Introduction
Myristoylated alanine rich C-kinase substrate
(MARCKS) has been described as having contradicting roles in cancer
biology. Studies of MARCKS in colon cancer, melanoma, prostate
cancer, hepatocellular carcinoma as well as glioblastoma multiforme
(GBM) have indicated that MARCKS behaves as a tumor suppressor
(1–5). In contrast, studies in breast cancer
and choloangiocarcinoma have suggested that MARCKS expression
correlates with poor prognosis (6,7).
Collectively, this information highlights the importance of the
cellular environment in driving MARCKS signaling. The significance
of MARCKS signaling with respect to growth and response to
treatment of lung cancer is under investigation.
Common drivers of lung cancer include the fusion
protein, anaplastic lymphoma kinase (ALK), and mutations and
hyperactivation of receptor tyrosine kinases (RTKs) such as:
epidermal growth factor receptor (EGFR), c-MET (hepatocyte growth
factor receptor, HGFR), insulin growth factor receptor 1 (IGF-1R)
and fibroblast growth factor receptor (FGFR) (8). RTKs phosphorylate and activate
phosphatidylinositol 3-kinase (PI3K) which then phosphorylates
phosphatidylinositol (4,5)-bisphosphate (PIP2)
producing phosphatidylinositol (3,4,5)-triphosphate (PIP3)
(9). PIP3 serves as an
anchoring and activation site for the pleckstrin homology (PH)
domain of the pro-growth, pro-survival kinase, Akt, among others
(10). Importantly, MARCKS
contains an effector domain (ED) capable of binding to the plasma
membrane through an electrostatic interaction with PIP2
molecules, thereby sequestering them (11,12)
and preventing their conversion to PIP3. This can
effectively attenuate Akt activity. However, when the MARCKS ED is
bound by calcium-calmodulin or phosphorylated by serine kinases
such as protein kinase C (PKC) or Rho-associated coiled-coil kinase
(ROCK), MARCKS releases PIP2 at the plasma membrane and
migrates into the cytoplasm due to electrostatic repulsion by the
phosphate groups (13,14), though this is reversible through
phosphatase action (15).
MARCKS is expressed in human lung tissue; however,
investigation of MARCKS involvement in lung cancer biology is
limited (16). Hanada et al
(17) found that MARCKS expression
may provide a possible biomarker and prognostic indicator for human
squamous cell carcinoma of the lung. MARCKS expression was
increased in tumor tissue compared to adjacent normal tissue, and
high MARCKS expression by immunohistochemistry (IHC) was associated
with poor prognosis. Additionally, Chen et al (18) described MARCKS being expressed in
higher levels in non-small cell lung cancer (NSCLC) compared to
normal tissue. Using a peptide mimetic targeting the N-terminal
region of MARCKS, they were able to reduce migration in in
vitro as well as in vivo models.
In the present study, we investigated MARCKS
expression in multiple lung tumor pathologies. Moreover, we
examined the importance of MARCKS and selected MARCKS mutants in
terms of lung cancer cell phenotype, particularly in response to
ionizing radiation. Our data suggest a critical dependence of the
phosphorylation status of the MARCKS effector domain (ED) with
respect to radiosensitization.
Materials and methods
Cell culture
Human lung cancer cell lines A549, H1299, H1792 and
H1975 (American Type Culture Collection, Manassas, VA, USA) were
cultured in RPMI-1640 with 10% fetal bovine serum (FBS), 1%
penicillin-streptomycin (pen-strep) and 1% GlutaMAX
(Invitrogen/Life Technologies, Grand Island, NY, USA). 293FT human
embryonic kidney cells (Invitrogen/Life Technologies) were cultured
in Dulbecco’s modified Eagle’s medium (DMEM) with 10% FBS and 1%
pen-strep. All cells were maintained at 37°C in 5%
CO2.
Immunohistochemical tissue array
A lung tumor tissue microarray (TMA) (cat.
#BC041114) was purchased from US Biomax, Inc. (Rockville, MD, USA).
The TMA was deparaffinized in xylene followed by 100, 95 and 70%
graded ethanol solutions and rehydrated in dH2O.
Endogenous peroxidase was blocked by a 5-min incubation with 3%
hydrogen peroxide solution in phosphate-buffered saline (PBS). The
TMA was then incubated with sodium citrate buffer (Dako,
Carpinteria, CA, USA) for 20 min for antigen retrieval.
