Introduction
Imidazole, an organic compound with the formula
C3H4N2, is classified as an
alkaloid. It is incorporated into many important biological
molecules; the most widespread is the amino acid histidine, which
has an imidazole side-chain. Imidazole is an important
pharmacophore in drug discovery. Its derivatives have diverse
pharmaceutical effects, including histamine-H3 antagonist (1), anti-inflammatory (2), gastroprotective (3), and antioxidant (4). Importantly, several imidazole
derivatives had anticancer activity against a variety of malignant
cells. Misonidazole can sensitize hypoxic tumor cells to radiation
therapy and directly kill hypoxic cancer cells (5,6).
Furthermore, N-fused imidazoles inhibited catalytic activity of
topoisomerase IIα and induced apoptosis at the G1/S phase in kidney
and breast cancer cell lines (7).
Temozolomide is an alkylating drug that induces apoptosis and
senescence in glioma cells (8).
Due to their therapeutic significance, the mechanism responsible
for the chemotherapeutic effects of imidazoles has received
considerable interest for future development of novel anticancer
drugs.
Autophagy is an evolutionarily conserved process
through which protein aggregates, damaged organelles and
intracellular pathogens are sequestered by autophagosomes and
degraded by lysosomes, resulting in nutrient recycling and energy
generation (9), enabling cells to
sustain metabolism under conditions of starvation or growth factor
withdrawal. Autophagy has been linked to a variety of pathological
processes such as neuronal degeneration, pathogen infection, aging
and tumorigenesis (10). It is
noteworthy that autophagy may facilitate survival of tumor cells
under stress conditions, e.g., nutrient limitation, hypoxia and
antineoplastic treatment. Furthermore, inhibition of autophagy
promotes cancer cell death (11–13)
and potentiates various anticancer therapies (14–17).
The critical role of autophagy in regulating cell survival and cell
death makes it be a potential therapeutic target for tumor
treatment. Autophagosome clearance depends on its fusion with a
lysosome (18). As a weak base,
imidazole can enter lysosomes and induce cytoplasmic vacuolization
(19). However, the effects of
imidazole on autophagy are still poorly documented.
Many anticancer treatments, including
chemotherapeutic drugs and radiation therapy, actually induce
apoptosis and thereby utilize apoptotic machinery to kill cancer
cells (20,21). The Bcl-2-homology domain 3 only
(BH3-only) proteins are essential for initiation of various
physiological apoptotic situations, including developmentally
programmed cell death and stress-induced apoptosis (22,23).
Pro-apoptotic protein Bim (Bcl-2-interacting modulator of cell
death) is one of these BH3-only proteins. Alternative splicing
generates three Bim isoforms, including BimS,
BimL and BimEL, with different variations in
pro-apoptotic activities (24). In
a variety of cell types, its upregulation can induce apoptosis via
promoting release of cytochrome c, which consequentially
induces formation of the apoptosome and the activation of caspases
(24,25). Moreover, Bim plays a key role in
the anoikis of many tumor cells, such as lung cancer, breast
cancer, osteosarcoma, and melanoma (26–28).
Thus, it has attracted increasing attention as a plausible target
for cancer treatment. It has been reported that imidazole can
induce cell apoptosis (29).
However, the mechanisms by which imidazole induces apoptosis remain
largely unknown.
In the present study, we carefully evaluated the
autophagic and apoptotic events induced by imidazole in human
endometrial adenocarcinoma cell line (HEC-1B). By systematically
studying imidazole induced alterations in autophagy, we were able
to characterize one potential autophagy modulator and explore the
roles of autophagy related drugs in cancer therapy. Simultaneously,
we also focused this study on the FoxO3a regulation of Bim
expression after imidazole treatment, in an attempt to gain further
mechanistic insights on the molecular pathways leading to
imidazole-induced apoptosis.
Materials and methods
Chemicals and antibodies
All chemicals were purchased from Sigma-Aldrich (St.
Louis, MO, USA), unless otherwise indicated. The LC3 antibody was
obtained from Sigma-Aldrich. Antibodies against Bim, FoxO3a,
cleaved-caspase 9, cleaved-caspase 3 and p62 were purchased from
Cell Signaling Technology (Danvers, MA, USA). Antibodies against
GFP and β-actin were obtained from Abmart (Shanghai, China). The
LAMP-1 antibody was obtained from Abcam (Cambridge, UK). The
HRP-conjugated goat anti-rabbit or anti-mouse secondary antibodies
were purchased from Santa Cruz Biotechnology (Santa Cruz, CA,
USA).
Cell culture
The HEC-1B cells were purchased from the Institute
of Basic Medical Sciences, Chinese Academy of Medical Sciences
(Beijing, China), MDA-MB-435S and 293T cells were obtained from
Peking University Laboratory Animal Center (Beijing, China). Cells
were cultured in Dulbecco’s modified Eagle’s medium: nutrient
mixture F-12 (DMEM/F12) supplemented with 10% fetal bovine serum
(Invitrogen, Carlsbad, CA, USA) and 100 U/ml penicillin and 100
μg/ml streptomycin. For imidazole treatment experiments, imidazole
dissolved in the phosphate-buffered saline (PBS; Invitrogen) was
added into culture medium (various doses and intervals). The HEC-1B
cells were treated with 50 nM rapamycin for 12 h or incubated in
Earle’s balanced salt solution (EBSS; Invitrogen) for 1 h to induce
autophagy. Bafilomycin A1 (Baf A1), a proton-ATPase inhibitor, was
used to inhibit the autophagic flux.
Live-cell imaging
The HEC-1B cells were treated with or without 5 mM
imidazole for 6 h, and then examined for vacuolization under an
inverted light microscope (x400 magnification; Olympus, Tokyo,
Japan). For acridine orange (AO) staining, cells grown on
coverslips were treated with vehicle or 5 mM imidazole for 6 h, and
then stained with the pH-sensitive fluorescent dye AO (5 μg/ml in
PBS) at 37°C for 10 min. After washing twice with PBS, samples were
viewed under a confocal fluorescence imaging microscope (Nikon
D-Eclipse C1; Nikon, Tokyo, Japan) equipped with an 60× oil
immersion objective, and confocal images were acquired using EZ-C1
software (version 3.90; Nikon).
Immunofluorescence
The HEC-1B cells were seeded on glass coverslips and
then treated with vehicle or 5 mM imidazole for 6 h, then fixed in
4% paraformaldehyde and permeabilized with 0.3% Triton X-100 in
PBS. For blockage of non-specific binding sites, cells were
incubated with 5% normal goat serum in PBS for 1 h at room
temperature. The anti-LAMP-1 antibody (1:100 dilution in the PBS
containing 5% bovine serum albumin) was incubated with treated
cells at 4°C overnight. After washing three times, cells were
incubated for 1 h with goat anti-rabbit secondary antibody
conjugated to TRITC (Santa Cruz Biotechnology). Counterstaining of
nuclei was done with Hoechst 33342 (Sigma-Aldrich). Cells were
imaged under the confocal microscope and image analysis was
performed using EZ-C1 FreeViewer software (Nikon).
Examination of AVs by transmission
electronic microscopy
The HEC-1B cells, with or without imidazole
treatment, were fixed in 2.5% glutaraldehyde in 0.1 M sodium
cacodylate buffer at 4°C overnight and postfixed with 1% osmium
tetroxide in 0.1 M sodium cacodylate buffer for 1 h at room
temperature. After fixation, cells were dehydrated in a gradient of
30–100% acetone and embedded in SPI-Pon 812 resin. Ultrathin
sections were obtained using a microtome (Leica UC6i; Leica
Microsystems, Wetzlar, Germany). After staining with lead
citrate/uranyl acetate, sections were examined under a transmission
electron microscope (JEM-1230; JEOL, Japan) at 80 kV.
