Introduction
Lung cancer is the leading cause of cancer-related
death worldwide (1). In general,
lung cancer is histologically classified as non-small cell lung
cancer (NSCLC) and small cell lung cancer (SCLC), which account for
~85 and 15% of all lung malignancies, respectively. To overcome
this deadly disease, considerable efforts have been made to
identify the molecular mechanisms of lung cancer initiation and
progression. Especially, the recent advance in comprehensive genome
analysis tools revolutionized our understanding of lung cancer
molecular biology (2–6). These advances led to the
identification of several critical genes for lung cancer. These
include the epidermal growth factor receptor (EGFR), Kirsten
rat sarcoma viral oncogene homolog (K-RAS), anaplastic
lymphoma kinase (ALK), and fibroblast growth factor receptor
(FGFR) (6–12). Somatic alterations of these genes,
including gene mutations or gene rearrangements, induce protein
activation, which subsequently induces the activation of downstream
pathways critical for lung cancer cell survival and proliferation.
These pathways include the phosphatidylinositol-3-kinase/protein
kinase B (PI3K/AKT) and the extracellular signal-regulated kinase
(ERK)/mitogen-activated protein kinase (MAPK) pathways. The effects
of receptor activation on these downstream pathways have been
clearly demonstrated in vitro (13,14).
Cancer cells are surrounded by the extracellular matrix,
extracellular fluid, and stroma cells. The aforementioned
cell-autonomous alterations of the receptors can affect cancer cell
biology, but we also speculate that non-cell-autonomous alterations
such as ligand concentration in the microenvironment can affect the
biology of lung cancer cells.
Recently, fibroblast growth factors (FGFs) and their
receptors, FGFRs, attracted the attention of lung cancer
researchers, because FGFR somatic activating mutations and
gene amplifications have been repeatedly reported in lung cancer
(5,15). Moreover, some FGFR inhibitors are
reported to be effective in NSCLC (16) and SCLC (17). FGFR activation induces the
activation of the downstream PI3K/AKT and MAPK pathways (18,19),
promoting lung cancer cell proliferation and inhibiting lung cancer
cell apoptosis. FGFs are known to induce the activation of FGFRs,
which subsequently induce downstream proteins activation. The FGF
family comprises 23 FGFs, and the roles of some FGFs, especially
FGF2, have been well studied in the cancer field (20,21).
Recently Wilson and colleagues reported that receptor tyrosine
kinase (RTK)-driven cancer cell sensitivity to the corresponding
RTK inhibitors was affected by the extracellular ligand
concentration (22). In this
study, they used several ligands, including FGF2 and found that
extracellular ligands can reduce cancer cell sensitivity to
specific RTK-inhibitors.
However, whether extracellular FGFs affect lung
cancer cell functions, such as, proliferation, treatment
sensitivity, and apoptosis, remain unclear. Moreover, which FGFs
are potent and how extracellular FGFs affect lung cancer cell
biology remain unclear.
The objective of this study was to determine whether
extracellular FGFs can affect lung cancer cell biology and to
understand how extracellular FGFs affect the biology of NSCLC and
SCLC. In this study, out of the 23 reported FGFs, we focused on
FGF2, FGF9, and FGF10. FGF2 and FGF10 were chosen because they have
been studied in some cancers, including lung cancer (21,23).
FGF9 was chosen because we have previously reported that patients
with NSCLC with high FGF9 expression present a worse prognosis than
patients with low FGF9 expression (24). We also reported that lung specific
expression of FGF9 induced the formation of lung adenocarcinoma in
a genetically engineered mouse model (25). In this study, we used NSCLC (PC9)
and SCLC (H69, H82 and H146) cells. We also examined FGF2 and FGF9
concentrations in the serum of patients with lung cancer.
Materials and methods
Cell culture
PC9 (NSCLC, EGFR exon 19 deletion,
EGFRdelE746-A750), H69 (SCLC), H82 (SCLC), and H146 (SCLC) cell
lines were used in this study. PC9 was obtained as previously
described (26). The other cell
lines were purchased from the American Type Culture Collection
(ATCC, Manassas, VA, USA). All cell lines were cultured in
RPMI-1640 growth medium, supplemented with 10% fetal bovine serum
(FBS) at 37°C in a humidified 5% CO2 incubator.
Reagents
Recombinant human FGF2, FGF9 and FGF10 were
purchased from Peprotech (Rocky Hill, NJ, USA). In this study, the
FGFs was used at 100 ng/ml as previously described (23,27,28).
Erlotinib was purchased from LC Laboratories (Boston, MA, USA).
Docetaxel was purchased from Wako (Osaka, Japan). Etoposide was
purchased from Sigma-Aldrich Japan (Tokyo, Japan). Phospho-p44/42
MAPK (MAPK Tyr202/Tyr204) (p-ERK1/2) antibody (no. 3126), total
p44/42 MAPK (ERK1/2) antibody (no. 3127), phospho-AKT (Ser473; D9E)
(p-AKT) antibody (no. 4060), and total AKT antibody (no. 9272) were
purchased from Cell Signaling Technologies (Danvers, MA, USA).
