Introduction
Lung cancer is the leading cause of cancer-related
death for both men and women worldwide (1). Non-small cell lung cancer (NSCLC)
accounts for ~80% of lung cancers and the prognosis of patients
with advanced NSCLC remains very poor despite advances in treatment
(2).
For most individuals with advanced NSCLC, cytotoxic
chemotherapy is the mainstay of treatment based on the moderate
improvement in survival that it confers. However, the outcome of
chemotherapy in such patients has reached a plateau in terms of the
response rate (25–35%) and overall survival period (OS; 8–10
months) (3,4). One promising treatment strategy
involves the further subdivision of NSCLC into clinically relevant
molecular subsets according to a classification schema based on
specific so-called oncogenic driver mutations. These mutations
occur in genes that encode signal proteins crucial for cellular
proliferation and survival. Thus, cancer might rely on the
expression of these single oncogenes for survival. This concept is
also called oncogene addiction (5). The most prevalent mutated or
rearranged oncogenes identified in NSCLC are KRAS, epidermal
growth factor receptor (EGFR), anaplastic lymphoma kinase
(ALK), and ROS1, among others (6). The identification of EGFR
mutations as one type of oncogenic driver mutation in a subset of
patients with NSCLC, coupled with the development of EGFR
tyrosine kinase inhibitors (EGFR-TKIs), has opened new ways to
treat this disease (7–10). Recently, a novel fusion transcript
with transforming activity that is formed by the translocation of
echinoderm microtubule-associated protein-like 4 (EML4)
(2p21) and ALK (2p23) has been described in a subset of
NSCLCs (11). The EML4-ALK
rearrangement has been identified in 5–10% of NSCLC cases and ALK
inhibitors have shown marked antitumor effects in such tumors
(11–14). However, some of these tumors are
resistant to inhibitors and acquired resistance to ALK inhibitors
has been already found to limit the therapeutic potential of these
agents (15–17); thus, investigations of more
effective strategies are warranted.
Crizotinib was the first clinically available ALK
inhibitor for ALK-positive NSCLC in the world. This drug,
however, was initially designed as a MET inhibitor and also
exhibits ROS1 and RON kinase inhibitory activity (18). In contrast, second-generation ALK
inhibitors (i.e., alectinib and ceritinib) are selective ALK
inhibitors that are effective against second-site mutations of the
ALK domain that induce resistance to crizotinib (19–21).
The MET signal, which is inhibited by crizotinib but not by
alectinib, is dysregulated in many human cancers and promotes tumor
growth, invasion and dissemination. Abnormalities in the MET signal
are reportedly correlated with poor clinical outcomes and drug
resistance in patients with cancer, including lung cancer (22–25).
Especially, MET amplification and its ligand, hepatocyte
growth factor (HGF), have been observed in EGFR-mutated
NSCLC cells that are resistant to EGFR-TKIs (26,27).
In ALK-positive NSCLC, however, the role of the MET signal
remains unclear. In the present study, the role of the MET signal
in ALK-positive NSCLC, especially its inhibition by
alectinib, was investigated.
Materials and methods
Cell cultures, ligands and reagents
The H3122 and H2228 cell lines (NSCLC cell lines
with EML4-ALK rearrangements) were maintained in RPMI-1640
medium with 10% FBS (Sigma-Aldrich, St. Louis, MO, USA). HGF was
purchased from R&D Systems (Minneapolis, MN, USA). Crizotinib
and alectinib (ALK inhibitors) and PHA-665752 (a MET inhibitor)
were purchased from Selleck Chemicals (Houston, TX, USA).
In vitro growth inhibition assay
The growth-inhibitory effects were examined using a
3, 4, 5-dimethyl-2H-tetrazolium bromide assay (MTT; Sigma-Aldrich),
as described previously (28).
Briefly, 180 μl/well of the cell suspension (2,000/well) was seeded
onto 96-well microculture plates and incubated in 10%
FBS-containing medium for 24 h. The cells were treated with
crizotinib or alectinib at various concentrations and were cultured
at 37°C in a humidified atmosphere for 72 h. When the influence of
HGF was investigated, the ligand (10 ng/ml) was added at the same
time. After the culture period, 20 μl of MTT reagent were added and
the plates were further incubated for 2 h. After centrifugation of
the plates, the culture medium was discarded and the wells were
filled with dimethyl-sulfoxide. The absorbance of the cultures at
570 nm was measured using VERSAmax (Japan Molecular Devices, Tokyo,
Japan). The experiment was performed in triplicate.
Antibody
Antibodies specific for ALK, phospho-ALK, MET,
phospho-MET, EGFR, phospho-EGFR, HER2, phospho-HER2, AKT,
phospho-AKT, ERK1/2, phospho-ERK1/2 and β-actin were obtained from
Cell Signaling (Beverly, MA, USA).
Western blot analysis
A western blot analysis was performed as described
previously (28). Briefly,
subconfluent cells were washed with cold phosphate-buffered saline
(PBS) and harvested with lysis A buffer containing 1% Triton X-100,
20 mM Tris-HCl (pH 7.0), 5 mM EDTA, 50 mM sodium chloride, 10 mM
sodium pyrophosphate, 50 mM sodium fluoride, 1 mM sodium
orthovanadate and the protease inhibitor mix Complete™ (Roche
Diagnostics; Basel, Switzerland). Whole-cell lyses were separated
using SDS-PAGE and were blotted onto a polyvinylidene fluoride
membrane. After blocking with 3% bovine serum albumin in a TBS
buffer (pH 8.0) with 0.1% Tween-20, the membrane was probed with
the primary antibody. After rinsing twice with TBS buffer, the
membrane was incubated with a horseradish peroxidase-conjugated
secondary antibody and washed, followed by visualization using an
ECL detection system and LAS-4000 (GE Healthcare, Buckinghamshire,
UK).
