Introduction
Lung cancer is the leading cause of cancer-related
deaths worldwide. Non-small cell lung cancer (NSCLC) accounts for
~85% of lung cancers (1). The
median survival time of patients with advanced NSCLC is <1 year
(2).
Epidermal growth factor receptor (EGFR)-targeted
therapy, using epidermal growth factor receptor-tyrosine kinase
inhibitors (EGFR-TKIs), for patients harboring EGFR
mutations such as exon 19 deletions or the L858R point mutation in
exon 21, is one of the most accepted and well-studied molecular
targeted therapies to date (3–5).
Despite a high proportion of response to EGFR-TKIs, many patients
with EGFR mutations will relapse and eventually develop resistance
to EGFR-TKIs. Recently, several studies have already elucidated the
mechanism underlying acquired resistance to EGFR-TKIs in NSCLC
harboring EGFR mutations. We have previously reported the role of
FGF2-FGFR1 activation as one of the mechanisms of acquired
resistance (6). However, the
mechanisms of acquired resistance are not yet fully understood.
Epigenetic modifications play an important role in
the control of gene expression in mammalian cells. Recently,
aberrant DNA hypermethylation of CpG islands in the gene promoter
region has become one of the major mechanisms for silencing tumor
suppressor or other cancer-associated genes such as
O6-methylguanine-DNA methyltransferase (MGMT), p16,
RARb, TIMP-3, and DAPK in lung cancer cells
(7,8).
Furthermore, DNA hypermethylation is often
associated with responses to chemotherapy (9). One classical example is that
MGMT promoter methylation in gliomas is a useful predictor
of tumor responsiveness to the alkylating agent carmustine as well
as a predictor of overall and disease-free survival in gliomas
(10).
In addition, drug-induced DNA hypermethylation could
be a mechanism that modifies tumor cell response to
chemotherapeutic agents (11–15).
Therefore, one possible reason for the development of
chemoresistance in NSCLC might be the epigenetic inactivation of
certain tumor suppressor genes due to chemotherapy treatment. Since
most studies have focused on a limited number of candidate genes,
many epigenetically silenced tumor suppressor genes involved in
cancer-chemoresistance remain unidentified (12,14).
Microarray-based screen pairing of the differential genetic profile
of chemosensitive and chemoresistant cell lines have been reported
(16–20). The association between PTEN
DNA methylation and acquired resistance to gefitinib in NSCLC cells
was previously reported (21).
However, only few reports focused on the relationship between
EGFR-TKI resistance and aberrant DNA methylation (22,23).
Furthermore, DNA demethylating agent, 5-azacytidine, might increase
the cellular sensitivity to gefitinib and control NSCLC cell growth
and apoptosis (24).
In this study, the cells described in a previous
report (6) were used. PC9
gefitinib-resistant (PC9 GR), gr1, and gr3 cells acquired
resistance to gefitinib through FGF2-FGFR1 activation. We further
sought to clarify the additional mechanisms involved in gefitinib
resistance, beside FGF2-FGFR1 activation.
To elucidate the key epigenetic regulation
mechanisms responsible for gefitinib resistance, the global DNA
methylation patterns of gefitinib-sensitive and -resistant lung
cancer cell lines were compared.
Materials and methods
Cell lines
The human NSCLC cell line PC9 [EGFR exon 19
deletion (delE746-A750)] was used. PC9 was kindly gifted by S.
Kobayashi (Beth Israel Deaconess Medical Center, Boston, MA, USA)
(25). Cells were cultured in
RPMI-1640 growth medium, supplemented with 10% fetal bovine serum
(FBS) at 37°C in a humidified 5% CO2 incubator.
Establishment of PC9 GR, gr1, and gr3
cells
We established gefitinib-resistant cell lines by
long-term exposure to gefitinib as characterized previously
(6). FGFR1 and FGF2 expression was
increased in PC9 GR cells compared to that in PC9 naïve (PC9 na)
cells. Gefitinib-resistant clones were also established from PC9 GR
cells, namely, PC9 GR1 and gr3. Proliferation of these PC9 GR cells
(PC9 GR, gr1, and gr3) was partly dependent on the activation of
the FGF2-FGFR1 pathway.
Reagents
The cell lines were treated with the following
inhibitors as single agents at various concentrations. Gefitinib
was a gift from AstraZeneca Pharmaceuticals (London, UK).
5-Azacitidine was purchased from Wako Pure Chemical Industries
(Osaka, Japan).
Cell proliferation assay
The MTS assay was performed according to the
manufacturer’s protocol by using CellTiter 96 AQueous One Solution
Assay (no. G3582; Promega Corporation, Madison, WI, USA). Briefly,
5×102 cells were seeded per well in 96-well plates and
allowed to attach for 24 h. The cells were then treated with
gefitinib at various concentrations. Control cells were treated
with the same concentration of the vehicle dimethyl sulfoxide
(DMSO). Forty-eight or 72 h after treatment with the drugs, cell
growth was analyzed.
Microarray analysis
Agilent SurePrint G3 Human Gene Expression 8×60K
Array (G4851A; Agilent Technologies, Inc., Santa Clara, CA, USA)
was used to monitor the expression profiles of the samples. Total
RNA was prepared using the RNeasy Mini kit (no. 74106; Qiagen,
Hilden, Germany), and labeled cRNA was prepared using standard
Agilent protocols. The log2 of the fold change to PC9 na
was calculated for each sample by using GeneSpring GX software
(Agilent Technologies, Inc.). These microarray data were deposited
in the Gene Expression Omnibus (GEO) database under dataset
accession no. GSE38302.
