Introduction
The decapeptide gonadotropin-releasing hormone
(GnRH, Glp-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2) was
first identified as the key regulator of the pituitary-gonadal axis
(1). It is synthesized in the
hypothalamic neurons and then released, in a pulsatile manner, into
the hypophyseal portal circulation to reach the anterior pituitary.
By binding to its specific receptors (GnRH-R) on the pituitary
gonadotropes, it stimulates the synthesis/release of the two
gonadotropins and, subsequently, the steroid production from the
gonads (2,3).
In order to be activated, pituitary GnRH-R require
pulsatile stimulation by GnRH. Moreover, it has been found that
their sustained stimulation by GnRH agonists, after an initial
flare event, leads to desensitization, with the consequent
suppression of gonadotropin and steroid secretion. On the basis of
this activity, GnRH agonists are successfully used for the
treatment of different hormone-related pathologies, such as
steroid-dependent tumors (prostate, breast and endometrial cancer)
(4,5).
It is now well established that GnRH-R are also
expressed in different human cancer cells and tissues, where they
are associated with a strong antitumor activity (antiproliferative/
antimetastatic/antiangiogenic) (6–10).
These data indicate that, when utilized for the treatment of
hormone-dependent cancers, in addition to their activity at the
pituitary level, GnRH analogs may also exert a direct antitumor
effect on cancer cells.
More recently, tumor GnRH-R have been considered as
a promising molecular target for novel treatment strategies.
Specifically, on the basis of their binding to the GnRH-R expressed
on cancer cells, GnRH derivatives could be employed not only as
anticancer agents, but could also serve as targeting moieties to
deliver chemotherapeutic agents directly to cancer cells. This
targeted drug delivery can be achieved by attaching a
chemotherapeutic drug to a GnRH analog, either directly or through
a chemically or enzymatically cleavable spacer. After binding to
the GnRH receptors on cancer cells, the bioconjugate is
internalized by receptor-mediated endocytosis and then processed at
the lysosomal level, leading to the release of the free drug or to
the formation of drug containing metabolites that further exert a
cytotoxic effect. Thus, these cytotoxic GnRH-based bioconjugates
act as drug delivery systems, specifically affecting the
proliferation of cancer cells expressing the GnRH-R, which is the
case of the majority of cancer cells, while sparing non-cancerous
cells from the unnecessary exposure (11). The major advantage of this targeted
chemotherapeutic approach is the increased selectivity of cytotoxic
agents as well as their decreased peripheral toxicity. A crucial
feature to be possessed by these compounds is their stability in
the blood circulation in order to prevent an early drug release
before targeting the cancer cells.
Cytotoxic GnRH-based bioconjugates were first
developed in Schally’s laboratory. The most potent compounds
consisted of the GnRH derivative [D-Lys6]-GnRH to which
the chemotherapeutic agents doxorubicin (Dox) (AN-152, also known
as AEZS-108) or 2-pyrrolino-Dox (AN-207) (11) were attached via an ester bond,
through a glutarate spacer.
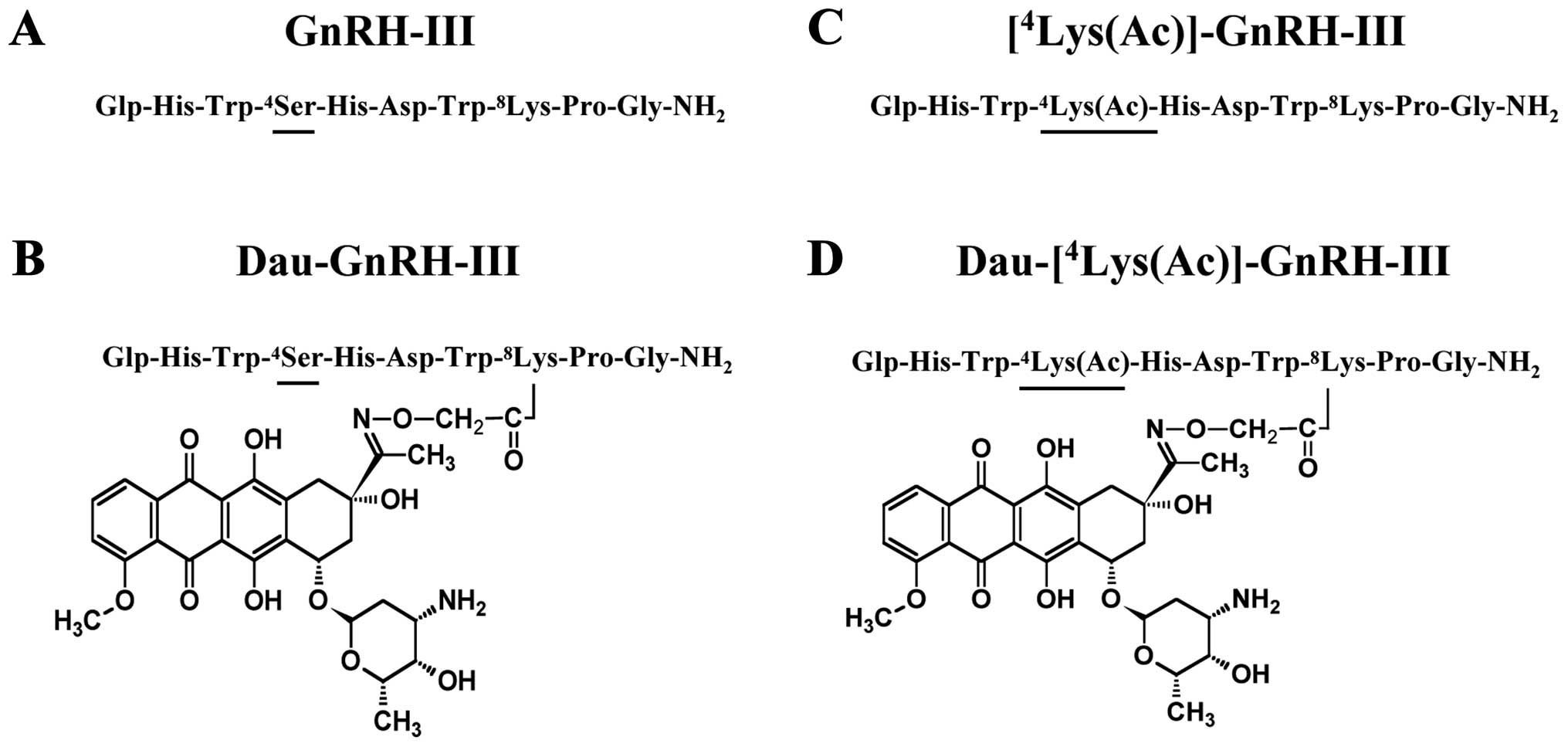
In addition to the classical GnRH, another isoform
of the neurohormone, designed as GnRH-III, has been discovered.
GnRH-III is a decapeptide
(Glp-His-Trp-Ser-His-Asp-Trp-Lys-Pro-Gly-NH2, Glp is
pyroglutamic acid) isolated from sea lamprey (Petromyzon
marinus) that has 60% homology with GnRH, with four different
amino acids in positions 5–8 (Fig.
1). This GnRH isoform has lower potency than GnRH in
stimulating gonadotropin secretion at the pituitary level and it
exerts an antiproliferative effect on many types of cancer cells
expressing the GnRH-R (12). On
the basis of these features, GnRH-III derivatives are considered
suitable tumor targeting moieties for the preparation of anticancer
drug delivery systems. Recently, a panel of anthracycline-GnRH-III
derivative bioconjugates has been designed, synthesized and
biochemically characterized (13–16).
The most promising cytotoxic compounds contained the anticancer
drug daunorubicin (Dau) attached via an oxime bond to the Lys in
position 8 of GnRH-III. Two of them, [4Ser,
8Lys(Dau=Aoa)]-GnRH-III (here denoted as Dau-GnRH-III),
in which daunorubicin was coupled to the 8Lys in the
native form of GnRH-III, and [4Lys(Ac),
8Lys(Dau=Aoa)]-GnRH-III (here denoted as
Dau-[4Lys(Ac)]-GnRH-III, in which daunorubicin was
attached to the 8Lys of a GnRH-III derivative in which
4Ser was replaced by an acetylated lysine
[4Lys(Ac)] (Fig. 1) are
of particular interest. They have been shown to exert significant
in vitro cytostatic effect on cancer cells expressing
GnRH-R, such as breast, colon and androgen-dependent prostate
cancer cells and to be stable in human serum at least for 24 h. In
the presence of digestive enzymes, in particular chymotrypsin,
Dau-[4Lys(Ac)]-GnRH-III proved to be more stable. This
compound also exerted significant in vivo tumor growth
inhibitory effect on colon carcinoma-bearing mice (14). Both bioconjugates were degraded by
lysosomal enzymes, and H-Lys(Dau=Aoa)-OH was identified as the
smallest drug-containing metabolite produced in the presence of rat
liver lysosomal homogenate. This metabolite was able to bind to DNA
in vitro, a result that could contribute to the
understanding of the antitumor effect of the bioconjugates
(14,15).
It is hypothesized that these GnRH-III bioconjugates
might exert their antitumor activity through the classical form of
the GnRH-R; however, this issue has not been addressed in previous
studies.
Prostate cancer is the most commonly diagnosed
cancer for men and the second leading cause of cancer-related
deaths among men in Western countries (17). Prostate cancer patients can be
initially cured by radiotherapy or surgery; however, most of them
will experience disease progression. In this phase, in which most
tumors are dependent on the presence of androgens,
androgen-deprivation therapy, aimed at blocking androgen
secretion/activity, represents the most effective treatment
(5). This therapy includes
chemical castration, which can be achieved by GnRH agonists, given
either alone or in combination with an antiandrogen (5,18).