Real-time reverse transcription PCR
(RT-PCR)
One microgram of total RNA from cultured cell lines
was converted to cDNA using the GeneAmp RNA-PCR kit (Applied
Biosystems, Foster City, CA, USA). Real-time PCR was performed
using SYBR Premix Ex Taq and Thermal Cycler Dice (Takara, Shiga,
Japan), as described previously (29). The glyceraldehyde 3-phosphate
dehydrogenase (GAPD, NM_002046) gene was used to normalize
the expression levels in subsequent quantitative analyses. The
experiment was performed in triplicate. To amplify the target genes
encoding HGF and MET, the following primers were used:
HGF-F, TTAAACTCCTGGCCT CAAGCAATC; HGF-R,
TCCTATCTTGGGCAAAGCAA CTG; MET-F, TGAGTACCGGAGACAGGTGCAG; and
MET-R, TAGCAGCTTCAACGGCAAAGTTC.
Statistical analysis
Continuous variables were analyzed using the
Student’s t-test and the results were expressed as the average and
standard deviations (SD). The statistical analyses were two-tailed
and were performed using Microsoft Excel (Microsoft, Redmond, WA,
USA). A P-value of <0.05 was considered statistically
significant.
Results
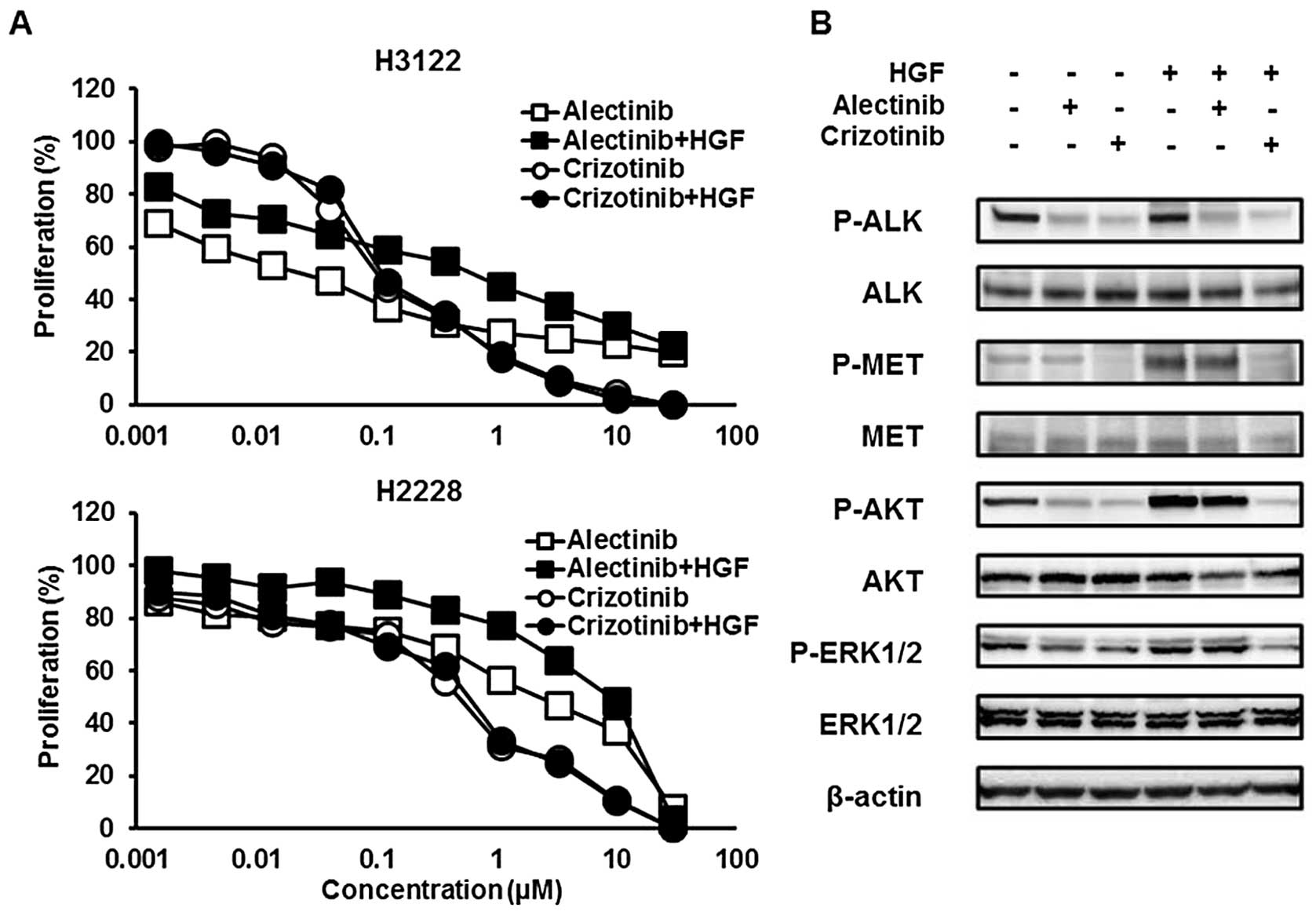
HGF induced resistance to alectinib but
not to crizotinib
Crizotinib exhibits MET inhibitory activity, but
alectinib does not. Then, HGF, a MET ligand, can be associated with
resistance to alectinib, but not to crizotinib, via the MET signal.
