purchased from Cell Signaling. Anti-tubulin and Flag
antibodies were purchased from Sigma-Aldrich. MG132 was purchased
from Calbiochem.
Cell culture and hypoxic condition
HeLa cells and human umbilical vein endothelial
cells (HUVECs) were grown in Dulbecco’s modified Eagle’s medium
supplemented with 10% fetal bovine serum (FBS) and EBM-2 medium
supplemented with growth factors (Lonza), respectively. Cells were
maintained at 37°C in a humidified atmosphere containing 5%
CO2. Hypoxic conditions were created by incubating cells
at 37°C in a chamber containing 5% CO2, 1%
O2, and the remainder N2.
Plasmid construction and
transfection
To construct expression vectors for ARD1 variants,
ARD1 cDNA was amplified by polymerase chain reaction (PCR) and
subcloned into GFP- or Myc-tagged pCS2+ vectors for cell
expression, and pGEX-4T for bacterial induction of the recombinant
protein. Mutations in ARD1 were created using the Muta-Direct™ Site
Directed Mutagenesis kit (Intron) according to the manufacturer’s
instructions. Cells were transfected with Lipofectamine (Life
Technology) or Polyfect (Qiagen) according to the manufacturer’s
instructions.
Immunoblotting and
immunoprecipitation
Cells were harvested and extracted in lysis buffer
(10 mM HEPES at pH 7.9, 40 mM NaCl, 0.1 mM
ethylenediaminetetraacetic acid (EDTA), 5% glycerol, 1 mM
dithiothreitol (DTT), and protease inhibitors). The concentrations
of the protein extracts were measured with the BCA assay. For
immunoprecipitations, relevant primary antibodies were added to 1
mg of the protein extracts and incubated overnight at 4°C. The
immunoprecipitates and total cell lysates were resolved in sodium
dodecyl-sulfate polyacrylamide gel electrophoresis gels and
transferred onto nitrocellulose membranes (Amersham Pharmacia
Bioscience). The membrane was probed with a primary antibody
followed by a secondary antibody conjugated with horseradish
peroxidase, and detected using an ECL system (Intron
Biotechnology).
In vitro acetylation assay
Recombinants of GST-ARD1 variants were freshly
prepared as described previously (11). These recombinants were incubated
with or without His-tagged oxygen-dependent degradation (ODD)
domain of HIF-1α recombinants in the reaction mixture (50 mM
Tris-HCl at pH 8.0, 0.1 mM EDTA, 1 mM DTT, 10% glycerol, and 10 mM
acetyl-CoA) at 37°C.
Reverse transcription (RT)-PCR
analysis
Total RNA was extracted using an RNA extraction kit
(Invitrogen). cDNA was synthesized from 2 μg of RNA using an
oligo(dT) primer. Primers used for PCR were as follows: human
VEGF, 5′-GAGAATTCGGCCTCCGAAACCATGAACTT TCTGCT-3′ (forward)
and 5′-GAGCATGCCCTCCTGCCC GGCTCACCGC-3′ (reverse); ARD1,
5′-ATGAACATCCGC AATGCGAG-3′ (forward) and 5′-CTCATATCATGGCT
CGAGAGG-3′ (reverse); cyclin D1, 5′-CTGGCCATGAA CTACCTGGA-3′
(forward) and 5′-GTCACACTTGATCAC TCTGG-3′ (reverse); GAPDH,
5′-ACCACAGTCCATGCCAT CAC-3′ (forward) and
5′-TCCACCACCCTGTTGCTGTA-3′ (reverse). The PCR amplification was
carried out for 25 cycles with ARD1, cyclin D1, and
GAPDH, and for 30 cycles with VEGF.
Tube formation assay
For the tube formation assay, 24-well plates were
coated with Matrigel (BD Biosciences) and allowed to polymerize at
37°C for 30 min. HUVECs were seeded (5×104 cells per
well) onto Matrigel with 500 μl conditioned medium from HeLa cells.
Tube formation was assessed after 4 h and quantified by determining
the number of rings.
Cell proliferation assay
The cell growth rate was measured using a
non-radioactive proliferation assay kit (Promega) according to the
manufacturer’s instructions. Briefly, cells were plated on 96-well
plates and grown for 3 days. Substrate solution (20 μl) was then
added and the cells were incubated for 1 h to allow color
development. The absorbance at 492 nm was measured as an index of
the number of proliferating cells.
Statistical analysis
Results are presented as means ± SD, and P-values
were calculated by applying the two-tailed Student’s t-test to data
from three independent experiments. Differences were considered
statistically significant when P<0.05.
