Introduction
Multiple myeloma (MM) is a kind of haematological
malignancy, which is caused by abnormal proliferation and
differentiation of plasma cells in bone marrow. However, there is
no ideal treatment for MM, particularly multidrug resistant and
refractory MM. As a proteasome inhibitor for biological target
therapy, bortezomib has been used to treat MM in recent years.
Although bortezomib obtains better curative effect in many MM
patients, some patients still relapse after several courses of
treatment or they are primary drug resistant to bortezomib. For
those patients, combined chemotherapy is indispensable. Multidrug
resistance (MDR) of tumor cells towards chemotherapeutic drugs is
the main reason for the failure of chemotherapy, so to find
selective MDR reversal agents has become important in tumor
treatment.
Several investigations have proved that the
chemotherapy resistance is related to enhanced DNA damage repair in
tumor cells (1,2), but the mechanism of this phenomenon
remains ambiguous and open to discussion. Alkylating agents (known
as DNA cross-linking agents) are major chemotherapeutic drugs for
many hematologic malignancies and solid tumors, such as melphalan
and cyclophosphamide are used for the treatment of MM as well as
lymphoma and leukemia. Alkylating agents target DNA and induce
interstrand cross-link (ICL) damage, thus the cellular replication
stops (3). ICLs can cause serious
replication forks blocking and DSBs are the result of a stalled
replication fork at an ICL (4,5).
Studies showed that repair of the ICLs depends upon base excision
repair (BER), nucleotide excision repair (NER) and homologous
recombination (HR) (4,6).
The Fanconi anemia (FA)/BRCA pathway is a
specialized DNA repair pathway that removes interstrand crosslinks
(ICLs), a particularly toxic lesion resulting from exposure to
alkylating agents such as mitomycin C and cisplatin (7). The FA/BRCA pathway is initiated when
the FA core complex (comprised of FANCA/B/C/E/F/G/L/M) recognizes
and binds an ICL through an interaction mediated by FANCM and its
binding partners (8). Following
lesion binding, the FA core complex monoubiquitinates FANCD2 and
FANCI via the E3 ubiquitin ligase activity of FANCL.
Monoubiquitinated FANCD2/FANCI interact with downstream FA proteins
(FANCD1/J/N/O), which form stable complexes with proteins
participating in homologous recombination (HR), such as BRCA1 and
RAD51 (9,10). The ubiquitinated FANCD2-I
heterodimer localizes to the ICL and recruits endonucleases that
incise the DNA on either side of the lesion to create a
double-stranded break (DSB). The complementary strand containing
the unhooked crosslink is replicated by a trans-lesion synthesis
(TLS) polymerase, and downstream FA proteins (FANCD1/J/N/O) assist
in coordination of HR to repair the DSB (8).
Several studies have proven that the FA/BRCA pathway
plays an important role in the process of DNA damage repair
(11–13). A variety of alkylating agents exert
their effects through the FA/BRCA pathway, so adjusting this
pathway may change the effects of the alkylating agents (14–20).
Phenylbutyrate had therapeutic utility as a cisplatin sensitizer in
head and neck cancer by inhibiting the FA/BRCA pathway through the
downregulation of BRCA1, and this sensitization correlated to a
significant decrease in the formation of cisplatin-induced FANCD2
nuclear foci, which is a functional read out of the FA/BRCA pathway
(21). This abrogation of the
FA/BRCA pathway by phenylbutyrate was not due to loss of FANCD2
monoubiquitylation but rather correlated to a
phenylbutyrate-mediated reduction in the expression of the BRCA1
protein. In addition, it was found that FANCD1/BRCA2 small
interference RNA efficiently enhanced cellular sensitivity toward
alkylating agents ACNU and TMZ in human glioblastoma A172 cells
(22). Using the proteasome
inhibitor bortezomib drastically reduced the FA/BRCA gene
expression in myeloma cells, resulting in diminished DNA damage
repair and enhanced melphalan sensitivity (23). The above findings suggest that the
downregulation of FA/BRCA pathway might be an effective strategy to
increase cellular sensitivity toward alkylating agents. These
results provide evidence for targeting Fanconi anemia-mediated DNA
repair to enhance chemotherapeutic response and circumvent drug
resistance in patients with a tumor.
According to related literature, Poly(ADP-ribose)
polymerase-1 (PARP-1), as an enzyme critical in BER, plays an
important role in DNA repair and perception of DNA damage (24–26).
PARP-1 is involved in the repair of single-strand DNA breaks
through the BER pathway and may also be involved in repair of
double-strand breaks, through the homologous recombination (HR)
pathway (27). Thus, its enzyme
activity is regulated by its binding to DNA, recognizing single-
and double-strand breaks and also by interacting with a wide
variety of chromatin-associated proteins, including components of
the transcription machinery, sequence-specific DNA-binding
transcription factors, chromatin modifying enzymes and histone
variants (28).
PARP-1 inhibition has been shown to block DNA
repair, so the inhibitors of PARP-1 are promising assistant drugs
in antitumour treatment (29,30).
Several studies have confirmed that PARP-1 inhibitors can obviously
improve the effects of alkylating agents used in chemotherapy
including cisplatin, cyclophosphamide, temozolomide and oxaliplatin
(31–34). Enhanced efficacy of PARP-1
inhibitors was most likely caused by disruption of DNA damage
repair pathways. In addition, when being applied independently,
PARP-1 inhibitors show tumor-selective killing effect (35) and strong inhibition of angiogenesis
(36) on tumors with homologous
recombination repair defects (such as BRCA1 and BRCA2 defects).
However, the role of PARP-1 in DNA damage is not well-understood,
and the mechanism of PARP-1 inhibitors increasing the sensitivity
of tumor cells to alkylating agents remains to be fully
elucidated.
A number of studies show that the FA/BRCA pathway is
involved in the repair process of ICL with a variety of components,
inhibiting the expression of certain factors in FA/BRCA pathway
seems to be capable of suppressing DNA repair and enhancing the
sensitivity of tumor cells to alkylating agents (11–23).
PARP-1 inhibitors can improve the efficacy of alkylating agents
such as cisplatin, and cyclophosphamide due to the inhibition of
BER after ICLs caused by alkylating agents (29–34).
Thus, we speculate that effects of PARP-1 inhibitors may be
associated with inhibition of the FA/BRCA pathway, and the drug
resistance to alkylating agent-based chemotherapy might be reversed
by inhibiting the activity of this pathway. Therefore, in the
present study, we investigated the effect of PJ34, a potent PARP-1
inhibitor, on the melphalan-induced cytotoxicity and DNA damage in
a multidrug-resistant cell line RPMI8226/R. We compared the
sensitivity of RPMI8226/R cells to melphalan before and after the
treatment with PJ34, and detected the expression level of related
factors in the FA/BRCA pathway. We attempted to demonstrate the
influence of PJ34 on DNA repair and drug resistance and the
possible relationship with the FA/BRCA pathway.
Materials and methods
Materials
The human multiple myeloma cell line RPMI8226 was
kindly gifted by Professor Jianfeng Zhou (Department of Hematology,
Tongji Hospital of Huazhong University of Science and Technology),
the original cell line was purchased from the American Type Culture
Collection (ATCC, Manassas, VA, USA). PJ34, melphalan, DMSO were
purchased from Sigma Chemical Co. CCK-8 Cell Counting kit was
purchased from Dojindo Laboratories (Kumamoto, Japan). RPMI-1640
and fetal calf serum were purchased from Invitrogen Life
Technologies. The primary and second antibodies of FANCD2, BRCA2,
RAD51, γH2AX and β-actin were purchased from Santa Cruz
Biotechnology.
Cell culture
RPMI8226 cells were cultured in RPMI-1640 medium
supplemented with 10% fetal calf serum, and incubated in a
humidified atmosphere containing of 5% CO2 at 37°C.