Non-specific interactions were blocked in 20-min incubation (room
temperature) in 5% bovine serum albumin (BSA), 100 mM Tris (pH 8.2
to 8.5) solution. MARCKS was stained with an anti-MARCKS primary
antibody (EP1446Y, 1 h at room temperature; Abcam, Cambridge, MA,
USA) followed by an anti-rabbit alkaline phosphatase (AP)
conjugated secondary antibody (Invitrogen; 1 h at room
temperature). Following the manufacturer’s protocols, Vector Red
(Vector Laboratories, Inc., Burlingame, CA, USA) was used to detect
MARCKS. Cells were counterstained with 1% methyl green solution.
Lastly, the TMA was dehydrated using 70% ethanol, 50/50
ethanol/xylene, 100% xylene gradations and coverslip mounted using
PROTOCOL SecureMount (cat. #23-022-208; Thermo Fisher Scientific,
Waltham, MA, USA). Images were captured using a Zeiss Observer A1
microscope using an AxioCam MRc 5 camera. A board certified
pathologist scored each tissue in a blinded fashion using a scale
of 0, 0.5, 1, 2 or 3 with 0 equaling no detection and 3 being the
highest intensity. The highest ethical standards were followed
while collecting and handling patient tumors and information.
Informed consent was obtained from each patient before tumor
collection.
Generation of lentiviral MARCKS
constructs and packaging plasmids
The ViraPower HiPerform T-REx Gateway Expression
System (cat. #A11141) and the pENTR221 entry vector
(pENTR221-MARCKS) containing the wild-type (WT) MARCKS sequence
were purchased from Invitrogen. The WT-MARCKS sequence from
pENTR221-MARCKS was cloned into the pLenti6.3/TO/V5-DEST
destination vector using Clonase from the Gateway expression system
per the manufacturer’s protocol to yield pLenti6.3/TO/V5-MARCKS-WT.
The non-phosphorylatable (NP) MARCKS construct, in which the 4
serines of the ED were changed to alanines, was synthesized by
GenScript (GenScript USA, Inc., Piscataway, NJ, USA) and cloned
into the pUC57 vector. The psPAX2 packaging plasmid (Addgene
plasmid 12260; Addgene, Cambridge MA, USA) and pCMV-VSV-G envelope
plasmid (Addgene plasmid 8454) were obtained from Addgene.
Viral vector production
293FT cells (1.5×106) were plated in 7 ml
of DMEM supplemented with 10% FBS without antibiotics onto a
poly-D-lysine (cat. #P7886; Sigma) coated 10-cm dish. Lipofectamine
2000 (27 μl) (cat. #11668; Invitrogen) was combined with 1.5 ml
Opti-MEM media (cat. #11058; Invitrogen) and incubated for 5 min at
room temperature. An additional aliquot of 1.5 ml Opti-MEM media
had 4 μg of psPAX2, PCMV-VSV-G, and appropriate lentiviral vector
plasmid added. The plasmid and Lipofectamine media were mixed
together and incubated for 20 min at room temperature. The
Lipofectamine-plasmid mixture was added to the 293FT cells and
incubated overnight. The following morning, media was replaced with
fresh DMEM supplemented with 10% FBS and 1% pen-strep. Lentiviral
supernatant was collected at 24 h, filtered through a 0.45-μm
filter, aliquoted and stored at −80°C. The QuickTiter p24 ELISA
(Cell Biolabs, Inc., San Diego, CA, USA) was used to quantify
lentivirus aliquots (3).
Stable cell line selection
A549 cells (5×105) were plated in 6-well
plates and allowed to adhere overnight. The following morning,
similar amounts of p24 quantified lentiviral particles containing
tetracycline-repressor (TetR) plasmid was used to infect the cells.
The cells were incubated with 500 μl of virus for 2 h, at 37°C, in
5% CO2 followed by 3-day incubation in growth media.
Following 3 passages with 200 μg/ml geneticin (G418; Life
Technologies), A549 TetR cells were re-plated on 6-well plates at
5×105 cells/well for MARCKS lentiviral infection. An
identical procedure as described above was performed for MARCKS
virus infection, except selection was with 8 μg/ml blasticidin
(Life Technologies). The tetracycline homologue, doxycycline (2
μg/ml) was used to induce expression.
MARCKS peptide design
MARCKS-ED TAT peptide was designed by conjugating
the cell permeable HIV Tat peptide (cat. #61214; Anaspec, Fremont,
CA, USA) via disulfide linkage to the 25 amino acid ED sequence
(KKKKKRFSFKKSFK LSGFSFKKNKK) (19). The control sequence was designed
using the Expasy random peptide generator (http://www.expasy.org/randseq) using the average amino
acid composition computed from Swiss-Prot (CEIEEHAWNTVEMFSSF
PGTQLYNDA) to control for size of the peptide. To eliminate the
positive charge effect, lysine and arginine residues were changed
to glutamates and then conjugated with the HIV Tat peptide. The
lyophilized peptides were re-suspended in sterile ddH2O
at a stock concentration of 5 mM to be used for the
experiments.
iCELLigence assay
The iCELLigence platform (ACEA Biosciences, Inc.,
CA, USA) was used to monitor the cell response to doxycycline and
radiation, by measuring real-time changes in cellular impedance
(20,21). Cells were plated in duplicate and
allowed to attach under identical conditions, either overnight or
for 7 h, before doxycycline addition. In the case of radiation,
both sham and radiation therapy (RT) plates were removed from the
incubator after exposure for the same duration. For studies with
MARCKS-ED peptide, lung cancer cells were plated for 6 h before the
addition of MARCKS-ED peptide mimetic or control peptide.