Results
Autoacetylation at the K136 residue is
conserved in ARD1 variants
To investigate the autoacetylation activity of ARD1,
three variants, GST-mARD1225, GST-mARD1235,
and GST-hARD1235, were purified and then subjected to an
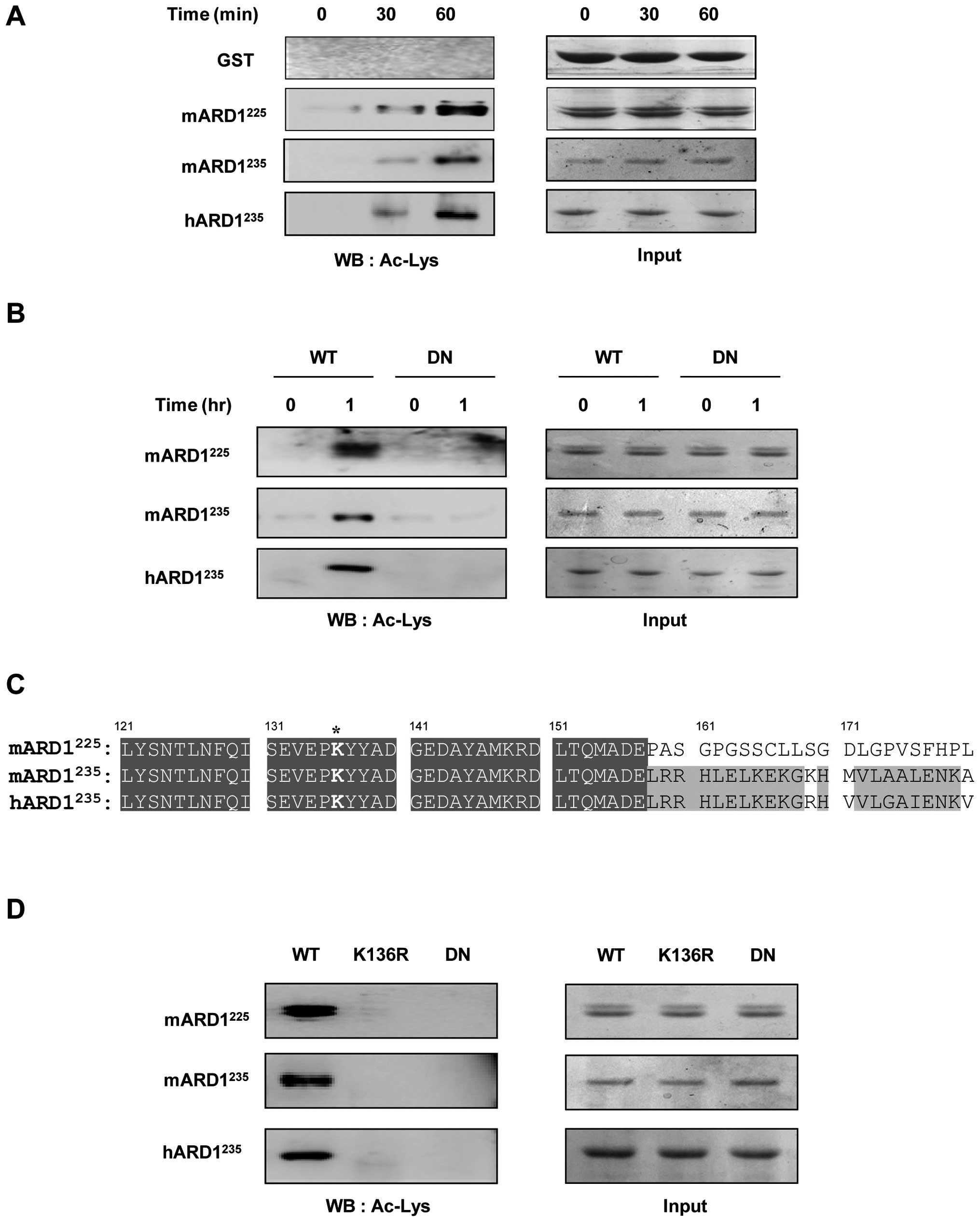
in vitro acetylation assay. As shown in Fig. 1A, GST-mARD1225,
GST-mARD1235, and GST-hARD1235 acetylated
themselves in a time-dependent manner, whereas the control GST
protein was not acetylated. To confirm the self-acetylation
activity of ARD1, a dominant negative ARD1 was constructed with
amino acid mutations at R82A and Y122F, blocking its binding to
acetyl-CoA, and then subjected it to the in vitro
acetylation reaction. Although wild-type mARD1225,
mARD1235, and hARD1235 acetylated themselves,
the dominant negative ARD1 mutants were resistant to this
acetylation, confirming that all ARD1 variants have self-activated
autoacetylation activities (Fig.
1B).
The target site of autoacetylation was predicted
using data from our previous study and sequence alignment (11). We have reported that
hARD1235 acetylation occurs at Lys136. Sequence
alignment revealed that this site is conserved in
mARD1225 and mARD1235 (Fig. 1C). To verify whether Lys136 is also
a target site for the autoacetylation of mARD1225 and
mARD1235, we constructed ARD1 mutants in which Lys136
was replaced with Arg (K136R), and then performed the in
vitro auto-acetylation assay. As expected, K136R mutation
abolished the autoacetylation activity of mARD1225 and
mARD1235, as well as hARD1235 (Fig. 1D). These results indicate that all
ARD1 variants have autoacetylation activity and the target site is
conserved at Lys136.
Autoacetylation of mARD1225,
but not mARD1235 and hARD1235, decreases
HIF-1α stability under hypoxic conditions
Autoacetylation is an important mechanism to
regulate the enzymatic activity and the biological functions of
acetyltransferase (17–20). Based on previous reports suggesting
that ARD1 variants might have different biological functions
(6), we hypothesized that even
though ARD1 variants have common autoacetylation activity, this
activity regulates each ARD1 variant separately. Thus, ARD1
variants have different biological functions depending on the
specific isoform and physiological conditions.