However, after an excellent initial response, relapse occurs in
most patients within a median of 2–3 years and the tumor progresses
towards a condition of resistance to castration
[castration-resistant prostate cancer (CRPC)]. For CRPC patients
the therapeutic options are still very limited, since taxane-based
(i.e., docetaxel) treatment can usually provide a progression-free
survival of a few months (19,20).
In Limonta’s laboratory it has been widely
demonstrated that GnRH receptors are expressed in CRPC cells (DU145
and PC3) and that their activation by means of GnRH agonists
significantly reduces cancer cell proliferation and metastatic
behavior (21–25). In line with these observations,
GnRH-R have also been reported to be expressed in prostate cancer
biopsies from patients who developed CRPC after orchiectomy and
antiandrogen therapy (26,27). These data strongly support the
notion that the GnRH-R can represent an effective target for
cytotoxic GnRH-based bioconjugates also in CRPC cells.
In the study reported here, we aimed to investigate
whether the two oxime bond-linked daunorubicin-GnRH-III derivative
bioconjugates, Dau-GnRH-III and Dau-[4Lys(Ac)]-GnRH-III,
might affect the growth of castration-resistant DU145 and PC3
prostate cancer cells to elucidate the involvement of the classical
GnRH-R (type I GnRH-R) in this antitumor activity.
Our results show that both bioconjugates
significantly decrease CRPC cell proliferation and that this effect
is completely counteracted by a classical GnRH-R antagonist
(Antide) as well as by silencing the type I GnRH-R.
Materiald and methods
Materials
The GnRH agonist Zoladex [Goserelin acetate,
D-Ser(tBu)6Aza-Gly10-GnRH] was kindly
provided by AstraZeneca Pharmaceuticals (Macclesfield, Cheshire,
UK). The GnRH antagonist Antide (A8802) and MTT
[3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide]
(M5655) were purchased from Sigma-Aldrich (Milan, Italy).
All amino acid derivatives,
benzotriazole-1-yloxytris-pyrrolidinophosphonium-hexafluoro-phosphate
(PyBOP), Bis-Boc-aminooxyacetic acid (Bis-Boc-Aoa-OH) and
Rink-Amide MBHA resin were purchased from NovaBiochem
(Läufelfingen, Switzerland). Scavengers, coupling agents and
cleavage reagents [triisopropylsilane (TIS), 4-methylmorpholine
(NMM), piperidine, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU),
trifluoroacetic acid (TFA)] were obtained from Sigma-Aldrich
(Taufkirchen, Germany). Solvents for peptide synthesis and
purification were purchased from Acros Organics (Geel, Belgium) and
Riedel deHaën (Seelze, Germany). All reagents and solvents were of
analytical grade or highest available purity.
Synthesis and chemical characterization
of GnRH-III and [4Lys(Ac)]-GnRH-III peptides and of
daunorubicin containing bioconjugates
The peptides GnRH-III
(Glp-His-Trp-Ser-His-Asp-Trp-Lys-Pro-Gly-NH2) and
[4Lys(Ac)]-GnRH-III
[Glp-His-Trp-Lys(Ac)-His-Asp-Trp-Lys-Pro-Gly-NH2] were
prepared by solid phase synthesis on a Rink-Amide MBHA resin,
according to Fmoc/tBu chemistry. For the synthesis of daunorubicin
containing bioconjugates, a combination of solid phase peptide
synthesis and chemoselective ligation in solution (oxime bond
formation) was employed. For this purpose, the aminooxyacetylated
derivatives of GnRH-III were first prepared:
Glp-His-Trp-Ser-His-Asp-Trp-Lys(Aoa)-Pro-Gly-NH2 and
Glp-His-Trp-Lys(Ac)-His-Asp-Trp-Lys(Aoa)-Pro-Gly-NH2
(Aoa, aminooxyacetyl). Previously reported synthetic protocols were
used (14,15), with the following modifications: i)
the removal of the Fmoc protecting group with 2% piperidine, 2% DBU
in DMF was performed for a shorter time (3+7 min) in order to avoid
the succinimide formation from 6Asp; ii) the acetylation
of the ɛ-amino group of 4Lys was not performed on the
resin as previously published; the Fmoc-Lys(Ac)-OH was incorporated
during the solid phase peptide synthesis in the presence of PyBOP
and NMM. The amino-oxyacetylation of the ɛ-amino group of
8Lys was performed on the resin using 5 equiv of
Bis-Boc-Aoa/PyBOP/NMM (1:1:2 v/v/v) in DMF for 60 min. After
completion of the synthesis of protected GnRH-III-based peptides,
they were cleaved from the resin, simultaneously with the removal
of the side chain protecting groups, using a mixture of 95% TFA,
2.5% water and 2.5% TIS, for 2.5 h at room temperature. Then, the
peptides were precipitated with ice-cold diethyl ether, washed
three times with diethyl ether and solubilized in 100% acetic acid
prior to freeze drying. The crude products were purified by
semi-preparative RP-HPLC and characterized by mass spectrometry, as
previously published (14,15). The attachment of daunorubicin to
the aminooxyacetyated derivatives of GnRH-III was carried out in
solution (0.2 M sodium acetate, pH 5.0) at a peptide concentration
of 10 μg/μl and using 30% excess of daunorubicin. The bioconjugates
Dau-GnRH-III
[Glp-His-Trp-Ser-His-Asp-Trp-Lys(Dau=Aoa)-Pro-Gly-NH2]
and Dau-[4Lys(Ac)]-GnRH-III
[Glp-His-Trp-Lys(Ac)-His-Asp-Trp-Lys(Dau=Aoa)-Pro-Gly-NH2]
(Fig. 1) were purified by
semi-preparative RP-HPLC and analyzed by ESI-ion trap mass
spectrometry (14,15).
Cell culture
The human DU145 and PC3 prostate cancer cell lines
were purchased from American Type Culture Collection (Rockville,
MD). Cells were grown in RPMI medium (Biochrom KG, Berlin, Germany)
supplemented with 5% (DU145) or 7.5% (PC3) fetal bovine serum (FBS;
Life Technologies, Inc., Paisley, Scotland, UK), glutamine (1 mM)
and antibiotics (100 IU/ml penicillin G sodium and 100 μg/ml
streptomycin sulfate) in a humidified atmosphere of 5%
CO2/95% air at 37°C.
Cell proliferation studies
Preliminary experiments were performed to evaluate
whether the peptides employed as targeting moieties, GnRH-III and
its 4Lys-acetylated derivative, exert a significant
antiproliferative effect on castration-resistant prostate cancer
cells expressing the type I GnRH-R. DU145 cells (5×104
cells/dish) were plated in 100-mm dishes. After 2 days, the cells
were treated with GnRH-III, [4Lys(Ac)]-GnRH-III and
Zoladex (a classical agonist of the type I GnRH-R, utilized as a
control) at a dose of 10−6 M. The treatment was
performed daily, for 6 days, as previously described (21). At the end of the treatments, cells
were harvested and counted using a hemocytometer. Each experimental
group consisted of four replicates, and each experiment was
repeated three times.
Cellular uptake of bioconjugates
determined by fluorescence microscopy
To evaluate the cellular uptake of the two
biocon-jugates, DU145 cells were seeded in 24-well plates in
complete medium. After 24 h of incubation at 37°C, cells were
treated with Dau, Dau-GnRH-III or
Dau-[4Lys(Ac)]-GnRH-III (100 μM) for 3.5 h. Taking
advantage of the autofluorescence properties of daunorubicin at an
excitation wavelength of 488 nm, the cellular uptake of the three
compounds was determined by fluorescence microscopy. At the end of
the incubation period, cells were examined under a Zeiss Axiovert
200 microscope with a ×20/1.4 objective lens linked to a Coolsnap
Es CCD camera (Roper Scientific-Crisel Instruments, Rome,
Italy).
In vitro cytostatic effect of
bioconjugates determined by MTT assay
To evaluate the in vitro cytostatic effect of
Dau-GnRH-III bioconjugates, DU145 cells (3×104
cells/well) were plated in 24-well plates and cultured in 5%
FBS-supplemented medium for 24 h. Cells were then treated with Dau
(10−10-10−6 M, for 48 or 72 h) or with the
bioconjugates Dau-GnRH-III and Dau-[4Lys(Ac)]-GnRH-III
(10−8-10−4 M, for 48 or 72 h). Cells treated
with serum-free medium were used as a control. After treatment, the
MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide]
assay was performed. Thus, MTT was added to each well (final
concentration 0.5 mg/ml) and during 1 h incubation at 37°C purple
crystals were formed by mitochondrial dehydrogenase enzyme present
in the living cells. After that, the supernatant was removed; the
crystals were dissolved in 300 μl of 2-Propanol (33539 Sigma) and
the optical density (OD) was determined at λ=550 using an ELx80
Absorbance Microplate Reader (BioTek, Bedfordshire, UK).
Similar experiments were performed on human
castration-resistant PC3 prostate cancer cells. To elucidate
whether the two daunorubicin containing bioconjugates exert their
cytostatic effect through the activation of the type I GnRH-R,
DU145 cells were treated with Dau-GnRH-III or
Dau-[4Lys(Ac)]-GnRH-III (5×10−6 M, for 48 h),
either in the presence or in the absence of the GnRH antagonist
Antide (a classical antagonist of the type I GnRH-R,
5×10−6 M). The MTT assay was then performed as described
above. Each experimental group consisted of four replicates and
each experiment was repeated three times.