Quantitative RT-PCR
Total RNA was prepared using an RNeasy Mini kit
(Qiagen, Hilden, Germany) and the RNA was then reverse transcribed
to cDNA using TaqMan Reverse Transcription reagents (Invitrogen,
Carlsbad, CA, USA). For quantitative RT-PCR analysis, we used the
ABI PRISM 7000 Sequence Detection system (Life Technologies,
Carlsbad, CA, USA). Human glyceraldehyde-3-phosphate dehydrogenase
(GAPDH) was used for normalization of input cDNA. The probes used
in this study were, Hs00915142 for FGFR1, Hs01552926 for FGFR2,
Hs00179829 for FGFR3, Hs01106908 for FGFR4, and H99999905 for
GAPDH.
MTS proliferation assay
Evaluation of the proliferation was done using an
MTS proliferation assay (CellTiter 96 AQueous One Solution Assay
kit, Promega, Madison, WI, USA). The MTS proliferation assay was
conducted according to the manufacturer’s protocol. Briefly,
2.0×103 cells were seeded per well in 96-well plates.
The cells were then treated with or without FGF2 (100 ng/ml), FGF9
(100 ng/ml), and FGF10 (100 ng/ml). Control cells were treated with
the same concentration of the vehicle, dimethyl sulfoxide (DMSO).
Seventy-two hours after treatment, the number of viable cells was
measured.
Western blotting
Proteins were extracted using a cell lysis buffer
(Cell Signaling Technologies). Protein concentrations were
quantified by BCA protein assay (Thermo Scientific, Waltham, MA,
USA) and equal amounts of protein were denatured and reduced with
sample buffer. After boiling, aliquots of the samples were
subjected to electrophoresis. The fractionated proteins were
transferred to polyvinylidene difluoride (PVDF) membranes. The
membrane was incubated with the diluted primary antibodies followed
by incubation with the secondary antibodies. For the protein
detection, the membrane was incubated with agitation in LumiGLO
reagent and peroxide (Cell Signaling Technologies) and exposed to
X-ray film.
Erlotinib sensitivity assay
Erlotinib sensitivity was evaluated using the MTS
proliferation assay with or without erlotinib and with or without
FGFs. Briefly, 3.0×103 cells were seeded per well in