Immunoblotting
Immunoblotting was performed as previously described
(3). Briefly, cells were lysed
using MPER lysis buffer supplemented with protease (cat. #P8340;
Sigma-Aldrich, St. Louis, MO, USA) and phosphatase inhibitors
(Sigma-Aldrich #P0044 and P5726). Samples were separated by
electrophoresis through either an 8 or 10% SDS-polyacrylamide gel
(SDS-PAGE) and transferred to a PVDF membrane (Immobilon; EMD
Millipore, Billerica, MA, USA). Blots were blocked in 5% milk, 1%
BSA and probed with the following antibodies at the manufacturer’s
recommended concentrations: phospho-MARCKS (Abcam ab81295), MARCKS
(Abcam ab52616), phospho-Akt (S473) (cat. #D9E, #4060; Cell
Signaling Technology, Danvers, MA, USA), phospho-Akt (T308) (Cell
Signaling Technology C31E5E, #2965), Akt (Cell Signaling Technology
C67E7, #4691), actin (Santa Cruz Biotechnology, sc-1616), and V-5
(Invitrogen, R96125).
Clonogenic assay
Cells were diluted and plated in defined numbers to
give six replicates per treatment condition. After 6 h, the media
was changed (with or without doxycycline) and incubated overnight.
The following morning, cells were treated with 0, 3, 5 or 8 Gy of
irradiation using a 320 kV X-ray irradiator (Kimtron, Inc.,
Woodbury, CT, USA). After one week, cells were fixed and stained
with 6.0% glutaraldehyde and 0.5% crystal violet. Colonies
consisting of 50 or more cells were counted for each condition. A
surviving fraction (S.F.) was calculated by using the equation
(number of colonies formed/number of cells plated)/(number of
colonies for sham irradiated group/number of cells plated). The
result was plotted as the mean and the standard error of the mean
in a semi-logarithmic format (22,23).
The dose enhancement ratio (DER) was calculated as the dose (Gy)
for the non-induced A549 cells (doxycycline absent) divided by the
dose for induced A549 cells (doxycycline supplemented) for which a
survival fraction of 0.2 was achieved.
Senescence associated β-galactosidase
staining
β-galactosidase was used as a marker for senescence
using the Cell Signal kit (cat. #9860). Cells were plated, attached
overnight, and then treated overnight with or without doxycycline.
Cells were exposed to 5 Gy and stained 1 week following
irradiation. The manufacturer’s protocol was followed and images
were captured on a Zeiss Observer A1 microscope using an AxioCam
MRc 5 camera. Five representative images per condition were
captured.
Mitotic catastrophe assay
Lung cancer cells were plated, attached overnight,
and then treated overnight with or without doxycycline. Cells were
exposed to 5 Gy of irradiation and fixed with 4% paraformaldehyde
0, 24, 48, 72 and 96 h following irradiation. Fixed cells were
stained with 300 ng/ml DAPI for 10 min and washed 3 times with PBS.
Nine representative images per condition were captured using a
Zeiss Observer A1 microscope using an AxioCam MRc 5 camera.
Double-strand DNA damage
quantification
Lung cancer cells were plated onto sterile
coverslips, attached overnight, and then treated overnight with or
without doxycycline. The following day cells were treated with 8 Gy
of irradiation. At the indicated time-points, cells were rinsed in
PBS and incubated for 5 min, at 4°C, in ice-cold buffer (10 mM
HEPES/KOH, pH 7.4, 300 mM sucrose, 100 mM NaCl, 3 mM
MgCl2) supplemented with 1% protease (Sigma #P8340) and
1% phosphatase inhibitors (Sigma P0044 and P5726) followed by
fixation in 70% ethanol for 15 min. The cells were blocked and
incubated with primary antibodies (1:500 dilution, phospho-γH2AX
Ser139, Millipore, cat. #MI-07-164). The secondary antibody was the
anti-rabbit Alexa Fluor 594-conjugated antibody (1:2,000 dilution;
Invitrogen). DAPI was used for nuclear staining. The coverslips
were subsequently mounted onto slides with mounting media
(Aqua-Poly/Mount, cat. #18606; Polysciences, Inc., Warrington, PA,
USA) and analyzed via an EVOS fldigital inverted fluorescence
microscope (AMG; Life Technologies). Positive and negative controls
were included on all experiments. A total of 500 cells were
assessed. For foci quantification, cells with >10 foci were
counted as positive according to the standard procedure (24–26).