Infinium assay
Genomic DNA was extracted from cell lines by using a
DNeasy Blood & Tissue kit (Qiagen). Aliquots (500 ng) of DNA
were subjected to bisulfite conversion by using an EZ DNA
Methylation-Gold kit (Zymo Research, Irvine, CA, USA). DNA
methylation status of 27,578 CpG loci was examined at a single-CpG
resolution by using the Infinium HumanMethylation27 Bead Arrays
(Illumina, San Diego, CA, USA). After hybridization, the
specifically hybridized DNA was fluorescence-labeled by a
single-base extension reaction and detected using a BeadScan reader
(Illumina), in accordance with the manufacturer’s protocols. The
data were then assembled using GenomeStudio methylation software
(Illumina). At each CpG site, the ratio of the fluorescence signal
was measured using a methylated probe relative to the sum of the
methylated and unmethylated probes, i.e., the so-called β-value,
which ranges from 0.00 to 1.00, reflecting the methylation level of
an individual CpG site.
Quantitative RT-PCR
Total cellular RNA was prepared from the cells by
using an RNeasy Mini kit, and 1.0 μg of the RNA was then reverse
transcribed to cDNA by using TaqMan Reverse Transcription Reagents
(N8080234; Invitrogen Life Technologies, Carlsbad, CA, USA). For
quantitative reverse transcription-polymerase chain reaction
(RT-PCR) analysis, we used an ABI Prism 7000 Sequence Detection
System (Invitrogen Life Technologies). Human glyceraldehyde
3-phosphate dehydrogenase (GAPDH) was used for normalization
of input cDNA. The probe ID and primer sequences are available on
request.
5-aza-dC treatment
PC9 cells were seeded at a density of
1.0×105 cells/well in 6-well plates on day 0 and then