Analysis of caspase-3 activation by
western blotting
DU145 cells were seeded in 100-mm dishes
(5×105 cells/dish). After 24 h, cells were treated with
Dau (5×10−8 M) or with the bioconjugates {Dau-GnRH-III
and Dau-[4Lys(Ac)]-GnRH-III} at a concentration of
10−5 M for 48 or 72 h. At the end of the treatment,
cells (both adherent and floating in the supernatant) were lysed in
RIPA buffer (0.05 M Tris-HCl pH 7.7, 0.15 M NaCl, 0.8% SDS, 10 mM
EDTA, 100 μM NaVO4, 50 mM NaF, 0.3 mM PMSF, 5 mM
iodoacetic acid) containing leupeptin (50 μg/ml), aprotinin (5
μl/ml) and pepstatin (50 μg/ml). Protein concentration was
determined by BCA assay, according to the manufacturer’s
instructions (Thermo Scientific, Rockford, IL). Protein extracts
(30 μg) were resuspended in sample buffer (0.5 M Tris-HCl pH 6.8,
20% glycerol, 10% SDS, 0.2% β-mercaptoethanol, 0.05% bromophenol
blue) and heated at 95°C for 5 min. Following electrophoretic
separation by SDS-PAGE, proteins were transferred onto
nitrocellulose membranes. Membranes were blocked with 5% non-fat
dry milk prior to the overnight incubation at 4°C with either the
rabbit monoclonal caspase-3 antibody (no. 9665, Cell Signaling
Technology, Danvers, MA) or the rabbit monoclonal cleaved caspase-3
antibody (no. 9664, Cell Signaling Technology). Detection was
performed using a horseradish peroxidase-conjugated anti-rabbit
secondary antibody and enhanced chemiluminescence reagents (GE
Healthcare Life Sciences, Milan, Italy).
Actin expression was evaluated as a loading control.
Goat anti-human actin (SC-1616) antibody was from Santa Cruz
Biotechnology (Santa Cruz, CA). Detection was performed using a
horseradish peroxidase-conjugated anti-rabbit secondary antibody
and enhanced chemiluminescence reagents (Supersignal
Chemiluminescence Detection System, Thermo Scientific). The
experiments were repeated three times.
Type I GnRH-R silencing
To investigate the role of the type I GnRH-R in the
cytostatic activity of Dau-GnRH-III derivative bioconjugates, siRNA
experiments were carried out. Type I GnRH-R small interfering RNAs
were purchased from Invitrogen (Burlington, ON, Canada). Their
sequences were as follows: type I GnRH-R-S (5′-UUGCAGAGUAACUCUCCAG
CAUACC-3′) and type I GnRH-R-AS (5′-GGUAUGCUGG AGAGUUACUCUGCAA-3′).
A non-specific scrambled siRNA was used as a control (Medium GC
Duplex, Invitrogen). Transfections were performed using
Lipofectamine 2000 (Invitrogen) as a transfectant (according to the
manufacturer’s instructions). The transfection efficiency was
assessed using the Block-it Alexa fluor red fluorescent oligo and
it was usually >70% of total cells. The effective knockdown of
type I GnRH-R after siRNA transfections was monitored by
RT-PCR.
For this purpose, DU145 cells were seeded in 60-mm
dishes (2×105 cells/dish). After three days, cells were
transfected with type I GnRH-R siRNA or with scrambled siRNA at a
final concentration of 150 nM, for 24 h. Reverse transcription was
performed on 2 μg total RNA, as previously described (25), and cDNA synthesis was performed
using the Gene AMP kit (Perkin-Elmer Cetus, Norwalk, CT) with an
oligo(dt)16 as a primer. PCR was carried out for 30
cycles (95°C for 60 sec, 58°C for 60 sec, 72°C for 120 sec), in the
presence of the following primers: upstream primer,
5′-GACCTTGTCTGGAAAGATCC-3′ (50 pmol) and downstream primer
5′-CAGGCTGATCACCACCATCA-3′ (50 pmol) (25). The amplified cDNA products were
separated on 1.5% agarose gels stained with ethidium bromide and
visualized under UV light. The experiments were repeated three
times.
Cytostatic assays after type I GnRH-R
silencing
DU145 cells were seeded (4.5×104
cells/well) in 24-well plates and after 24 h the cells were
transfected with either type I GnRH-R siRNA or with scrambled
siRNA. After 24 h, cells were treated with Dau-GnRH-III or
Dau-[4Lys(Ac)]-GnRH-III (10−5 M) for 48 h.
The evaluation of cell growth was done by MTT assay, as described
above. Each experimental group consisted of four replicates and
each experiment was repeated three times.
Statistical analysis
Results of proliferation and MTT studies were
analyzed by one-way ANOVA followed by Bonferroni’s test.
Results
Synthesis and chemical characterization
of GnRH-III and [4Lys(Ac)]-GnRH-III peptides and of
daunorubicin containing bioconjugates
The peptides GnRH-III and
[4Lys(Ac)]-GnRH-III were prepared by solid phase peptide
synthesis according to Fmoc/tBu chemistry, purified by RP-HPLC and
analyzed by ESI-ion trap mass spectrometry {GnRH-III:
MW(calc)=1259.35; MW(exp)=1259.10; [4Lys(Ac)]-GnRH-III:
MW(calc)=1342.48; MW(exp)=1342.0}.
The bioconjugates Dau-GnRH-III and
Dau-[4Lys(Ac)]-GnRH-III were synthesized by a
combination of solid phase peptide synthesis and chemoselective
ligation in solution. The oxime bond was formed between the oxo
group at the C-13 position of daunorubicin and the
aminooxyacetylated 8Lys of GnRH-III derivative peptides.
The bioconjugates were purified by RP-HPLC and analyzed by ESI-ion
trap mass spectrometry {Dau-GnRH-III: MW(calc)=1841.91;
MW(exp)=1841.65; Dau - [4Lys(Ac)] - GnRH - III: MW(calc)
=1925.05; MW(exp)=1924.70}.
Antiproliferative effect of GnRH-III and
[4Lys(Ac)]-GnRH-III peptides on DU145 prostate cancer
cells
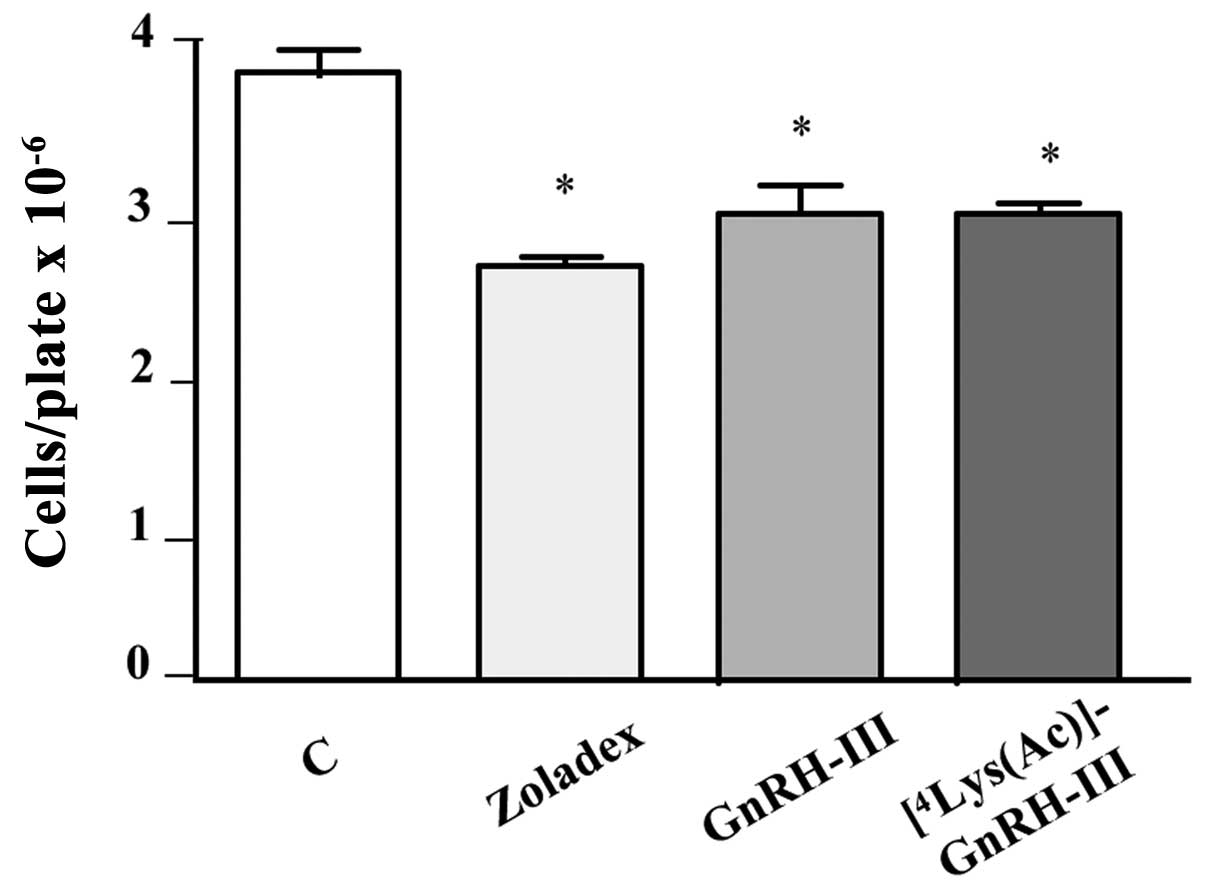
First experiments were performed to evaluate the
antitumor activity, on castration-resistant prostate cancer cells,
of the carrier decapeptides used for the synthesis of daunorubicin
containing bioconjugates. DU145 cells were treated daily, for 6
days, with GnRH-III, [4Lys(Ac)]-GnRH-III or with Zoladex
(a specific agonist of the classical form of GnRH, utilized as a
positive control), at a dose of 10−6 M. At the end of
the treatments, cells were counted using a hemocytometer. As shown
in Fig. 2, both GnRH-III and its
derivative [4Lys(Ac)]-GnRH-III exerted a significant
antiproliferative effect on DU145 cells; in agreement with our
previous data, similar results were obtained after treating the
cells with the GnRH agonist Zoladex (21). When the treatment of the cells with
the three decapeptides was performed for a shorter time interval (3
days) the proliferation of the cells was not affected (data not
shown). These observations are in line with our previous data
demonstrating that, in DU145 cells, GnRH agonists can exert a
significant antiproliferative effect only when the treatments are
performed for long time intervals (4–7 days) (21). The cytostatic activity of GnRH-III
on castration-resistant PC3 prostate cancer cells has previously
been demonstrated (28).