To examine the influence of HGF, the sensitivities of H3122 and
H2228 cell lines to crizotinib or alectinib were tested with or
without HGF (10 ng/ml) using an MTT assay. Both cell lines were
sensitive to crizotinib, even in the presence of HGF, whereas these
cell lines were resistant to alectinib in the presence of HGF
(Fig. 1A). The 50% inhibitory
concentrations (IC50) of crizotinib were 0.10 without
HGF and 0.11 with HGF in the H3122 cell line and 0.48 without HGF
and 0.58 μM with HGF in the H2228 cell line, respectively (Table I). The IC50 values of
alectinib were 0.023 without HGF and 0.61 with HGF in the H3122
cell line and 2.26 without HGF and 8.95 μM with HGF in the H2228
cell line, respectively.
 | Table IIC50 values of alectinib
and crizotinib with or without HGF. |
Table I
IC50 values of alectinib
and crizotinib with or without HGF.
| H3122 | H2228 |
|---|
|
|
|
|---|
| HGF (−) | HGF (+) | HGF (−) | HGF (+) |
|---|
| Crizotinib
(μM) | 0.10 | 0.11 | 0.48 | 0.58 |
| Alectinib (μM) | 0.023 |
0.61a | 2.26 |
8.95a |
Crizotinib, but not alectinib, inhibited
the MET signal activated by HGF
Next, to investigate the influence of HGF on the MET
signal, western blot analyses were performed. The cells were
treated with ALK inhibitors for 3 h and HGF was added 30 min before
sample collection. Alectinib (0.05 μM) did not inhibit the
phosphorylation of MET activated by HGF (10 ng/ml), whereas
crizotinib (0.1 μM) inhibited HGF-induced MET phosphorylation
(Fig. 1B). HGF also phosphorylated
two downstream signals, AKT and ERK and this phosphorylation was
cancelled by crizotinib, but not by alectinib. These findings
indicated that HGF is associated with resistance to a selective ALK
inhibitor, alectinib, but not to crizotinib in ALK-positive
NSCLC via the MET signal.
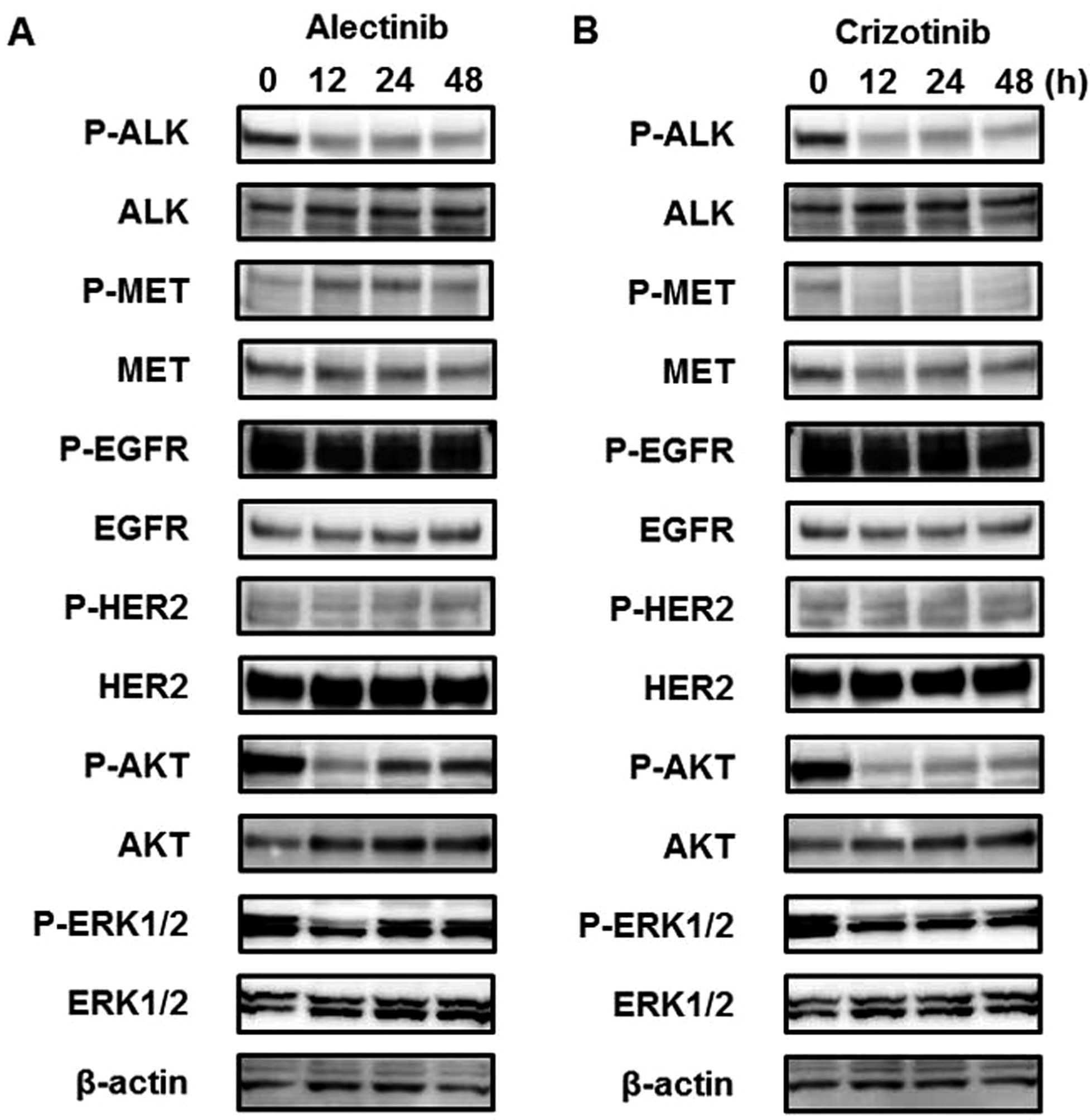
Alectinib activates the MET signal even
without HGF
To investigate the long-term influence of ALK
inhibitors on the MET signal, western blot analyses were performed
in a time-dependent manner. Three hours after treatment with
alectinib (0.05 μM), the phosphorylation of MET was not changed
(Fig. 1B), whereas longer
alectinib treatment periods (12, 24 or 48 h) activated the MET
signal (Fig. 2). Furthermore,
downstream signals, such as AKT and ERK, were also activated along
with the activated MET signal. In contrast to alectinib, crizotinib
(0.1 μM) did not activate but instead inhibited the MET signal
(Fig. 2B). These findings suggest
that the MET signal is activated after treatment with alectinib,
even in the absence of HGF. The mRNA expression levels of
HGF and MET examined using real-time RT-PCR did not
change after alectinib treatment (data not shown). Previous studies
have shown that HER signals are related to resistance to ALK
inhibitors (30,31) and these signals exhibit crosstalk
with the MET signal (22–24). Therefore, the phosphorylation of
EGFR and HER2 were investigated, but no changes were observed
(Fig. 2).