allowed to attach for a 24-h period. 5-Aza-2′-deoxycytidine
(5-aza-dC) was then added at a final concentration of 1 μM. After
48 h, the medium was changed, and 5-aza-dC was added again at the
same final concentration. Total RNA was extracted from all cells on
day 5.
KL and S100P silencing by siRNAs
PC9 na cells were transfected with a final
concentration of 20 nM of S100P siRNA, KL siRNA, or negative
control siRNA (no. s194780 for S100P siRNA and no. s225120 for KL
siRNA; Ambion, Grand Island, NY, USA). Silencer Select and negative
control mix Silencer Select (Ambion) were used according to the
manufacturer’s instructions. For transfection, siLentFect (no.
170-3361; Bio-Rad, Hercules, CA, USA) was used according to the
manufacturer’s protocol. Knockdown of KL and S100P
expression was confirmed using quantitative RT-PCR. For the
viability assay, cells were seeded in 6-well plates at a density of
100,000 cells/well 24 h after transfection with S100P siRNA, KL
siRNA, or negative siRNA control. The following day, the cells
transfected with siRNAs were seeded in 96-well plates at a density
of 500 cells/well and were incubated with four different doses of
gefitinib for an additional 72 h. Cell viability was measured using
the MTS assay. Cells treated with DMSO were used as the
control.
Results
Analysis of DNA methylation changes
associated with gefitinib resistance by Infinium assay
To identify DNA methylation changes associated with
differential gefitinib sensitivity, we used PC9 na and isogenic
gefitinib-resistant cell lines, PC9 GR, gr1, and gr3. We performed
genome-wide DNA methylation profiling of PC9 GR, gr1, gr3, and PC9