Because it was reported that mARD1225
decreases HIF-1α stability by triggering protein degradation under
hypoxic conditions (2,21), we compared the effect of
acetyltransferase activities of ARD1 variants on the stability of
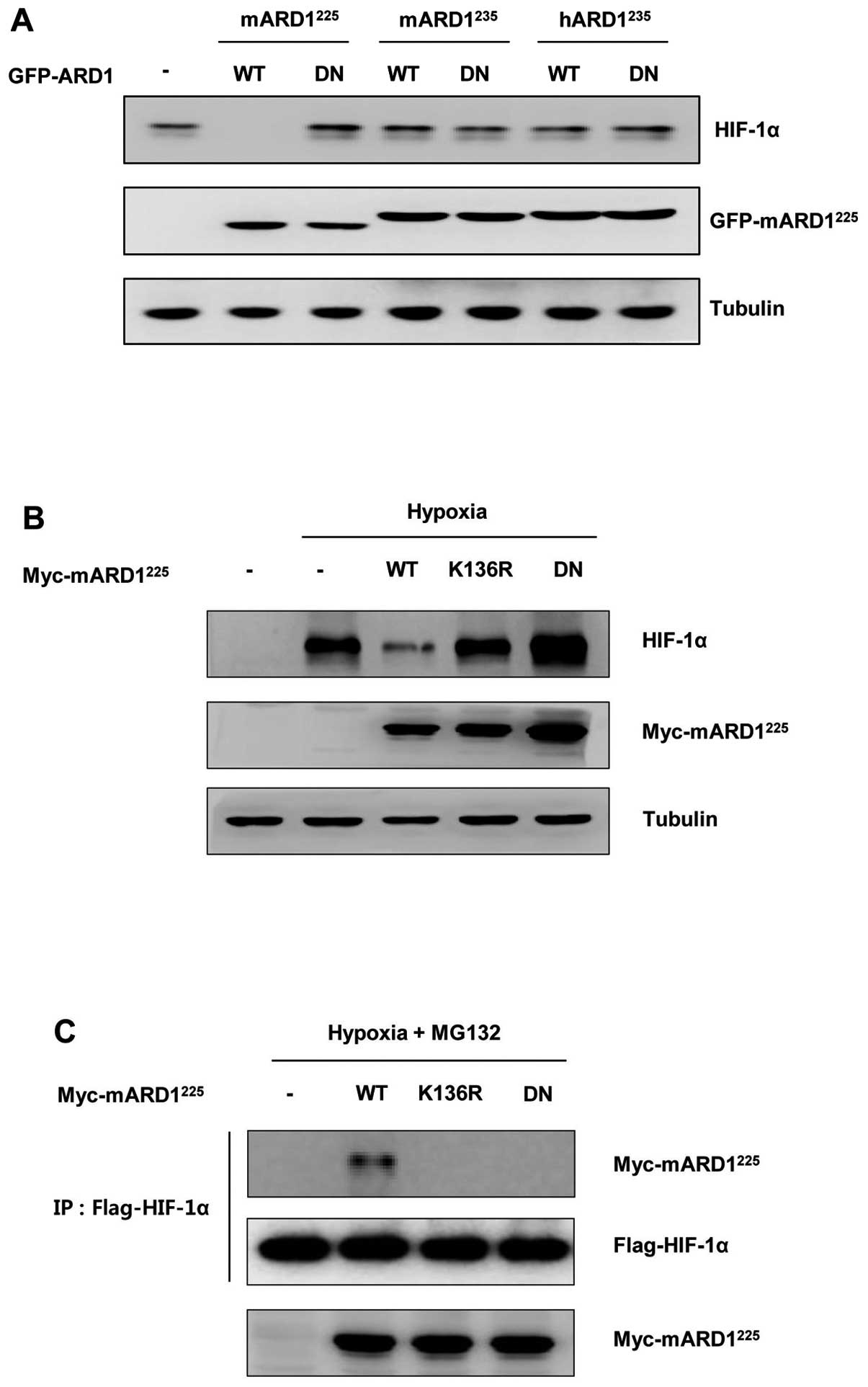
HIF-1α. HeLa cells were transfected with plasmids for wild-type or
dominant negative ARD1 variants and incubated under hypoxic
conditions. Consistent with the previous study, HIF-1α protein was
decreased in wild-type mARD1225 transfected cells, but
not in dominant negative mARD1225 transfected cells
(Fig. 2A). In addition,
mARD1235 and hARD1235 did not change HIF-1α
protein levels regardless of whether wild-type or dominant negative
mutants were used. These results not only confirm distinct
functions of ARD1 variants, but also suggest a specific role of
mARD1225 in the regulation of HIF-1α under hypoxic
conditions.
To clarify the effect of mARD1225
autoacetylation on the stability of HIF-1α, the K136R mutant
mARD1225 plasmid was transfected into HeLa cells and the
HIF-1α protein level was determined. As shown in Fig. 2B, wild-type mARD1225
reduced HIF-1α protein levels under hypoxic conditions while the
K136R mutation inhibited the ability of mARD1225 to
decrease HIF-1α protein levels. This indicates that
mARD1225 autoacetylation plays an indispensable role in
the down-regulation of HIF-1α stability under hypoxic conditions.
Interestingly, we also observed that neither the K136R mutant nor
the dominant negative mARD1225 could bind to HIF-1α,
while the wild-type mARD1225 binds to HIF-1α under
hypoxic conditions (Fig. 2C).
These data suggest that mARD1225 binds to HIF-1α only
after the acquisition of enzymatic activity through its
autoacetylation.