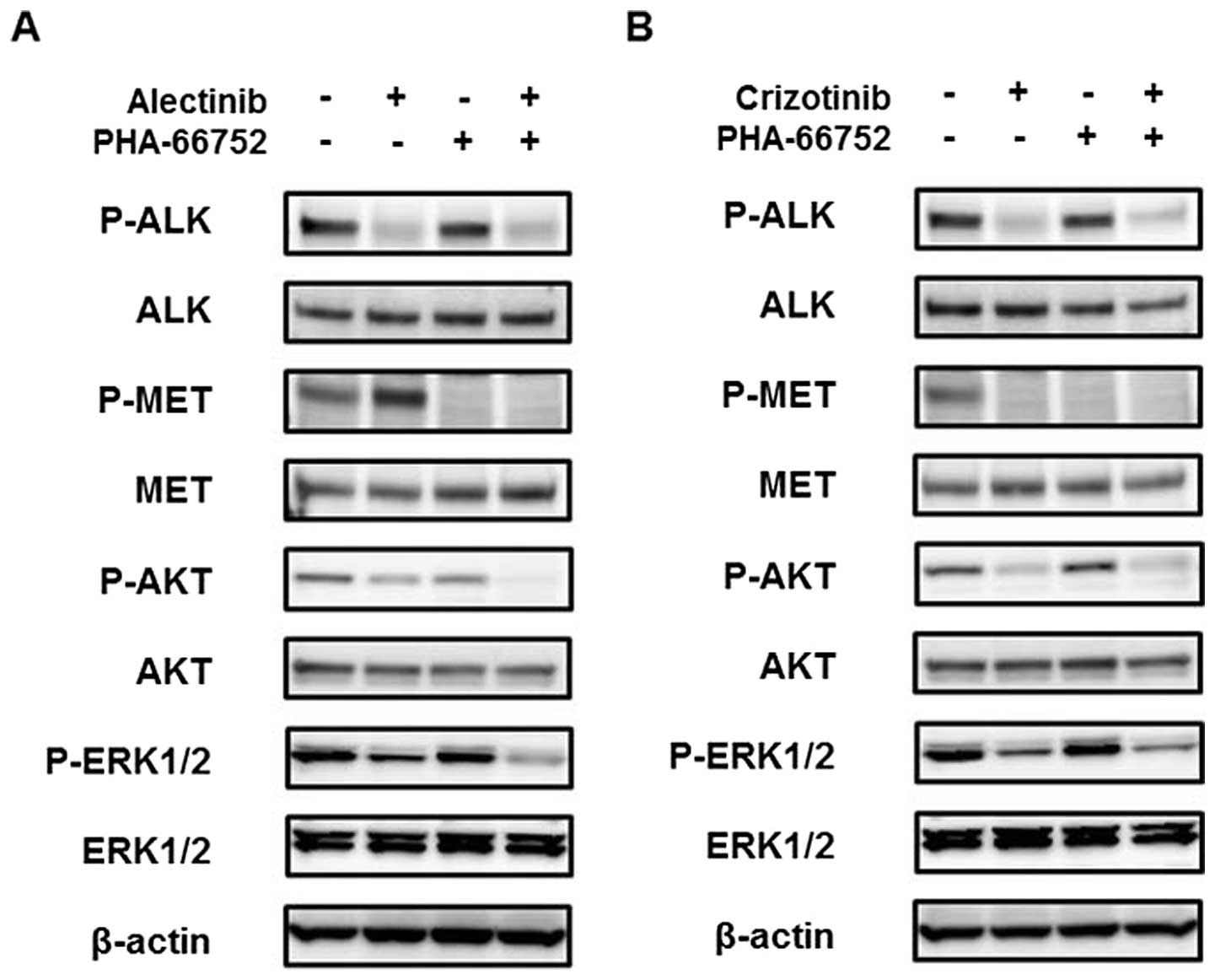
A MET inhibitor enhances the efficacy of
alectinib
To determine whether the inhibition of the activated
MET signal enhances the efficacy of alectinib, combination therapy
consisting of alectinib and a MET inhibitor, PHA-66752, was
evaluated. Neither of the cell lines was sensitive to PHA-66752,
with IC50 values of 5.83 and 3.68 μM for the H3122 and
H2228 cell lines, respectively (Fig.