Statistics
Statistical calculations and data graphing were
performed using the GraphPad Prism (GraphPad Software, Inc., La
Jolla, CA, USA). ANOVA was used for assessing results in the
clonogenic assay, senescence associated β-galactosidase staining
assay, mitotic catastrophe assay and double-strand DNA damage
quantification. All statistical tests were two-sided and were
performed using a significance level of 5%.
Results
Lung tumor TMA staining for MARCKS
expression
Previous reports of MARCKS in lung cancer looked at
the broad group of NSCLC or squamous cell carcinomas but did not
perform a comprehensive analysis (17,18).
We purchased a lung tumor TMA from US Biomax, Inc. containing a
total of 200 lung tissue samples comprised of 62 adenocarcinoma, 12
atypical carcinoid, 4 adenosquamous carcinoma, 8 bronchoalveolar
carcinoma, 8 large cell carcinoma, 2 mucinous adenocarcinoma, 20
normal lung, 4 papillary adenocarcinoma, 16 small cell carcinoma,
and 64 squamous cell carcinoma tissue samples using Biomax
nomenclature (Table I). The TMA
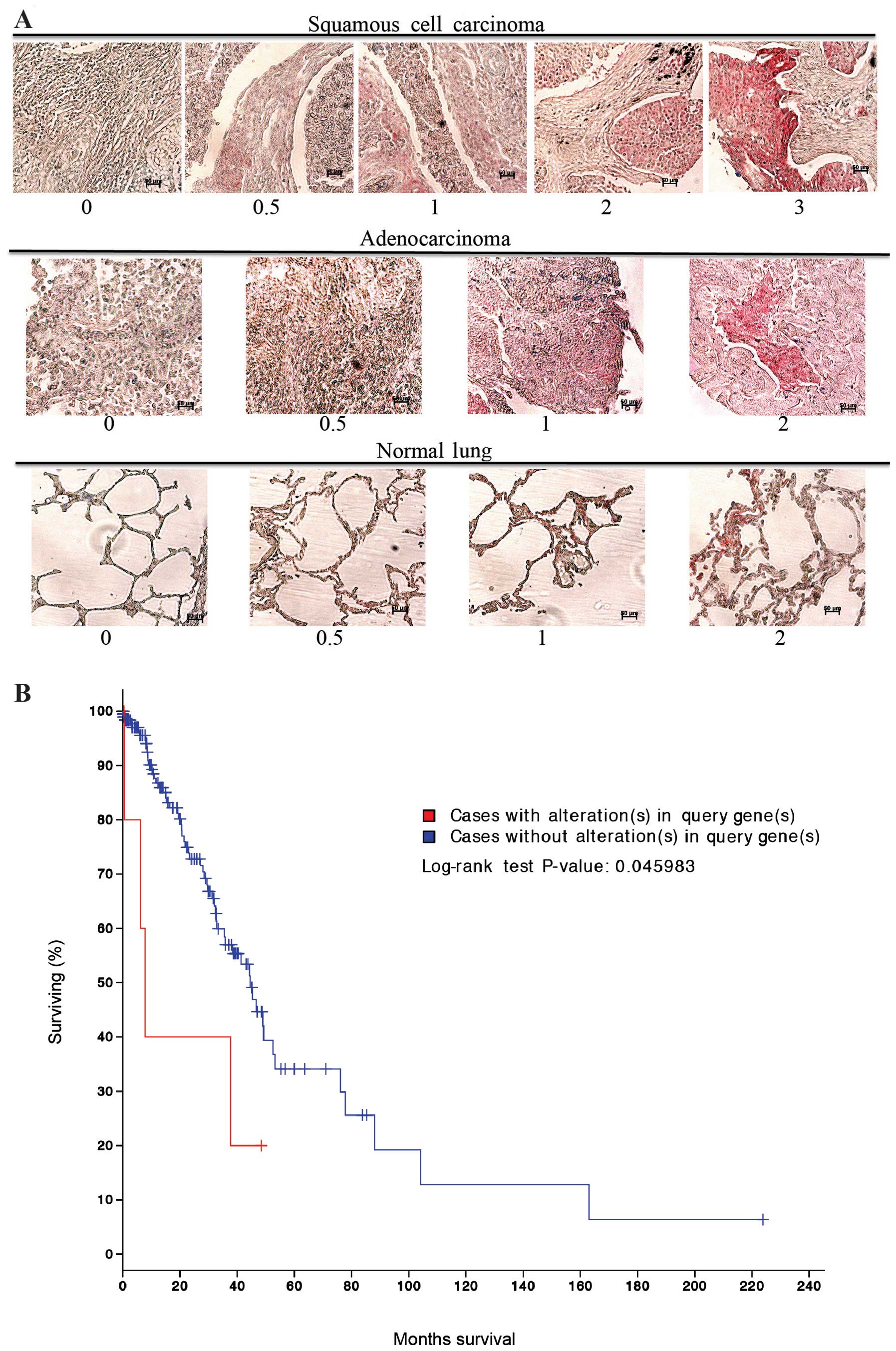
was stained for MARCKS and scored in a blinded fashion using a 0–3
scale as indicated in Fig. 1A.