Autoacetylation of mARD1225 is
required for HIF-1α acetylation
When mARD1225 regulates the stability of
HIF-1α under hypoxic conditions, the acetylation of the Lys532
residue in the ODD domain of HIF-1 is a critical step triggering
HIF-1α degradation (2,21). Thus, we hypothesized that
mARD1225 autoacetylation stimulates the ability of
mARD1225 to acetylate the Lys532 residue in the ODD
domain of HIF-1α. Because many studies have reported conflicting
data on HIF-1α acetylation in vitro (2,22),
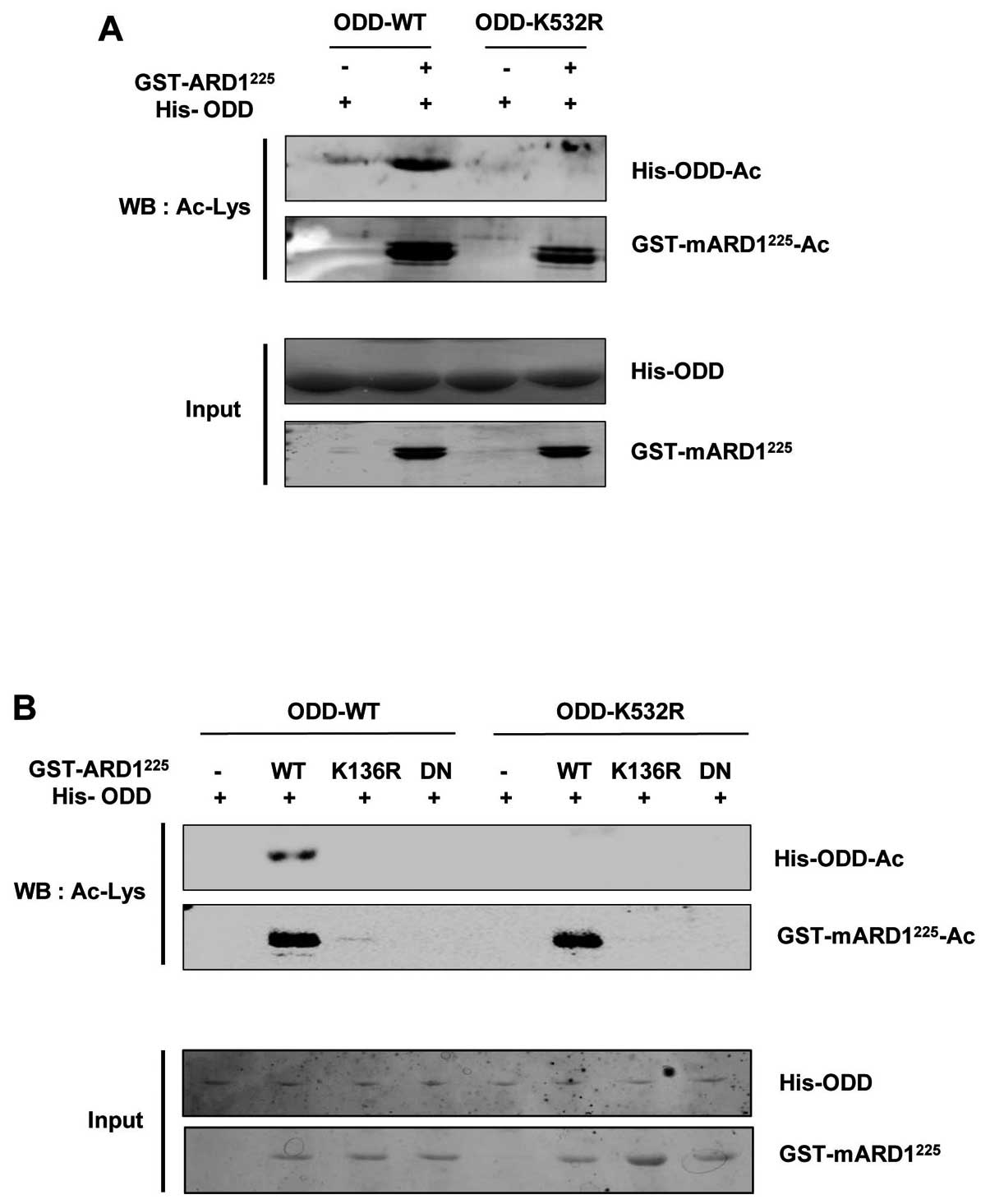
we first determined whether mARD1225 directly acetylated
the Lys532 residue in the ODD domain in vitro. Purified
recombinants for the His-tagged ODD domain in HIF-1α and the
GST-tagged mARD1225 were prepared and subjected to
acetylation in vitro. As shown in Fig. 3A, the ODD domain recombinant was
successfully acetylated by the wild-type mARD1225
recombinant, while the acetylation of the ODD domain of HIF-1α was
abrogated when the Lys532 residue was substituted with Arg (K532R).
This demonstrated that mARD1225 directly acetylates the
Lys532 residue in HIF-1α.
To evaluate the effect of mARD1225
autoacetylation on the acetylation of the ODD domain of HIF-1α, we
subjected the K136R mutant mARD1225 recombinant to the
in vitro ODD domain acetylation assay. As expected, the
K136R mutant mARD1225 recombinant failed to acetylate
the ODD domain of HIF-1α in vitro, whereas the wild-type
mARD1225 recombinant successfully acetylated this domain
(Fig. 3B). These results indicate
that autoacetylation is the critical step to stimulate the
catalytic activity of mARD1225 that is required for the
acetylation of the Lys532 residue in the ODD domain of HIF-1α.
Autoacetylation of mARD1225
inhibits angiogenesis
When the HIF-1α protein is stabilized under hypoxic
conditions, it upregulates the expression level of several genes
that promote angiogenesis (23–25).