Plasmids construction and
transfection
The full length of human microtubule-associated
protein 1 light chain 3 (LC3; GenBank accession no. NM_022818.4)
was amplified by PCR using forward (5′-TTCTCGAGCTATGCCGTCGGAGA
AGA-3′) and reverse (5′-AAGGATCCTTAC ACTGACAATTT CATCC-3′) primers,
and the HEC-1B cDNA library as template. The PCR fragment of LC3
digested with XhoI and BamHI (New England Biolabs,
Ipswich, MA, USA) was cloned into pEGFP-C1 plasmid (Clontech, Palo
Alto, CA, USA) for generation of enhanced green fluorescent
protein-LC3 (EGFP-LC3) transfection vector. A plasmid that encoded
human FoxO3a (GenBank accession no. NM_001455.3) tagged with EGFP
(FoxO3a-EGFP) was constructed into pEGFP-N1 plasmid (Clontech) at
XhoI and BamHI sites using the same cDNA library and
specific forward (5′-AACT CGAGATGGCAGAGGCACCGGCT-3′) and reverse
(5′-TTG GATCCTTGCCTGGCACCCAGCTCTG-3′) primers in PCR. All
constructs were verified by sequencing. The mRFP-GFP-LC3 expression
plasmid was a generous gift from Dr Hou (Key Laboratory of
Zoonosis, College of Veterinary Medicine, China Agricultural
University, Beijing, China). For transient transfection, all
vectors described above were transfected to the cells using
Lipofectamine 2000 (Invitrogen) according to the manufacturer’s
protocols. Twenty-four hours after transfection, cells were treated
with vehicle or imidazole for indicated intervals and viewed with
confocal fluorescence microscopy.
Cell viability assay
The HEC-1B cells were seeded in 96-well plates at a
density of 1×104 cells per well. After imidazole
treatment, cell viability was assessed using
3-(4,5-dimethyl-thiazol- 2-yl)-2,5-diphenyl tetrazolium bromide
(MTT) dye (Sigma-Aldrich) absorbance and expressed as the
percentage of non-treated cells. The MTT assay was done as
described (30). Briefly, after
exposure of cells to imidazole, culture media were changed by
serum-free culture media. Then, MTT dissolved in PBS was added to
each well for 4 h at 37°C. After this interval, the culture media
containing MTT were discarded and DMSO was added to each well,
dissolving the precipitate. The optical density was measured at 570
nm spectral wavelength using a microplate reader (Bio-Rad,
Hercules, CA, USA).
Reverse transcriptase-polymerase chain
reaction (RT-PCR)
After imidazole treatment, total RNA was obtained
with a TRIzol reagent (Invitrogen) and quantified by
spectrophotometry (Nanodrop 1000; Thermo Scientific, Waltham, MA,
USA). RevertAid First Strand cDNA Synthesis kit (K1622; Thermo
Scientific) was used to reverse-transcribe RNA into cDNA. PCR was
performed using the Veriti 96-well thermal cycler (Applied
Biosystems, Carlsbad, CA, USA) for 30 cycles. Primer sequences were
as follows: Bim, 5′-ATGGCAAA GCAACCTTCTGA-3′ (forward) and
5′-CGCATATCTGCA GGTTCAGCC-3′ (reverse); GAPDH, 5′-CAAGGTCATCC
ATGACAACTTTG-3′ (forward) and 5′-GTCCACCACCCT GTTGCTGTAG-3′
(reverse).
Gene silencing with siRNA
The HEC-1B cells were grown to 50–60% confluence in
6-well cell culture plates and then transfected with FoxO3a small
interfering RNA (siRNA) (GenePharma, Shanghai, China) using
Lipofectamine 2000, according to the manufacturer’s protocols. The
plates were incubated at 37°C in a CO2 incubator for 6
h, and mixtures were replaced with fresh complete medium and were
incubated for an additional 24 h before silencing efficiency was
measured (western blotting). A non-targeting siRNA was used as a
negative control.
Western blot analysis
Cells lysates were prepared by extracting proteins
with RIPA buffer (Sigma-Aldrich) containing protease cocktail
inhibitor (Roche, Basel, Switzerland). Equal amounts of protein
samples (20 μg) were subjected to sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and transferred to a
nitrocellulose membrane (Millipore, Billerica, MA, USA). After
blocking with 5% non-fat milk in Tris-buffered saline containing
0.1% Tween-20 (TBST), the membrane was incubated with primary
antibodies diluted in blocking solution (1:1,000) at 4°C overnight.
After washing with TBST, the membrane was incubated for an
additional 1 h with the appropriate secondary antibodies conjugated
to horseradish peroxidase at a dilution of 1:5,000. The protein
bands were visualized using an enhanced chemiluminescence detection
system (GE Healthcare, Little Chalfont, Buckinghamshire, UK).
Images of western blotting were processed using ImageJ software
(Wayne Rasband, NIH, Bethesda, MD, USA).
Statistical analyses
Data were presented as mean ± standard deviation of
at least three independent experiments. Statistical significance
was analyzed using the Student’s t-test for comparison between the
means or one-way analysis of variance with post hoc
Dunnett’s test (SAS version 9.1; SAS Institute Inc., Cary, NC,
USA). Differences were considered statistically significant when
P<0.05.
Results
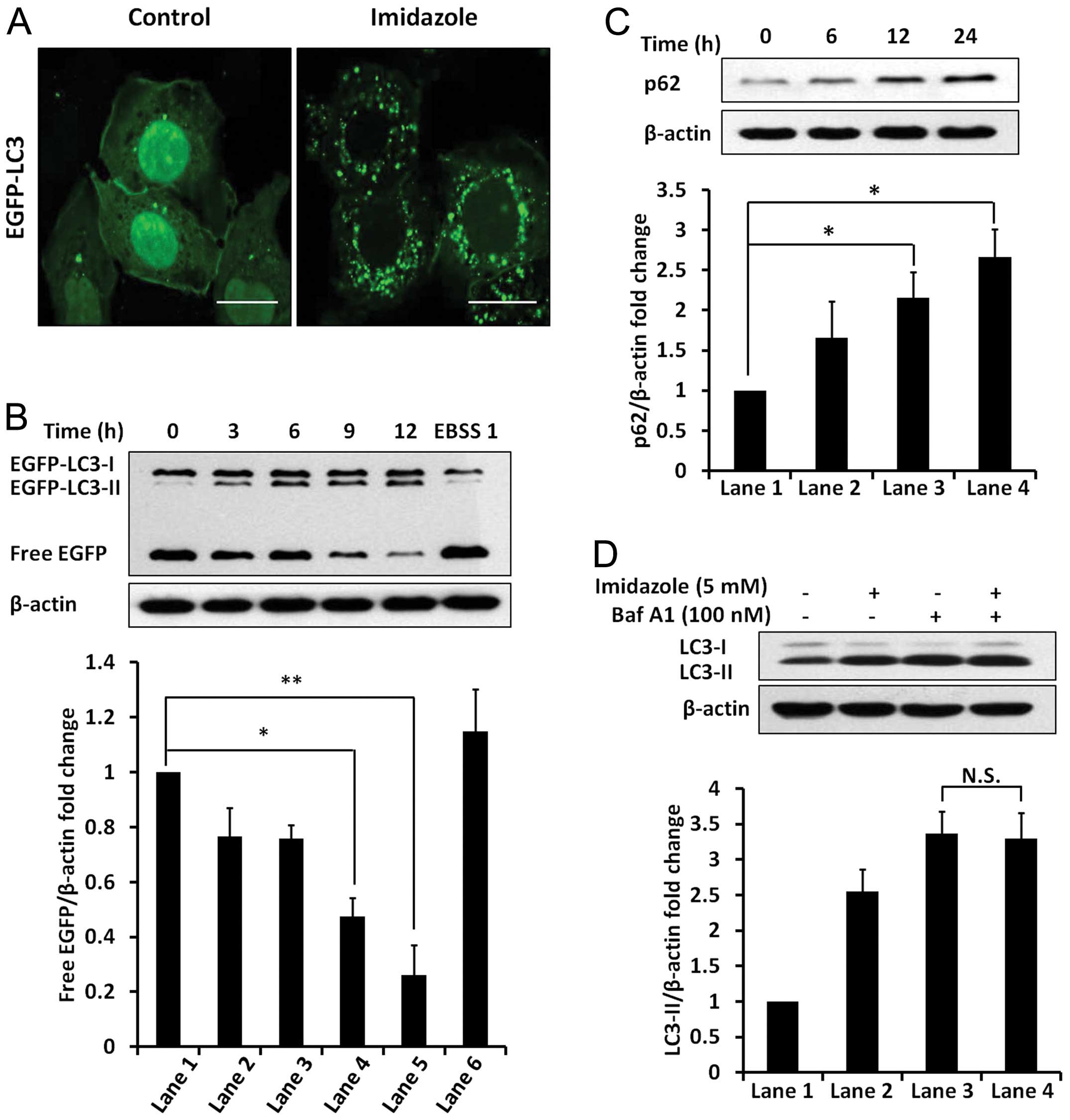
Imidazole induced cytoplasmic
vacuolization and accumulation of AVs in HEC-1B cells
The HEC-1B cells had large vacuoles in their
cytoplasm after addition of 5 mM imidazole (Fig. 1A, top row). Staining the
imidazole-treated cells with AO was consistent with an increase of
the acidic organelle (Fig. 1A,
middle row). Further, lysosomes were stained using lysosome
associated membrane protein (LAMP)-1 directed antibody, which
revealed an increase in number and in size of lysosomes following
imidazole treatment (Fig. 1A).