MARCKS could be detected in most of the lung cancer histologies
represented, but was positive in a minority of the samples overall
(Table I). Squamous cell carcinoma
had the highest percentage of samples stain for MARCKS and there
was a wide range of staining intensities. For the squamous cell
carcinoma samples: 6 tumors had the highest intensity scoring of 3,
4 had an intensity score of 2, 8 scored an intensity of 1, and 17
cases scored a 0.5. In total, 35 out of 64 (54.7%) squamous cell
carcinoma cases stained positive for MARCKS expression. In
contrast, adenocarcinomas had lower staining percentage and lower
staining intensity compared to squamous cell carcinoma. Overall, 17
out of 62 (27.4%) adenocarcinoma samples stained positive for
MARCKS though most of them were of 0.5 score (Table I and Fig. 1A). The remaining tumor histologies
had a much lower rate of MARCKS staining. Notably, in normal lung
tissue 2 samples had a staining intensity of 2, 1 sample had a
staining intensity of 1, and 3 samples had a staining intensity of
0.5. In total, 6 out of 20 (30.0%) normal lung samples stained
positive for MARCKS (Table I and
Fig. 1A). cBioPortal (www.cbioportal.org) (27,28)
analysis of The Cancer Genome Atlas (TCGA) dataset for lung
adenocarcinomas showed that alterations in the MARCKS gene were
associated with a significant (P=0.046) decrease in survival
(Fig. 1B).
 | Table IPatient and immunohistochemical
characteristics for selective lung cancer tissue staining for
anti-MARCKS. |
Table I
Patient and immunohistochemical
characteristics for selective lung cancer tissue staining for
anti-MARCKS.
|
Characteristics | Data | | | | |
|---|
| Average age,
years | 54 | | | | |
| Gender, n (%) |
| Male | 138 (69.7) | | | | |
| Female | 62 (31.3) | | | | |
| | Intensity |
| |
|
| Histology | Positives per group
(%) | 3
n (%) | 2
n (%) | 1
n (%) | 0.5
n (%) |
|
|
Adenocarcinoma | 17/62 (27.4) | 0 (0) | 1 (1.6) | 3 (4.8) | 13 (21.3) |
| Atypical
carcinoid | 2/12 (16.7) | 0 (0) | 0 (0) | 0 (0) | 2 (16.7) |