na by using Infinium HumanMethylation27 Bead Arrays that comprise
27,578 CpG sites across >14,000 genes.
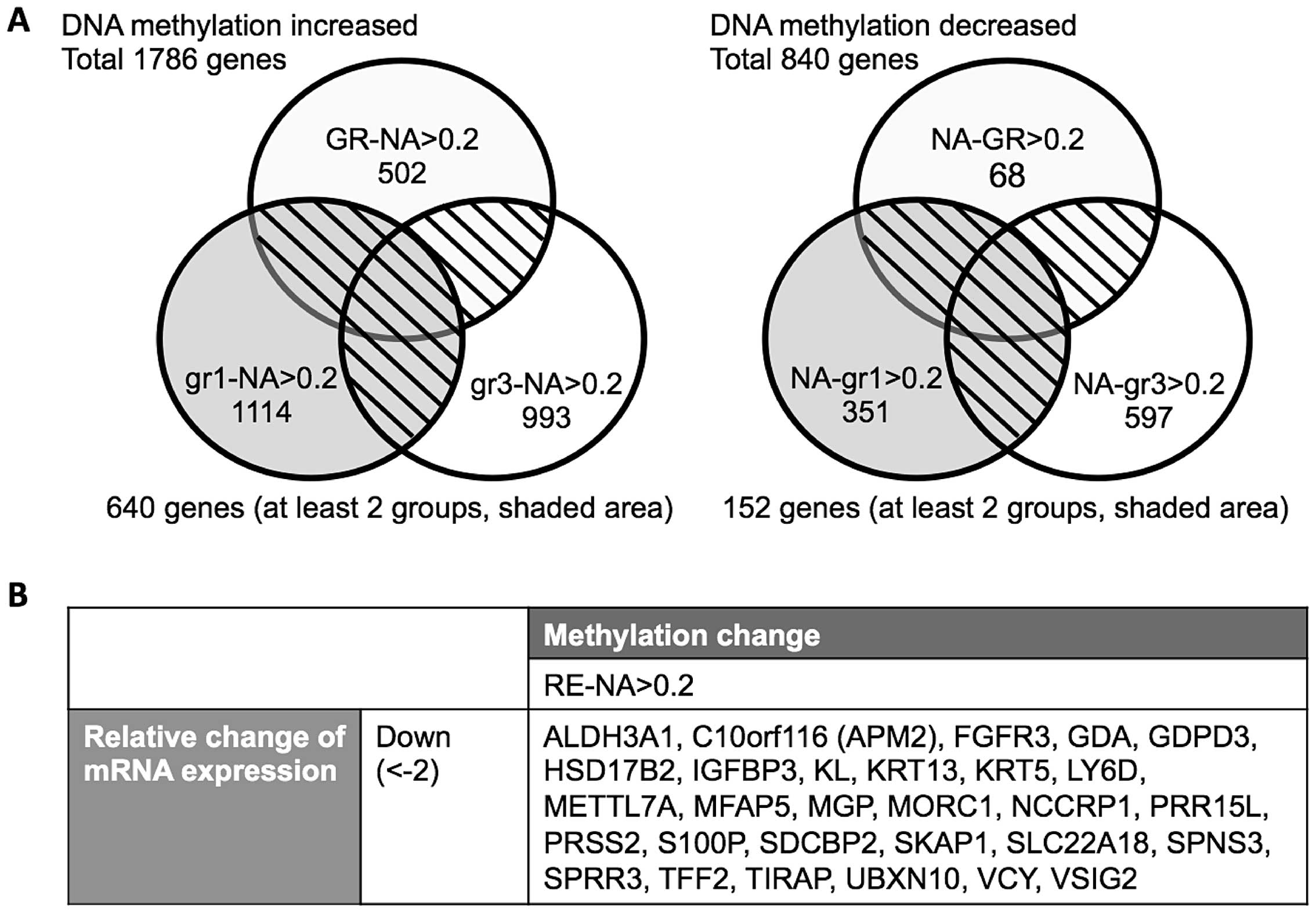
First, differentially CpG-methylated genes were
extracted based on a significant increase or decrease of their beta
score between PC9 GR, gr1, or gr3 and PC9 na. Following exposure to
gefitinib, we identified 1,786 genes, which were hypermethylated in
at least one of the PC9 GR cell lines compared to PC9 na cells,
while only 840 genes were hypomethylated in at least one of the
resistant cell lines compared with PC9 na cells, suggesting that
hypermethylation occurs more frequently than hypomethylation during
the selection process for acquired gefitinib resistance (Fig. 1A).
DNA methylation increased in 640 genes in at least
two of the three gefitinib-resistant cell lines compared to PC9 na
cells (Fig. 1A, left, shaded
area). In contrast, DNA methylation decreased in 152 genes in at
least two of the three gefitinib-resistant cell lines compared to
PC9 na cells (Fig. 1A, right,
shaded area).
Association of DNA methylation and gene
expression
In a second step, mRNA expression profiles of
sensitive PC9 na cells and PC9 GR cell lines were analyzed using
Agilent SurePrint G3 Human Gene Expression 8×60K Array. For each
CpG on the Infinium HumanMethylation27 Bead Arrays, the
corresponding gene was matched to the Agilent cDNA Microarray data
by using the Entrez Gene ID. Signal intensity in the gene
expression data was used as an indicator of the expression
level.
We further screened for the genes of interest by
identifying those that presented a decreased mRNA expression in at
least two of the three resistant cell lines.
Finally, we compared our candidate genes to those