Furthermore, imidazole treatment resulted in a significant increase
in the abundance of LC3-II in a dose- and time-dependent manner
(Fig. 1B). Two additional cell
lines, MDA-MB-435S and 293T, displayed a similar increase in the
abundance of LC3-II following treatment with imidazole (Fig. 1C). These results suggested that the
number of autophagosome increased in response to imidazole. This
observation was further supported by TEM analyses showing that the
number of autophagic vacuoles (AVs) was markedly increased in
cytoplasm following treatment with imidazole (Fig. 1D). Taken together, these data
indicated that the accumulation of AVs was induced in
imidazole-treated cells.
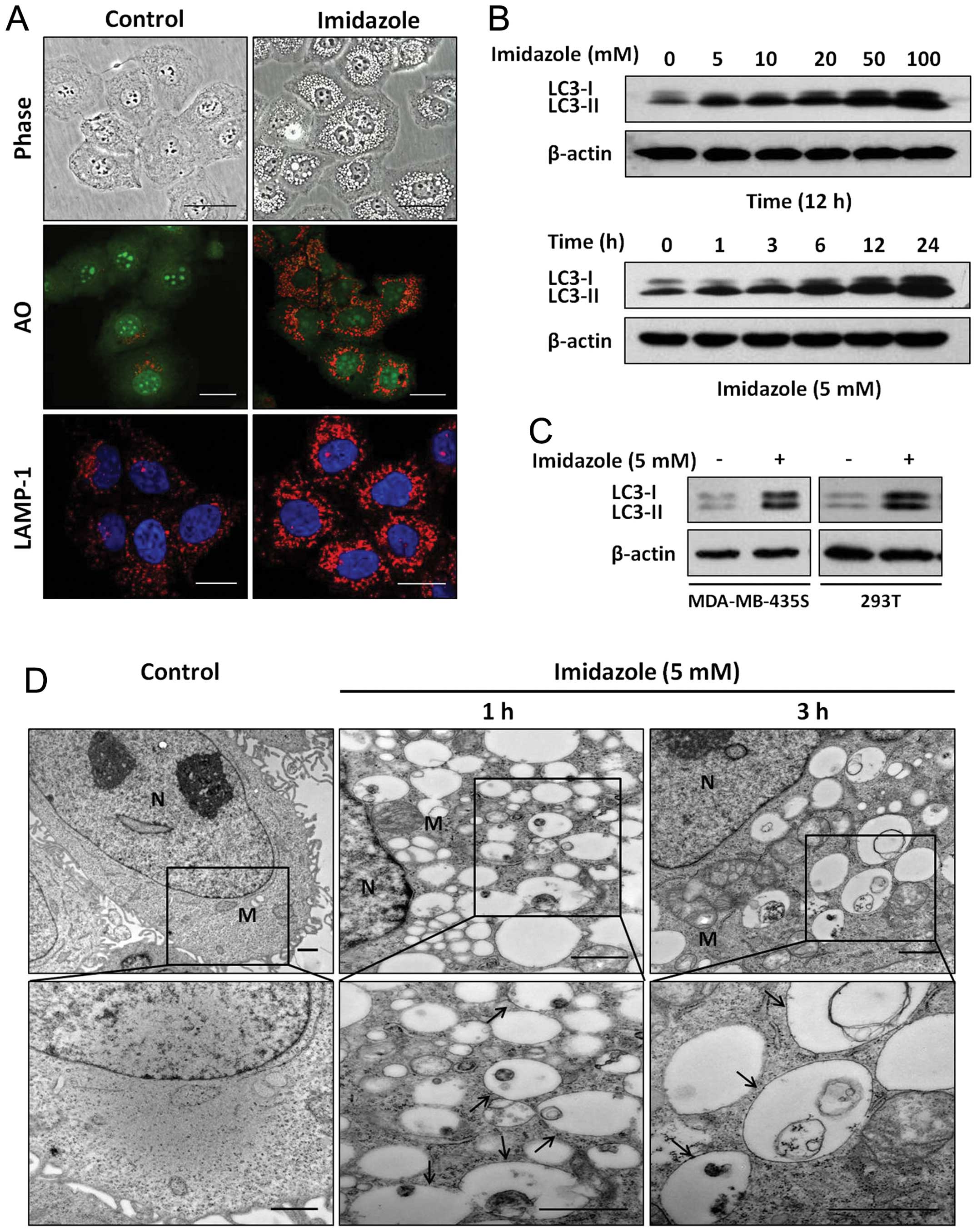 | Figure 1Imidazole treatment induces cellular
vacuolization and accumulation of AVs. (A) Vacuoles formation in
imidazole-treated cells. The HEC-1B cells with or without 5 mM
imidazole treatment for 6 h were observed (phase-contrast
microscopy, ×400 magnification). The distribution of acidic
organelles was examined by AO staining and visualized with various
channels using confocal microscopy (x400 magnification). Cells were
fixed, permeabilized and analyzed using polyclonal antibody against
LAMP-1 to recognize lysosomes. Hoechst staining was used to
identify nuclei. AO, acridine orange. Bars, 20 μm. (B) Imidazole
increased accumulation of LC3-II in a dose- and time-dependent
manner. Cells that were treated with increasing amounts of
imidazole or in a certain concentration of imidazole for indicated
intervals were examined by western blotting with anti-LC3 antibody,
and β-actin was used as loading control. (C) MDA-MB-435S and 293T
cell lines were treated with or without imidazole for 12 h, and the
conversion of LC3-I to LC3-II was monitored by western blotting.
(D) Ultrastructures of HEC-1B cells with or without imidazole
treatment. Representative TEM images are presented at ×12,000 or
×25,000 magnification. N, nucleus; M, mitochondrion; arrows
indicate AVs. Bars, 1 μm. Comparable results were obtained in at
least three separate experiments. |
Imidazole blocked autophagic
degradation
Autophagy flux was monitored using EGFP-LC3 in
HEC-1B cells treated with imidazole. The addition of imidazole
significantly increased the number of EGFP-LC3 puncta (Fig. 2A) and in the abundance of
EGFP-LC3-II protein (Fig. 2B).
These results further indicated the accumulation of AVs in response
to imidazole. Based on immunoblot analysis of the free EGFP
fragment, there was a time-dependent reduction in imidazole-treated
cells (Fig. 2B). In contrast,
induction of autophagy in response to EBSS increased free EGFP
fragment. Furthermore, the p62 levels were markedly increased in
response to imidazole treatment (Fig.
2C). In addition, dual treatment with imidazole and Baf A1 did
not result in any significant change in the abundance of LC3-II,
compared to Baf A1 treatment alone (Fig. 2D), suggesting that imidazole
functions were similar to Baf A1 by blocking autophagic flux.
Therefore, we attributed the imidazole-induced accumulation of AVs
to the blockage of autophagic degradation rather than increased
induction of autophagy.