3A). PHA-66752 (0.5 μM) alone did not inhibit the cellular
growth of the H3122 and H2228 cell lines, but its combination with
alectinib enhanced the efficacy of alectinib (Fig. 3B). In contrast to alectinib, the
combination of PHA-66752 and crizotinib did not enhance the
efficacy of crizotinib (Fig. 3B).
Western blot analyses revealed that the phosphorylation of MET at
12 h after treatment with alectinib was inhibited by PHA-66752, and
the AKT and ERK downstream signals were also inhibited (Fig. 4A). Because the phosphorylation of
MET was inhibited by crizotinib, the addition of PHA-66752 had no
effect on the MET and downstream signals (Fig. 4B). These findings indicated that
the inhibition of the activated MET signal enhanced the efficacy of
alectinib, but not of crizotinib.
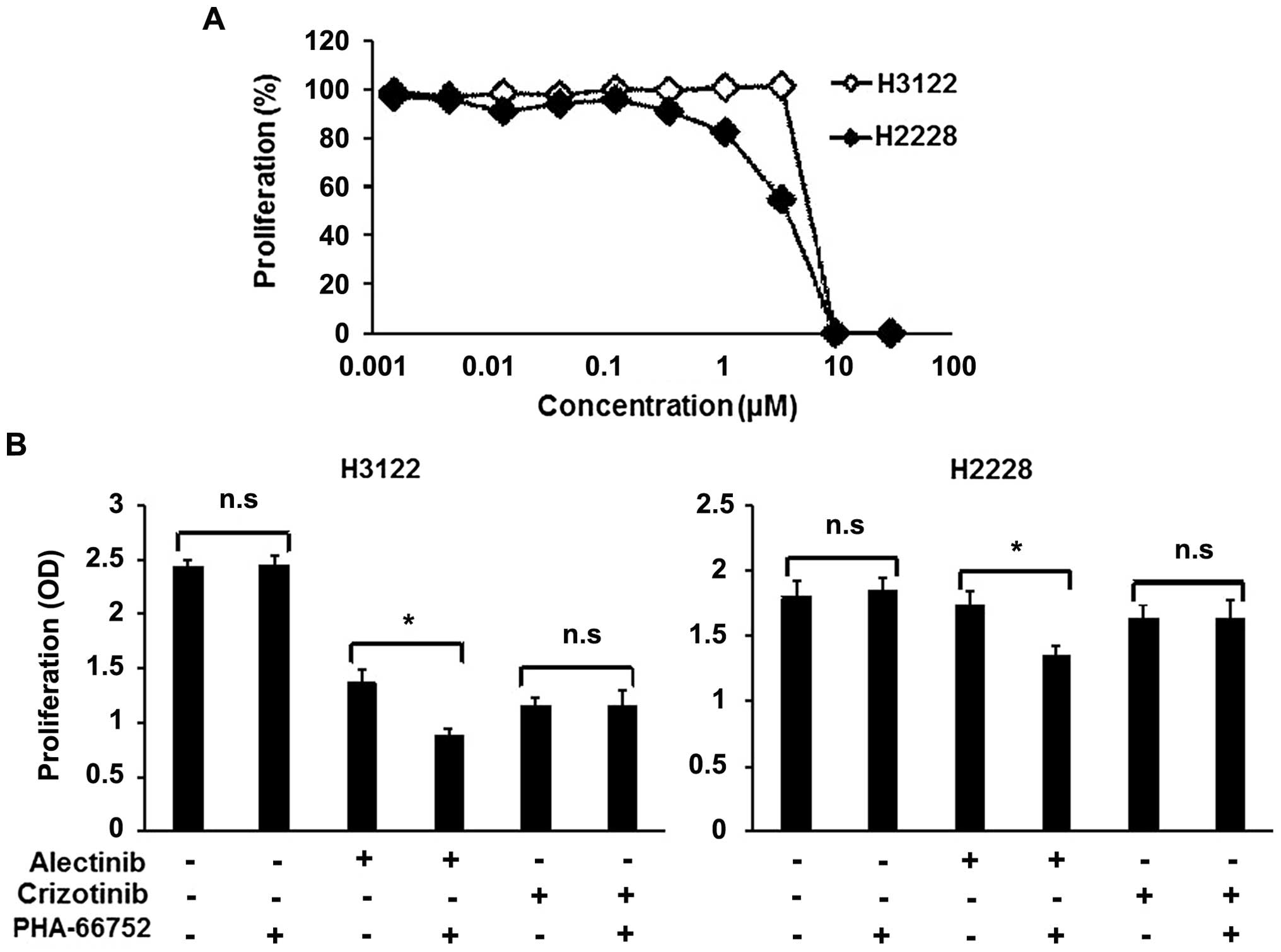 | Figure 3Combination therapy with a MET
inhibitor in ALK-positive cell lines. To examine the
synergistic effect of a MET inhibitor (PHA-66752), we used an MTT
assay. (A) Sensitivity of the ALK-positive cell lines to
PHA-66752. Both the H3122 and H2228 cell lines were not sensitive
to PHA-66752. The IC50 values of H3122 and H2228 were
5.83 and 3.68 μM, respectively. Lines, mean of independent
triplicate experiments. (B) Synergistic effect of ALK inhibitors
and a MET inhibitor. Alectinib, crizotinib and PHA-66752 were used
at concentrations of 0.05, 0.1 and 0.5 μM, respectively. PHA-66752
alone did not influence proliferation in either the H3122 or H2228
cell line. In contrast, PHA-66752 enhanced the efficacy of
alectinib, but not of crizotinib, in both cell lines. Columns, mean
of independent triplicate experiments; error bars, SD;
*P<0.05. |
Discussion
Alectinib has been identified as a potent,
selective, ALK inhibitor that is effective against most second-site
mutations of the ALK domain, notably L1196M and C1156Y, with
a 10-fold stronger potency than crizotinib (19,20).