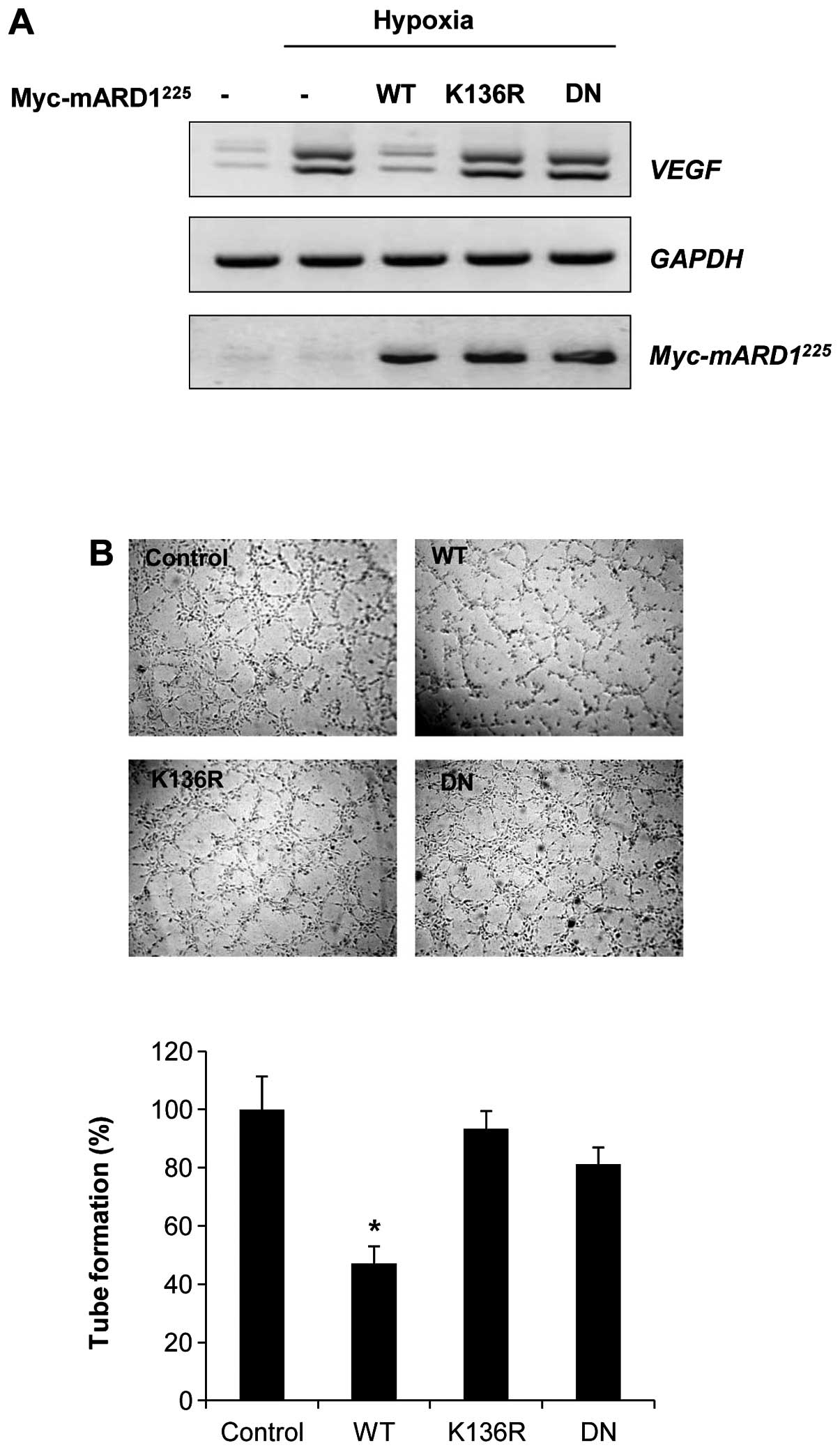
To determine the effect of mARD1225 on hypoxia-induced
angiogenic activity, we examined the expression of VEGF
mRNA, a potent downstream target of HIF-1α for promoting
angiogenesis. Consistent with the data shown in Fig. 2B, wild-type mARD1225
significantly decreased the mRNA level of VEGF. However, the
K136R or dominant negative mARD1225 had no influence on
the expression of VEGF (Fig. 4A).
In addition, conditioned media from cells transfected with
wild-type mARD1225 showed a strong inhibitory effect on
endothelial tube formation, whereas conditioned media from cells
transfected with K136R or dominant negative mARD1225 had
no effect on tube formation (Fig.
4B). These results indicate that the ability of
mARD1225 to inhibit tumor angiogenesis might be
regulated by autoacetylation.
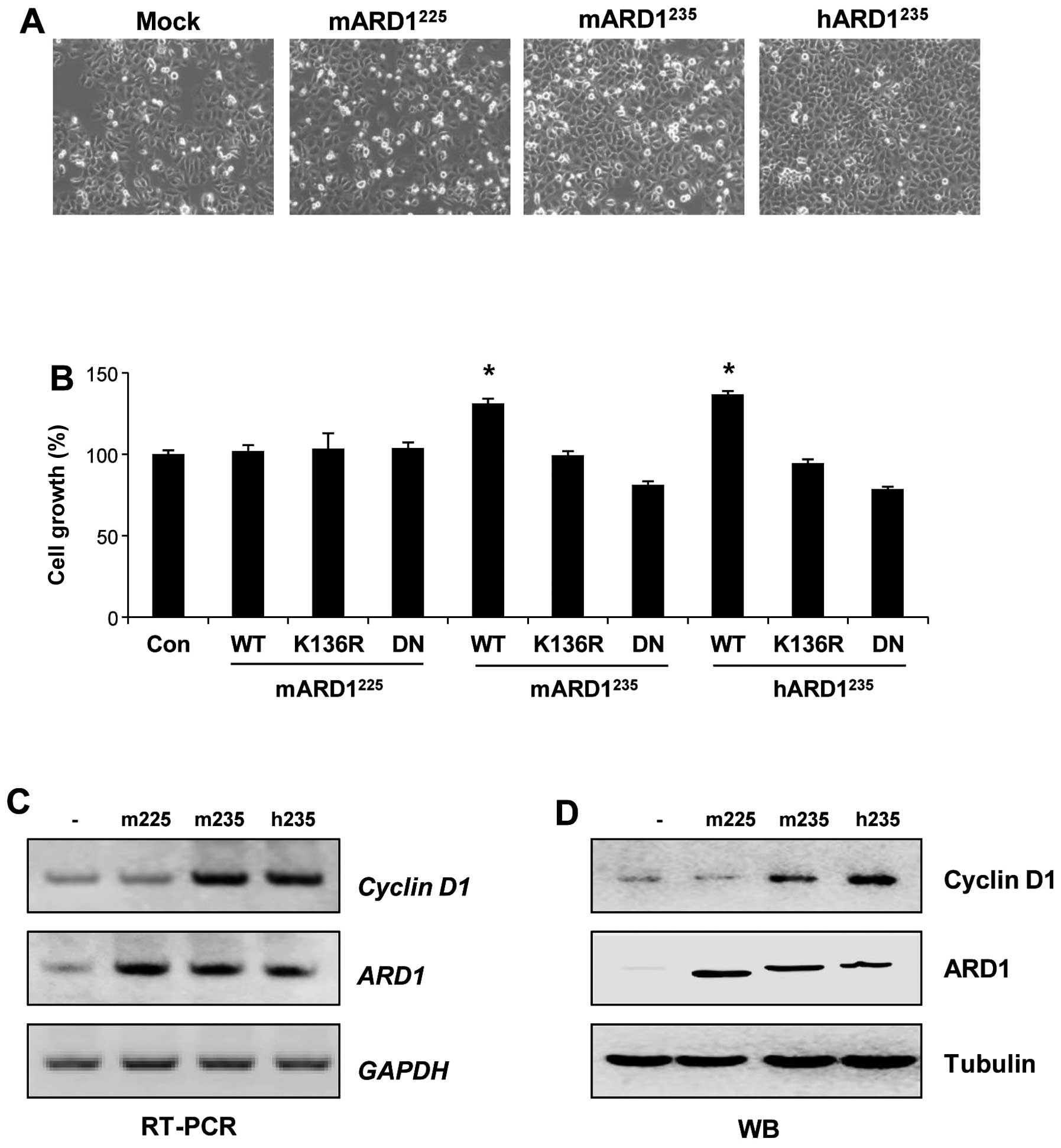
Autoacetylation of mARD1235
and hARD1235 but not mARD1225 promotes cell
proliferation
The distinct roles of autoacetylated ARD1 variants
in regulating tumor growth were investigated under normoxic
conditions. Because hARD1235 promotes cell proliferation
(3,26), the effects of the ARD1 variants on
cell growth were compared. Cell proliferation was analyzed after
HeLa cells were transfected with plasmids for ARD1 variants. As
shown in Fig. 5A and B, wild-type
mARD1235 and hARD1235 significantly increased
cell growth. However, wild-type mARD1225 did not alter
cell growth, indicating distinct roles of ARD1 variants in the
regulation of cell proliferation under normoxic conditions.