96-well plates. The cells were then treated with erlotinib and with
or without FGF2 (100 ng/ml), FGF9 (100 ng/ml), and FGF10
(100 ng/ml). Control cells were treated with the same concentration
of DMSO. Seventy-two hours after treatment, the number of viable
cells was measured.
Apoptosis assay
PC9 cells were seeded in 6-well plates at 100,000
cells per well for flow cytometric analysis using Gallios (Beckman
Coulter, Brea, CA, USA). The cells were then treated with or
without erlotinib at 1 μmol/l for 48 h. As a control, cells were
treated with the same concentration of DMSO. We analyzed the cell
apoptotic status using TACS Annexin V-FITC (R&D Systems,
Minneapolis, MN, USA) according to the manufacturer’s protocol.
Human serum sample collection and FGF2
and FGF9 quantification
All human samples were obtained with written
informed consent. This study was reviewed and approved by the
Institutional Review Board of Keio University School of Medicine.
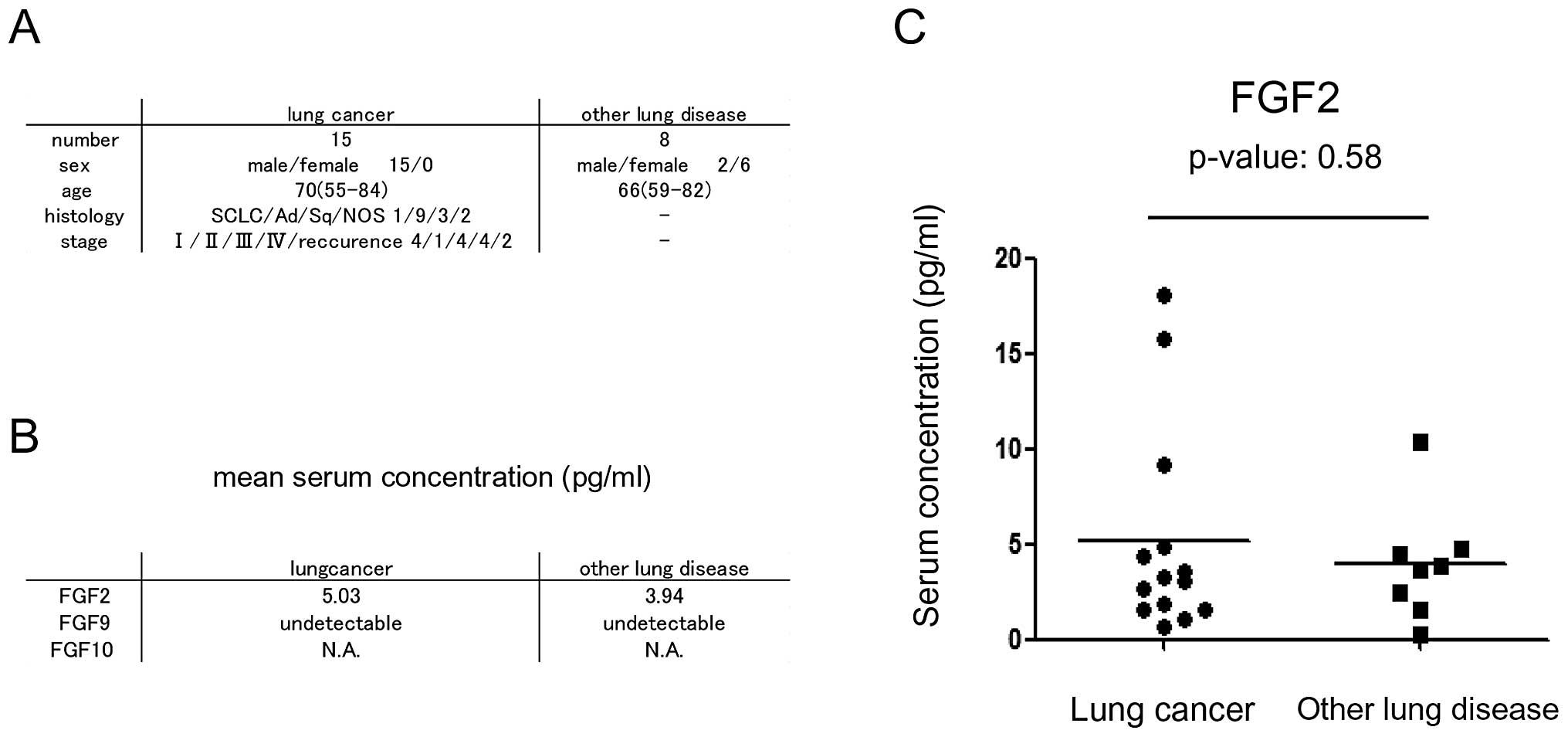
Fifteen patients with lung cancer and 8 patients with other lung
disease, including infectious lung disease, interstitial lung
disease and chronic obstructive pulmonary disease (COPD) were
included in this study. All serum samples were obtained before
chemotherapy. FGF2 and FGF9 concentrations were measured using
enzyme-linked immunosorbent assay (ELISA). FGF2 and FGF9 ELISA were
purchased from R&D Systems and Ray Biotech Inc. (Norcross, GA,
USA), respectively.
Statistical analysis
All p-values are two-sided. Student’s t-test was
used for comparison in this study. The p-values <0.05 were
considered statistically significant.
Results
FGFs promotes SCLC cell proliferation in
a cell-specific manner
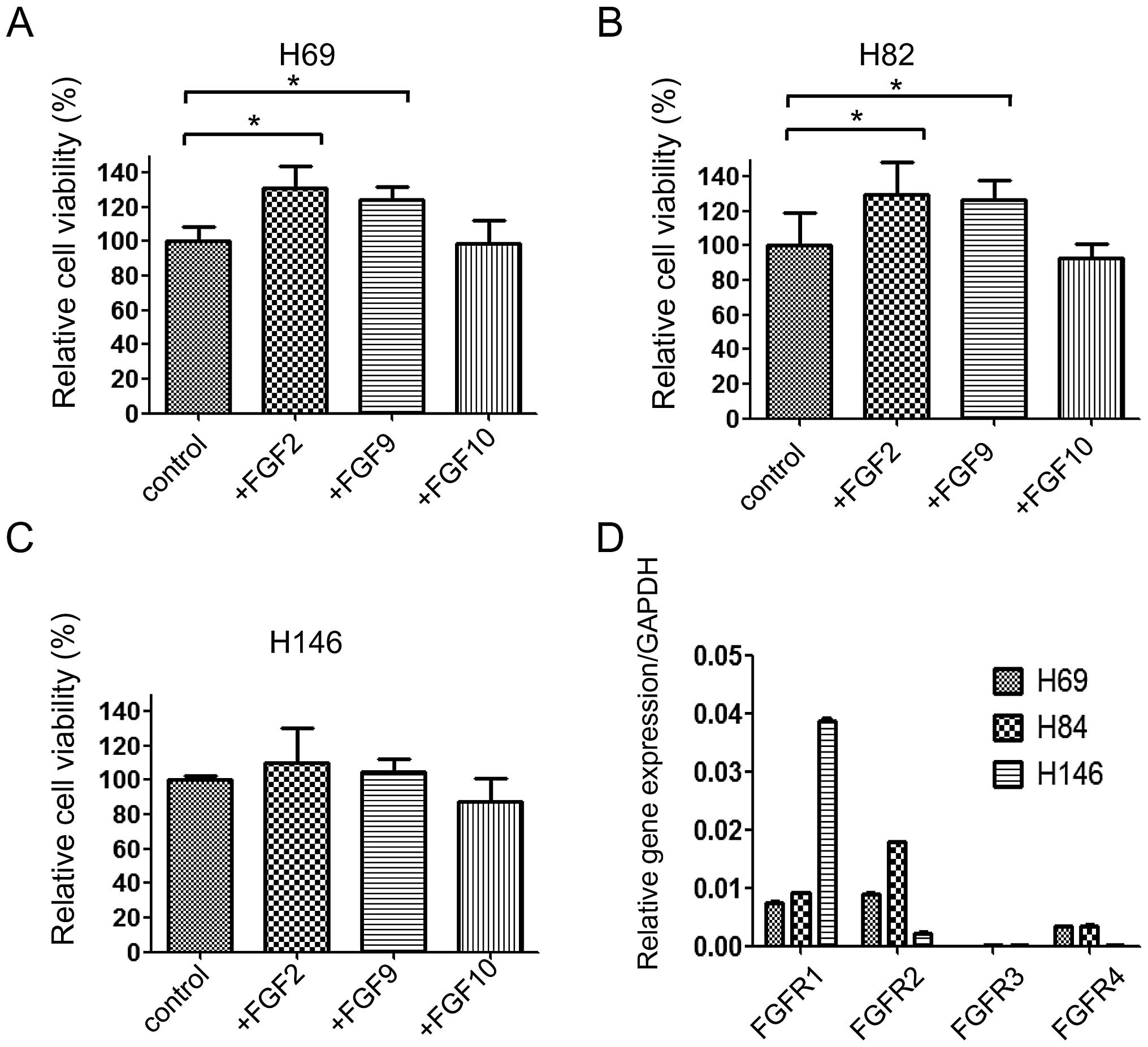
First, to determine whether FGFs promoted lung
cancer cell proliferation, the lung cancer cells were incubated
with or without FGFs. None of the three FGFs promoted NSCLC PC9
cell proliferation. However, FGF2 and FGF9 significantly increased
the proliferation of H82 and H69 cells, two of the SCLC cell lines.