This drug was tested in a phase I/II study that enrolled 46
ALK-positive crizotinib-naive Japanese patients; a response
rate of 94% was reported and the drug seemed to have fewer adverse
events than those associated with crizotinib (14). Therefore, in the future, this drug
might play a central role in the treatment of ALK-positive
NSCLC. Crizotinib, the first available ALK inhibitor, exhibits MET
inhibitory activity, whereas alectinib does not exert such an
activity (18). In this study, we
found that HGF mediated resistance to alectinib, but not to
crizotinib, via the MET signal, that alectinib activated the MET
signal even in the absence of HGF and that the inhibition of MET
enhanced the efficacy of alectinib in ALK-positive NSCLC
cell lines. To the best of our knowledge, this is the first study
to discuss the role of the MET signal in ALK-positive
NSCLC.
Uncontrolled cell survival, growth, angiogenesis and
metastasis are essential hallmarks of cancer (32). HGF and its receptor, MET, have a
causal role in all of these processes, thus providing a strong
rationale for targeting these molecules in cancer. In addition, the
MET signal has been frequently implicated in resistance to targeted
therapies (22–25). Especially, MET amplification
and its ligand, HGF, are found in EGFR-mutated NSCLC cells
that are resistant to EGFR-TKIs (26,27).
ALK-positive NSCLC resistant to crizotinib includes secondary
ALK mutations, ALK gene amplification, the activation
of other kinases and the epithelial-mesenchymal transition
(15–17,30,33).
To overcome these mechanisms, second-generation ALK inhibitors,
HSP90 inhibitors and combinations with other kinase inhibitors have
been tried. In contrast to EGFR-TKI or alectinib, crizotinib
exhibits MET inhibitory activity; therefore, resistance associated
with the MET signal has not been reported. Because of the absence
of MET inhibitory activity, the resistance to alectinib may be
associated with the MET signal and we found that HGF mediated the
resistance to alectinib, but not to crizotinib, via the MET signal.
Similar to our present study, a previous study revealed that
fibroblast-derived HGF-induced MET activation caused resistance to
TEA658 (another ALK inhibitor without MET inhibitory activity) but
not to crizotinib (34). Most
human cancers are composed of cancer cells that coexist with a
variety of extracellular matrix components and cell types,
including fibroblasts. Therefore, alectinib might be less effective
in such environments. Moreover, without HGF stimulation, the MET
signal was activated after the treatment of alectinib and the
inhibition of the activated MET signal enhanced the efficacy of
alectinib. Indeed, a case of alectinib resistance induced by
MET amplification has been reported and a recent study
showed that ALK-positive NSCLC exhibited a high level of MET
expression (35,36). These findings suggest that the MET
signal can salvage the growth and survival of ALK-positive
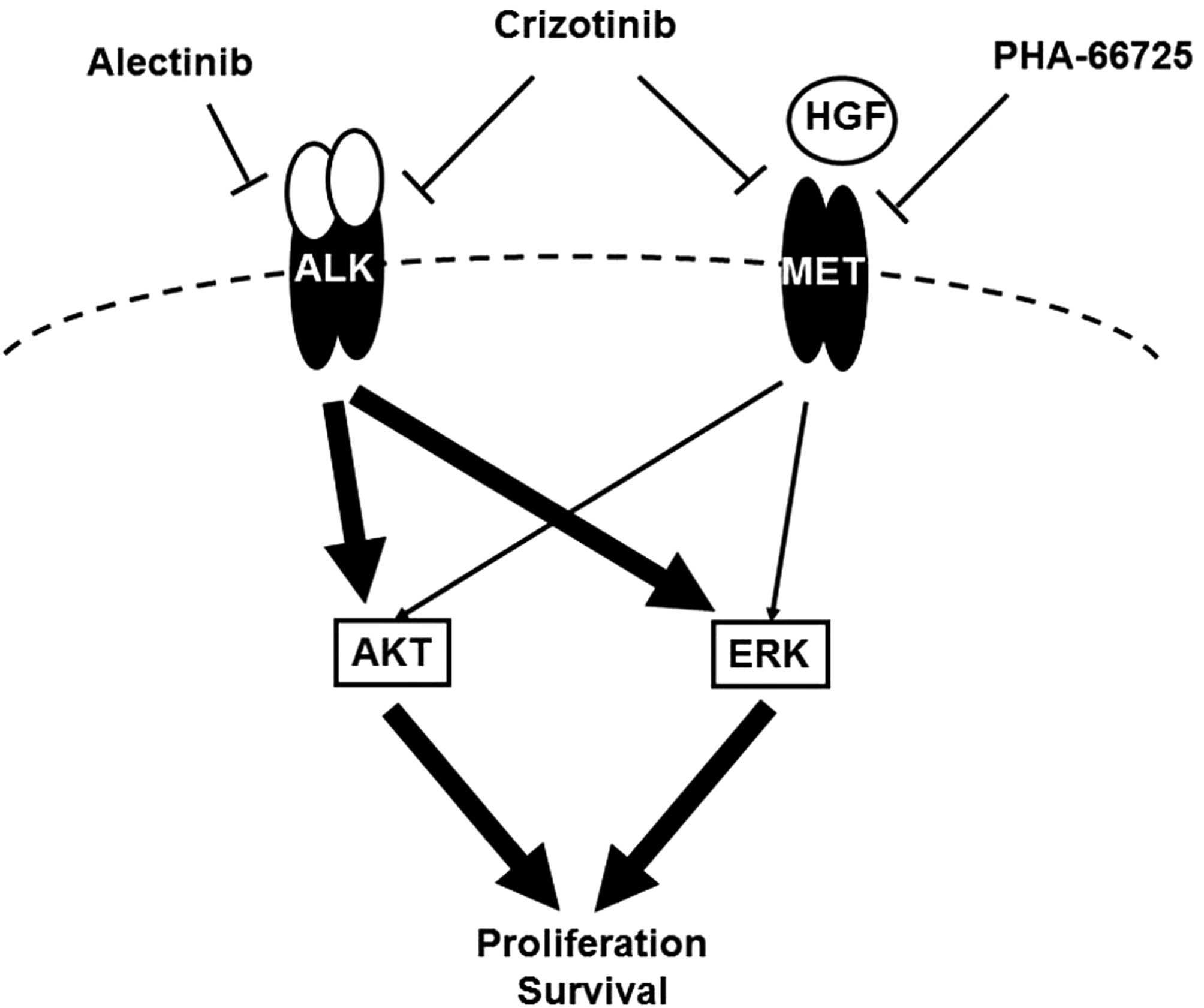
NSCLC after the inhibition of the ALK signal (Fig. 5). The expressions of HGF and MET
after treatment with alectinib were unchanged. Several crosstalk
mechanisms between MET and other signals have been reported
(22–24) and the phosphorylation of HER family
was investigated, but no changes were observed. MET interaction
with many other membrane receptors has been reported and a recent
study demonstrated that EGR1, a transcription factor, could
sustained MET signal after ALK inhibition (24,37).