Multidrug-resistant RPMI8226/R cells were cultured in the same
environment as above, but they were selected by stepwise exposure
of parental sensitive RPMI8226 cells to increasing concentrations
of melphalan. The RPMI8226/R cells were maintained in the presence
of 4.5 μmol/l melphalan and grown in drug-free medium 2 weeks
before the experiments.
Cytotoxicity and chemosensitivity
assay
RPMI8226 and RPMI8226/R cells in the exponential
proliferation period were seeded into 96-well plates
(1×105 cells/well). Twenty-four hours after plating,
PJ34, melphalan and melphalan plus PJ34 were added to 3 test groups
with increasing drug concentrations. Each concentration was added
in quadruplicate. The reversal reagent of the control group was
replaced by RMPI-1640. The reversal effect of PJ34 was determined
by CCK-8 assay according to the manufacturer’s instructions. Drug
effects were determined at the level of 50% inhibition
(IC50) compared to controls.
Western blot analysis
Cell lysates were prepared by suspending cell
pellets in lysis buffer. The proteins were separated by SDS-PAGE
and western blot analysis was performed as previously described
(37). A total of 50 μg protein
was used for the western blotting unless otherwise indicated.
β-actin was used as the loading control.
Reverse transcription-polymerase chain
reaction (RT-PCR)
Total RNA was extracted from RPMI8226/R cells after
treatment with PJ34 at different concentrations with TRIzol reagent
(Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s
instruction. cDNA was synthesized using the RevertAid™ First Strand
cDNA Synthesis kit (Fermentas). The final cDNA was used for the
subsequent PCR. FANCD2, BRCA2 and RAD51 gene expressions were
quantified by semi-quantitative RT-PCR using SYBR-Green/Fluorescein
qPCR Master Mix (Fermentas). β-actin was used as the endogenous
control.
Alkaline comet assays
To assess DNA DSBs repair, the comet assay was
performed under alkaline conditions using CometSlide assay kits
(Trevigen) following the protocol. RPMI8226/R cells were incubated
at 37°C for 24 h in three groups (PBS, melphalan or melphalan plus
PJ34) to allow for DNA damage repair. Cells were embedded in
agarose, lysed, and subjected to alkaline electrophoresis.
Immediately before image analysis, cells were stained with ethidium
bromide and visualized under a fluorescence microscope. The program
CometScore™ Version 1.5 was utilized to analyze the comet images.
The tail length, comet length, tail moment and olive tail moment
induced by different treatment were the analysis parameters. A
total of 75 cells/sample were scored to determine the average
percentage of DNA damaged.
Immunofluorescence microscopy and
quantification of γH2AX foci
RPMI8226/R cells were incubated at 37°C for 24 h in
three groups (PBS, melphalan or melphalan plus PJ34). After
treatment, cells were fixed with 4% paraformaldehyde, permeabilized
and blocked by incubation in PBS containing 0.5% Triton X-100, 15%
goat serum, 0.2% fish skin gelatin and 0.03% NaN3. Next,
cells were stained with mouse monoclonal anti-γH2AX antibody,
followed by FITC-conjugated goat anti-mouse secondary antibody and
counterstained with DAPI to visualize the cell nucleus. The
percentage of cells containing more than 10 nuclear fluorescent
foci per total cell number was calculated by examining a minimum of
120 cells for each experimental group.
Apoptosis assay
As previously described (37), to detect apoptosis, cells of the
four groups were washed in PBS, resuspended in 500 μl binding
buffer containing Annexin V-FITC/PI and analyzed by flow
cytometry.
Statistical analysis
SPSS 13.0 was used to perform the statistical
analysis. Data are presented as mean ± SD, and analyzed by the
Student’s t-test. P<0.05 was considered statistically
significant.
Results
Establishment and characterization of the
RPMI8226/R cells
The multidrug resistant RPMI8226/R cell line was
established with a 4.5 μmol/l final concentration of melphalan. In
order to identify the establishment of resistance to different
anti-cancer agents, the sensitivities of the RPMI8226/R and the
RPMI8226 cells were compared. According to the CCK-8 assay results,
the RPMI8226/R cells exhibited resistance not only to melphalan,
but also to six other anticancer agents: adriamycin (ADM),
cyclophosphamide (CTX), cisplatin (DDP), Ara-C, vincristine (VCR)
and VP-16. The relative resistance of RPMI8226/R cells, as compared
to the RPMI8226, is demonstrated in Table I. The IC50 values for
RPMI8226/R and RPMI8226 were 20.43±0.21 and 4.79±0.18 μmol/l,
respectively. The degree of resistance was evaluated in terms of
resistance index (RI) which is calculated according to the
relation: RI =
IC50(RPMI8226/R)/IC50(RPMI8226). Therefore,
RPMI8226/R cell line was ~4.27-fold more resistant to melphalan
than the original cell line. Moreover, RPMI8226/R cell line was
8.70-fold more resistant to CTX, another alkylating agent. When
RPMI8226/R cell line was cultured in RPMI-1640 without melphalan
for 10 weeks, there was no drug resistance decrease with the
similar RI as before (RI = 4.10), showing the cell line had stable
drug resistance, but the RI decreased to 3.05 after 12 weeks and
recovered to 4.21 by adding melphalan (4.5 μM) into the medium
again.
 | Table ICross-resistance patterns of RPMI8226
and RPMI8226/R cell lines to various chemotherapeutic agents
(μmol/l, mean ± SD). |
Table I
Cross-resistance patterns of RPMI8226
and RPMI8226/R cell lines to various chemotherapeutic agents
(μmol/l, mean ± SD).