reported by other investigators to be downregulated through DNA
methylation in chemoresistant cell lines (15,19,26).
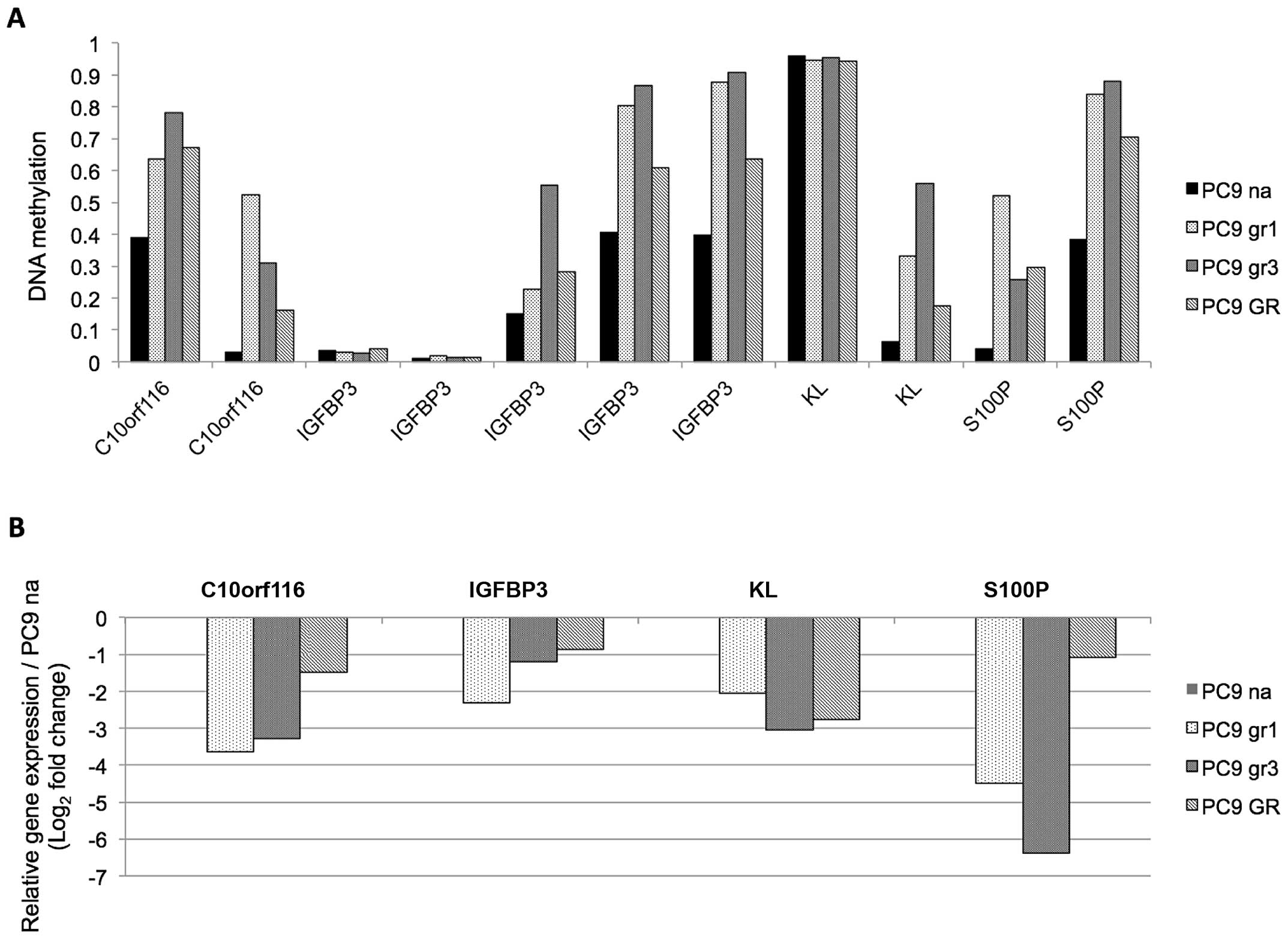
This approach allowed us to identify 29 genes that were
downregulated in PC9 GR cell lines and presented a change in DNA
methylation (Fig. 1B). Genes with
increased CpG methylation and concomitant decreased gene expression
included ALDH3A1, C10orf116 (APM2), FGFR3,
GDA, GDPD3, HSD17B2, IGFBP3, KL,
KRT13, KRT5, LY6D, METTL7A,
MFAP5, MGP, MORC1, NCCRP1,
PRR15L, PRSS2, S100P, SDCBP2,
SKAP1, SLC22A18, SPNS3, SPRR3,
TFF2, TIRAP, UBXN10, VCY, and
VSIG2.
In this study, we focused on four genes
(C10orf116, IGFBP3, KL, and S100P)
based on the expression pattern and previous reports, for further
examination. The methylation change and expression of mRNA from the
array data are presented in Fig.
2.
Confirmation of mRNA expression by
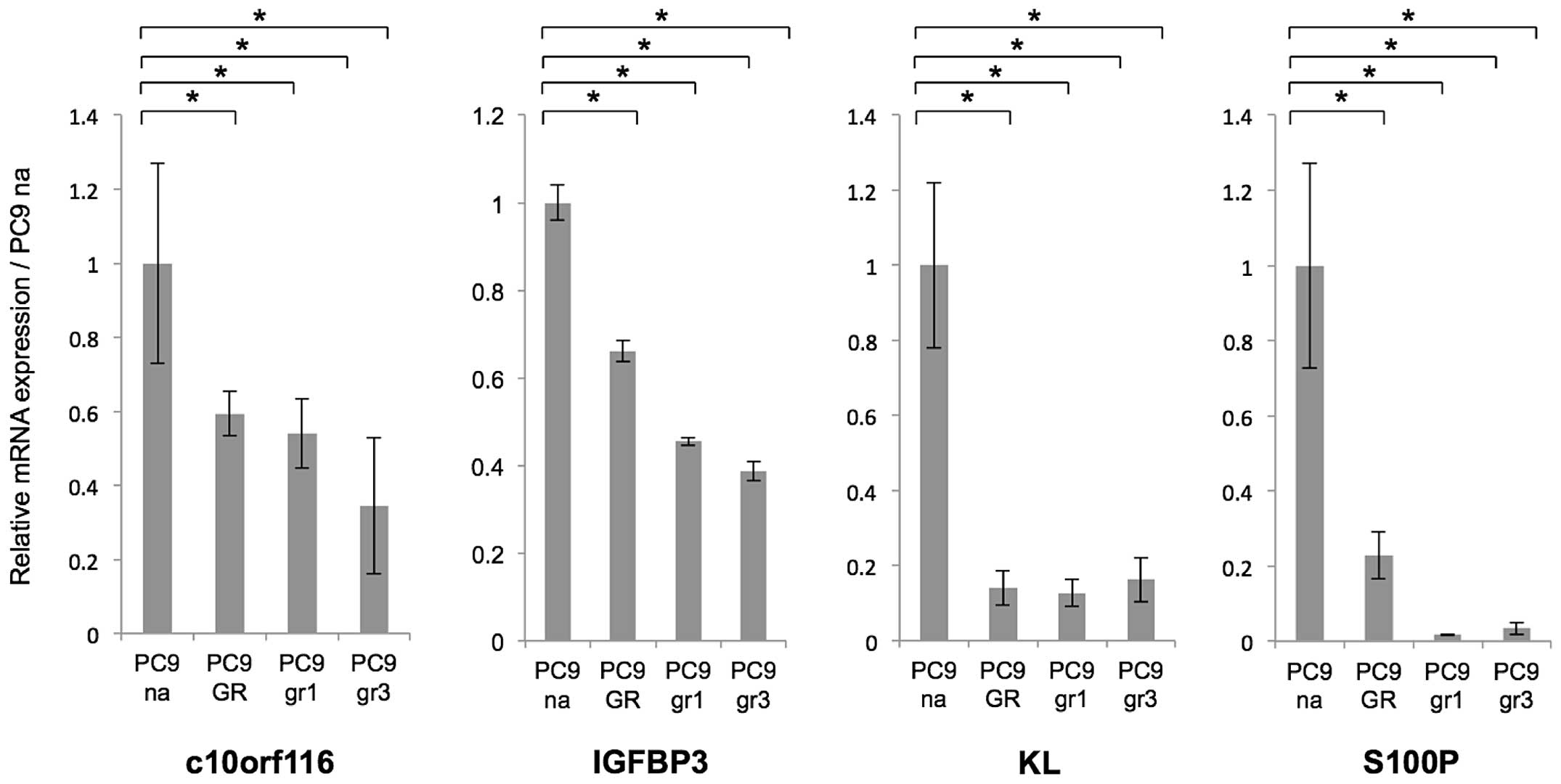
quantitative RT-PCR
Quantitative RT-PCR confirmed the observed
microarray expression changes in the selected four genes
(KL, S100P, C10orf116 and IGFBP3)
(Fig. 3). These data indicated
that the expression of the four genes was decreased in PC9
resistant cells.
Re-expression of target genes by 5-aza-dC
treatment
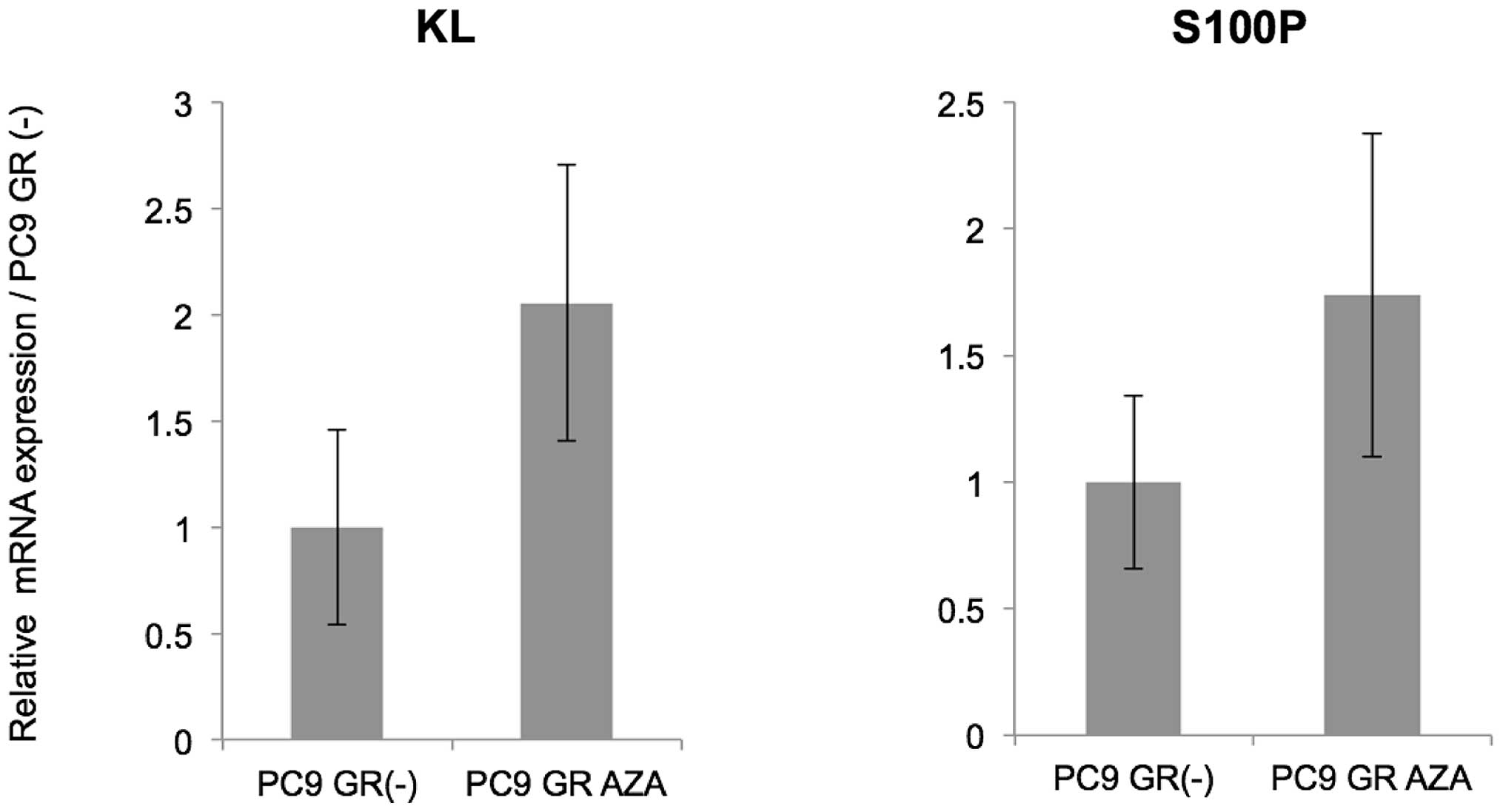
To make sure that the gene expression of the four
genes was regulated by DNA methylation, we performed DNA
demethylating experiments. Treatment with 5-aza-dC of PC9 GR cells
caused the re-expression of two genes (KL and S100P)
(Fig. 4). These findings indicated
that S100P and KL were in part epigenetically
silenced by methylation of CpG sites during gefitinib exposure.