Moreover, the abilities of mARD1235 and
hARD1235 to enhance cell proliferation were abolished by
K136R or dominant negative mutation of ARD1. This indicates that
the autoacetylation activity of mARD1235 and
hARD1235 is required for cell proliferation (Fig. 5B).
Based on our previous report showing that
hARD1235-induced cell proliferation is mediated by
cyclin D1 (11), the effects of
ARD1 variants on cyclin D1 levels were compared. After HeLa cells
were transfected with plasmids for ARD1 variants, mRNA and protein
expression levels of cyclin D1 were analyzed by RT-PCR and western
blot analysis, respectively. Consistent with the data shown in
Fig. 5A, mARD1235 and
hARD1235 increased the expression level of cyclin D1.
However, expression levels were unchanged by mARD1225
(Fig. 5C and D). These results
indicate that ARD1 variants have different effects on the
expression of cyclin D1, demonstrating distinct functions of ARD1
variants in the regulation of cell proliferation under normoxic
conditions.
Discussion
A number of acetyltransferases are known to be
self-activated by autoacetylation (17–20).
The present study provides data demonstrating that there is a
conserved autoregulatory mechanism in ARD1 variants and shows how
autoacetylation differentially regulates the enzymatic activities
and biological functions of ARD1 variants, depending on the
specific isoforms and physiological conditions.
We previously identified several ARD1 variants and
suggested that they have distinct biological functions (6,7). We
also reported that hARD1235 undergoes autoacetylation
that enhances its cell proliferative activity (11). Because mARD1225,
mARD1235, and hARD1235 have a conserved
acetyltransferase domain, we hypothesized that these three ARD1
variants have common autoacetylation activities. Consistent with
this prediction, mARD1225, mARD1235, and
hARD1235 were observed to self-acetylate in
vitro. In addition, the target site for autoacetylation was
found to be conserved at the Lys136 residue.
Based upon differences in the amino acid sequences
of the C-terminal region, the role of mARD1225 could be
different from that of mARD1235 or hARD1235.
While the effects of hARD1235 are related to cellular
growth, mARD1225 was originally found to inhibit
angiogenesis (2). Thus, we
speculated that, even though ARD1 variants share autoacetylation
activity to acquire their acetylation activity, their biological
functions might be distinct.
When mARD1225 modulates angiogenesis
under hypoxic conditions, it directly interacts with and acetylates
the Lys532 residue in the ODD domain of HIF-1α, triggering
degradation of the HIF-1α protein (2). The significance of
mARD1225 autoacetylation was clearly revealed by our
observation that the K136R mutant mARD1225 could not
acetylate the Lys532 residue in the ODD domain of HIF-1α in
vitro, whereas wild-type mARD1225 acetylated it.
Accordingly, stability of the HIF-1α protein was reduced in
wild-type mARD1225-expressing cells, but not in K136R
mutant mARD1-expressing cells. Furthermore, blocking
autoacetylation diminished the ability of mARD1225 to
inhibit VEGF expression and endothelial tube formation. From these
results, we conclude that autoacetylation serves as a key switch
for regulating the anti-angiogenic function of mARD1225
under hypoxic conditions.