| Agent | RPMI8226
(IC50) | RPMI8226/R
(IC50) | RI |
|---|
| Melphalan | 4.79±0.18 | 20.43±0.21 | 4.27 |
| Ara-C | 1.99±0.25 | 6.11±0.13 | 3.07 |
| VP-16 | 7.54±0.08 | 18.04±0.12 | 2.39 |
| DDP | 1.30±0.03 | 5.55±0.01 | 4.26 |
| ADM | 0.73±0.16 | 3.07±0.32 | 4.20 |
| CTX | 2.89±0.09 | 25.14±0.02 | 8.70 |
| VCR | 1.63±0.30 | 4.41±0.41 | 2.71 |
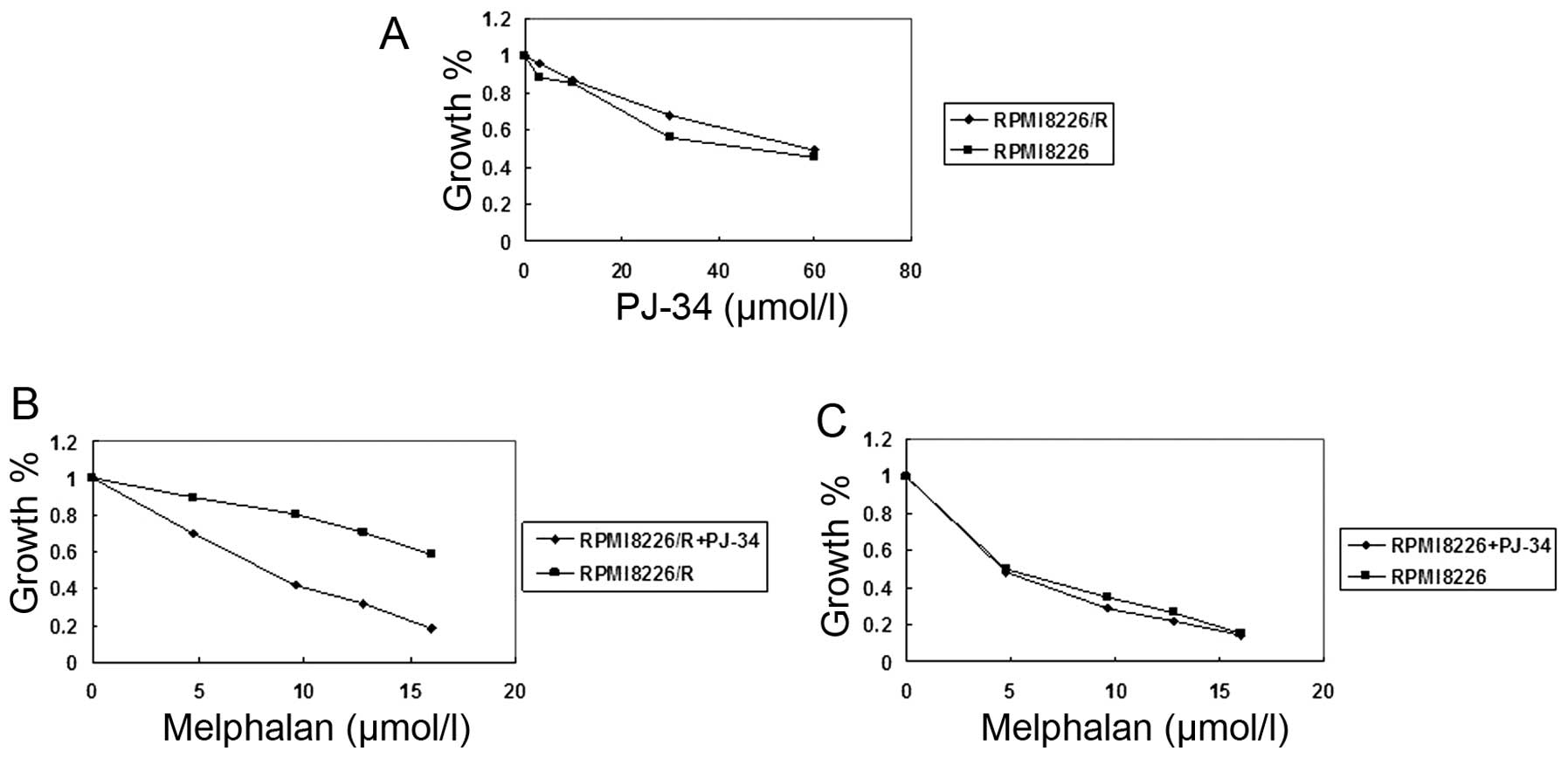
PJ34 enhanced the toxicity of melphalan
in RPMI8226/R cells but not in RPMI8226 cells
In the present study, we investigated the effect of
PJ34 combined with melphalan on the viability of RPMI8226 and
RPMI8226/R cells by CCK-8 assay. As shown in Fig. 1A and Table IIA, PJ34 alone did not exhibit a
cytotoxic effect on RPMI8226/R cells at doses up to 60 μM. Compared
with DMSO-pretreated cells, pretreatment with 60 μM PJ34 resulted
in significant inhibition of cell growth in RPMI8226/R cells after
melphalan treatment (Fig. 1B,
Table IIB). Similar result was
not observed in RPMI8226 cells (Fig.
1C, Table IIC). Taken
together, the pretreatment of PJ34 at 60 μM consistently enhanced
the toxicity of melphalan in RPMI8226/R cells, but not in RPMI8226
cells. In RPMI8226/R cells, IC50 of melphalan with 60 μM
of PJ34 reduced from 20.43 to 7.82 μM. Therefore, 60 μM of PJ34 was
selected for further experiments. The use of PJ34 combined with
melphalan led to a more significant reduction in viability
(increased toxicity) than the use of either PJ34 or melphalan alone
in RPMI8226/R cells (P<0.05). These results all clearly indicate
that PJ34 is a potential chemosensitizer of melphalan.
| Table IIThe effects of PJ34 on cell growth in
RPMI8226 and RPMI8226/R. |
Table II
The effects of PJ34 on cell growth in
RPMI8226 and RPMI8226/R.
| A, The effects of
PJ34 on cell growth of RPMI8226 and RPMI8226/R |
|---|
|
|---|
| Cell growth
(%) |
|---|
|
|
|---|
| PJ-34 (μM) | RPMI8226 | RPMI8226/R | P-value |
|---|
| 3 | 88.64±2.00 | 95.90±2.62 | 0.055 |
| 10 | 84.93±1.00 | 87.02±1.08 | 0.068 |
| 30 | 55.95±1.94 | 67.80±1.51 | 0.017 |
| 60 | 45.57±1.51 | 48.95±1.22 | 0.109 |
|
| B, The effects of
PJ34 on RPMI8226/R growth inhibited by melphalan |
|
| Cell growth
(%) |
|
|
| Melphalan (μM) | RPMI8226/R |
RPMI8226/R+PJ34 | P-value |
|
| 4.8 | 89.47±0.50 | 69.92±0.33 | 0.000 |
| 9.6 | 80.19±0.45 | 41.81±0.13 | 0.000 |
| 12.8 | 70.41±0.50 | 31.87±0.22 | 0.000 |
| 16 | 58.42±0.47 | 18.13±0.28 | 0.000 |
|
| C, The effects of
PJ34 on RPMI8226 growth inhibited by melphalan |