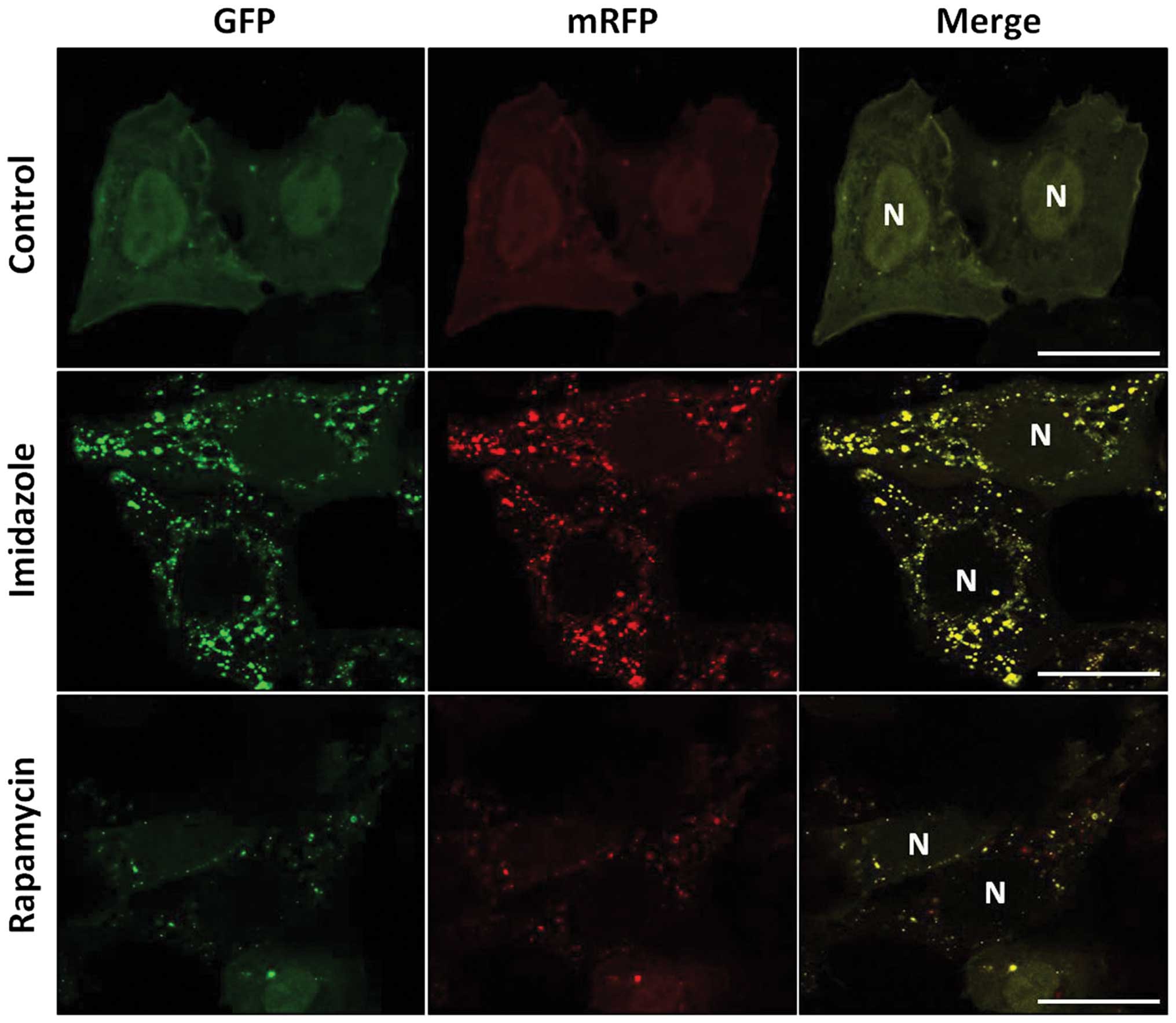
Imidazole impaired the maturation of
autophagosome into autolysosome
To address whether imidazole treatment affects
maturation of autophagosome, autolysosome formation was measured by
an mRFP-GFP-LC3 tandem construct. The low pH inside the lysosome
quenched the fluorescent signal of GFP; however, RFP has more
stable fluorescence in acidic compartments. Thus, autophagosomes
and autolysosomes are labeled with yellow (mRFP and GFP) and red
(mRFP only) signals, respectively (31). In the present study, imidazole
considerably increased the yellow puncta numbers without a
concomitant increase in red puncta (Fig. 3). In contrast, both yellow and red
puncta were increased in cells treated with rapamycin, a known
autophagy inductor. We inferred that autophagosome maturation into
autolysosome was blocked in the presence of imidazole.
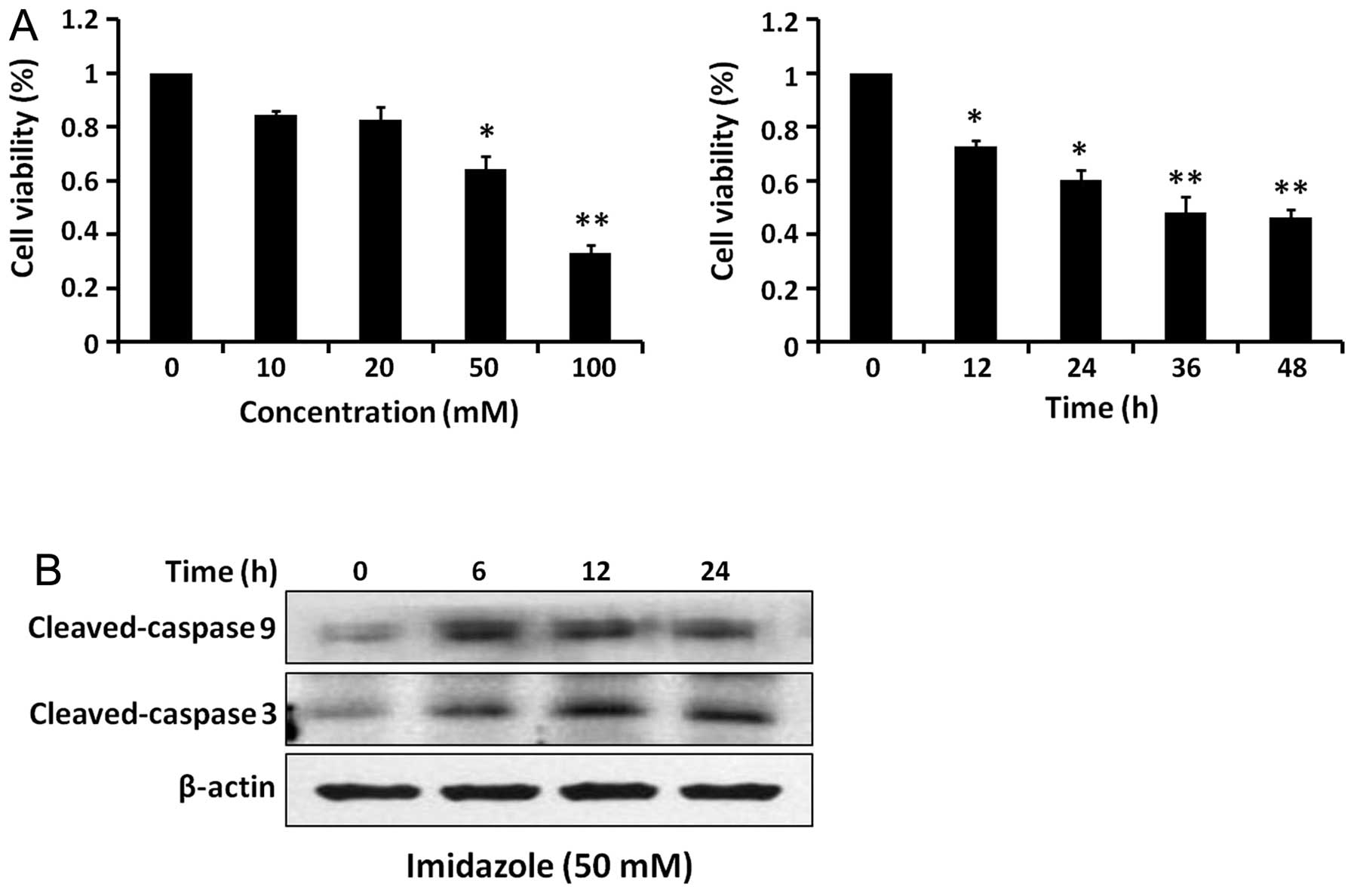
Effect of imidazole treatment on cell
viability
Imidazole reduced HEC-1B cell viability in a dose-
and time-dependent manner (Fig.
4A). Based on western blot analysis, both cleaved-caspase 9 and
cleaved-caspase 3 levels increased after imidazole treatment
(Fig. 4B). Therefore, imidazole
treatment induced intrinsic apoptotic cell death in HEC-1B
cells.