Cellular uptake of Dau-GnRH-III and
Dau-[4Lys(Ac)]-GnRH-III bioconjugates into DU145
cells
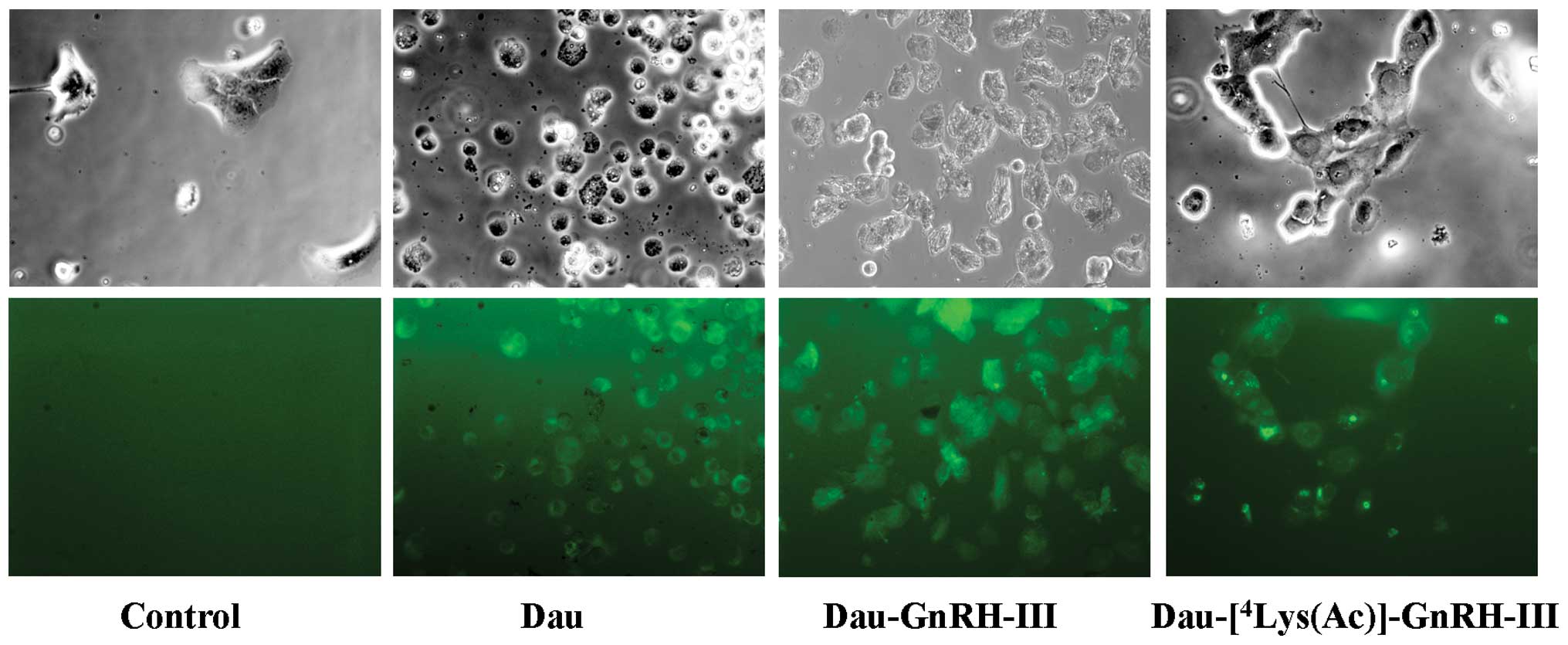
The cellular uptake of the daunorubicin-GnRH-III
derivative bioconjugates was investigated by fluorescence
microscopy, taking advantage of the fluorescence properties of
daunorubicin. DU145 cells were treated with Dau, Dau-GnRH-III or
Dau-[4Lys(Ac)]-GnRH-III, at a concentration of 100 μM,
for 3.5 h. At the end of the treatment, cells were examined under a
Zeiss Axiovert fluorescence microscope. As shown in Fig. 3, cells treated with Dau,
Dau-GnRH-III or Dau-[4Lys(Ac)]-GnRH-III displayed
intense fluorescent staining, indicating that not only Dau but also
the two daunorubicin-GnRH-III bioconjugates are internalized into
the cells.
Cytostatic effect of Dau-GnRH-III and
Dau-[4Lys(Ac)]-GnRH-III bioconjugates on DU145
cells
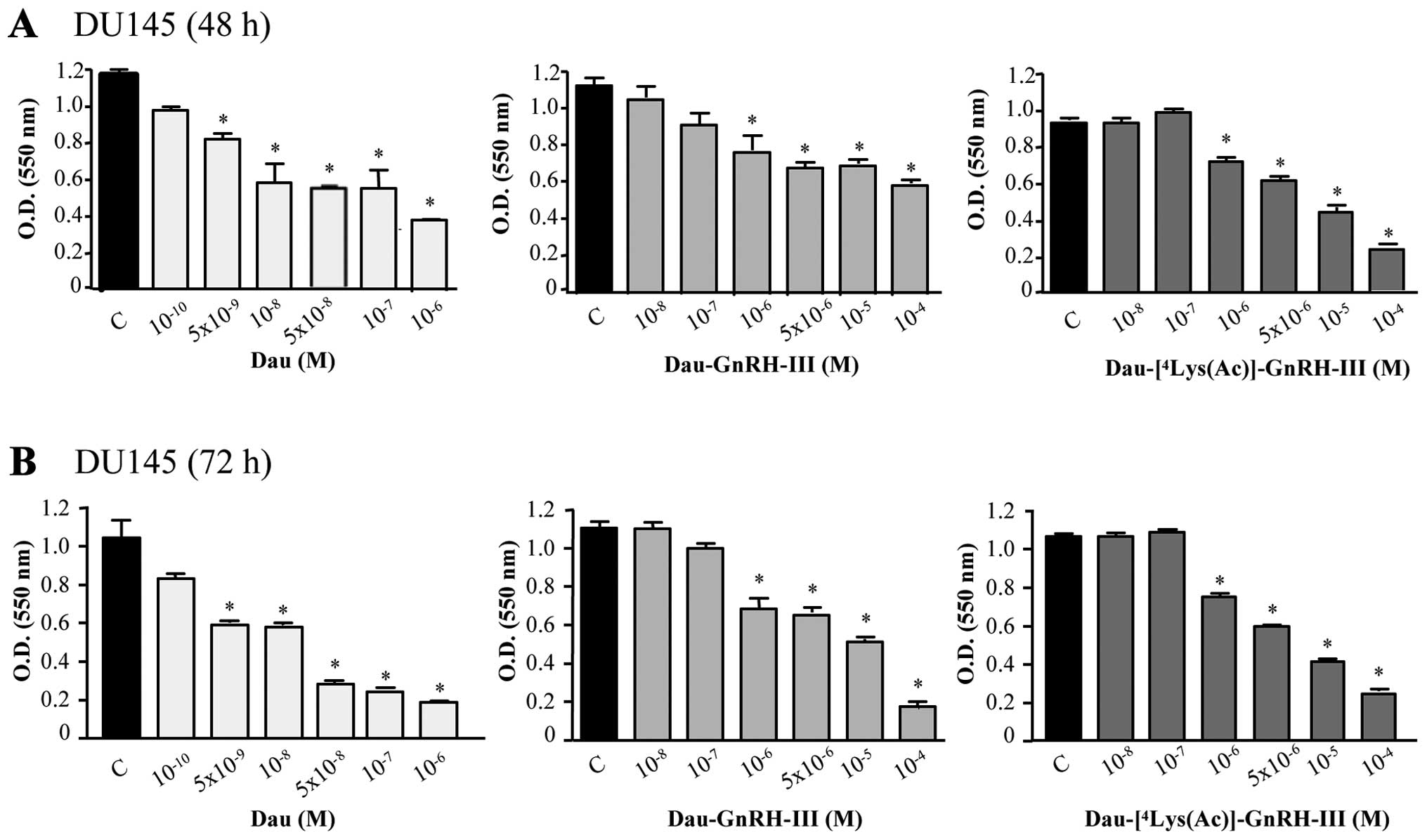
The cytostatic effect of the daunorubicin-GnRH-III
derivative bioconjugates was determined by MTT assay. DU145 cells
were treated with daunorubicin (Dau,
10−10-10−6 M) or with the bioconjugates
Dau-GnRH-III and Dau-[4Lys(Ac)]-GnRH-III
(10−8-10−4 M) for 48 or 72 h. Daunorubicin
exerted a significant and dose-dependent cytostatic effect on DU145
cells even at low doses (5×10−9 M), this result being in
line with the ability of the drug to freely diffuse into the cells.