|
| Cell growth
(%) |
|
|
| Melphalan (μM) | RPMI8226 | RPMI8226+PJ34 | P-value |
|
|
| 4.8 | 50.08±1.33 | 48.56±1.10 | 0.28 |
| 9.6 | 34.41±2.05 | 28.65±2.30 | 0.053 |
| 12.8 | 26.12±2.50 | 21.57±2.02 | 0.061 |
| 16 | 14.84±0.75 | 14.24±1.40 | 0.094 |
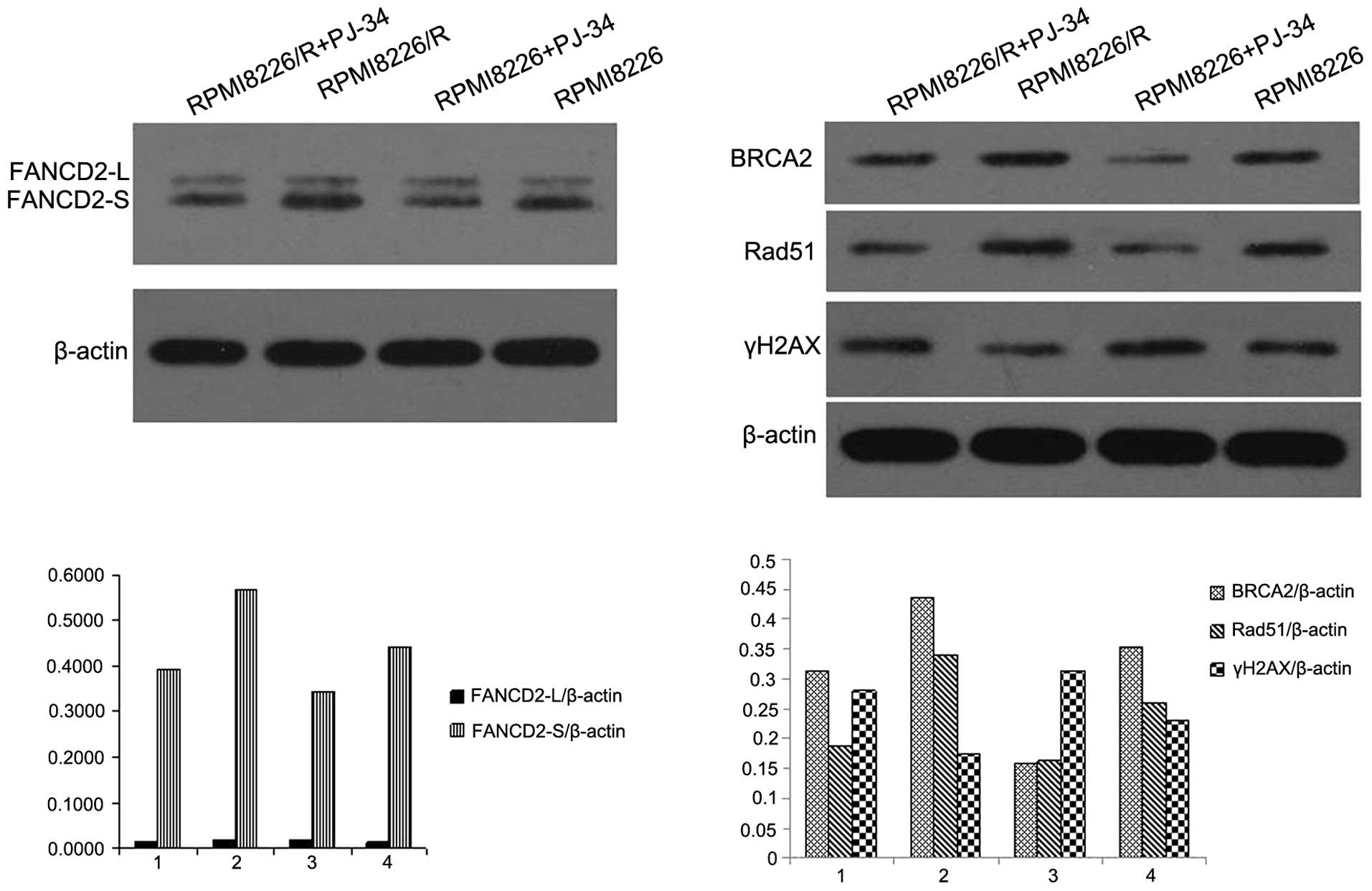
PJ34 suppressed the mRNA and protein
expressions of the factors in FA/BRCA pathway in RPMI8226/R
cells
The two cell lines, RPMI8226/R and RPMI8226, were
exposed to various concentrations of PJ34 for 24 h. The protein
expression of FANCD2, BRCA2 and Rad51 in RPMI8226/R cells was
higher than that in RPMI8226 cells (Fig. 2). In contrast, the expression of
γH2AX in RPMI8226/R cells was lower than that in RPMI8226 cells
before PJ34 treatment. It was found that PJ34 (60 μM) treatment
decreased the protein levels of FANCD2, BRCA2, Rad51 and the effect
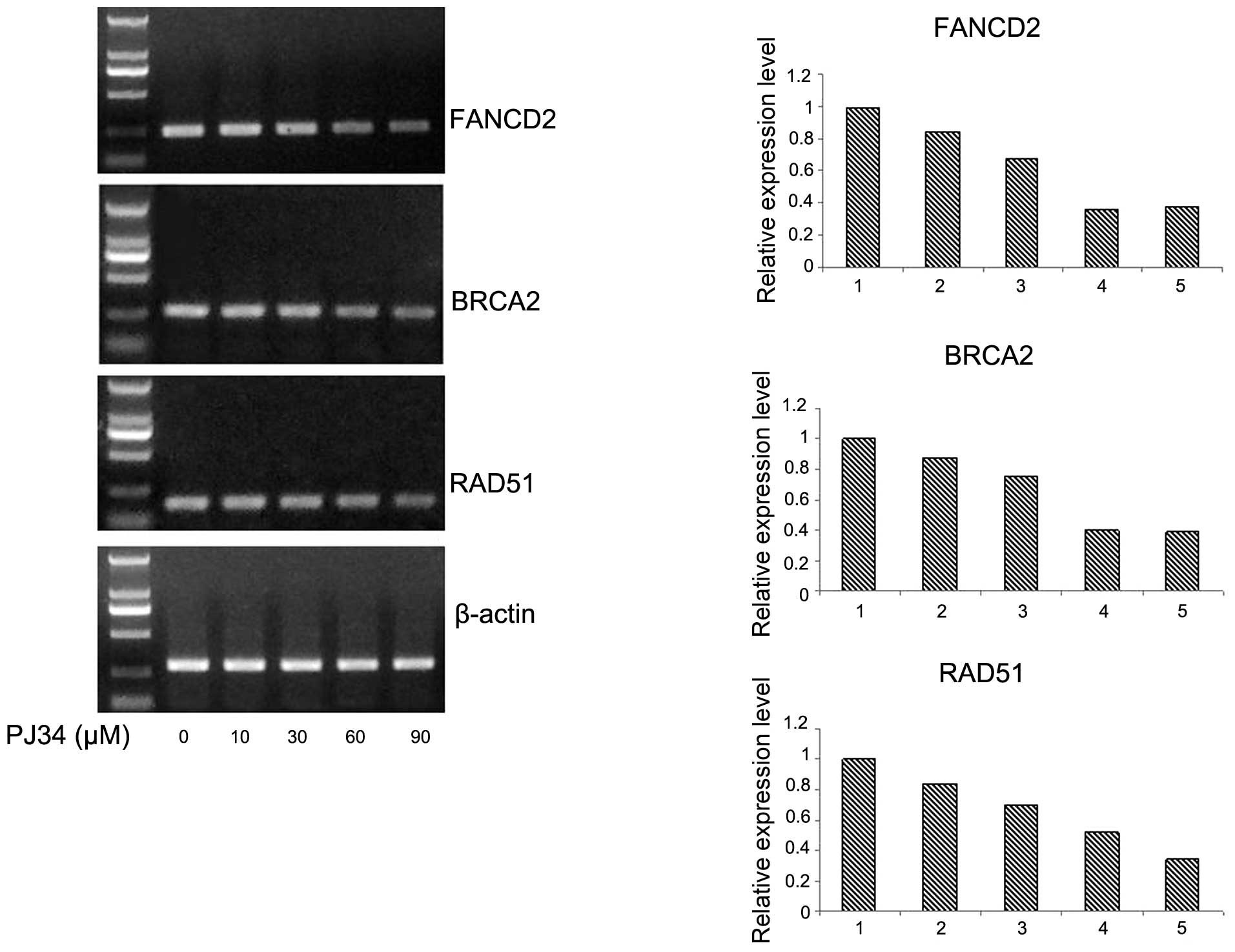
was the opposite on γH2AX expression in RPMI8226/R cells (Fig. 2; P<0.05). To elucidate whether
the observed effects of PJ34 on the above protein expression
occurred at the transcriptional level, total RNA was isolated and
subjected to RT-PCR analysis for FANCD2, BRCA2 and Rad51
transcripts. Exposure to PJ34 decreased FANCD2, BRCA2 and Rad51
mRNA levels dose-dependently (Fig.
3). The above results showed that the activity of FA/BRCA
pathway was enhanced in the resistant cell line RPMI8226/R, which
resulted in intensive repair to the DNA damage induced by
melphalan. Whereas the DNA damage of γH2AX protein expression
declined. The antagonism effects of PJ34 led to suppressed mRNA and
protein expressions of the factors in the FA/BRCA pathway and
enhanced DNA damage caused by melphalan in RPMI8226/R cells. In
summary, inhibition of the FA/BRCA pathway activity by PJ34 could
reduce DNA repair to reverse the drug resistance in
melphalan-exposed RPMI8226/R cells.
PJ34 plus melphalan enhance cell apoposis
in RPMI8226/R cells
To investigate the effects of PJ34 on cell apoptosis
in RPMI8226/R cells treated with melphalan, cells were given
differential treatment followed by flow cytometric analysis. As
shown in Table III, when cells
were treated with melphalan or PJ34 alone, the apoptosis percentage
of RPMI8226 was significantly higher than that of RPMI8226/R
(P<0.05). On the contrary, the apoptosis percentage of
RPMI8226/R cells was significantly higher than that of RPMI8226
(P<0.01) with the treatment of melphalan 9.6 μM plus PJ34 60 μM.