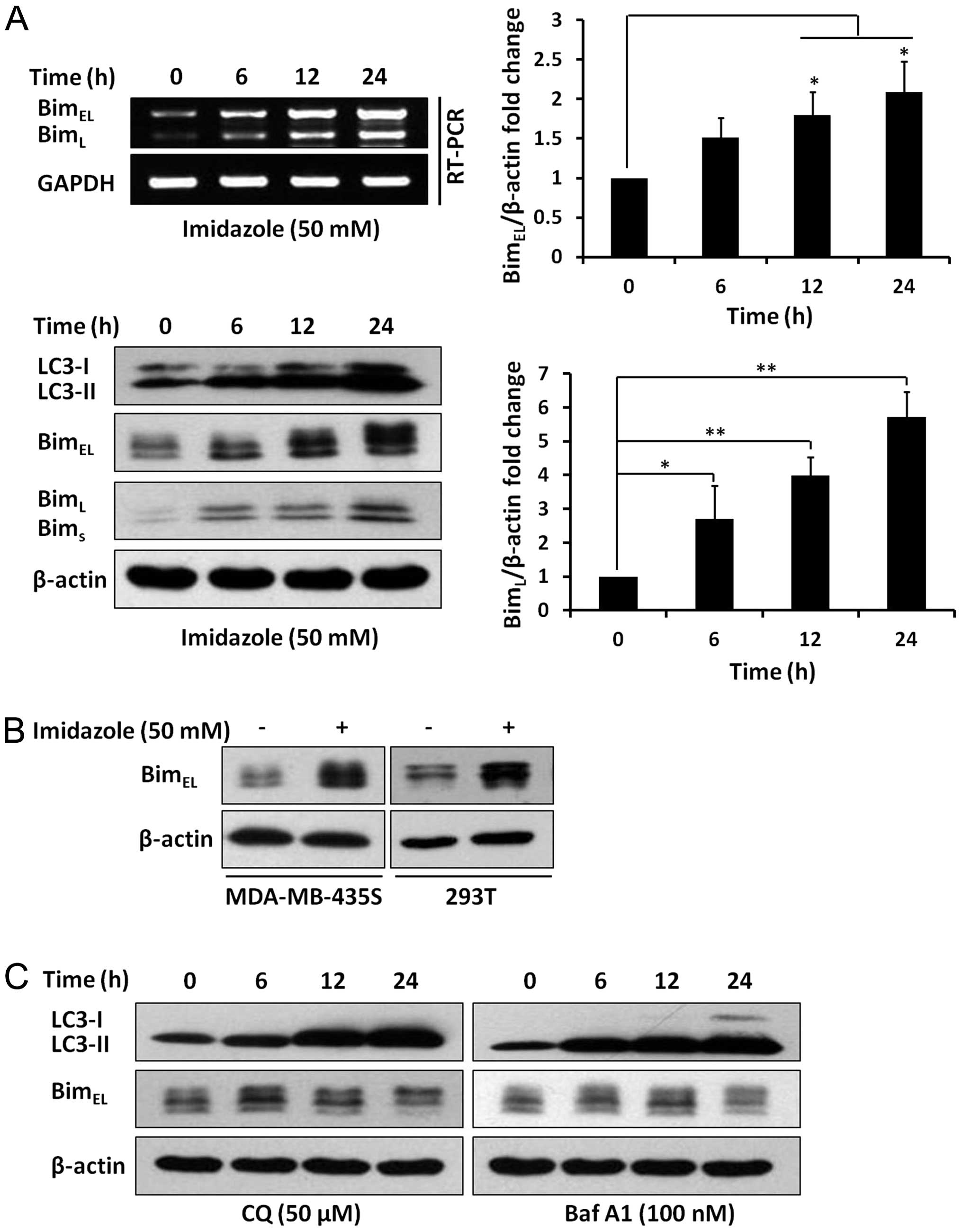
Imidazole treatment increased Bim
expression
BimEL and BimL protein levels
were dramatically increased after imidazole treatment (Fig. 5A). Western blotting results were
consistent with RT-PCR data (Fig.
5A), demonstrating that imidazole treatment increased Bim
expression, both at transcriptional and translational levels.
Furthermore, two additional cell lines, MDA-MB-435S and 293T,
displayed a similar increase in the abundance of BimEL
following treatment with imidazole (Fig. 5B). In addition, LC3-II levels were
markedly increased in response to CQ or Baf A1; however, the
BimEL levels were not obviously affected by CQ or Baf A1
treatment (Fig. 5C). Taken
together, we concluded that imidazole-induced Bim upregulation was
independent of the blockage of autophagic degradation.
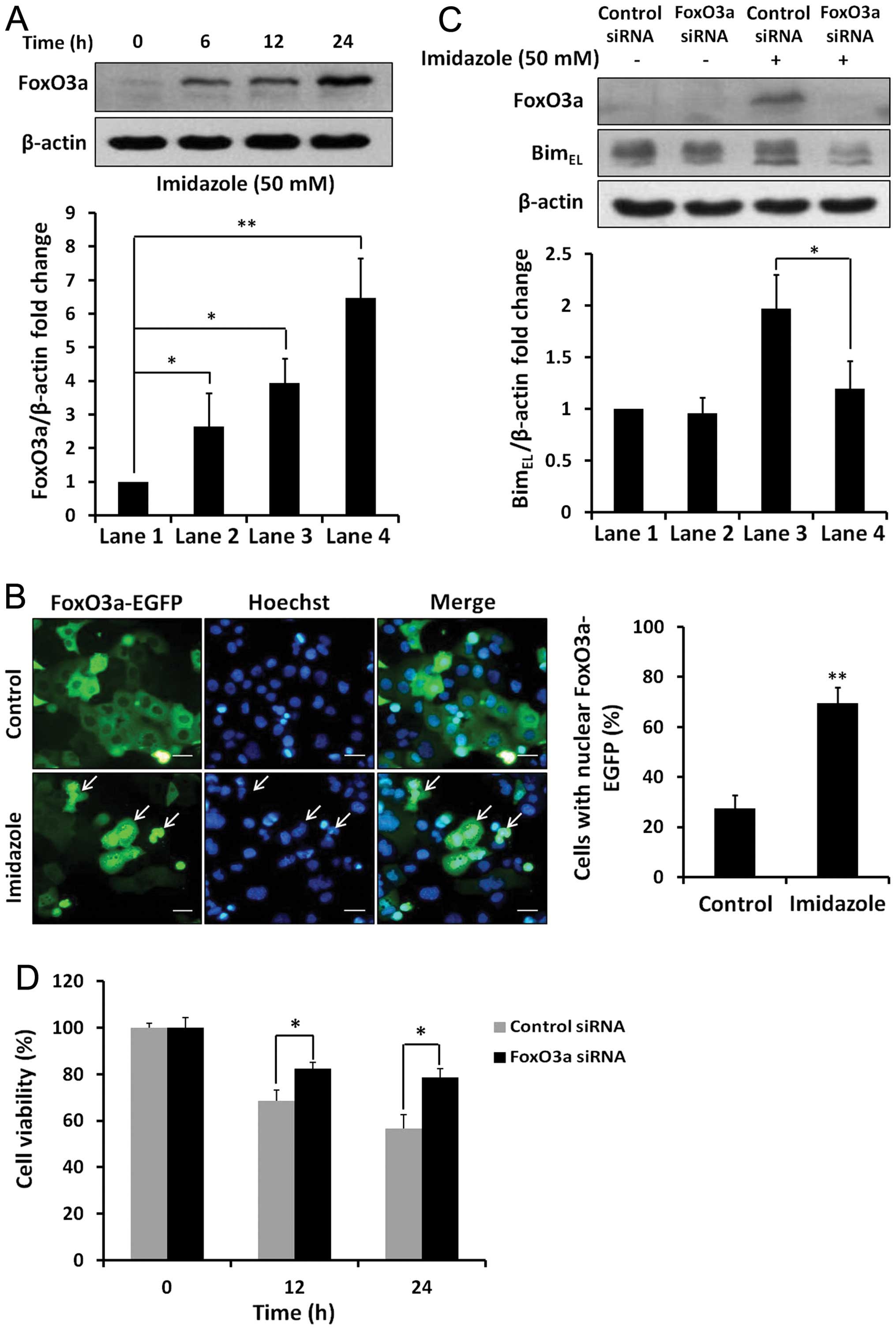
FoxO3a was involved in regulation of Bim
expression in respone to imidazole
Imidazole treatment increased FoxO3a protein levels
in HEC-1B cells (Fig. 6A). Using
fluorescence microscopy, the dynamic translocation of FoxO3a-EGFP
to the nucleus after imidazole treatment was visualised, whereas
FoxO3a-EGFP was located in the cytosol in control cells (Fig. 6B). Further, imidazole treatment for
24 h increased BimEL protein levels in negative control
cells, whereas FoxO3a knockdown abrogated the imidazole-induced
expression of Bim protein (Fig.
6C). Thus, FoxO3a was involved in regulating the transcription
of Bim in imidazole-treated cells. In addition, imidazole-induced
cell death was obviously attenuated by siRNA-mediated inhibition of
FoxO3a (Fig. 6D). Taken together,
we inferred that the transcription factor FoxO3a contributed to the
imidazole-induced Bim upregulation and apoptosis in HEC-1B
cells.