The two bioconjugates displayed a dose-dependent cytostatic effect
at both tested time intervals; however, at higher doses than those
of free daunorubicin, both at 48 h (Fig. 4A) and at 72 h (Fig. 4B). This provides an indication of
the mechanism of action of the bioconjugates that require binding
to the GnRH receptors before being internalized and further
processed in the cells, at lysosomal level. The IC50
values corresponding to these cytostatic effects at 72 h were as
follows: 0.002±1.45 μM for daunorubicin, 7.18±1.12 μM for
Dau-GnRH-III and 3.796±1.20 μM for
Dau-[4Lys(Ac)]-GnRH-III, respectively. These data
indicate that the replacement of 4Ser by Lys(Ac) led to
an increased cytostatic effect in these cells.
Dau-GnRH-III and
Dau-[4Lys(Ac)]-GnRH-III bioconju-gates induce apoptosis
in DU145 cells
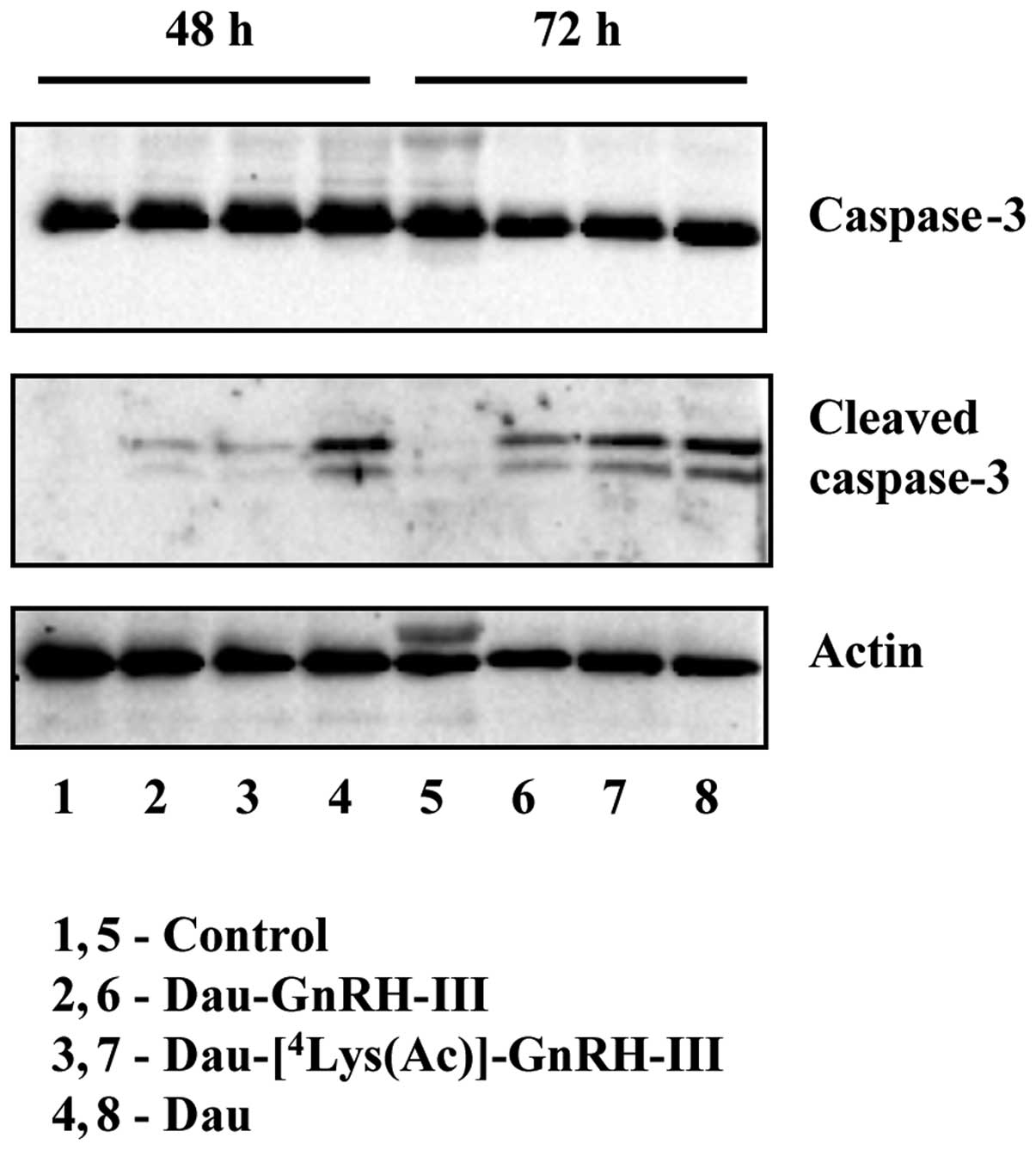
To gain insight into the involvement of apoptosis in
the antitumor activity of Dau-GnRH-III and
Dau-[4Lys(Ac)]-GnRH-III on DU145 cells, we investigated
the effects of the two bioconjugates on the activation of
caspase-3. Cells were treated with free daunoru-bicin
(5×10−8 M), Dau-GnRH-III or
Dau-[4Lys(Ac)]-GnRH-III (10−5 M) for 48 or 72
h. The protein expression of pro-caspase-3 and cleaved (active)
caspase-3 was then assessed by western blotting. As shown in
Fig. 5, the total levels of
pro-caspase-3 did not change throughout the treatments.
Daunorubicin remarkably induced pro-caspase-3 activation at both
time intervals; the two bioconjugates conspicuously increased the
levels of cleaved caspase-3 after 72 h of treatment (Fig. 5).
The antitumor effect of Dau-GnRH-III and
Dau-[4Lys(Ac)]-GnRH-III bioconjugates on DU145 cells is
mediated by the type I GnRH-R
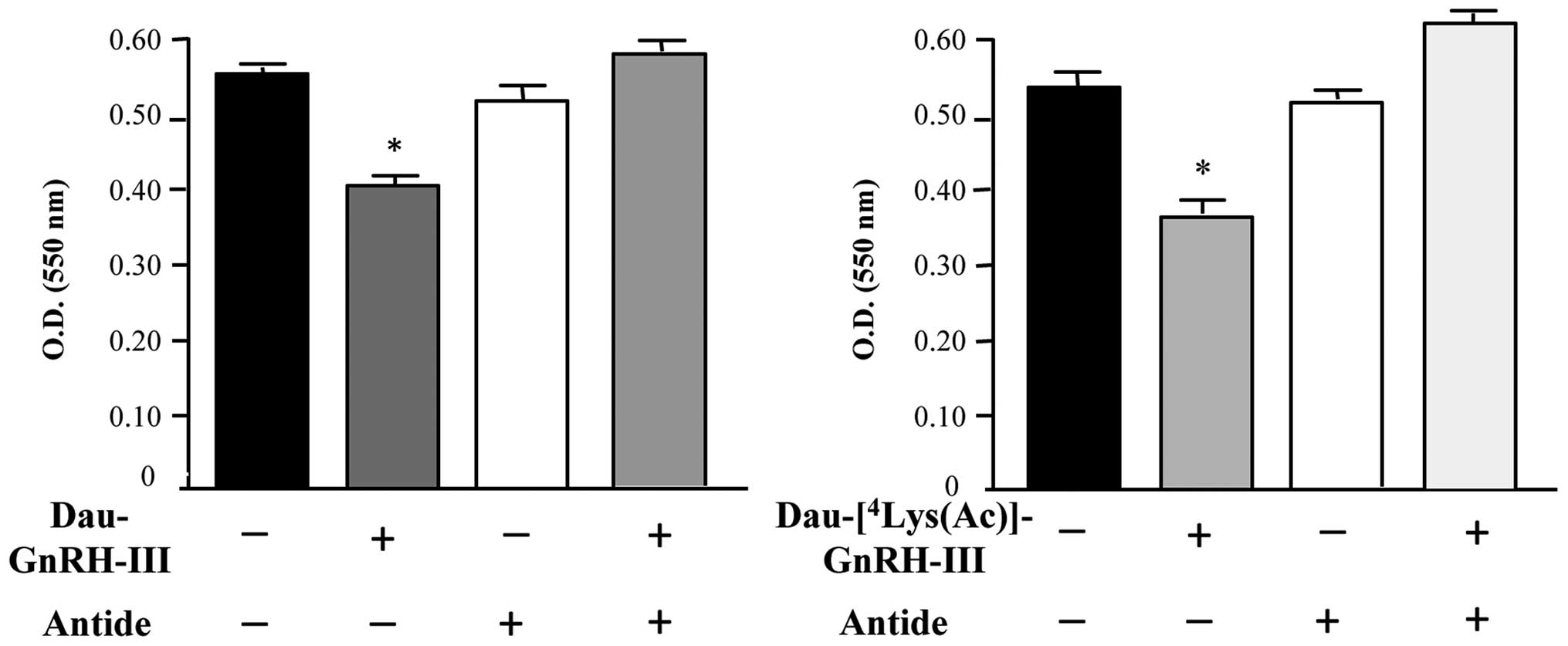
Experiments were performed to investigate whether
the two bioconjugates Dau-GnRH-III and
Dau-[4Lys(Ac)]-GnRH-III might exert their antitumor
effect through the activation of the type I GnRH-R. For this
purpose, we first treated DU145 cells with the two bioconjugates
(5×10−6 M), either alone or in the presence of the GnRH
antagonist Antide (a classical antagonist of the type I GnRH-R).
The effect of the treatment was then evaluated by MTT assay, which
showed that both bioconjugates significantly reduced the growth of
DU145 cells. When administered alone, the GnRH antagonist Antide
did not affect cell proliferation, as previously reported (21). On the other hand, Antide completely
counteracted the antiproliferative effect of the two bioconjugates
(Fig. 6).