Interestingly, FGFs exerted their effects on lung cancer cell
proliferation in a cell-specific manner. In H69 and H82 cells, FGF2
and FGF9 significantly increased the proliferation, while FGF10 did
not (Fig. 1A and B). In H146
cells, none of the FGFs affected cell proliferation (Fig. 1C). To investigate this discrepancy,
quantitative RT-PCR was performed to determine the expression of
FGFRs. We found that FGFR1 expression was observed in all three
cell lines, while FGFR3 expression was not. FGFR2 and FGFR4 were
preferentially expressed in H69 and H82 cells, which were affected
by FGF2 and FGF9 in terms of proliferation (Fig. 1D). Thus, we speculate that FGF2 and
FGF9 effects on proliferation are mediated through FGFR2 and
FGFR4.
FGFs activate the PI3K/AKT and MAPK
pathways
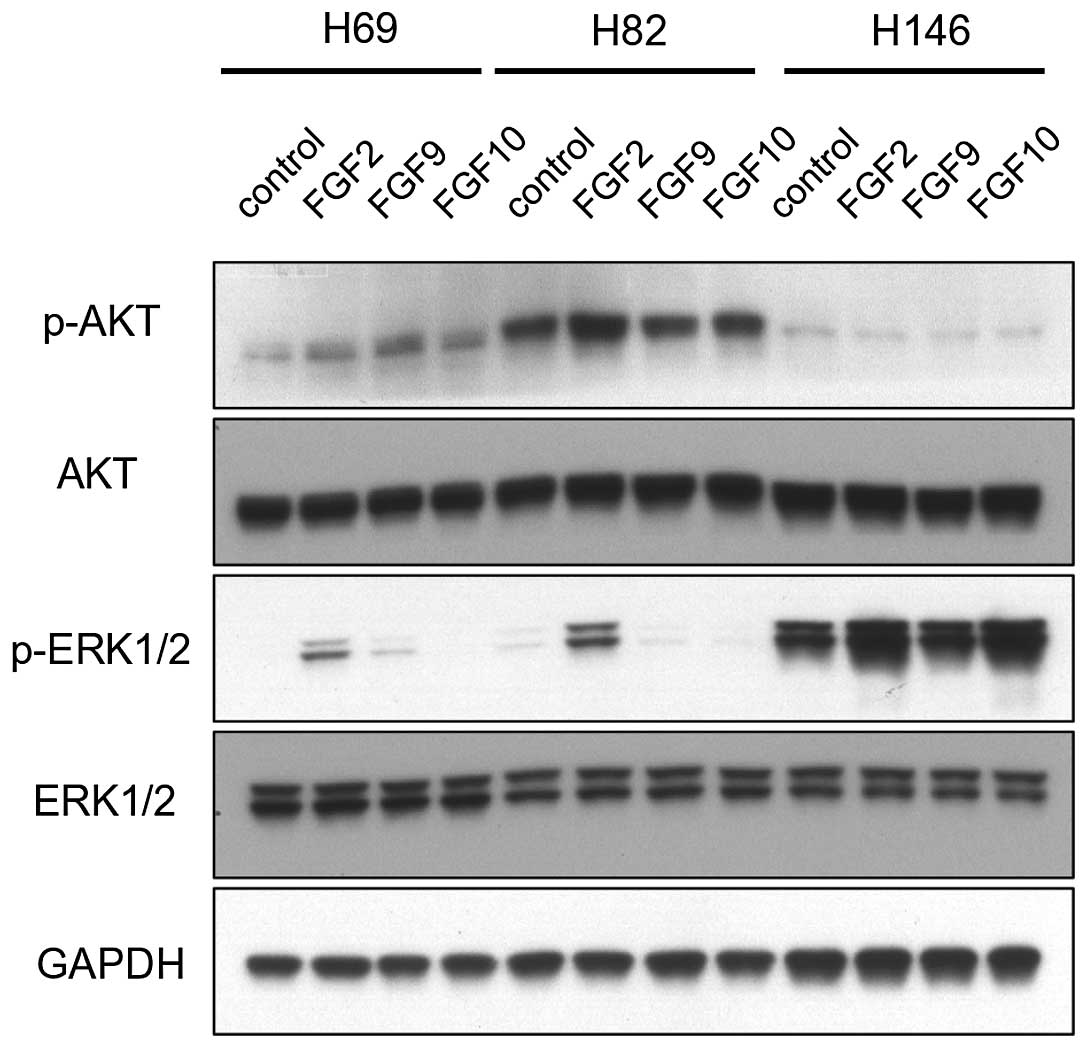
Western blot analysis was performed to understand
how FGF signal is transduced to cellular downstream pathways,
including the PI3K/AKT and MAPK pathways. To evaluate the
activation of the PI3K/AKT and MAPK pathways, the phosphorylated
form of AKT (p-AKT) and ERK1/2 (p-ERK1/2) were detected by western
blot analysis (Fig. 2). FGF
stimulation only modestly increased the p-AKT signal. In H69 cells,
FGF2, FGF9, and FGF10 slightly increased the p-AKT signal. In H82
cells, only FGF2 slightly increased the p-AKT signal.