We speculate that these might be associated with our findings.
Extensive preclinical work has been done on MET
inhibitors, including monoclonal antibodies and kinase inhibitors
and this work has led to further clinical trials examining these
agents (22–25). Clinical trials of combination
therapy with EGFR-TKIs in patients with acquired resistance to
EGFR-TKIs have also been performed. Our experiments showed that the
MET signal salvaged the growth and survival of ALK-positive
NSCLC after treatment with a selective ALK inhibitor, which might
play a central role in the treatment of ALK-positive NSCLC.
In addition, the inhibition of the MET signal enhanced the efficacy
of a selective ALK inhibitor. These findings are expected to
promote novel clinical trials of MET inhibitors.
In conclusion, we found that HGF mediated resistance
to alectinib, but not to crizotinib, via the MET signal, that
alectinib activated the MET signal even in the absence of HGF and
that the inhibition of MET enhanced the efficacy of alectinib in
ALK-positive NSCLC cell lines. These findings suggest that
activated MET acts as a salvage signal in ALK-positive
NSCLC. This novel role of the MET signal in ALK-positive
NSCLC may pave the way for further clinical trials examining MET
inhibitors.
Acknowledgements
We thank Mr. Shinji Kurashimo, Mr. Yoshihiro Mine,
Ms. Eiko Honda, Ms. Tomoko Kitayama, and Ms. Ayaka Kurumatani for
their technical assistance. We thank Dr P.A. Jänne (Department of
Medical Oncology, Dana-Farber Cancer Institute, Boston, MA, USA)
for providing the H3122 cell line. This study was supported by the
Third-Term Comprehensive 10-Year Strategy for Cancer Control and
Grant-in Aid for Japan Society for Promotion of Science
Fellows.
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel R, DeSantis C, Virgo K, et al:
Cancer treatment and survivorship statistics, 2012. CA Cancer J
Clin. 62:220–241. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schiller JH, Harrington D, Belani CP, et
al: Comparison of four chemotherapy regimens for advanced
non-small-cell lung cancer. N Engl J Med. 346:92–98. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ohe Y, Ohashi Y, Kubota K, et al:
Randomized phase III study of cisplatin plus irinotecan versus
carboplatin plus paclitaxel, cisplatin plus gemcitabine, and
cisplatin plus vinorelbine for advanced non-small-cell lung cancer:
Four-Arm Cooperative Study in Japan. Ann Oncol. 18:317–323. 2007.
View Article : Google Scholar
|
|
5
|
Weinstein IB: Cancer: addiction to
oncogenes - the Achilles heal of cancer. Science. 297:63–64. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pao W and Girard N: New driver mutations
in non-small-cell lung cancer. Lancet Oncol. 12:175–180. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Paez JG, Janne PA, Lee JC, et al: EGFR
mutations in lung cancer: correlation with clinical response to
gefitinib therapy. Science. 304:1497–1500. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lynch TJ, Bell DW, Sordella R, et al:
Activating mutations in the epidermal growth factor receptor
underlying responsiveness of non-small-cell lung cancer to
gefitinib. N Engl J Med. 350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pao W, Miller V, Zakowski M, et al: EGF
receptor gene mutations are common in lung cancers from ‘never
smokers’ and are associated with sensitivity of tumors to gefitinib
and erlotinib. Proc Natl Acad Sci USA. 101:13306–13311. 2004.