Acquisition of gefitinib resistance after
siRNA-mediated knockdown of KL and S100P in PC9 na cells
To determine whether decreased expression of
KL and S100P affects the sensitivity of PC9 cells to
gefitinib, we performed gene knockdown experiments by using siRNA.
Knockdown of KL and S100P were confirmed by
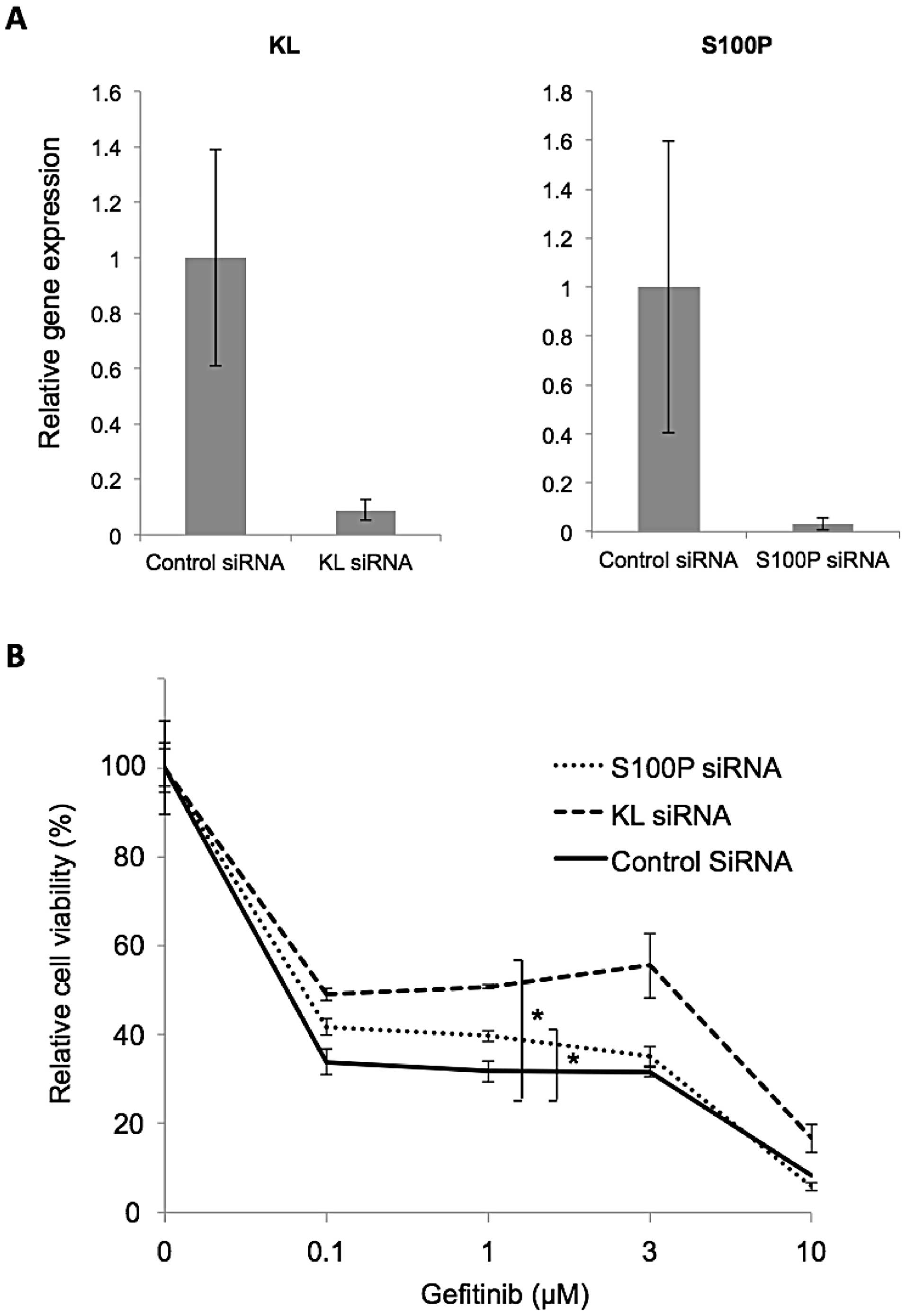
quantitative RT-PCR (Fig. 5A).
As expected, KL or S100P knockdown
resulted in a partial gain of gefitinib resistance in PC9 na cells
(Fig. 5B).
Taken together, these results demonstrated that the
loss of KL and S100P expression is involved in the
acquired gefitinib resistance of PC9 na cells.