| Adenosquamous | 1/4 (25.0) | 0 (0) | 0 (0) | 0 (0) | 1 (25.0) |
|
Bronchoalveolar | 1/8 (12.5) | 0 (0) | 0 (0) | 0 (0) | 1 (12.5) |
| Large cell | 0/8 (0.0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Mucinous
adenocarcinoma | 1/2 (50.0) | 0 (0) | 0 (0) | 0 (0) | 1 (50.0) |
| Papillary
adenocarcinoma | 0/4 (0.0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Normal lung | 6/20 (30.0) | 0 (0) | 2 (10.0) | 1 (5.0) | 3 (15.0) |
| Small cell | 2/16 (12.5) | 0 (0) | 0 (0) | 1 (6.3) | 1 (6.3) |
| Squamous | 35/64 (54.7) | 6 (9.5) | 4 (6.3) | 8 (12.7) | 17 (26.6) |
| Grade |
| 1 | 8/19 (42.1) | 0 (0) | 0 (0) | 2 (10.5) | 6 (31.6) |
| 2 | 32/85 (37.6) | 6 (7.1) | 4 (4.7) | 6 (7.1) | 16 (18.8) |
| 3 | 12/23 (52.2) | 0 (0) | 1 (4.3) | 3 (13.0) | 8 (34.8) |
| 4 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Stage |
| I | 26/74 (35.1) | 4 (5.4) | 3 (4.1) | 4 (5.4) | 15 (20.3) |
| II | 9/38 (23.7) | 0 (0) | 1 (2.6) | 0 (0) | 8 (21.1) |
| III | 23/62 (37.1) | 2 (3.2) | 1 (1.6) | 8 (12.9) | 12 (19.4) |
| IV | 1/4 (25.0) | 0 (0) | 0 (0) | 0 (0) | 1 (25.0) |
| T |
| 1 | 6/24 (25.0) | 0 (0) | 3 (12.5) | 0 (0) | 3 (12.5) |
| 2 | 32/98 (32.7) | 6 (6.1) | 1 (1.0) | 4 (4.1) | 21 (21.4) |
| 3 | 17/48 (35.4) | 0 (0) | 0 (0) | 6 (14.6) | 10 (20.8) |
| 4 | 4/8 (50.0) | 0 (0) | 1 (12.5) | 1 (12.5) | 2 (25.0) |
| N |
| 0 | 30/92 (32.6) | 4 (4.3) | 3 (3.3) | 6 (6.5) | 17 (18.5) |
| 1 | 23/68 (33.8) | 0 (0) | 2 (2.9) | 5 (7.4) | 16 (23.5) |
| 2 | 6/20 (30.0) | 2 (10.0) | 0 (0) | 1 (5.0) | 3 (15.0) |
| M |
| 0 | 58/176 (33.0) | 6 (3.4) | 5 (2.8) | 12 (6.8) | 35 (19.9) |
| 1 | 1/4 (25.0) | 0 (0) | 0 (0) | 0 (0) | 1 (25.0) |
MARCKS manipulation in lung cancer
cells
In order to study the importance of MARCKS in lung
cancer, we analyzed several lung cancer cell lines for MARCKS
protein expression. Western blotting was performed and revealed
that A549 and H1299 human lung cancer cell lines expressed low
levels of MARCKS. Alternatively, the human lung cancer cell lines
H1792 and H1975 expressed higher levels of MARCKS (Fig. 2A). Therefore, we selected A549 for