Table III also showed
significantly increased population of apoptotic cells in
concomitant treatment group (55.68±0.78%) compared with untreated
control (6.73±0.28%) and melphalan (23.88±1.38%) or PJ34
(17.82±1.12%) single treatment groups (P<0.05). Therefore, PJ34
improved the apoptosis percentage of RPMI8226/R cells
significantly.
| Table IIIThe effects of PJ34 on cell apoptosis
induced by melphalan. |
Table III
The effects of PJ34 on cell apoptosis
induced by melphalan.
| Cell apoptosis
(%) | |
|---|
|
| |
|---|
| Group | RPMI8226 | RPMI8226/R | P-value |
|---|
| Control | 15.28±2.84 | 6.73±0.28 | 0.144 |
| PJ-34 | 22.70±2.16 | 17.82±1.12 | 0.105 |
| Melphalan | 31.08±0.47 | 23.88±1.38 | 0.02 |
| PJ-34+
melphalan | 45.38±4.53 | 55.68±0.78 | 0.003 |
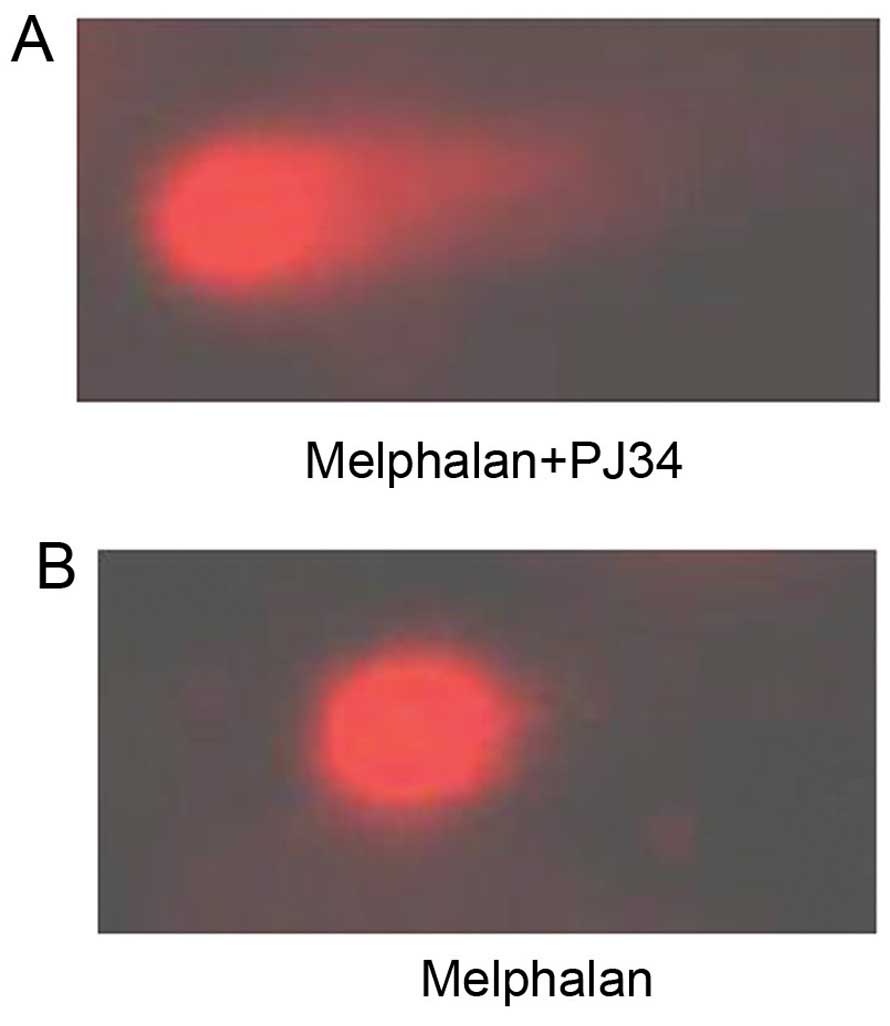
PJ34 increases DNA strand breaks induced
by melphalan in RPMI8226/R cells
Treatment with melphalan led to the breakage of DNA
strands. Following unwinding, DNA fragments left the nuclear zone
and moved to the positive pole under the effect of the electric
field in the electrophoresis liquid, forming the distinctive comet
tail formation. Thus, damage to cellular DNA of the RPMI8226/R
cells due to exposure to melphalan combined with PJ34 or without
PJ34 were revealed by alkaline comet assay (Fig. 4). A manifest difference of
chemosensitivity to melphalan was observed in RPMI8226/R cells
following exposure to PJ34 at 60 μM, as shown in Table IV. The differences of tail length,
comet length, tail moment, olive tail and percentage of comet-like
cell between PJ34 treated group and untreated group were all
significant (P<0.05) (Table
IV). Increased DNA damage of RPMI8226/R cells with PJ34
treatment was detected. The data revealed that ~80% of cells did
not exhibit any DNA damage in PJ34 untreated group compared to 55%
in PJ34 treated group. The data also showed a significant increase
(P=0.018) in Olive tail moment for PJ34 treated group compared to
the control one, and the length of comet tail was 2.40-fold
increased. The results suggested that the DNA damage might be
corrected by natural repair mechanisms after incubation without
PJ34 and PJ34 was able to enhance DNA damage induced by melphalan
in RPMI8226/R cells.
| Table IVThe effects of PJ34 on DNA damage in
RPMI8226/R cells examined by the comet assay. |
Table IV
The effects of PJ34 on DNA damage in
RPMI8226/R cells examined by the comet assay.
| Group | Tail length | Comet length | Tail moment | Olive tail
moment | Comet-like cell
(%) |
|---|
| RPMI8226/R | 12.97±3.31 | 40.81±8.97 | 1.53±0.40 | 1.89±0.37 | 20.27±2.22 |
|
RPMI8226/R+PJ-34 | 31.08±10.13 | 66.39±12.81 | 7.58±4.70 | 6.35±3.02 | 45.38±2.91 |
| P-value | 0.01 | 0.014 | 0.027 | 0.018 | 0.003 |
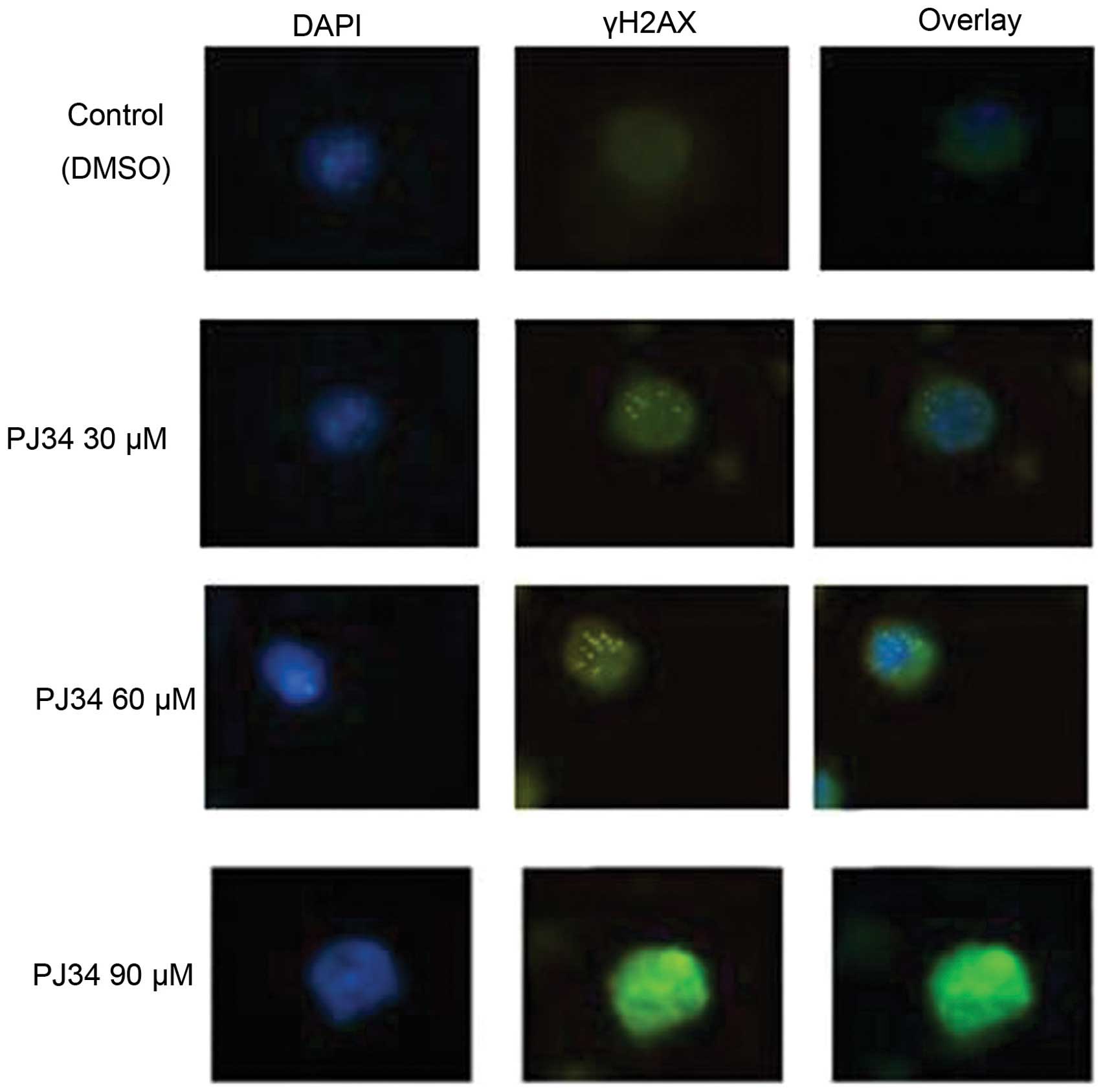
PJ34 induces the formation of γH2AX foci
and blocks the repair of melphalan-induced DNA damage in RPMI8226/R
cells
Among the many factors that may affect cell
sensitivity to DNA damage, DNA repair is the most critical one.