Discussion
In this study, we demonstrated that imidazole
potently inhibited autophagy by blocking autophagic degradation in
HEC-1B cells. Simultaneously, imidazole treatment induced apoptosis
in HEC-1B cells by activation of caspase 9 and 3. The proapoptotic
effect was mediated by increased expression of the BH3-only protein
Bim. Furthermore, imidazole upregulated the protein level of
transcription factor Foxo3a and induced its increased nuclear
localisation. Silencing experiments with FoxO3a siRNA prevented Bim
upregulation and cell death.
Autophagy is a cell-survival pathway involving
degradation and recycling of long-lived proteins, protein
aggregates, damaged cytoplasmic organelles, and intracellular
pathogens (9). Normally, it is a
tumor suppressor pathway, which may facilitate degradation of
oncogenic molecules, thereby preventing development of cancers.
However, autophagy appears to have a dual role in cancer, as it
also promotes survival of tumor cells under stress conditions,
e.g., hypoxic or low-nutrition environments (32). Genetic or pharmacological
inhibition of autophagy can increase cancer cellular sensitivity to
various anticancer therapies, such as DNA-damaging agents,
antihormones and radiation (33).
Therefore, inhibition of autophagy is therapeutically beneficial
for anticancer treatments. In our experiments, we systematically
studied the effects of imidazole on autophagic events in HEC-1B
cells; in these studies, the number of AVs was markedly increased
in imidazole-treated HEC-1B cells. Based on examination of several
endpoints related to the autophagy flux (31), we demonstrated that accumulation of
AVs was due to inhibition of autophagic degradation rather than
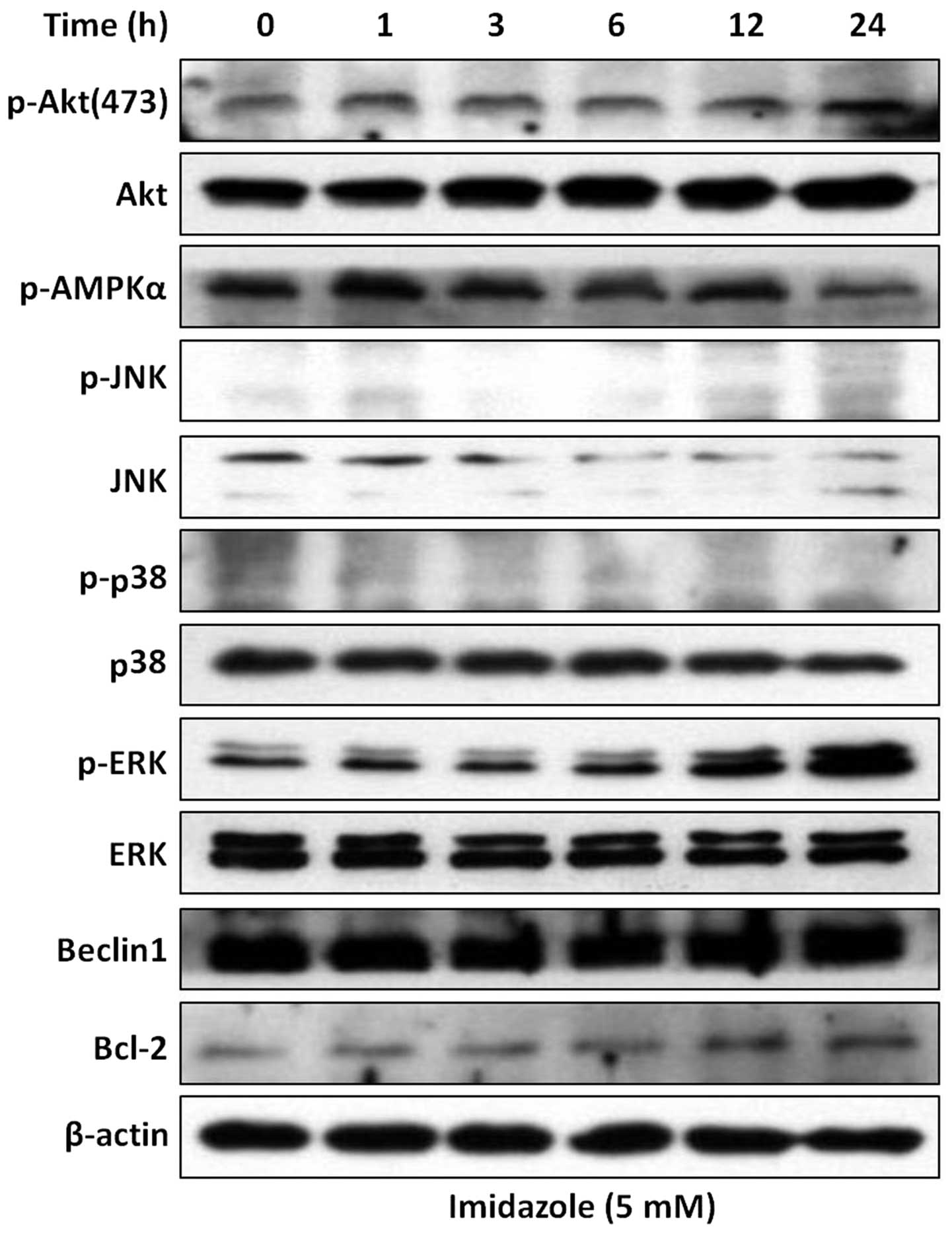
induction of autophagic flux. In addition, several protein kinases
and core molecular proteins, required for induction of autophagy
such as Akt, AMPK, mitogen-activated protein kinase (ERK, p38 and
JNK) (34), Beclin 1 and Bcl-2
(35), were not obviously changed
after imidazole treatment (Fig.
7). Although the levels of phospho-ERK were increased in
response to imidazole treatment, ERK pathway is involved in the
control of autophagy process at the maturation step rather than the
initiation step (36). These
results further supported the concept that imidazole functions as
an inhibitor of autophagy by blocking autophagic degradation. Thus,
as a potent autophagy inhibitor, imidazole may be expected to have
therapeutic effects on cancers with high basal autophagy.
It has been reported that lysosomotropic agents
induce accumulation of AVs by blocking autophagic degradation, such
as chloroquine (CQ) (37) and
matrine (38). They can enter into
lysosomes and block trafficking and proteolytic activation of
lysosomal proteases. As a weak base, imidazole may enter into acid
organelles and alter the intralysosomal pH (19,39).
Thus, we propose that entrapment of imidazole in the lysosomes
elevated their pH, which caused lysosome dysfunction and inhibition
of autophagic degradation. This conclusion was supported by the
important observation that the autolysosome maturation, depending
on the acidification and degradation capacity of the lysosome, was
inhibited after imidazole treatment.
Conversely, it has been reported that imidazole can
induce cell apoptosis (29), and
its derivatives have anticancer activity in a variety of malignant
cells (40). However, the
molecular mechanisms underlying imidazole-induced cell apoptosis
are not fully elucidated. It is known that BH3-only protein Bim can
trigger cytochrome c release and activation of caspase 9,
which consequentially causes cellular apoptosis (24). In our experiments, imidazole
treatment caused HEC-1B cell apoptosis accompanied by the
activation of caspase 9 and 3. Importantly, the pro-apoptotic
molecule Bim was obviously increased both at mRNA and protein
levels after imidazole treatment. It is well established that Bim
has an important role in the anoikis of a variety of cancer cells,
such as lung cancer, breast cancer, osteosarcoma and melanoma
(26–28). The absence of Bim leads to the
occurrence of tumor metastasis and acquisition of chemotherapy
resistance (24). Therefore, Bim
has attracted increasing attention as a plausible target for tumor
therapy. Various chemotherapeutic agents use Bim as a mediating
executioner of cell death (e.g., imatinib, gefitinib and
bortezomib) (24). Based on the
effects of Bim in tumorigenesis and tumor treatment, our findings
provided evidence that imidazole may be used as a Bim-targeting
agent for tumor therapies in the future, especially tumor
metastasis and chemoresistance.