Discussion
Previously, we demonstrated that the main mechanism
underlying acquired resistance to gefitinib in the resistant cells
PC9 GR, gr1, and gr3 involved the activation of the FGF2-FGFR1
pathway (6). However, the
inhibition of FGF2-FGFR1 pathway by a FGFR inhibitor or siRNA could
not fully recover the sensitivity to gefitinib in the resistant
cells. Thus, we hypothesized that other mechanisms may be involved
in the acquired resistance to gefitinib. In this study, we focused
on epigenetics as a potential mechanism.
Epigenetics is defined as heritable genetic changes,
which do not change the DNA sequence itself (26). These changes include DNA
methylations and chromatin modifications. Epigenetics regulate
multiple gene expression and play multiple important roles in
various biological processes, including differentiation,
imprinting, and oncogenesis (9).
In this study, we sought to clarify whether epigenetics have
functional roles in acquired resistance to cancer treatments. We
focused on aberrant DNA methylations occurring through acquired
resistance to gefitinib, one of the clinically available EGFR-TKIs.
Although there are many reports on the relationship between
cisplatin sensitivity or resistance and DNA methylation (19,20),
only few reports are available regarding the relationship between
EGFR-TKI resistance and DNA methylation, and none of them used a
comprehensive approach (21–23).
In this study, two comprehensive analysis tools, cDNA microarray
for gene expression and Infinium assay for DNA methylation, were
used. Epigenetic gene regulation is a complex biological process
that can change the expression of numerous genes. Thus, we expected
that the combination of the two comprehensive analyses could be a
powerful tool to appropriately detect a number of subtle, but
meaningful, epigenetic changes. As a result, 29 genes, which
presented a decreased mRNA expression and an increased DNA
methylation level, were identified in PC9 GR cells compared to
parental PC9 na cells. We speculated that changes in these gene
expressions might have some functional roles in acquired resistance
to gefitinib. Of these 29 genes, two genes, KL and
S100P, were found to be controlled by DNA methylation and
have functional roles in acquired gefitinib resistance.
Interestingly, the relationship between FGF2-FGFR1 pathway and
KL was reported (22,27).
Although we were unable to identify a direct relationship between
them in this study, decreased KL expression might influence
the FGF2-FGFR1 pathway. The contribution of KL and
S100P in the acquired resistance was relatively small
compared to that of the FGF2-FGFR1 pathway (6), but significant. We expect that a
collection of ‘small’ effects on acquired resistance can induce a
biologically or clinically meaningful effect.
Further studies aiming at elucidating the mechanisms
involved in acquired resistance are warranted because these can
help lung cancer patients who suffer from acquired resistance to
EGFR-TKI.
Acknowledgements
The authors thank Ms. Miyuki Yamamoto and Ms. Mikiko
Shibuya for their expert technical assistance. This study was
supported in part by Grants-in-Aid for Scientific Research on
Priority Areas from the Ministry of Education, Culture, Sports,
Science, and Technology of Japan (Tokyo, Japan) to K.S. (Grant no.
22590870), H.Y. (Grant no. 25860656), H.T. (Grant no. 24790822, no.
26860616), and K.N. (Grant no. 23501311).
References
|
1
|
Herbst RS, Heymach JV and Lippman SM: Lung
cancer. N Engl J Med. 359:1367–1380. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schiller JH, Harrington D, Belani CP,
Langer C, Sandler A, Krook J, Zhu J and Johnson DH; Eastern
Cooperative Oncology Group. Comparison of four chemotherapy
regimens for advanced non-small-cell lung cancer. N Engl J Med.
346:92–98. 2002. View Article : Google Scholar
|
|
3
|
Mok TS, Wu YL, Thongprasert S, Yang CH,
Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y,
Nishiwaki Y, Ohe Y, Yang JJ, Chewaskulyong B, Jiang H, Duffield EL,
Watkins CL, Armour AA and Fukuoka M: Gefitinib or
carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med.
361:947–957. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Paez JG, Jänne PA, Lee JC, Tracy S,
Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ,
Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE and
Meyerson M: EGFR mutations in lung cancer: correlation with
clinical response to gefitinib therapy. Science. 304:1497–1500.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lynch TJ, Bell DW, Sordella R,
Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat
SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J and
Haber DA: Activating mutations in the epidermal growth factor
receptor underlying responsiveness of non-small-cell lung cancer to
gefitinib. N Engl J Med. 350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Terai H, Soejima K, Yasuda H, Nakayama S,
Hamamoto J, Arai D, Ishioka K, Ohgino K, Ikemura S, Sato T, Yoda S,
Satomi R, Naoki K and Betsuyaku T: Activation of the FGF2-FGFR1
autocrine pathway: a novel mechanism of acquired resistance to
gefitinib in NSCLC. Mol Cancer Res. 11:759–767. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cheung HH, Lee TL, Rennert OM and Chan WY:
DNA methylation of cancer genome. Birth Defects Res C Embryo Today.
87:335–350. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Heller G, Zielinski CC and
Zöchbauer-Müller S: Lung cancer: from single-gene methylation to
methylome profiling. Cancer Metastasis Rev. 29:95–107. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wilting RH and Dannenberg JH: Epigenetic
mechanisms in tumorigenesis, tumor cell heterogeneity and drug
resistance. Drug Resist Updat. 15:21–38. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Esteller M, Garcia-Foncillas J, Andion E,
Goodman SN, Hidalgo OF, Vanaclocha V, Baylin SB and Herman JG:
Inactivation of the DNA-repair gene MGMT and the clinical
response of gliomas to alkylating agents. N Engl J Med.