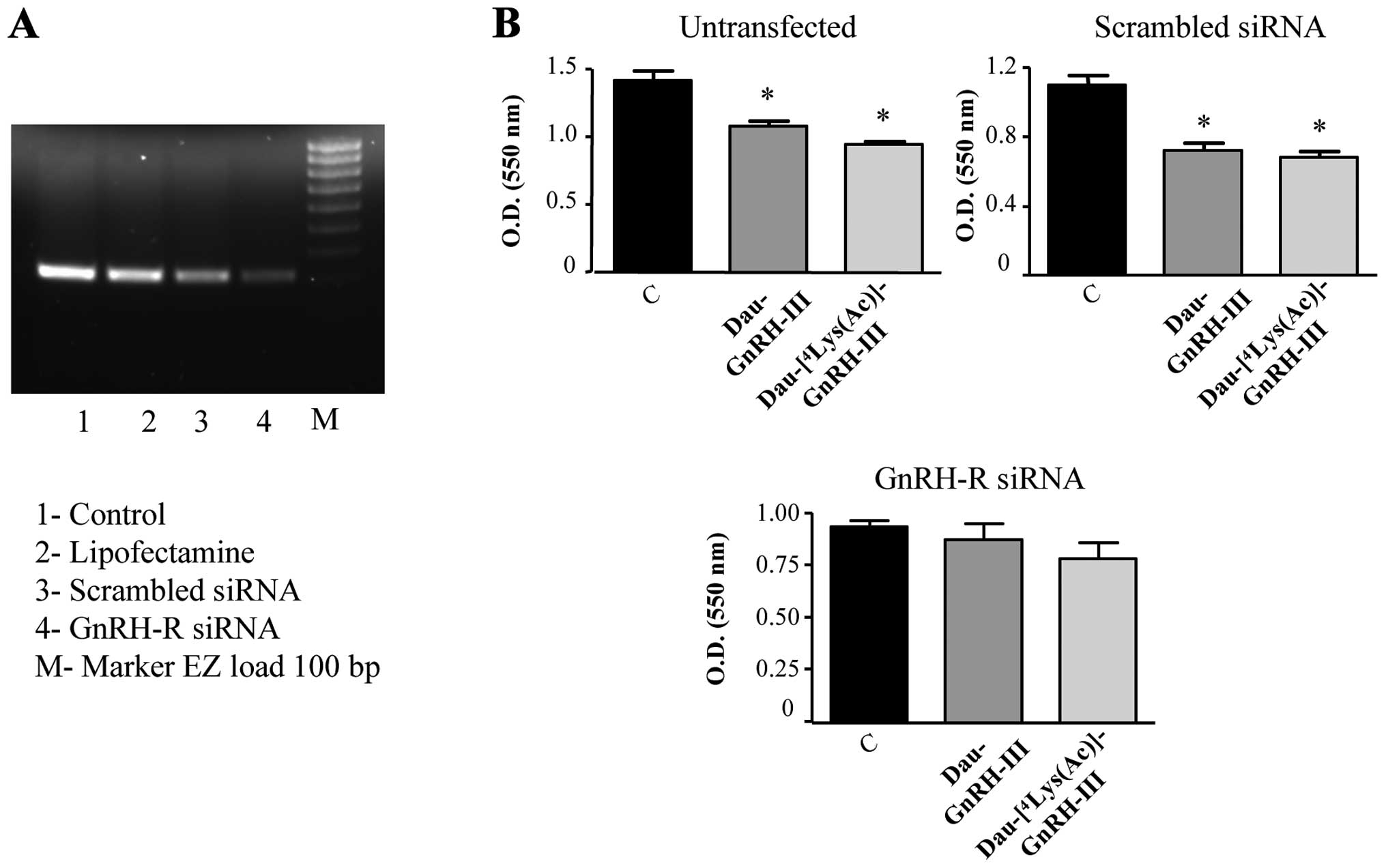
To unambiguously confirm the crucial role of type I
GnRH-R in mediating the antiproliferative activity of the two
daunorubicin-GnRH-III bioconjugates, the siRNA technique was
employed. DU145 cells were transfected with a specific siRNA (or
with a scrambled siRNA, as a negative control), as previously
reported (25). Transfection of
the specific siRNA resulted in a significant decrease of type I
GnRH-R mRNA levels at 24 h (Fig.
7A). On the basis of these results, DU145 cells (untransfected,
transfected with a scramble siRNA or with the specific siRNA) were
then treated with Dau-GnRH-III or with
Dau-[4Lys(Ac)]-GnRH-III for 48 h; the effect of the
treatments on the cell growth was evaluated by MTT assay. As
depicted in Fig. 7, panel B, in
untransfected control cells (in the presence of the transfectant
lipofectamine alone) as well as in cells transfected with the
scrambled siRNA, both bioconjugates significantly reduced the cell
growth. The antiproliferative effect of both compounds was
completely abrogated in DU145 cells transfected with the specific
type I GnRH-R siRNA (Fig. 7B).
Cytostatic effect of Dau-GnRH-III and
Dau-[4Lys(Ac)]-GnRH-III bioconjugates on PC3 cells
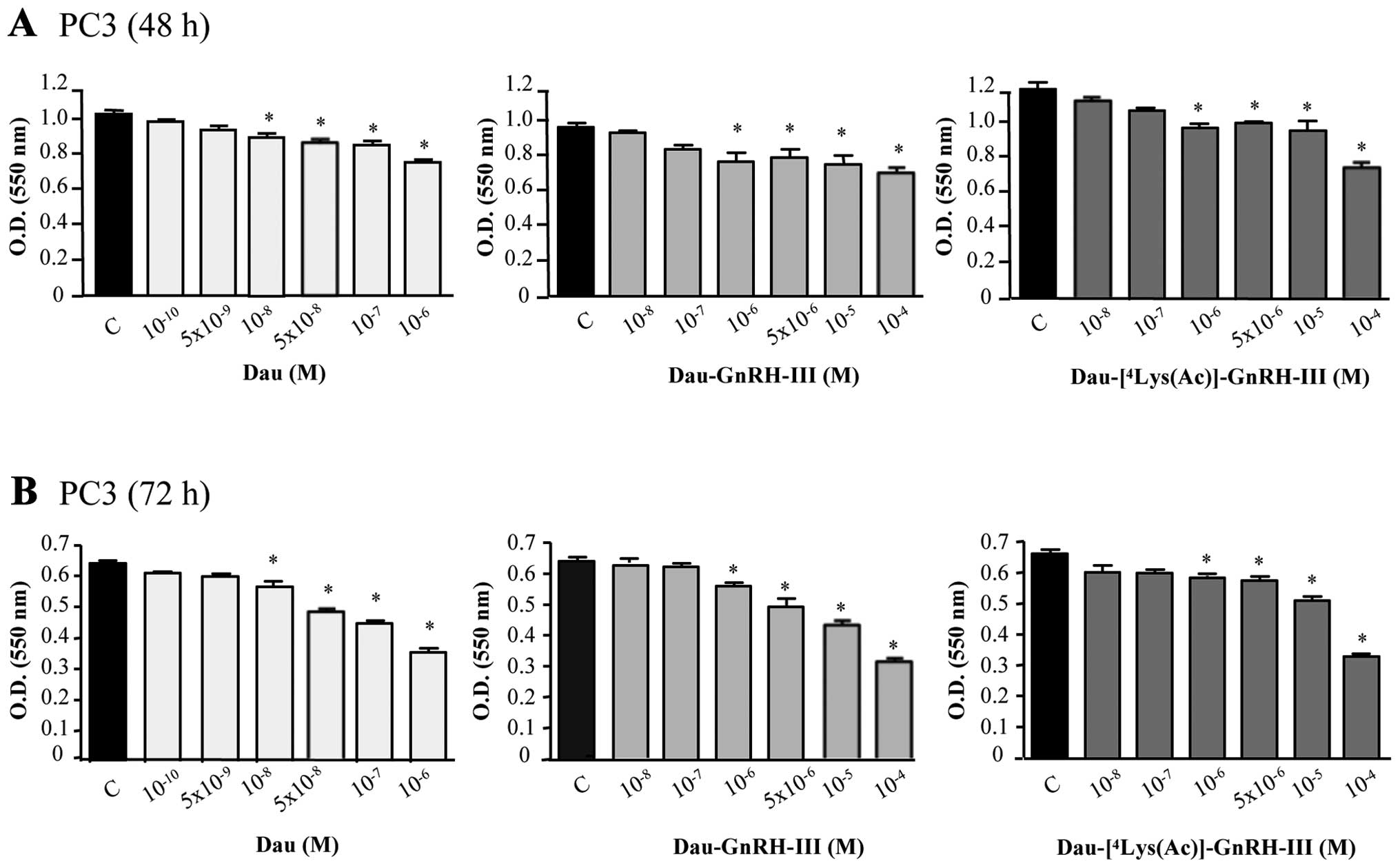
These experiments were performed with the aim of
confirming the cytostatic activity of oxime bond-linked
daunorubicin-GnRH-III derivative bioconjugates on
castration-resistant prostate cancer cells. For this purpose, PC3
cells were treated with free daunorubicin (Dau,
10−10-10−6 M) or with the two bioconjugates
Dau-GnRH-III and Dau-[4Lys(Ac)]-GnRH-III
(10−8-10−4 M, for 48 or 72 h). The effect of
the treatments on the cell growth was determined by MTT assay.