On the contrary, the effect of FGFs on the
activation of the MAPK pathway was significant. FGF2 dramatically
increased p-ERK1/2 in H69 and H82 cells. FGF9 increased p-ERK1/2 in
H69 cells. However, FGF10 only slightly increased p-ERK1/2 in H146
cells.
These data indicate that FGF signal is mainly
transduced through the MAPK pathway rather than the PI3K/AKT
pathway. Of the three FGFs tested, FGF2 was the most potent in
activating the MAPK pathway. Although there is some discrepancy
between the aforementioned cell proliferation data and the MAPK
pathway activation, we speculate that the effect of FGFs on cell
proliferation is mediated through the MAPK pathway.
FGFs induce erlotinib resistance in PC9
cells
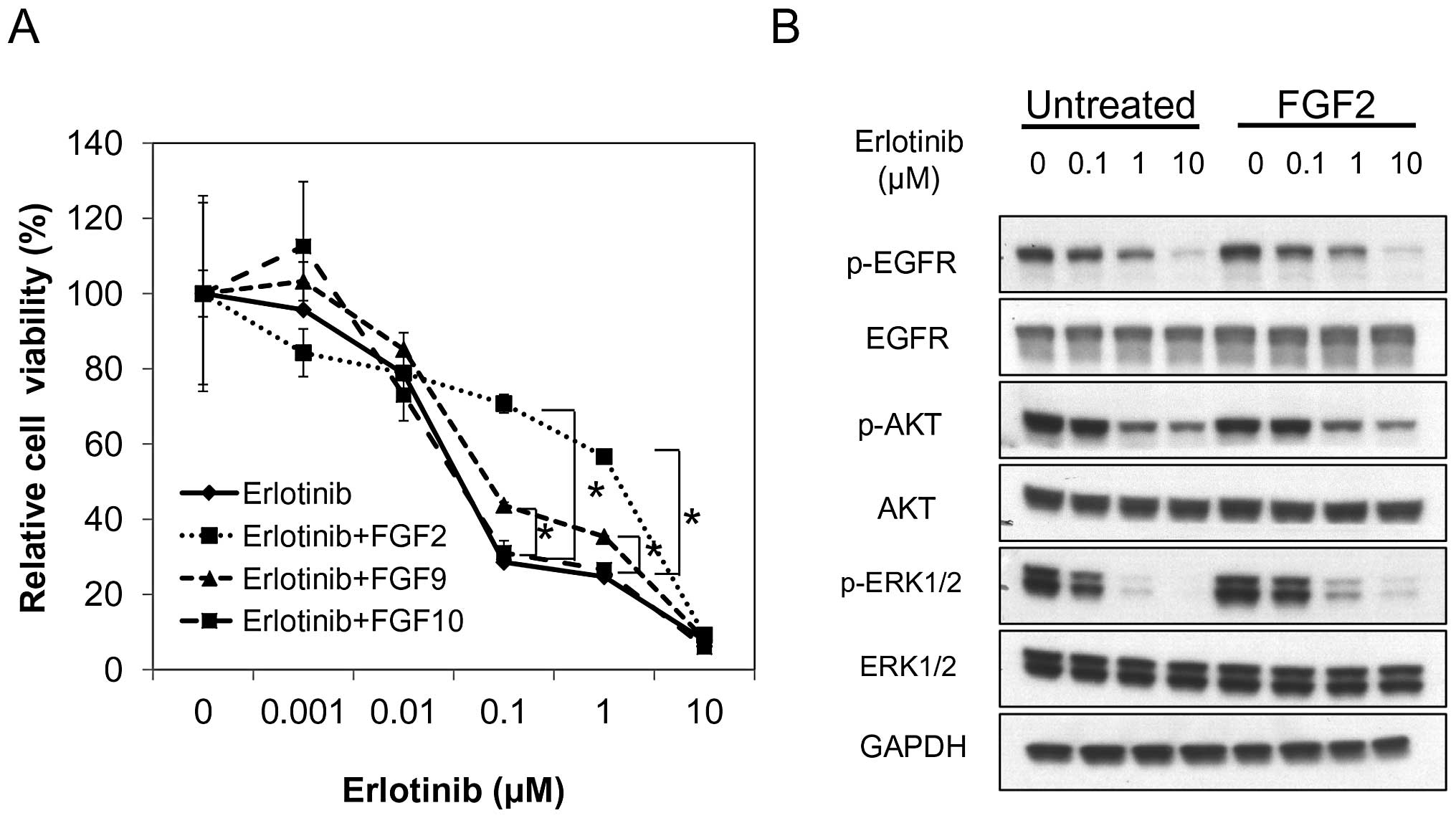
To investigate whether extracellular FGFs can affect
the sensitivity of cancer cells to anticancer drugs, we performed
the MTS proliferation assay with or without several anticancer
drugs. For SCLC cells, we used etoposide and docetaxel. However,
the three FGFs did not affect H69, H82, and H146 sensitivity to
these drugs (data not shown).