overexpression of wild-type (WT) and non-phosphorylatable (NP)
MARCKS (shown schematically in Fig.
2B). Lentiviral particles were used to establish WT-MARCKS and
NP-MARCKS overexpression in A549 cells under the regulation of a
tetracycline-inducible promoter as shown by western blotting
(Fig. 2C). Because MARCKS
phosphorylation has been associated with its cytoplasmic
localization (29), the NP-MARCKS
construct (shown schematically in Fig.
2B) should remain membrane bound and more effectively sequester
PIP2 than the WT-MARCKS. As such, we used confocal
immunofluorescence (Fig. 2D) to
determine the subcellular localization of WT-MARCKS vs. NP-MARCKS
by probing for the V5 epitope. Indeed confocal imaging shows that
WT-MARCKS was expressed predominately in the cytoplasm while the
NP-MARCKS construct was predominately localized to the plasma
membrane region of the cell.
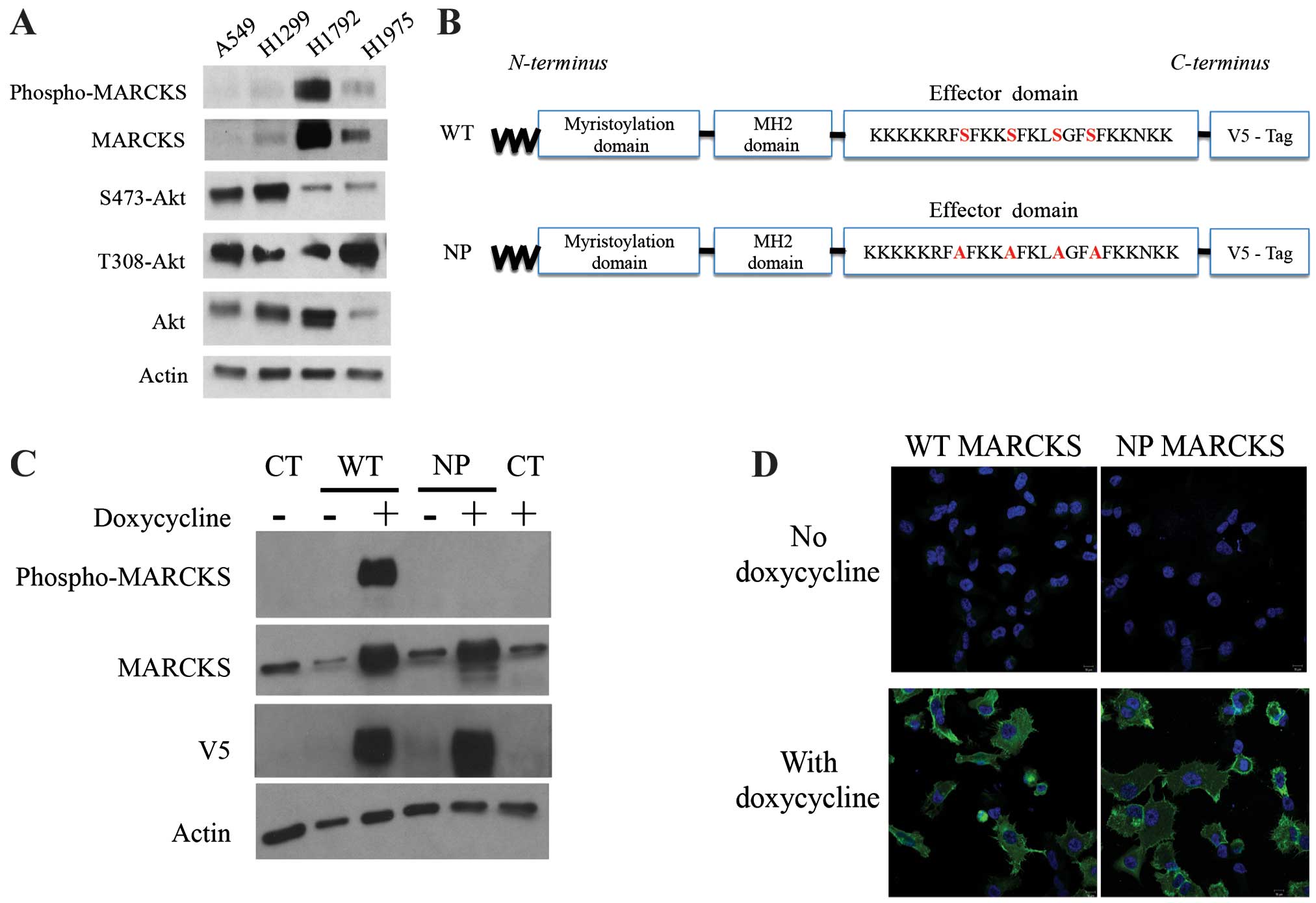 | Figure 2MARCKS and MARCKS mutant
overexpression in lung cancer cell lines. (A) A549, H1299, H1792
and H1975 lung cancer cell lines were probed by western blot
analysis for phospho-MARCKS, MARCKS, Ser473-Akt, Thr308-Akt, Akt
and actin. (B) A schematic diagram depicts the mutant MARCKS
constructs. MARCKS contains an N-terminal myristoylation domain, an
MH2 domain and an effector domain (ED). The wild-type (WT)-MARCKS
and non-phosphorylatable (NP)-MARCKS constructs as designed also
contained a C-terminal V5 tag for differentiation from endogenous
protein. The NP-MARCKS four serine residues in the ED region were
mutated to alanines (indicated in red). (C) Western blot analysis
of stably selected A549 cells with control lentiviral (CT), WT or
NP MARCKS doxycycline-inducible expression. Cells were cultured
with (+) or without (−) doxycycline for 18 h before western
blotting. MARCKS, phospho-MARCKS, V5 and actin antibodies were
used. (D) V5 tagged wild-type MARCKS (WT MARCKS) or
non-phosphorylatable MARCKS (NP MARCKS) stably infected (by
lentivirus) A549 cells were treated without (upper panel) or with