Histone H2AX phosphorylation (γH2AX) forms nuclear foci that can be
visualized by immunostaining using anti-γH2AX antibody. These foci
correspond 1:1 with unrepaired DSBs. So the presence of γH2AX
nuclear foci is regarded as a sensitive indicator for the presence
of DSB in chromosomal DNA, and the number of γH2AX foci is believed
to be closely related to the number of DSB in the cells. To assess
the role of PJ34 in DSB repair, we used immunofluorescence analysis
and quantification of γH2AX positive RPMI8226/R cells after
exposure to various concentrations of PJ34 combined with melphalan
(9.6 μM) for 24 h. It was found that although majority of the
control DMSO-treated cells had no γH2AX foci in the nuclei, there
were still around 15% of cells containing more than 10 foci. On the
other hand, various concentrations of PJ34 all induced increases in
the percentage of γH2AX positive cells (Table V). As shown in Fig. 5 and Table V, the percentage of γH2AX positive
cells increased significantly after exposure to increased
concentrations of PJ34 combined with melphalan (P<0.05),
indicating that there is more DNA damage. The percentage of γH2AX
positive cells decreased in control group with melphalan treatment
alone revealed that DSBs were repaired with time, while there was
significantly higher number of residual γH2AX foci in RPMI8226/R
cells when PJ34 was added with melphalan (Table V). PJ34 alone did not induce γH2AX
(data not shown). To further confirm the role of PJ34 in DSB
repair, we also evaluated the effects of PJ34 on γH2AX protein
levels after melphalan treatment in RPMI8226/R cells by western
blotting. As shown in Fig. 2,
after exposure to 60 μM of PJ34, the RPMI8226/R and RPMI8226 cells
both retained higher levels of γH2AX than controls. Moreover, after
exposure to 90 μM of PJ34, a sub-population of cells even displayed
high levels of uniform pan-nuclear γH2AX staining without
identifiable γH2AX foci (Fig. 2).
These data strongly supported that PJ34 slowed down the repair of
DSB.
| Table VQuantification of γH2AX foci
formation in RPMI8226/R cells. |
Table V
Quantification of γH2AX foci
formation in RPMI8226/R cells.
| Group | γH2AX positive
cells (%) |
|---|
| 1.
Melphalan+DMSO | 15.1±1.4 |
| 2. Melphalan+PJ34
30 μM | 26.5±3.3 |
| 3. Melphalan+PJ34
60 μM | 53.4±5.6 |
| 4. Melphalan +PJ34
90 μM | 64.3±6.4 |
Our results suggested that the effect of melphalan
on initial DNA damage was enhanced by PJ34 in RPMI8226/R cells.
Despite the more rapid removal of the DSB, more DSBs remained in
RPMI8226/R cells due to the excessive level of initial DSB. The net
outcome was that RPMI8226/R cells consistently bear more DNA damage
after 24-h treatment with melphalan combined with PJ34, which
likely contributed to the increased cellular sensitivity to
melphalan.
Taken together, all the above results indicated that
the PARP-1 inhibitor PJ34 increased melphalan-induced activation of
the FA/BRCA pathway and caused persistent melphalan-induced DNA
damage.
Discussion
MM accounts for ~10% of hematologic malignant
tumors. At first MM patients respond well to alkylating agent-based
chemotherapy regimens, but almost all patients will relapse after
achieving a degree of remission, even those with complete remission
(CR). Repeated chemotherapies are more likely to cause multidrug
resistance, MDR will result in refractory multiple myeloma. In
recent years, although the targeted therapeutic drugs (thalidomide,
lenalidomide and bortezomib) have made certain curative effects in
cancer clinical treatment, there are still some patients who
relapse or die within a year after therapy. Currently autologous
hematopoietic stem cell transplantation (Auto-HSCT) can improve
survival rate in patients with MM, but most of the patients due to
old age are not suitable for Auto-HSCT. Therefore, looking for more
effective chemotherapy drugs, improving the curative effects of
advanced stage patients, is still the topic needing further
exploration.
The alkylating agent-based chemotherapy regimens
play important roles in treatment of MM, multidrug resistance is
the main impact factor for the survival of patients with MM.
Research has suggested that the mechanism of chemotherapy drug
resistance mainly includes: increased drug efflux, changes in drug
resistance related genes, enhanced DNA damage repair and suppressed
apoptosis (38,39). Among them, DNA damage repair is a
hotspot of research on the alkylating agent resistance mechanism
and MM individualized treatment. The FA/BRCA pathway plays an
important role in the regulation of DNA damage repair, it can
adjust intracellular reactions to DDP and other alkylating agents
(14–23). We speculate that the FA/BRCA
pathway may be associated with the chemotherapy drug resistance.
Recent research of MDR is focused on searching for potential
targets for chemical sensitization agents. Many agents have been
confirmed as inhibitors of MDR, but the effective one without toxic
side effects has not been found yet. Thus, we need to continue
seeking for MDR inhibitors with low toxicity. PARP-1 inhibitor PJ34
is such a candidate drug, its antitumor activity is known, and it
can reverse drug resistance by infiltration and enhanced gathering
the target cells (35,36,40–42).