As previously described, PC9 cells are sensitive to
EGFR tyrosine kinase inhibitors, such as gefitinib and erlotinib.
In our study, we used erlotinib. Erlotinib alone inhibited PC9 cell
proliferation as previously described. FGF2 and FGF9 induced a
significant resistance to erlotinib in PC9 cells when compared with
erlotinib alone, while FGF10 did not (Fig. 3A). FGF2 was more potent in inducing
resistance to erlotinib than FGF9. These data indicate that FGFs,
especially FGF2, can affect NSCLC cell sensitivity to
erlotinib.
To determine how FGFs, especially FGF2, affected
erlotinib sensitivity, western blot analysis was performed to
assess the effect on downstream pathways. PC9 cells were treated
with erlotinib and with or without FGF2. Erlotinib effectively
inhibited the phosphorylation of EGFR irrespective of the presence
of FGF2. The effect of FGF2 on p-AKT was modest. However, FGF2
induced an increase in p-ERK1/2 in the presence of 0.1, 1.0, and 10
μM erlotinib (Fig. 3B). These data
indicate that FGF2 induced erlotinib resistance through activation
of the MAPK pathway rather than the PI3K/AKT pathway.
FGFs inhibit erlotinib-induced apoptosis
in PC9 cells
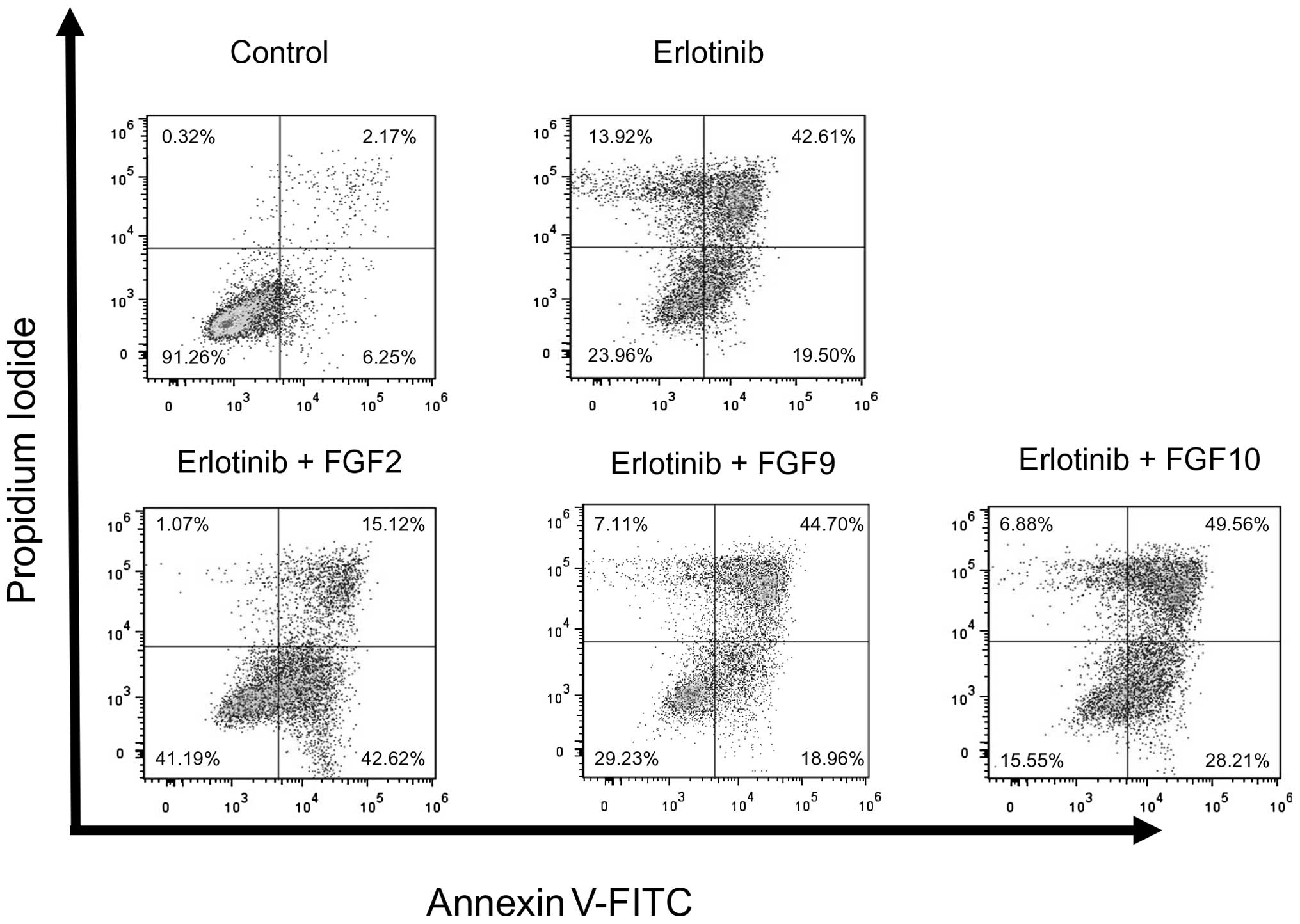
To determine whether FGFs affect erlotinib-induced
apoptosis in PC9 cells, apoptosis was analyzed using Annexin V-FITC
and propidium iodide (PI) double staining and flow cytometry. PC9
cells were incubated with erlotinib and with or without FGFs for 48
h. Erlotinib alone induced early phase apoptosis in 19.5% of the
cells, and late phase apoptosis in 42.61% of the cells. FGF2, but
not FGF9 and FGF10, inhibited erlotinib-induced apoptosis in PC9
cells. FGF2 reduced the proportion of cells in the late phase
apoptosis from 42.61 to 15.21% and increased the proportion of
non-apoptotic cells. When compared to erlotinib alone, the
proportion of double negative cells, PI-negative and Annexin
V-FITC-negative cells, increased from 23.69 to 41.19% when the
cells were treated with erlotinib and FGF2 (Fig. 4). These data indicate that FGF2
affect erlotinib-induced apoptosis in PC9 cells.