Daunorubicin exerted a significant and dose-dependent
antiproliferative effect on PC3 cells. Moreover, both
daunorubicin-GnRH-III derivative bioconjugates dose-dependently
inhibited PC3 cell growth both at 48 (Fig. 8A) and 72 h (Fig. 8B) of treatment. The IC50
values at 72 h were as follows: 1.54±1.25 μM for daunorubicin,
93.4±1.3 μM for Dau-GnRH-III and 60.4±1.53 μM for
Dau-[4Lys(Ac)]-GnRH-III, respectively. These data
confirm that Dau-GnRH-III and Dau-[4Lys(Ac)]-GnRH-III
exert a significant antiproliferative activity on
castration-resistant prostate cancer cells. However, it must be
noted that PC3 cells seem to be less responsive to the treatments
than DU145; this may be due to the fact that PC3 cells are more
resistant to apoptosis, due to the lack of expression of the two
oncosuppressor proteins p53 and PTEN (29).
Discussion
It is now well established that GnRH-R (type I, the
classical form of GnRH-R) are expressed in several types of cancer
cells and tumors, both related and unrelated to the reproductive
system, and are associated with a significant antitumor activity
(6,7,9,10).
These observations strongly support the notion that this receptor
might represent an important molecular target for cytotoxic
anticancer drug-GnRH derivative bioconjugates. The rationale for
this treatment strategy is that the GnRH analog will act as a
targeting moiety and specifically deliver the cytotoxic agents to
malignant tissues, thus increasing their local efficacy, while
limiting the peripheral toxicity. In line with this concept, the
doxorubicin-[D-Lys6]-GnRH bioconjugate, AEZS-108
(formerly known as AN-152), in which the chemotherapeutic drug was
attached to the GnRH derivative through an ester bond, via a
glutarate spacer, was developed. This bioconjugate was shown to
inhibit the growth of breast, endometrial and ovarian cancer cells
both in vitro and in preclinical studies (30); moreover, phase I/II clinical trials
reported the efficacy and safety of AEZS-108 in patients with
gynecological cancers (11,31).
However, mild side effects such as leukopenia and neutropenia as
well as a reduction in the plasma levels of LH and FSH were also
reported. These side effects were mainly attributed to the cleavage
of the ester bond in circulation, leading to the release of free
Dox and [D-6Lys(glutarate)]-GnRH-I; the half-life of
AESZ-108 was estimated to be about 2 h. AEZS-108 is at present in
phase III studies in advanced endometrial cancer positive for
GnRH-R (30). Recently, this
compound has been shown to exert a cytotoxic effect on human
urinary bladder, glioblastoma and pancreatic cancer cells, in
vitro and in vivo (32–34).
At present, various research studies are focused on
the development of cytotoxic bioconjugates with increased chemical
and enzymatic stability in circulation (i.e., in human serum) and
with enhanced antitumor activity. A promising peptide to be used as
a targeting moiety for the preparation of anticancer drug delivery
systems is lamprey GnRH-III. It has been shown that this natural
isoform of GnRH has a low potency at the pituitary level, while it
exerts a strong anti-tumor activity on cancer cells expressing the
GnRH-R (12). Thus, in our
studies, GnRH-III decapeptide was employed as a targeting moiety,
to which the chemotherapeutic agent daunorubicin was attached via
an oxime bond to the Lys in position 8 (13,15).
Furthermore, the replacement of 4Ser by Lys(Ac) led to a
daunorubicin containing bioconjugate with enhanced cellular uptake,
in vitro and in vivo antitumor activity (especially
on colon carcinoma) and increased stability against digestive
enzymes, in particular chymotrypsin (14). Both compounds, Dau-GnRH-III and
Dau-[4Lys(Ac)]-GnRH-III, were stable in human serum for
at least 24 h, but degradable in the presence of rat liver
lysosomal homogenate. Thus, these cytotoxic bioconjugates are
promising drug delivery systems for targeted cancer
chemotherapy.
The aim of the study reported here was to
investigate whether cytotoxic GnRH-III-based bioconjugates might
affect the growth of CRPC cells and to elucidate whether the type I
GnRH-R (the classical form of the GnRH-R in humans) might mediate
their effect. At present, the therapeutic options for CRPC patients
are still very limited. Therefore, the identification of novel
molecular markers as well as the development of more effective
targeted treatment strategies, such as cytotoxic compounds
specifically delivered at the level of the tumor, might help
increasing the treatment options for these patients (19,20).
Limonta’s laboratory has widely reported that GnRH-R
are expressed in human CRPC cells (DU145 and PC3), and their
activation significantly reduces cell
proliferation/invasiveness/migration (21–25).
By fluorescence microscopy, taking advantage of the
fluorescence properties of daunorubicin, we first demonstrated that
both Dau-GnRH-III derivative bioconjugates were internalized into
DU145 prostate cancer cells. Treatment of these cells with both
compounds induced a dose-dependent and significant
antiproliferative effect, with Dau-[4Lys(Ac)]-GnRH-III
being more effective that Dau-GnRH-III, as indicated by the
IC50 values. A cytostatic effect of the two
bioconjugates was also observed in human PC3 CRPC cells. In both
cell lines, the IC50 values for the cytostatic effect of
free daunorubicin were much lower than those calculated for the
bioconjugates, confirming the notion that the chemotherapeutic drug
could freely diffuse into the cells, while the bioconjugates
required to be internalized through receptor-mediated endocytosis.
Furthermore, our results showed that apoptosis was also involved in
the antitumor activity of the two bioconjugates, as indicated by
the expression levels of the cleaved (active) form of caspase-3 in
DU145 cells.
Taken together, these results demonstrate that the
Dau-GnRH-III and Dau-[4Lys(Ac)]-GnRH-III bioconjugates
exert a significant antitumor activity on human
castration-resistant prostate cancer cells. Similar results with
these two bioconjugates were previously reported in MCF-7 human
breast cancer, HT-29 human colon and LNCaP human androgen-dependent
prostate cancer cells in vitro, as well as on colon
carcinoma-bearing mice (14,35).
The observation that GnRH-R targeted bioconjugates are effective in
reducing the proliferation of CRPC cells is in line with data
previously reported with two different cytotoxic GnRH-derivatives:
AEZS-108 (doxorubicin-[D-Lys6]-GnRH) (36) and
gemcitabine-[D-Lys6]-GnRH derivative (37).
The efficacy of peptide-based drugs is strongly
dependent on their stability against enzyme-catalyzed hydrolysis
during transport (e.g., blood circulation, digestive tract) as well
as on their degradation by lysosomal enzymes. As underlined in
Introduction, the oxime bond-linked daunorubicin-GnRH-III
derivative bioconjugates have been previously shown to be stable in
human serum at least for 24 h. Moreover, the replacement of
4Ser by Lys(Ac) led to enhanced stability in the
presence of digestive enzymes (in particular, against
chymotrypsin). On the other hand, they were degraded by rat liver
lysosomal enzymes; the smallest drug containing metabolite obtained
in the presence of lysosomal enzymes was H-Lys(Dau=Aoa)-OH, which
was shown to bind to DNA in vitro (14,15).
These properties clearly support the suitability of these two
bioconjugates as targeted chemotherapeutics for tumors expressing
the GnRH-R, including castration-resistant prostate cancer.
It is still unclear through which receptor GnRH-III
might exert its antitumor activity on human cells. Here we provide
evidence that, in DU145 cells, the cytostatic effect of both
Dau-GnRH-III and Dau-[4Lys(Ac)]-GnRH-III is completely
counteracted by the simultaneous treatment of the cells with
Antide, a well known antagonist of the type I GnRH-R, the classical
form of the GnRH-R. More importantly, we show that the antitumor
activity of the two bioconjugates is completely abrogated in cells
in which the type I GnRH-R has been silenced by siRNA technique.
This clearly demonstrates that oxime bond-linked
daunorubicin-GnRH-III derivative bioconjugates exert their strong
antitumor activity on CRPC cells through the activation of the
classical isoform of the GnRH-R. In agreement with our
observations, Hegedüs and coworkers have reported that both these
compounds inhibit the binding of [125I]GnRH-I to the
membranes of human pituitary and human prostate cancer specimens
(38).
The existence of a GnRH-R isoform different from the
classical type I isoform in humans is still a matter of debate.
Since the discovery of a second form of the GnRH peptide (GnRH-II)
in most vertebrates, including humans, the existence of a putative
cognate type II GnRH-R has been speculated. However, the human type
II GnRH-R gene carries a frameshift and a premature stop codon,
indicating that a functional full-length receptor protein does not
exist in humans (3,39). The results reported here, together
with these observations, further sustain the notion that it is only
the classical form of the GnRH-R that mediates the biological
effects of the different GnRH isoforms (including GnRH-III) in
humans.
In conclusion, our results demonstrate that
cytotoxic GnRH-III-based bioconjugates can exert a specific
antiproliferative/proapoptotic activity on castration-resistant
prostate cancer cells and that this antitumor effect is
specifically mediated by the type I GnRH-R. These data provide a
basis for further development of targeted cancer
chemotherapeutics.
Of particular interest are the so-called
‘multi-functional’ or ‘multi-target’ bioconjugates, in which two
anticancer drugs with different mechanisms of action are attached
to the same GnRH-III-based targeting moiety. Such compounds
containing both methotrexate and daunorubicin attached to the same
GnRH-III targeting moiety have recently been reported. They were
shown to exert a higher in vitro cytostatic effect on
breast, colon and hormone-dependent prostate cancer cells than the
‘mono-functional’ bioconjugates containing only one anticancer drug
(40). The combination of two
anticancer drugs in one bioconjugate targeting GnRH-R could be
advantageous in the treatment of hormone-independent tumors such as
castration-resistant prostate cancer; this issue will be addressed
in further studies.
Acknowledgements
This study was supported by a research grant from
the Zukunftskolleg, University of Konstanz, (Project 879/08).
References
|
1
|
Schally AV, Arimura A, Baba Y, Nair RM,
Matsuo H, Redding TW and Debeljuk L: Isolation and properties of
the FSH and LH-releasing hormone. Biochem Biophys Res Commun.