As in our previous study (37), we proposed that the activation of
FA/BRCA pathway might contribute to enhanced DNA ICL repair
capacity in drug-resistant cells, this pathway should contribute to
the acquired drug resistance of tumor cells to DNA crosslinking
agents, so the FA/BRCA pathway represents a new target for
preventing acquired drug resistance. It is our intention to further
enhance the effectiveness of chemotherapy in multidrug-resistant MM
patients by identifying other relevant drugs that synergize with
alkylating agents. Based on previous reports that the PARP-1
inhibitors can exert synergism with various conventional
chemotherapeutic agents including cisplatin, cyclophosphamide,
temozolomide and oxaliplatin in tumors (31–34),
we reasoned that the PARP-1 inhibitor PJ34 in conjunction with
melphalan, might enhance antitumor effect of both agents in
multidrug resistant MM cells. To test this hypothesis, we performed
a synergy test for the PJ34 and melphalan and found a significant
enhancement in the antitumor effect compared with that of either
agent as a single treatment. Our results demonstrated that when
used in combination to treat multidrug resistant RPMI8226/R cells,
the IC50 of melphalan could be reduced by 2.6-fold. Flow
cytometry also showed that combination treatment of PJ34 and
melphalan caused a marked increase in apoptosis. Western blot
analysis and RT-PCR demonstrated that the exposure of RPMI8226/R
cells to combined treatment induced a marked decrease in FANCD2,
BRCA2 and Rad51 protein and mRNA expression levels, which causing
downregulation of the activity of the FA/BRCA pathway, suggesting
the suppression of this signaling pathway by PJ34 and melphalan
co-treatment. Moreover, significant differences in DNA damage
repair were observed between treatment with melphalan alone and
treatment with melphalan and PJ34 in RPMI8226/R cells, as observed
using the comet assay and γH2AX foci analysis, which demonstrated
that the degree of DNA damage caused by melphalan. Histone H2AX is
the sensor of DNA damage, one of the early occurred events after
DNA DSBs is the formation of phosphorylated histone H2AX (γH2AX),
γH2AX is the gold standard for detecting DSB (43). Therefore, the level of γH2AX can
reflect the DNA damage caused by chemotherapy drugs in tumor cells,
and also can be the index of chemotherapy sensitivity. The present
study also found that the protein expression level of γH2AX
increased after exposure to PJ34 in RPMI8226/R cells, indicating
increasing DSBs and enhanced drug sensitivity. Our results
demonstrate for the first time that PJ34 can decrease the
expression of the factors in the FA/BRCA pathway upregulated by
melphalan in multidrug resistant cell line RPMI8226/R and indicate
PJ34 can sensitize MDR cells with low concentration of melphalan,
thus PJ34 can be used to treat refractory mutidrug resistant MM
patients with low concentrations of melphalan.
Rad51 is the key recombination protein promoting the
pairing and exchange of strands between homologous DNA molecules
during HRR (homologous recombination repair) (44). Rad51 interacts with many accessory
proteins in the FA/BRCA pathway and is involved in the repair of
DNA cross-links (9,10). Increased levels of Rad51 correlate
with increased erroneous recombination and resistance to
DNA-damaging agents in tumor cells (45). Rad51 overexpression leads to a
worse clinical outcome in lung cancer (46). In the present study, PJ34 enhanced
melphalan-induced cytotoxicity via downregulation of Rad51
expression in human multidrug resistant RPMI8226/R cells. Our study
has shown that the upregulation of Rad51 expression via the FA/BRCA
pathway in melphalan-treated RPMI8226/R cells was required for cell
survival and downregulation of Rad51 expression by PJ34 resulted in
enhancement of melphalan sensitivity in resistant RPMI8226/R cells.
However, a recent report showed that an MYC-dependent decrease in
Rad51 expression is not sufficient to sensitize cells to MMC in
H1299 lung cancer cells (47).
Therefore, we suppose that Rad51 must be regulated together with
other factors in the FA/BRCA pathway to maintain genomic stability
in cells responding to DNA damage.
The sensitivity of cancer cells to chemotherapeutic
drug-induced DNA damage depends on the balance between DNA damage
and repair. Therefore, targeting of DNA repair that promote cell
survival is proposed as a promising strategy to enhance the
efficacy of conventional chemotherapeutic agents. One signaling
pathway that has recently drawn much attention for this purpose is
the FA/BRCA pathway and abnormal activation of this pathway has
been reported to play an important role in chemoresistance in a
variety of tumors (11–23). These findings ignited enthusiasm
for targeting DNA repair pathway as an anticancer modality and
provide a superior strategy for overcoming development of
resistance of cancer cells to targeted therapy. We found that PJ34
sensitized the multidrug resistant RPMI8226/R cells to melphalan
and the basal levels of DSB repair proteins γ-H2AX increased when
cells were treated with PJ34 which inhibited the FA/BRCA pathway
activity. Thus, the possible mechanism is PJ34 might interfere with
the DNA repair process by the suppression of the FA/BRCA pathway
and cause damaged DNA to accumulate by slowing down the removal of
DNA damage induced by melphalan, resulting in improved sensitivity
to melphalan.
It has been reported that activation of the FA/BRCA
pathway is supposed to be a resistance mechanism in alkylating
agent-based chemotherapy (14–16)
and PARP-1 inhibitors can improve the antitumor effect of
alkylating agents (31–34). Thus, our findings suggest that
concomitant targeting of the FA/BRCA pathway is a promising
strategy to enhance anti-tumor effect of PJ34 in MM patients.
Although our present study has several limitations
such as in vitro nature of the design and only one cell line
tested, it demonstrated synergistic interaction between PJ34 and
melphalan in multidrug resistant human MM cells. While PJ34 by
itself showed only a limited antitumor effect, it synergistically
potentiated melphalan-mediated apoptosis and DNA damage with
activation of the FA/BRCA pathway in multi-drug resistant
RPMI8226/R cells. These findings suggest that targeting of DNA
repair pathway can be a promising strategy for the patients with
multidrug resistant multiple myeloma. Further comprehensive
molecular studies should be performed to test the safety and in
vivo synergistic antitumor effect of PJ34 and melphalan
combination therapy for the clinical application in multidrug
resistant multiple myeloma.
Acknowledgements
We thank Professor Jianfeng Zhou for providing the
human multiple myeloma cell line RPMI8226. The present study was
supported by the National Natural Science Foundation of China
(grant no. 81001053) and the Fundamental Research Funds for the
Central Universities (grant No. 4101041).
References
|
1
|
Spanswick VJ, Lowe HL, Newton C, et al:
Evidence for different mechanisms of ‘unhooking’ for melphalan and
cisplatin-induced DNA interstrand cross-links in vitro and in
clinical acquired resistant tumour samples. BMC Cancer. 12:4362012.
View Article : Google Scholar
|
|
2
|
Bouwman P and Jonker J: The effects of
deregulated DNA damage signalling on cancer chemotherapy response
and resistance. Nat Rev Cancer. 12:587–598. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Altieri F, Grillo C, Maceroni M, et al:
DNA damage and repair: from molecular mechanisms to health
implications. Antioxid Redox Signal. 10:891–937. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hlavin EM, Smeaton MB and Miller PS:
Initiation of DNA interstrand cross-link repair in mammalian cells.
Environ Mol Mutagen. 51:604–624. 2010.PubMed/NCBI
|
|
5
|
Bessho T: Induction of DNA
replication-mediated double strand breaks by psoralen DNA
interstrandcross-links. J Biol Chem. 278:5250–5254. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sczepanski JT, Jacobs AC, Van Houten B, et
al: Double-strand break formation during nucleotide excision repair
of a DNA interstrand cross-link. Biochemistry. 48:7565–7567. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim H and D’Andrea AD: Regulation of DNA
cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev.
26:1393–1408. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mouw KW and D’Andrea AD: Crosstalk between
the nucleotide excision repair and Fanconi anemia/BRCA pathways.
DNA Repair. 19:130–134. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Levitus M, Joenje H and de Winter JP: The
Fanconi anemia pathway of genomic maintenance. Cell Oncol. 28:3–29.