FGF2 and FGF9 concentrations in human
serum samples
To determine the clinical relevance of extracellular
FGFs in patients with lung cancer, we measured FGF serum
concentrations in 15 patients with lung cancer and 8 patients with
other lung disease. Of the three FGFs tested in this study, FGF2
and FGF9 presented some functional effects on the cancer cell
biology, while FGF10 did not. Thus, we focused on FGF2 and FGF9.
FGF2 and FGF9 specific ELISAs were used to quantify FGF2 and FGF9
serum concentrations. The clinical characteristics of the patients
are presented in Fig. 5A. The mean
serum FGF2 concentration was 5.03 pg/ml in patients with lung
cancer, while it was 3.94 pg/ml in patients with other lung disease
(Fig. 5B). Although the
concentration was higher in patients with lung cancer than in
patients with other lung disease, the difference was not
statistically significant (p=0.58). The mean serum FGF9
concentration was less than the detectable range in patients with
lung cancer and other lung disease (Fig. 5B).
Discussion
To date, many ‘cell autonomous’ cancer-specific
alterations such as EGFR gene mutation, FGFR gene mutation, or
ALK translocation have been characterized. The
characterization enabled us to develop molecular targeted therapy
for the altered gene itself or its downstream pathways. However,
the tumor microenvironment may affect the biology of lung cancer
cells. Characterization of the effect of extracellular
microenvironment on cancer cells may help us to develop new
therapies targeting ‘non-cell autonomous’ alteration of the cancer
cell microenvironment. In this study, among the various
microenvironment components, we focused on FGF ligands.
The FGF family include 23 proteins, although only 18
are FGFR ligands (19). The FGFs,
which function as FGFR ligands, activate FGFR, which, in turn,
phosphorylates the FGFR substrate 2 (FRS2) and recruits growth
factor receptor-bound 2 (GRB2), finally resulting in the activation
of the MAPK and PI3K/AKT pathways (18). Of all FGFs, FGF2 is the most
studied in the cancer field. However, little is known about the
roles of other FGFs. In this study, we attempted to determine which
FGF among FGF2, FGF9, and FGF10, was the most potent in promoting
lung cancer cell proliferation or in inhibiting lung cancer cell
apoptosis.
Using cell line models, we found that FGFs can
affect the biology of NSCLC and SCLC cells. We determined that some
FGFs can activate the MAPK pathway, promote lung cancer cell
proliferation, and change lung cancer cell sensitivity to
erlotinib.
Using human serum samples, we found the
concentration of FGF2 was higher in patients with lung cancer
compared with that in patients with other lung disease, although it
was not statistically significant. We cannot determine whether the
concentration of FGF2 is higher in patients with lung cancer
compared with that in patients with other lung disease, because the
human study was incomplete with small patients number and several
confounding factors. Moreover, we cannot determine whether serum
FGF2 in patients with lung cancer was derived from lung cancer
tissue, because FGFs are not specifically secreted from lung cancer
cells.
We found that concentration of FGF2 was higher than
that of FGF9. The FGF9 serum concentration was below the detectable
limit by ELISA assay. Although FGF serum concentrations may not
directly reflect FGF concentration in the microenvironment of lung
cancer cells, combining the in vitro and human serum
results, we speculate that, out of the three FGFs tested, FGF2 is
the most potent FGF in promoting proliferation and in inhibiting
apoptosis of lung cancer cells. When comparing FGF9 and FGF10, FGF9
is more likely to promote proliferation and inhibit apoptosis than
FGF10.
In conclusion, our study demonstrates that FGFs have
multiple roles in the biology of lung cancer cells. To thoroughly
understand lung cancer cell biology, further studies of not only
cell autonomous alterations, but also non-cell autonomous
alterations are mandatory. Further studies on the role of
extracellular ligands may help us develop novel treatments
targeting extracellular ligands.
Acknowledgements
We thank Mikiko Shibuya for her excellent technical
assistance. This study was supported in part by Grants-in-Aid for
Scientific Research on Priority Areas from the Ministry of
Education, Culture, Sports, Science, and Technology of Japan to
K.S. (grant no. 22590870), H.Y. (grant no. 25860656), K.N. (grant
no. 23501311), and T.B. (grant no. 23390218).
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar
|
|
2
|
Cancer Genome Atlas Research Network.