43:393–399. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kakar SS, Malik MT, Winters SJ and
Mazhawidza W: Gonadotropin-releasing hormone receptors: structure,
expression, and signaling transduction. Vitam Horm. 69:151–207.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Millar RP: GnRHs and GnRH receptors. Anim
Reprod Sci. 88:5–28. 2005. View Article : Google Scholar
|
|
4
|
Emons G and Schally AV: The use of
luteinizing hormone releasing hormone agonists and antagonists in
gynaecological cancers. Hum Reprod. 9:1364–1379. 1994.PubMed/NCBI
|
|
5
|
Labrie F, Belanger A, Luu-The V, et al:
Gonadotropin-releasing hormone agonists in the treatment of
prostate cancer. Endocr Rev. 26:361–379. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Imai A and Tamaya T: GnRH receptor and
apoptotic signaling. Vitam Horm. 59:1–33. 2000. View Article : Google Scholar
|
|
7
|
Grundker C, Gunthert AR, Westphalen S and
Emons G: Biology of the gonadotropin-releasing hormone system in
gynecological cancers. Eur J Endocrinol. 146:1–14. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Montagnani Marelli M, Moretti RM,
Januszkiewicz-Caulier J, Motta M and Limonta P:
Gonadotropin-releasing hormone (GnRH) receptors in tumors: a new
rationale for the therapeutical application of GnRH analogs in
cancer patients? Curr Cancer Drug Targets. 6:257–269.
2006.PubMed/NCBI
|
|
9
|
Limonta P, Montagnani Marelli M, Mai S,
Motta M, Martini L and Moretti RM: GnRH receptors in cancer: from
cell biology to novel targeted therapeutic strategies. Endocr Rev.
33:784–811. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Limonta P and Manea M:
Gonadotropin-releasing hormone receptors as molecular therapeutic
targets in prostate cancer: Current options and emerging
strategies. Cancer Treat Rev. 39:647–663. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Engel J, Emons G, Pinski J and Schally AV:
AEZS-108 : a targeted cytotoxic analog of LHRH for the treatment of
cancers positive for LHRH receptors. Expert Opin Investig Drugs.
21:891–899. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kovacs M, Seprodi J, Koppan M, Horvath JE,
Vincze B, Teplan I and Flerko B: Lamprey gonadotropin
hormone-releasing hormone-III has no selective follicle-stimulating
hormone-releasing effect in rats. J Neuroendocrinol. 14:647–655.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Szabo I, Manea M, Orban E, et al:
Development of an oxime bond containing
daunorubicin-gonadotropin-releasing hormone-III conjugate as a
potential anticancer drug. Bioconjug Chem. 20:656–665. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Manea M, Leurs U, Orban E, et al: Enhanced
enzymatic stability and antitumor activity of daunorubicin-GnRH-III
bioconjugates modified in position 4. Bioconjug Chem. 22:1320–1329.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Orban E, Mezo G, Schlage P, et al: In
vitro degradation and antitumor activity of oxime bond-linked
daunorubicin-GnRH-III bioconjugates and DNA-binding properties of
daunorubicin-amino acid metabolites. Amino Acids. 41:469–483. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schlage P, Mezo G, Orban E, Bosze S and
Manea M: Anthracycline-GnRH derivative bioconjugates with different
linkages: synthesis, in vitro drug release and cytostatic effect. J
Control Release. 156:170–178. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar
|
|
18
|
Akaza H: Combined androgen blockade for
prostate cancer: review of efficacy, safety and cost-effectiveness.
Cancer Sci. 102:51–56. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fitzpatrick JM: Management of
castration-resistant prostate cancer: a call to urologists. BJU
Int. 110:772–774. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee DJ, Cha EK, Dubin JM, et al: Novel
therapeutics for the management of castration-resistant prostate
cancer (CRPC). BJU Int. 109:968–985. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dondi D, Limonta P, Moretti RM, Montagnani
Marelli M, Garattini E and Motta M: Antiproliferative effects of
lutein-izing hormone-releasing hormone (LHRH) agonists on human
androgen-independent prostate cancer cell line DU 145: evidence for
an autocrine-inhibitory LHRH loop. Cancer Res. 54:4091–4095.
1994.
|
|
22
|
Dondi D, Moretti RM, Montagnani Marelli M,
et al: Growth-inhibitory effects of luteinizing hormone-releasing
hormone (LHRH) agonists on xenografts of the DU 145 human
androgen-independent prostate cancer cell line in nude mice. Int J
Cancer. 76:506–511. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Limonta P, Moretti RM, Montagnani Marelli
M, Dondi D, Parenti M and Motta M: The luteinizing
hormone-releasing hormone receptor in human prostate cancer cells:
messenger ribonucleic acid expression, molecular size, and signal
transduc-tion pathway. Endocrinology. 140:5250–5256. 1999.
|
|
24
|
Montagnani Marelli M, Moretti RM, Mai S,
Procacci P and Limonta P: Gonadotropin-releasing hormone agonists
reduce the migratory and the invasive behavior of
androgen-independent prostate cancer cells by interfering with the
activity of IGF-I. Int J Oncol. 30:261–271. 2007.PubMed/NCBI
|
|
25
|
Montagnani Marelli M, Moretti RM, Mai S,
Januszkiewicz-Caulier J, Motta M and Limonta P: Type I
gonadotropin-releasing hormone receptor mediates the
antiproliferative effects of GnRH-II on prostate cancer cells. J
Clin Endocrinol Metab. 94:1761–1767. 2009.PubMed/NCBI
|
|
26
|
Gnanapragasam VJ, Darby S, Khan MM, Lock
WG, Robson CN and Leung HY: Evidence that prostate
gonadotropin-releasing hormone receptors mediate an
anti-tumourigenic response to analogue therapy in hormone
refractory prostate cancer. J Pathol. 206:205–213. 2005. View Article : Google Scholar
|
|
27
|
Liu SV, Schally AV, Hawes D, et al:
Expression of receptors for luteinizing hormone-releasing hormone
(LH-RH) in prostate cancers following therapy with LH-RH agonists.
Clin Cancer Res. 16:4675–4680. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Palyi I, Vincze B, Lovas S, et al:
Gonadotropin-releasing hormone analogue conjugates with strong
selective antitumor activity. Proc Natl Acad Sci USA. 96:2361–2366.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hong SW, Shin JS, Moon JH, et al:
NVP-BEZ235, a dual PI3K/mTOR inhibitor, induces cell death through
alternate routes in prostate cancer cells depending on the PTEN
genotype. Apoptosis. 19:895–904. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Engel JB, Schally AV, Buchholz S, Seitz S,
Emons G and Ortmann O: Targeted chemotherapy of endometrial,
ovarian and breast cancers with cytotoxic analogs of luteinizing
hormone-releasing hormone (LHRH). Arch Gynecol Obstet. 286:437–442.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Emons G, Gorchev G, Harter P, et al:
Efficacy and safety of AEZS-108 (LHRH agonist linked to
doxorubicin) in women with advanced or recurrent endometrial cancer
expressing LHRH receptors: a multicenter phase 2 trial (AGO-GYN5).
Int J Gynecol Cancer. 24:260–265. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Szepeshazi K, Schally AV, Keller G, et al:
Receptor-targeted therapy of human experimental urinary bladder
cancers with cytotoxic LH-RH analog AN-152 [AEZS-108]. Oncotarget.
3:686–699. 2012.PubMed/NCBI
|
|
33
|
Jaszberenyi M, Schally AV, Block NL, Nadji
M, Vidaurre I, Szalontay L and Rick FG: Inhibition of U-87 MG
glioblastoma by AN-152 (AEZS-108), a targeted cytotoxic analog of
luteinizing hormone-releasing hormone. Oncotarget. 4:422–432.
2013.PubMed/NCBI
|
|
34
|
Szepeshazi K, Schally AV, Block NL, et al:
Powerful inhibition of experimental human pancreatic cancers by
receptor targeted cytotoxic LH-RH analog AEZS-108. Oncotarget.
4:751–760. 2013.PubMed/NCBI
|
|
35
|
Manea M, Tovari J, Tejeda M, Schulcz A,
Kapuvari B, Vincze B and Mezo G: In-vivo antitumour effect of
daunorubicin-GnRH-III derivative conjugates on colon
carcinoma-bearing mice. Anticancer Drugs. 23:90–97. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Letsch M, Schally AV, Szepeshazi K, Halmos
G and Nagy A: Preclinical evaluation of targeted cytotoxic
luteinizing hormone-releasing hormone analogue AN-152 in
androgen-sensitive and insensitive prostate cancers. Clin Cancer
Res. 9:4505–4513. 2003.
|
|
37
|
Karampelas T, Argyros O, Sayyad N, et al:
GnRH-gemcitabine conjugates for the treatment of
androgen-independent prostate cancer: pharmacokinetic enhancements
combined with targeted drug delivery. Bioconjug Chem. 25:813–823.
2014. View Article : Google Scholar
|
|
38
|
Hegedüs R, Manea M, Orbán E, et al:
Enhanced cellular uptake and in vitro antitumor activity of
short-chain fatty acid acylated daunorubicin-GnRH-III
bioconjugates. Eur J Med Chem. 56:155–165. 2012.PubMed/NCBI
|
|
39
|
Cheng CK and Leung PC: Molecular biology
of gonadotropin-releasing hormone (GnRH)-I, GnRH-II, and their
receptors in humans. Endocr Rev. 26:283–306. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Leurs U, Lajko E, Mezo G, et al: GnRH-III
based multifunctional drug delivery systems containing daunorubicin
and methotrexate. Eur J Med Chem. 52:173–183. 2012. View Article : Google Scholar : PubMed/NCBI
|