2006.PubMed/NCBI
|
|
10
|
Cohn MA and D’Andrea AD: Chromatin
recruitment of DNA repair proteins: lessons from the fanconi anemia
and double-strand break repair pathways. Mol Cell. 32:306–312.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rodríguez A, Sosa D, Torres L, et al: A
Boolean network model of the FA/BRCA pathway. Bioinformatics.
28:858–866. 2012.
|
|
12
|
Karanja KK, Cox SW, Duxin JP, et al: DNA2
and EXO1 in replication-coupled, homology-directed repair and in
the interplay between HDR and the FA/BRCA network. Cell Cycle.
11:3983–3996. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Stecklein SR and Jensen RA: Identifying
and exploiting defects in the Fanconi anemia/BRCA pathway in
oncology. Transl Res. 160:178–197. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jacquemont C, Simon JA, D’Andrea AD, et
al: Non-specific chemical inhibition of the Fanconi anemia pathway
sensitizes cancer cells to cisplatin. Mol Cancer. 11:262012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chirnomas D, Taniguchi T, de la Vega M, et
al: Chemosensitization to cisplatin by inhibitors of the Fanconi
anemia/BRCA pathway. Mol Cancer Ther. 5:952–961. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Taniguchi T and D’Andrea AD: Molecular
pathogenesis of Fanconi anemia: recent progress. Blood.
107:4223–4233. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mori R, Yoshida K, Tanahashi T, et al:
Decreased FANCJ caused by 5FU contributes to the increased
sensitivity to oxaliplatin in gastric cancer cells. Gastric Cancer.
16:345–354. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Deans AJ and West SC: DNA interstrand
crosslink repair and cancer. Nat Rev Cancer. 11:467–480. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hegi ME, Sciuscio D, Murat A, et al:
Epigenetic deregulation of DNA repair and its potential for
therapy. Clin Cancer Res. 15:5026–5031. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Palagyi A, Neveling K, Plinninger U, et
al: Genetic inactivation of the Fanconi anemia gene FANCC
identified in the hepatocellular carcinoma cell line HuH-7 confers
sensitivity towards DNA-interstrand crosslinking agents. Mol
Cancer. 9:1272010. View Article : Google Scholar
|
|
21
|
Burkitt K and Ljungman M: Phenylbutyrate
interferes with the Fanconi anemia and BRCA pathway and sensitizes
head and neck cancer cells to cisplatin. Mol Cancer. 7:242008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kondo N, Takahashi A, Mori E, et al:
FANCD1/BRCA2 plays predominant role in the repair of DNA damage
induced by ACNU or TMZ. PLoS One. 6:e196592011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yarde DN, Oliveira V, Mathews L, et al:
Targeting the Fanconi anemia/BRCA pathway circumvents drug
resistance in multiple myeloma. Cancer Res. 69:9367–9375. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Adhikari S, Choudhury S, Mitra PS, et al:
Targeting base excision repair for chemosensitization. Anticancer
Agents Med Chem. 8:351–357. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
David KK, Andrabi SA, Dawson TM, et al:
Parthanatos, a messenger of death. Front Biosci. 14:1116–1128.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang Y, Dawson VL and Dawson TM:
Poly(ADP-ribose) signals to mitochondrial AIF: a key event in
parthanatos. Exp Neurol. 218:193–202. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bryant HE, Petermann E, Schultz N, et al:
PARP is activated at stalled forks to mediate Mre11-dependent
replication restart and recombination. EMBO J. 28:2601–2615. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Krishnakumar R and Kraus WL: The PARP side
of the nucleus: molecular actions, physiological outcomes, and
clinical targets. Mol Cell. 39:8–24. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Węsierska-Gądek J, Zulehner N, Ferk F, et
al: PARP inhibition potentiates the cytotoxic activity of C-1305, a
selective inhibitor of topoisomerase II, in human BRCA1-positive
breast cancer cells. Biochem Pharmacol. 84:1318–1331.
2012.PubMed/NCBI
|
|
30
|
Lavarone E, Puppin C, Passon N, et al: The
PARP inhibitor PJ34 modifies proliferation, NIS expression and
epigenetic marks in thyroid cancer cell lines. Mol Cell Endocrinol.
365:1–10. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Davidson D, Wang Y, Aloyz R, et al: The
PARP inhibitor ABT-888 synergizes irinotecan treatment of colon
cancer cell lines. Invest New Drugs. 31:461–468. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cheng H, Zhang Z, Borczuk A, et al: PARP
inhibition selectively increases sensitivity to cisplatin in
ERCC1-low non-small cell lung cancer cells. Carcinogenesis.
34:739–749. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cavallo F, Graziani G, Antinozzi C, et al:
Reduced proficiency in homologous recombination underlies the high
sensitivity of embryonal carcinoma testicular germ cell tumors to
cisplatin and poly (ADP-ribose) polymerase inhibition. PLoS One.
7:e515632012. View Article : Google Scholar
|
|
34
|
Donawho CK, Luo Y, Luo Y, et al: ABT-888,
an orally active poly(ADP-ribose) polymerase inhibitor that
potentiates DNA-damaging agents in preclinical tumor models. Clin
Cancer Res. 13:2728–2737. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ahel I, Ahel D, Matsusaka T, et al:
Poly(ADP-ribose)-binding zinc finger motifs in DNA
repair/checkpoint proteins. Nature. 451:81–85. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pyriochou A, Olah G, Deitch EA, et al:
Inhibition of angiogenesis by the poly(ADP-ribose) polymerase
inhibitor PJ-34. Int J Mol Med. 22:113–118. 2008.PubMed/NCBI
|
|
37
|
Xiao H, Xiao Q, Zhang K, et al: Reversal
of multidrug resistance by curcumin through FA/BRCA pathway in
multiple myeloma cell line MOLP-2/R. Ann Hematol. 89:399–404. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Vinod BS, Maliekal TT and Anto RJ:
Phytochemicals as chemosensitizers: from molecular mechanism to
clinical significance. Antioxid Redox Signal. 18:1307–1348. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tomicic MT and Kaina B: Topoisomerase
degradation, DSB repair, p53 and IAPs in cancer cell resistance to
camptothecin-like topoisomerase I inhibitors. Biochim Biophys Acta.
1835:11–27. 2013.PubMed/NCBI
|
|
40
|
Liu X, Luo X, Shi Y, et al: Poly
(ADP-ribose) polymerase activity regulates apoptosis in HeLa cells
after alkylating DNA damage. Cancer Biol Ther. 7:934–941. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gambi N, Tramontano F and Quesada P:
Poly(ADPR) polymerase inhibition and apoptosis induction in
cDDP-treated human carcinoma cell lines. Biochem Pharmacol.
75:2356–2363. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yang YG, Cortes U, Patnaik S, et al:
Ablation of PARP-1 does not interfere with the repair of DNA
double-strand breaks, but compromises the reactivation of stalled
replication forks. Oncogene. 23:3872–3882. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mah LJ, Orlowski C, Ververis K, et al:
Utility of γH2AX as a molecular marker of DNA double-strand breaks
in nuclear medicine: applications to radionuclide therapy employing
auger electron-emitting isotopes. Curr Radiopharm. 4:59–67. 2011.
View Article : Google Scholar
|
|
44
|
Richardson C: RAD51, genomic stability,
and tumorigenesis. Cancer Lett. 218:127–139. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Klein HL: The consequences of Rad51
overexpression for normal and tumor cells. DNA Repair. 7:686–693.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Qiao GB, Wu YL, Yang XN, et al: High-level
expression of Rad51 is an independent prognostic marker of survival
in non-small-cell lung cancer patients. Br J Cancer. 93:137–143.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Luoto KR, Meng AX, Wasylishen AR, et al:
Tumor cell kill by c-MYC depletion: role of MYC-regulated genes
that control DNA double-strand break repair. Cancer Res.
70:8748–8759. 2010. View Article : Google Scholar : PubMed/NCBI
|