In contrast to autoacetylation of
mARD1225, the auto-acetylation of mARD1235
and hARD1235 had no effect on angiogenesis. Under
hypoxic conditions, the stability of the HIF-1α protein was
unchanged by either wild-type or mutant mARD1235 and
hARD1235. However, autoacetylation of
mARD1235 and hARD1235 played an important
role in cell growth under normoxic conditions. Consistent with our
previous report (11), cell
proliferation was remarkably increased by wild-type
mARD1235 and hARD1235, but not by K136R
mutants. However, in terms of cell growth, autoacetylation of
mARD1225 appeared unrelated to cell proliferation under
normoxic conditions. Neither wild-type nor the K136R mutant of
mARD1225 had any effect on cell growth. Cyclin D1 was
increased by mARD1235 and hARD1235, but not
by mARD1225. These data support our previous suggestion
about distinct roles of ARD1 variants in tumor angiogenesis and
cell growth. In addition, data from the present study also suggest
that the distinct role of ARD1 variants is selectively regulated by
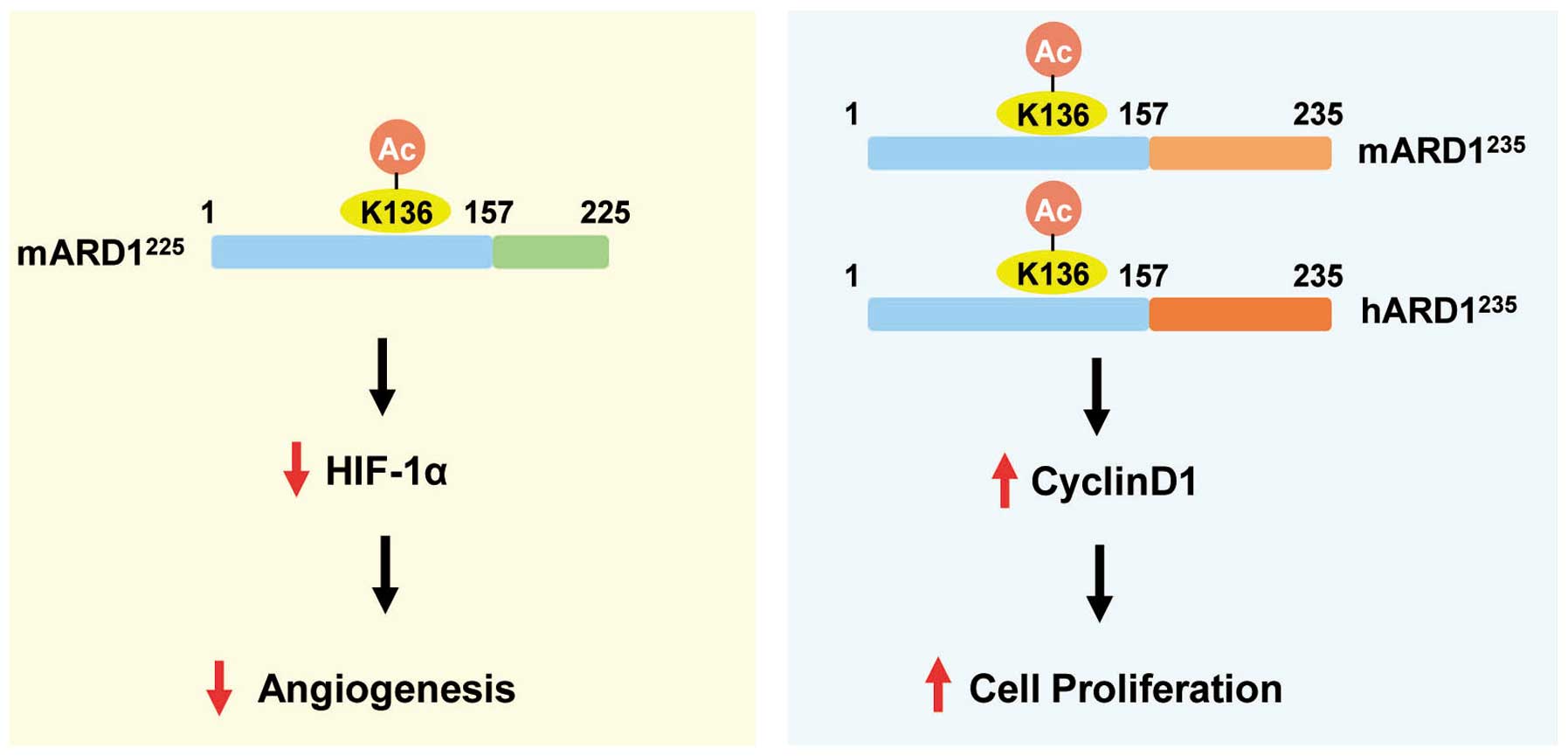
autoacetylation in an isoform-specific manner (Fig. 6).
Alternative splicing is a widespread process
generating multiple transcripts from a single mRNA precursor. This
process commonly occurs during gene expression and contributes to
protein diversity (27). Indeed,
more than half of all mammalian genes are alternatively spliced,
and diverse transcripts produced from alternative splicing often
have distinct functions (28,29).
Alternative splicing of exon 8 of mouse ARD1 leads to the
production of discrete ARD1 isoforms (mARD1225 and
mARD1235) that have distinct functions in tumorigenesis
(7). Different subcellular
localizations of mARD1225 and mARD1235 may
correlate with their distinct functions (7). In contrast to mice, alternative
splicing of ARD1 exon 8 does not occur in humans. Thus, only
hARD1235 is present in humans, indicating that
alternative splicing of ARD1 is a species-specific event. To
understand the evolutionary events leading to species-specific ARD1
isoforms, it might be necessary not only to identify diverse ARD1
variants in other species such as rat, rabbit, and monkey but also
to define the detailed individual functions of ARD1 variants in
each species.
In conclusion, the present study reveals different
roles of ARD1 variants in angiogenesis and cell proliferation. ARD1
variants use a common regulatory system called autoacetylation to
regulate their individual roles. Although autoacetylation is a
conserved mechanism that ARD1 variants use to regulate their
enzymatic activities, depending on physiological conditions,
autoacetylation selectively regulates the biological functions of
ARD1 in an isoform-specific manner. These findings offer new
insight into the distinct functions of ARD1 isoforms in cancer
development, and provide a clue as to how ARD1 variants could be
selectively targeted in cancer treatment.
Acknowledgements
We thank Dr Gregg L. Semenza for providing the
Flag-tagged HIF-1α plasmid. This study was supported by the Global
Research Laboratory Program (2011-0021874), Global Core Research
Center (GCRC) Program (2011-0030001), National Research Foundation
(NRF) grant (2013-036038) funded by the Ministry of Science, ICT,
and Future Planning (MSIP) and Basic Science Research Program
through the NRF of Korea funded by the Ministry of Education
(2013R1A1A2058956).
References
|
1
|
Driessen HP, de Jong WW, Tesser GI and
Bloemendal H: The mechanism of N-terminal acetylation of proteins.