View Article : Google Scholar
|
|
10
|
Mok TS, Wu YL, Thongprasert S, et al:
Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N
Engl J Med. 361:947–957. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Soda M, Choi YL, Enomoto M, et al:
Identification of the transforming EML4-ALK fusion gene in
non-small-cell lung cancer. Nature. 448:561–566. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kwak EL, Bang YJ, Camidge DR, et al:
Anaplastic lymphoma kinase inhibition in non-small-cell lung
cancer. N Engl J Med. 363:1693–1703. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shaw AT, Kim DW, Nakagawa K, et al:
Crizotinib versus chemotherapy in advanced ALK-positive lung
cancer. N Engl J Med. 368:2385–2394. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Seto T, Kiura K, Nishio M, et al:
CH5424802 (RO5424802) for patients with ALK-rearranged advanced
non-small-cell lung cancer (AF-001JP study): a single-arm,
open-label, phase 1–2 study. Lancet Oncol. 14:590–598. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Choi YL, Soda M, Yamashita Y, et al:
EML4-ALK mutations in lung cancer that confer resistance to ALK
inhibitors. N Engl J Med. 363:1734–1739. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Katayama R, Shaw AT, Khan TM, et al:
Mechanisms of acquired crizotinib resistance in ALK-rearranged lung
Cancers. Sci Transl Med. 4:120ra172012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Katayama R, Khan TM, Benes C, et al:
Therapeutic strategies to overcome crizotinib resistance in
non-small cell lung cancers harboring the fusion oncogene EML4-ALK.
Proc Natl Acad Sci USA. 108:7535–7540. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cui JJ, Tran-Dubé M, Shen H, et al:
Structure based drug design of crizotinib (PF-02341066), a potent
and selective dual inhibitor of mesenchymal-epithelial transition
factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J Med
Chem. 54:6342–6363. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sakamoto H, Tsukaguchi T, Hiroshima S, et
al: CH5424802, a selective ALK inhibitor capable of blocking the
resistant gatekeeper mutant. Cancer Cell. 19:679–690. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kodama T, Tsukaguchi T, Yoshida M, Kondoh
O and Sakamoto H: Selective ALK inhibitor alectinib with potent
antitumor activity in models of crizotinib resistance. Cancer Lett.
351:215–221. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Friboulet L, Li N, Katayama R, et al: The
ALK inhibitor ceritinib overcomes crizotinib resistance in
non-small cell lung cancer. Cancer Discov. 4:662–673. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Peters S and Adjei AA: MET: a promising
anticancer therapeutic target. Nat Rev Clin Oncol. 9:314–326. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gherardi E, Birchmeier W, Birchmeier C and
Vande Woude G: Targeting MET in cancer: rationale and progress. Nat
Rev Cancer. 12:89–103. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Corso S and Giordano S: Cell-autonomous
and non-cell-autonomous mechanisms of HGF/MET-driven resistance to
targeted therapies: from basic research to a clinical perspective.
Cancer Discov. 3:978–992. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sadiq AA and Salgia R: MET as a possible
target for non-small-cell lung cancer. J Clin Oncol. 31:1089–1096.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Engelman JA, Zejnullahu K, Mitsudomi T, et
al: MET amplification leads to gefitinib resistance in lung cancer
by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yano S, Wang W, Li Q, et al: Hepatocyte
growth factor induces gefitinib resistance of lung adenocarcinoma
with epidermal growth factor receptor-activating mutations. Cancer
Res. 68:9479–9487. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Arao T, Fukumoto H, Takeda M, Tamura T,
Saijo N and Nishio K: Small in-frame deletion in the epidermal
growth factor receptor as a target for ZD6474. Cancer Res.
64:9101–9104. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Matsumoto K, Arao T, Hamaguchi T, et al:
FGFR2 gene amplification and clinicopathological features in
gastric cancer. Br J Cancer. 106:727–327. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tanizaki J, Okamoto I, Okabe T, et al:
Activation of HER family signaling as a mechanism of acquired
resistance to ALK inhibitors in EML4-ALK-positive non-small cell
lung cancer. Clin Cancer Res. 18:6219–6226. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yamaguchi N, Lucena-Araujo AR, Nakayama S,
et al: Dual ALK and EGFR inhibition targets a mechanism of acquired
resistance to the tyrosine kinase inhibitor crizotinib in ALK
rearranged lung cancer. Lung Cancer. 83:37–43. 2014. View Article : Google Scholar :
|
|
32
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim HR, Kim WS, Choi YJ, Choi CM, Rho JK
and Lee JC: Epithelial-mesenchymal transition leads to crizotinib
resistance in H2228 lung cancer cells with EML4-ALK translocation.
Mol Oncol. 7:1093–1102. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yamada T, Takeuchi S, Nakade J, et al:
Paracrine receptor activation by microenvironment triggers bypass
survival signals and ALK inhibitor resistance in EML4-ALK lung
cancer cells. Clin Cancer Res. 18:3592–3602. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Feng Y, Minca EC, Lanigan C, et al: High
MET receptor expression but not gene amplification in ALK 2p23
rearrangement positive non-small-cell lung cancer. J Thorac Oncol.
9:646–653. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gouji T, Takashi S, Mitsuhiro T and Yukito
I: Crizotinib can overcome acquired resistance to CH5424802: is
amplification of the MET gene a key factor? J Thorac Oncol.
9:e27–e28. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Voena C, Di Giacomo F, Panizza E, et al:
The EGFR family members sustain the neoplastic phenotype of
ALK+ lung adenocarcinoma via EGR1. Oncogenesis.
2:e432013. View Article : Google Scholar
|