Comprehensive genomic characterization of squamous cell lung
cancers. Nature. 489:519–525. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Imielinski M, Berger AH, Hammerman PS, et
al: Mapping the hallmarks of lung adenocarcinoma with massively
parallel sequencing. Cell. 150:1107–1120. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wistuba II, Gazdar AF and Minna JD:
Molecular genetics of small cell lung carcinoma. Semin Oncol.
28:3–13. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Peifer M, Fernandez-Cuesta L, Sos ML, et
al: Integrative genome analyses identify key somatic driver
mutations of small-cell lung cancer. Nat Genet. 44:1104–1110. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Weir BA, Woo MS, Getz G, et al:
Characterizing the cancer genome in lung adenocarcinoma. Nature.
450:893–898. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Paez JG, Janne PA, Lee JC, et al: EGFR
mutations in lung cancer: correlation with clinical response to
gefitinib therapy. Science. 304:1497–1500. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yasuda H, Kobayashi S and Costa DB: EGFR
exon 20 insertion mutations in non-small-cell lung cancer:
preclinical data and clinical implications. Lancet Oncol.
13:e23–31. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yasuda H, Park E, Yun CH, et al:
Structural, biochemical, and clinical characterization of epidermal
growth factor receptor (EGFR) exon 20 insertion mutations in lung
cancer. Sci Transl Med. 5:216ra1772013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Soda M, Choi YL, Enomoto M, et al:
Identification of the transforming EML4-ALK fusion gene in
non-small-cell lung cancer. Nature. 448:561–566. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Choi YL, Takeuchi K, Soda M, et al:
Identification of novel isoforms of the EML4-ALK transforming gene
in non-small cell lung cancer. Cancer Res. 68:4971–4976. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Weiss J, Sos ML, Seidel D, et al: Frequent
and focal FGFR1 amplification associates with therapeutically
tractable FGFR1 dependency in squamous cell lung cancer. Sci Transl
Med. 2:62ra932010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Costa DB, Halmos B, Kumar A, et al: BIM
mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung
cancers with oncogenic EGFR mutations. PLoS Med. 4:1669–1680. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yasuda H, de Figueiredo-Pontes LL,
Kobayashi S and Costa DB: Preclinical rationale for use of the
clinically available multitargeted tyrosine kinase inhibitor
crizotinib in ROS1-translocated lung cancer. J Thorac Oncol.
7:1086–1090. 2012. View Article : Google Scholar
|
|
15
|
Liao RG, Jung J, Tchaicha J, et al:
Inhibitor-sensitive FGFR2 and FGFR3 mutations in lung squamous cell
carcinoma. Cancer Res. 73:5195–5205. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dutt A, Ramos AH, Hammerman PS, et al:
Inhibitor-sensitive FGFR1 amplification in human non-small cell
lung cancer. PLoS One. 6:e203512011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pardo OE, Latigo J, Jeffery RE, et al: The
fibroblast growth factor receptor inhibitor PD173074 blocks small
cell lung cancer growth in vitro and in vivo. Cancer Res.
69:8645–8651. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dieci MV, Arnedos M, Andre F and Soria JC:
Fibroblast growth factor receptor inhibitors as a cancer treatment:
from a biologic rationale to medical perspectives. Cancer Discov.
3:264–279. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Turner N and Grose R: Fibroblast growth
factor signalling: from development to cancer. Nat Rev Cancer.
10:116–129. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Donnem T, Al-Shibli K, Al-Saad S, Busund
LT and Bremnes RM: Prognostic impact of fibroblast growth factor 2
in non-small cell lung cancer: coexpression with VEGFR-3 and PDGF-B
predicts poor survival. J Thorac Oncol. 4:578–585. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rades D, Seibold ND, Gebhard MP, Noack F,
Bruchhage KL and Schild SE: Fibroblast growth factor 2 is of
prognostic value for patients with locally advanced squamous cell
carcinoma of the head and neck. Strahlenther Onkol. 190:68–74.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wilson TR, Fridlyand J, Yan Y, et al:
Widespread potential for growth-factor-driven resistance to
anticancer kinase inhibitors. Nature. 487:505–509. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nomura S, Yoshitomi H, Takano S, et al:
FGF10/FGFR2 signal induces cell migration and invasion in
pancreatic cancer. Br J Cancer. 99:305–313. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ohgino K, Soejima K, Yasuda H, et al:
Expression of fibroblast growth factor 9 is associated with poor
prognosis in patients with resected non-small cell lung cancer.
Lung Cancer. 83:90–96. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yin Y, Betsuyaku T, Garbow JR, Miao J,
Govindan R and Ornitz DM: Rapid induction of lung adenocarcinoma by
fibro-blast growth factor 9 signaling through FGF receptor 3.
Cancer Res. 73:5730–5741. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Terai H, Soejima K, Yasuda H, et al:
Activation of the FGF2-FGFR1 autocrine pathway: a novel mechanism
of acquired resistance to gefitinib in NSCLC. Mol Cancer Res.
11:759–767. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lefevre G, Babchia N, Calipel A, et al:
Activation of the FGF2/FGFR1 autocrine loop for cell proliferation
and survival in uveal melanoma cells. Invest Ophthalmol Vis Sci.
50:1047–1057. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fillmore CM, Gupta PB, Rudnick JA, et al:
Estrogen expands breast cancer stem-like cells through paracrine
FGF/Tbx3 signaling. Proc Natl Acad Sci USA. 107:21737–21742. 2010.
View Article : Google Scholar : PubMed/NCBI
|