343:1350–1354. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nyce JW: Drug-induced DNA
hypermethylation: a potential mediator of acquired drug resistance
during cancer chemotherapy. Mutat Res. 386:153–161. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Strathdee G, MacKean MJ, Illand M and
Brown R: A role for methylation of the hMLH1 promoter in loss of
hMLH1 expression and drug resistance in ovarian cancer. Oncogene.
18:2335–2341. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Taniguchi T, Tischkowitz M, Ameziane N,
Hodgson SV, Mathew CG, Joenje H, Mok SC and D’Andrea AD: Disruption
of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian
tumors. Nat Med. 9:568–574. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shen DW, Su A, Liang XJ, Pai-Panandiker A
and Gottesman MM: Reduced expression of small GTPases and
hypermethylation of the folate binding protein gene in
cisplatin-resistant cells. Br J Cancer. 91:270–276. 2004.PubMed/NCBI
|
|
15
|
Ibanez de Caceres I, Cortes-Sempere M,
Moratilla C, Machado-Pinilla R, Rodriguez-Fanjul V, Manguán-García
C, Cejas P, López-Ríos F, Paz-Ares L, de CastroCarpeño J, Nistal M,
Belda-Iniesta C and Perona R: IGFBP-3 hypermethylation-derived
deficiency mediates cisplatin resistance in non-small-cell lung
cancer. Oncogene. 29:1681–1690. 2010.PubMed/NCBI
|
|
16
|
Stone A, Valdés-Mora F, Gee JM, Farrow L,
McClelland RA, Fiegl H, Dutkowski C, McCloy RA, Sutherland RL,
Musgrove EA and Nicholson RI: Tamoxifen-induced epigenetic
silencing of oestrogen-regulated genes in anti-hormone resistant
breast cancer. PLoS One. 7:e404662012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zeller C, Dai W, Steele NL, Siddiq A,
Walley AJ, Wilhelm-Benartzi CS, Rizzo S, van der Zee A, Plumb JA
and Brown R: Candidate DNA methylation drivers of acquired
cisplatin resistance in ovarian cancer identified by methylome and
expression profiling. Oncogene. 31:4567–4576. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yu W, Jin C, Lou X, Han X, Li L, He Y,
Zhang H, Ma K, Zhu J, Cheng L and Lin B: Global analysis of DNA
methylation by Methyl-Capture sequencing reveals epigenetic control
of cisplatin resistance in ovarian cancer cell. PLoS One.
6:e294502011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chang X, Monitto CL, Demokan S, Kim MS,
Chang SS, Zhong X, Califano JA and Sidransky D: Identification of
hypermethylated genes associated with cisplatin resistance in human
cancers. Cancer Res. 70:2870–2879. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang X, Li W, Li H, Ma Y, He G and Tan G:
Genomic methylation profiling combined with gene expression
microarray reveals the aberrant methylation mechanism involved in
nasopharyngeal carcinoma taxol resistance. Anticancer Drugs.
23:856–864. 2012. View Article : Google Scholar
|
|
21
|
Noro R, Gemma A, Miyanaga A, Kosaihira S,
Minegishi Y, Nara M, Kokubo Y, Seike M, Kataoka K, Matsuda K, Okano
T, Yoshimura A and Kudoh S: PTEN inactivation in lung cancer cells
and the effect of its recovery on treatment with epidermal growth
factor receptor tyrosine kinase inhibitors. Int J Oncol.
31:1157–1163. 2007.PubMed/NCBI
|
|
22
|
Ogawa T, Liggett TE, Melnikov AA, Monitto
CL, Kusuke D, Shiga K, Kobayashi T, Horii A, Chatterjee A, Levenson
VV, Koch WM, Sidransky D and Chang X: Methylation of
death-associated protein kinase is associated with cetuximab and
erlotinib resistance. Cell Cycle. 11:1656–1663. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhu J, Wang Y, Duan J, Bai H, Wang Z, Wei
L, Zhao J, Zhuo M, Wang S, Yang L, An T, Wu M and Wang J: DNA
Methylation status of Wnt antagonist SFRP5 can predict the response
to the EGFR-tyrosine kinase inhibitor therapy in non-small cell
lung cancer. J Exp Clin Cancer Res. 31:802012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li XY, Wu JZ, Cao HX, Ma R, Wu JQ, Zhong
YJ and Feng JF: Blockade of DNA methylation enhances the
therapeutic effect of gefitinib in non-small cell lung cancer
cells. Oncol Rep. 29:1975–1982. 2013.PubMed/NCBI
|
|
25
|
Costa DB, Halmos B, Kumar A, Schumer ST,
Huberman MS, Boggon TJ, Tenen DG and Kobayashi S: BIM mediates EGFR
tyrosine kinase inhibitor-induced apoptosis in lung cancers with
oncogenic EGFR mutations. PLoS Med. 4:1669–1680. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sharma S, Kelly TK and Jones PA:
Epigenetics in cancer. Carcinogenesis. 31:27–36. 2010. View Article : Google Scholar
|
|
27
|
Wang Y, Chen L, Huang G, He D, He J, Xu W,
Zou C, Zong F, Li Y, Chen B, Wu S, Zhao W and Wu J: Klotho
sensitizes human lung cancer cell line to cisplatin via PI3k/Akt
pathway. PLoS One. 8:e573912013. View Article : Google Scholar : PubMed/NCBI
|