Inhibiting maturation of autolysosome by LAMP-2
knockdown caused accumulation of AVs and subsequent apoptosis in
HeLa cells (41). Similarly,
hydroxychloroquine, a derivate of CQ, induced AVs accumulation and
triggered mitochondria-mediated apoptosis in HeLa cells (42). As described above, imidazole
treatment also induced accumulation of AVs by blocking autophagic
degradation in HEC-B cells. The blockage of autophagic degradation
might have triggered the apoptotic procedure by upregulation of
Bim. In this study, two kinds of autophagy inhibitors CQ and Baf
A1, both known to inhibit autophagic degradation (37,43),
were used to investigate the role of blockage of autophagic
degradation in the regulation of Bim expression. We observed that
there were no significant changes in the Bim protein levels after
CQ or Baf A1 treatment in HEC-1B cells. Therefore, we inferred that
imidazole-induced Bim expression was independent of the blockage of
autophagic degradation. Furthermore, the reciprocal influence of
AVs accumulation and apoptotic cell death still require further
investigation.
The pro-apoptotic activity of Bim is tightly
controlled by transcriptional and post-transcriptional systems
(24). The forkhead-like
transcription factor FoxO3a (forkhead box O3a) is a key
transcriptional regulator of Bim (44). Cytokine withdrawal or apoptotic
stimuli cause upregulation of Bim through activation of FoxO3a in
various cell types, including osteoblasts, hepatocytes and neurons
(45–47). It has been demonstrated that the
FoxO3a-Bim pathway participates in apoptotic processes in response
to many chemotherapeutic agents. For example, paclitaxel can induce
BimEL expression and subsequently cell apoptosis via
increasing FoxO3a expression in MCF-7 breast cancer cells (48). Similarly, melatonin can induce
apoptosis in HepG2 hepatocarcinoma cells through the upregulation
of Bim mediated by nuclear translocation and activation of the
transcription factor FoxO3a (49).
In the present study, imidazole upregulated the protein level of
Foxo3a and induced its increased nuclear localisation, suggesting a
possible association between FoxO3a as a transcription factor of
Bim in HEC-1B cells. Furthermore, FoxO3a accumulation and Bim
protein expression were greatly reduced upon silencing of FoxO3a by
siRNA, validating that FoxO3a functions as a transcriptional
regulator of Bim expression after imidazole treatment. Moreover,
depletion of FoxO3a by siRNA significantly reduced
imidazole-mediated cell death. Therefore, our findings provide
evidence that FoxO3a-Bim pathway has a critical role in
imidazole-induced apoptosis in HEC-1B cells. However, the knockdown
of FoxO3a did not completely abolish imidazole-induced cell death.
Perhaps imidazole might induce other mechanisms to promote
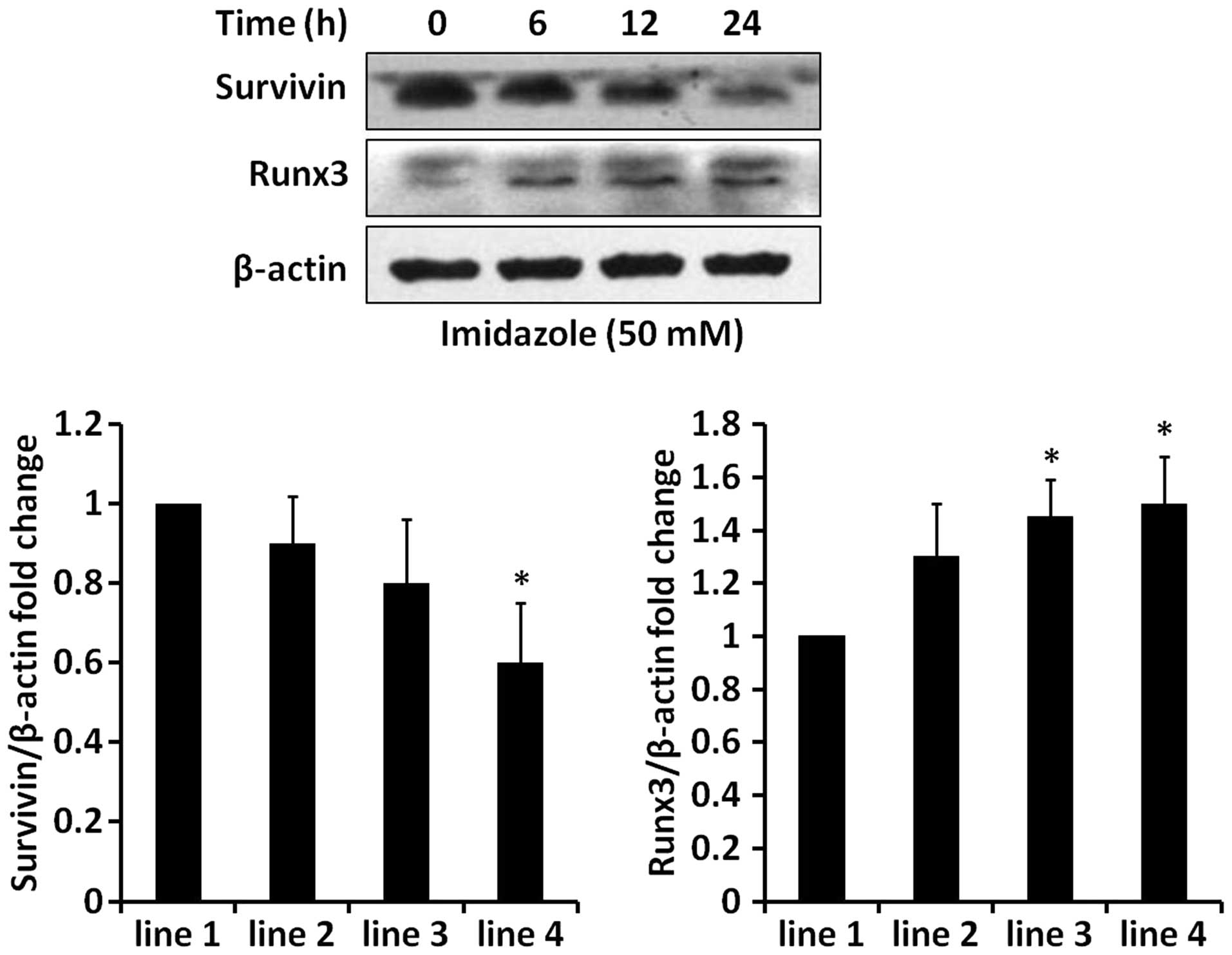
apoptosis in HEC-1B cells. Indeed, we also observed that imidazole
treatment caused an obvious change in several apoptosis-related
molecules, e.g., tumor suppressor Runx3 and anti-apoptotic protein
survivin (Fig. 8). These changes
of apoptosis-related molecules probably contributed to
imidazole-induced cell death.
In conclusion, this is the first report that
imidazole is a potent inhibitor of autophagy flux by blocking
autophagic degradation; simultaneously, imidazole induced apoptosis
by FoxO3a-Bim pathway in HEC-1B cells. Our data provided a
molecular link between imidazole drugs and anticancer therapies.
These findings could be an important advance for understanding the
oncostatic effects of imidazoles in vitro; however,
additional studies are required to further elucidate the
therapeutic value and clinical applications of imidazole
compounds.
Acknowledgements
We thank Dr John P. Kastelic of the University of
Calgary for useful discussion and suggestions on this
investigation. This study was supported by the National Science
& Technology Pillar Program during the 12th Five-year Plan
Period (2012BAI32B05).
References
|
1
|
Grassmann S, Sadek B, Ligneau X, et al:
Progress in the proxifan class: heterocyclic congeners as novel
potent and selective histamine H(3)-receptor antagonists. Eur J
Pharm Sci. 15:367–378. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Labanauskas L, Brukstus A, Udrenaite E,
Gaidelis P, Bucinskaite V and Dauksas V: Synthesis of
6,7-dialkoxy-2-arylmethylidene-2,3-dihydrobenzo[4,5]imidazo[2,1-b][1,3]thiazol-3-ones
exhibiting anti-inflammatory activity. Pharmazie. 55:429–431.