CRC Crit Rev Biochem. 18:281–325. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jeong JW, Bae MK, Ahn MY, et al:
Regulation and destabilization of HIF-1alpha by ARD1-mediated
acetylation. Cell. 111:709–720. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lim JH, Park JW and Chun YS: Human arrest
defective 1 acetylates and activates beta-catenin, promoting lung
cancer cell proliferation. Cancer Res. 66:10677–10682. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ohkawa N, Sugisaki S, Tokunaga E, et al:
N-acetyltransferase ARD1-NAT1 regulates neuronal dendritic
development. Genes Cells. 13:1171–1183. 2008.PubMed/NCBI
|
|
5
|
Shin DH, Chun YS, Lee KH, Shin HW and Park
JW: Arrest defective-1 controls tumor cell behavior by acetylating
myosin light chain kinase. PLoS One. 4:e74512009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kim SH, Park JA, Kim JH, et al:
Characterization of ARD1 variants in mammalian cells. Biochem
Biophys Res Commun. 340:422–427. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chun KH, Cho SJ, Choi JS, Kim SH, Kim KW
and Lee SK: Differential regulation of splicing, localization and
stability of mammalian ARD1235 and ARD1225 isoforms. Biochem
Biophys Res Commun. 353:18–25. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Arnesen T, Kong X, Evjenth R, et al:
Interaction between HIF-1 alpha (ODD) and hARD1 does not induce
acetylation and destabilization of HIF-1 alpha. FEBS Lett.
579:6428–6432. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fisher TS, Etages SD, Hayes L, Crimin K
and Li B: Analysis of ARD1 function in hypoxia response using
retroviral RNA interference. J Biol Chem. 280:17749–17757. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bilton R, Mazure N, Trottier E, et al:
Arrest-defective-1 protein, an acetyltransferase, does not alter
stability of hypoxia-inducible factor (HIF)-1alpha and is not
induced by hypoxia or HIF. J Biol Chem. 280:31132–31140. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Seo JH, Cha JH, Park JH, et al: Arrest
defective 1 autoacetylation is a critical step in its ability to
stimulate cancer cell proliferation. Cancer Res. 70:4422–4432.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Arnesen T, Gromyko D, Pendino F, Ryningen
A, Varhaug JE and Lillehaug JR: Induction of apoptosis in human
cells by RNAi-mediated knockdown of hARD1 and NATH, components of
the protein N-alpha-acetyltransferase complex. Oncogene.
25:4350–4360. 2006. View Article : Google Scholar
|
|
13
|
Ren T, Jiang B, Jin G, et al: Generation
of novel monoclonal antibodies and their application for detecting
ARD1 expression in colorectal cancer. Cancer Lett. 264:83–92. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shim JH, Chung YH, Kim JA, et al: Clinical
implications of arrest-defective protein 1 expression in
hepatocellular carcinoma: a novel predictor of microvascular
invasion. Dig Dis. 30:603–608. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang ZH, Gong JL, Yu M, et al:
Up-regulation of human arrest-defective 1 protein is correlated
with metastatic phenotype and poor prognosis in breast cancer.
Asian Pac J Cancer Prev. 12:1973–1977. 2011.PubMed/NCBI
|
|
16
|
Yu M, Gong J, Ma M, et al:
Immunohistochemical analysis of human arrest-defective-1 expressed
in cancers in vivo. Oncol Rep. 21:909–915. 2009.PubMed/NCBI
|
|
17
|
Thompson PR, Wang D, Wang L, et al:
Regulation of the p300 HAT domain via a novel activation loop. Nat
Struct Mol Biol. 11:308–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Santos-Rosa H, Valls E, Kouzarides T and
Martinez-Balbas M: Mechanisms of P/CAF auto-acetylation. Nucleic
Acids Res. 31:4285–4292. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Blanco-Garcia N, Asensio-Juan E, de la
Cruz X and Martinez-Balbas MA: Autoacetylation regulates P/CAF
nuclear localization. J Biol Chem. 284:1343–1352. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Stavropoulos P, Nagy V, Blobel G and Hoelz
A: Molecular basis for the autoregulation of the protein acetyl
transferase Rtt109. Proc Natl Acad Sci USA. 105:12236–12241. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee MN, Lee SN, Kim SH, et al: Roles of
arrest-defective protein 1(225) and hypoxia-inducible factor 1alpha
in tumor growth and metastasis. J Natl Cancer Inst. 102:426–442.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Murray-Rust TA, Oldham NJ, Hewitson KS and
Schofield CJ: Purified recombinant hARD1 does not catalyse
acetylation of Lys532 of HIF-1alpha fragments in vitro. FEBS Lett.
580:1911–1918. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Semenza GL: Targeting HIF-1 for cancer
therapy. Nat Rev Cancer. 3:721–732. 2003. View Article : Google Scholar
|
|
24
|
Hong SS, Lee H and Kim KW: HIF-1alpha: a
valid therapeutic target for tumor therapy. Cancer Res Treat.
36:343–353. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lee JW, Bae SH, Jeong JW, Kim SH and Kim
KW: Hypoxia-inducible factor (HIF-1)alpha: its protein stability
and biological functions. Exp Mol Med. 36:1–12. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Arnesen T, Thompson PR, Varhaug JE and
Lillehaug JR: The protein acetyltransferase ARD1: a novel cancer
drug target? Curr Cancer Drug Targets. 8:545–553. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nilsen TW and Graveley BR: Expansion of
the eukaryotic proteome by alternative splicing. Nature.
463:457–463. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Brett D, Pospisil H, Valcarcel J, Reich J
and Bork P: Alternative splicing and genome complexity. Nat Genet.
30:29–30. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mironov AA, Fickett JW and Gelfand MS:
Frequent alternative splicing of human genes. Genome Res.
9:1288–1293. 1999. View Article : Google Scholar : PubMed/NCBI
|