2000.PubMed/NCBI
|
|
3
|
Sevak R, Paul A, Goswami S and Santani D:
Gastroprotective effect of beta3 adrenoreceptor agonists ZD 7114
and CGP 12177A in rats. Pharmacol Res. 46:351–356. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Can-Eke B, Puskullu MO, Buyukbingol E and
Iscan M: A study on the antioxidant capacities of some
benzimidazoles in rat tissues. Chem Biol Interact. 113:65–77. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hoskin PJ, Saunders MI and Dische S:
Hypoxic radiosensitizers in radical radiotherapy for patients with
bladder carcinoma: hyperbaric oxygen, misonidazole, and accelerated
radiotherapy, carbogen, and nicotinamide. Cancer. 86:1322–1328.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Brown JM: The hypoxic cell: a target for
selective cancer therapy - eighteenth Bruce F. Cain Memorial Award
lecture. Cancer Res. 59:5863–5870. 1999.PubMed/NCBI
|
|
7
|
Baviskar AT, Madaan C, Preet R, et al:
N-fused imidazoles as novel anticancer agents that inhibit
catalytic activity of topoisomerase IIalpha and induce apoptosis in
G1/S phase. J Med Chem. 54:5013–5030. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gunther W, Pawlak E, Damasceno R, Arnold H
and Terzis AJ: Temozolomide induces apoptosis and senescence in
glioma cells cultured as multicellular spheroids. Br J Cancer.
88:463–469. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Levine B and Klionsky DJ: Development by
self-digestion: molecular mechanisms and biological functions of
autophagy. Dev Cell. 6:463–477. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rubinsztein DC, Gestwicki JE, Murphy LO
and Klionsky DJ: Potential therapeutic applications of autophagy.
Nat Rev Drug Discov. 6:304–312. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lum JJ, Bauer DE, Kong M, et al: Growth
factor regulation of autophagy and cell survival in the absence of
apoptosis. Cell. 120:237–248. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Degenhardt K, Mathew R, Beaudoin B, et al:
Autophagy promotes tumor cell survival and restricts necrosis,
inflammation, and tumorigenesis. Cancer Cell. 10:51–64. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Paglin S, Hollister T, Delohery T, et al:
A novel response of cancer cells to radiation involves autophagy
and formation of acidic vesicles. Cancer Res. 61:439–444.
2001.PubMed/NCBI
|
|
15
|
Kanzawa T, Germano IM, Komata T, Ito H,
Kondo Y and Kondo S: Role of autophagy in temozolomide-induced
cytotoxicity for malignant glioma cells. Cell Death Differ.
11:448–457. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gorka M, Daniewski WM, Gajkowska B,
Lusakowska E, Godlewski MM and Motyl T: Autophagy is the dominant
type of programmed cell death in breast cancer MCF-7 cells exposed
to AGS 115 and EFDAC, new sesquiterpene analogs of paclitaxel.
Anticancer Drugs. 16:777–788. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Apel A, Herr I, Schwarz H, Rodemann HP and
Mayer A: Blocked autophagy sensitizes resistant carcinoma cells to
radiation therapy. Cancer Res. 68:1485–1494. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mizushima N: Autophagy: process and
function. Genes Dev. 21:2861–2873. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ohkuma S and Poole B: Cytoplasmic
vacuolation of mouse peritoneal macrophages and the uptake into
lysosomes of weakly basic substances. J Cell Biol. 90:656–664.
1981. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cotter TG: Apoptosis and cancer: the
genesis of a research field. Nat Rev Cancer. 9:501–507. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bold RJ, Termuhlen PM and McConkey DJ:
Apoptosis, cancer and cancer therapy. Surg Oncol. 6:133–142. 1997.
View Article : Google Scholar
|
|
22
|
Strasser A: The role of BH3-only proteins
in the immune system. Nat Rev Immunol. 5:189–200. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bouillet P and Strasser A: BH3-only
proteins - evolutionarily conserved proapoptotic Bcl-2 family
members essential for initiating programmed cell death. J Cell Sci.
115:1567–1574. 2002.PubMed/NCBI
|
|
24
|
Akiyama T, Dass CR and Choong PF:
Bim-targeted cancer therapy: a link between drug action and
underlying molecular changes. Mol Cancer Ther. 8:3173–3180. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ren D, Tu HC, Kim H, et al: BID, BIM, and
PUMA are essential for activation of the BAX- and BAK-dependent
cell death program. Science. 330:1390–1393. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Simpson CD, Anyiwe K and Schimmer AD:
Anoikis resistance and tumor metastasis. Cancer Lett. 272:177–185.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Uehara N, Matsuoka Y and Tsubura A:
Mesothelin promotes anchorage-independent growth and prevents
anoikis via extracellular signal-regulated kinase signaling pathway
in human breast cancer cells. Mol Cancer Res. 6:186–193. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Woods NT, Yamaguchi H, Lee FY, Bhalla KN
and Wang HG: Anoikis, initiated by Mcl-1 degradation and Bim
induction, is deregulated during oncogenesis. Cancer Res.
67:10744–10752. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Iguchi K, Usui S, Ishida R and Hirano K:
Imidazole-induced cell death, associated with intracellular
acidification, caspase-3 activation, DFF-45 cleavage, but not
oligonucleosomal DNA fragmentation. Apoptosis. 7:519–525. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Denizot F and Lang R: Rapid colorimetric
assay for cell growth and survival. Modifications to the
tetrazolium dye procedure giving improved sensitivity and
reliability. J Immunol Methods. 89:271–277. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Su M, Mei Y and Sinha S: Role of the
crosstalk between autophagy and apoptosis in cancer. J Oncol.
2013:1027352013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang ZJ, Chee CE, Huang S and Sinicrope
FA: The role of autophagy in cancer: therapeutic implications. Mol
Cancer Ther. 10:1533–1541. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sridharan S, Jain K and Basu A: Regulation
of autophagy by kinases. Cancers (Basel). 3:2630–2654. 2011.
View Article : Google Scholar
|
|
35
|
Sinha S and Levine B: The autophagy
effector Beclin 1: a novel BH3-only protein. Oncogene. 27(Suppl 1):
S137–S148. 2008. View Article : Google Scholar
|
|
36
|
Corcelle E, Djerbi N, Mari M, et al:
Control of the autophagy maturation step by the MAPK ERK and p38:
lessons from environmental carcinogens. Autophagy. 3:57–59. 2007.
View Article : Google Scholar
|
|
37
|
Geng Y, Kohli L, Klocke BJ and Roth KA:
Chloroquine-induced autophagic vacuole accumulation and cell death
in glioma cells is p53 independent. Neuro Oncol. 12:473–481.
2010.PubMed/NCBI
|
|
38
|
Wang Z, Zhang J, Wang Y, et al: Matrine, a
novel autophagy inhibitor, blocks trafficking and the proteolytic
activation of lysosomal proteases. Carcinogenesis. 34:128–138.
2013. View Article : Google Scholar
|
|
39
|
Poole B and Ohkuma S: Effect of weak bases
on the intralysosomal pH in mouse peritoneal macrophages. J Cell
Biol. 90:665–669. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Krezel I: New derivatives of imidazole as
potential anticancer agents. Farmaco. 53:342–345. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gonzalez-Polo RA, Boya P, Pauleau AL, et
al: The apoptosis/ autophagy paradox: autophagic vacuolization
before apoptotic death. J Cell Sci. 118:3091–3102. 2005. View Article : Google Scholar
|
|
42
|
Boya P, Gonzalez-Polo RA, Casares N, et
al: Inhibition of macro-autophagy triggers apoptosis. Mol Cell
Biol. 25:1025–1040. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yamamoto A, Tagawa Y, Yoshimori T,
Moriyama Y, Masaki R and Tashiro Y: Bafilomycin A1 prevents
maturation of autophagic vacuoles by inhibiting fusion between
autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E
cells. Cell Struct Funct. 23:33–42. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zanella F, Link W and Carnero A:
Understanding FOXO, new views on old transcription factors. Curr
Cancer Drug Targets. 10:135–146. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kawamura N, Kugimiya F, Oshima Y, et al:
Akt1 in osteoblasts and osteoclasts controls bone remodeling. PLoS
One. 2:e10582007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Barreyro FJ, Kobayashi S, Bronk SF,
Werneburg NW, Malhi H and Gores GJ: Transcriptional regulation of
Bim by FoxO3A mediates hepatocyte lipoapoptosis. J Biol Chem.
282:27141–27154. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Gilley J, Coffer PJ and Ham J: FOXO
transcription factors directly activate bim gene expression and
promote apoptosis in sympathetic neurons. J Cell Biol. 162:613–622.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sunters A, Fernandez de Mattos S, Stahl M,
et al: FoxO3a transcriptional regulation of Bim controls apoptosis
in paclitaxel-treated breast cancer cell lines. J Biol Chem.
278:49795–49805. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Carbajo-Pescador S, Steinmetz C, Kashyap
A, et al: Melatonin induces transcriptional regulation of Bim by
FoxO3a in HepG2 cells. Br J Cancer. 108:442–449. 2013. View Article : Google Scholar :
|