(lower panel) 2 μg/ml doxycycline to induce expression. Shown are
confocal immunofluorescent images of anti-V5 (green) and DAPI
nuclear staining (blue) as merged images. |
NP-MARCKS decreases survival after
radiation
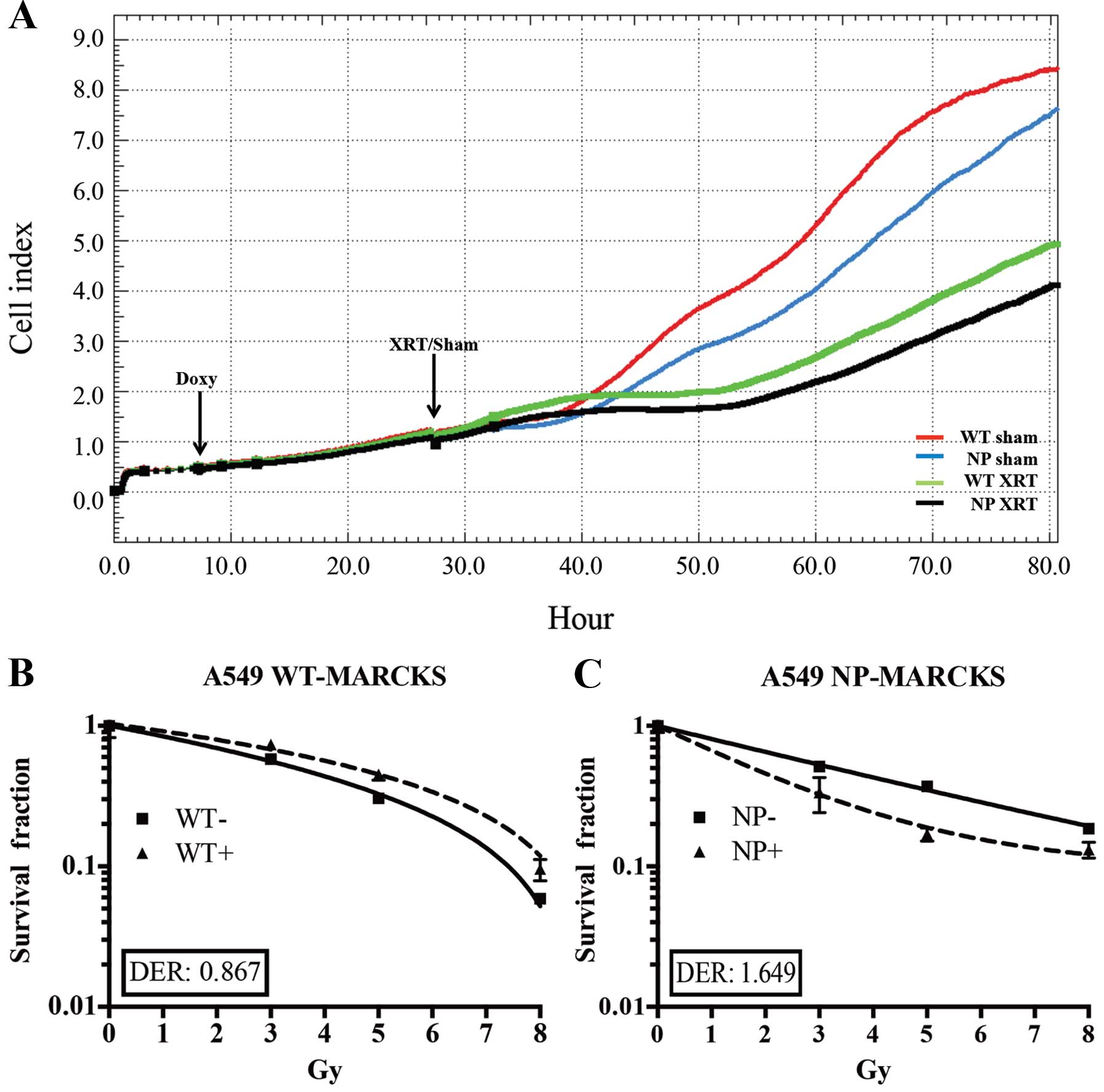
The iCELL-igence real-time impedance assay system
(ACEA) was used to screen for cell physiological differences
between WT-MARCKS and NP-MARCKS (Fig.
3A), particularly after irradiation as MARCKS knockdown has
been shown to promote radiation resistance in GBM (3). Both WT-MARCKS and NP-MARCKS samples
were plated and had very similar Cell index (impedance)
measurements for the first 6 h. After 6 h, all WT-MARCKS and
NP-MARCKS cells were dosed with or without doxycycline overnight.
The following morning sham or 5 Gy of irradiation was administered
to the cells. After two days of monitoring, we observed that both
the WT-MARCKS and NP-MARCKS irradiated groups had lower Cell index
measurements compared to their sham counterparts (Fig. 3A). In particular, the NP-MARCKS
irradiated cells had the lowest Cell index measurements overall. To
confirm these findings, A549 WT-MARCKS were cultured in standard
culture media without doxycycline (WT− or
NP−) or standard culture media with 2 μg/ml of
doxycycline (WT+ or NP+) for 18 h prior to
performance of a clonogenic assay, the gold standard assay for
determining radiation sensitivity (30). Two-way ANOVA testing calculated no
significant difference between the WT-MARCKS (−) and WT-MARCKS (+)
groups (Fig. 3B). We postulated
the lack of radiation enhancement could be due to lack of membrane
binding (needed for PIP2 sequestration) of WT-MARCKS. In
contrast to WT-MARCKS, we observed a significant decrease in
survival with NP-MARCKS overexpression (P=0.01; two-way ANOVA)
(Fig. 3C). Overexpression of
NP-MARCKS had a radiation sensitizing effect on the A549 cells with
a DER of 1.649. This experiment was repeated five times and similar
trends were observed.
Impaired DNA damage repair with NP-MARCKS
overexpression
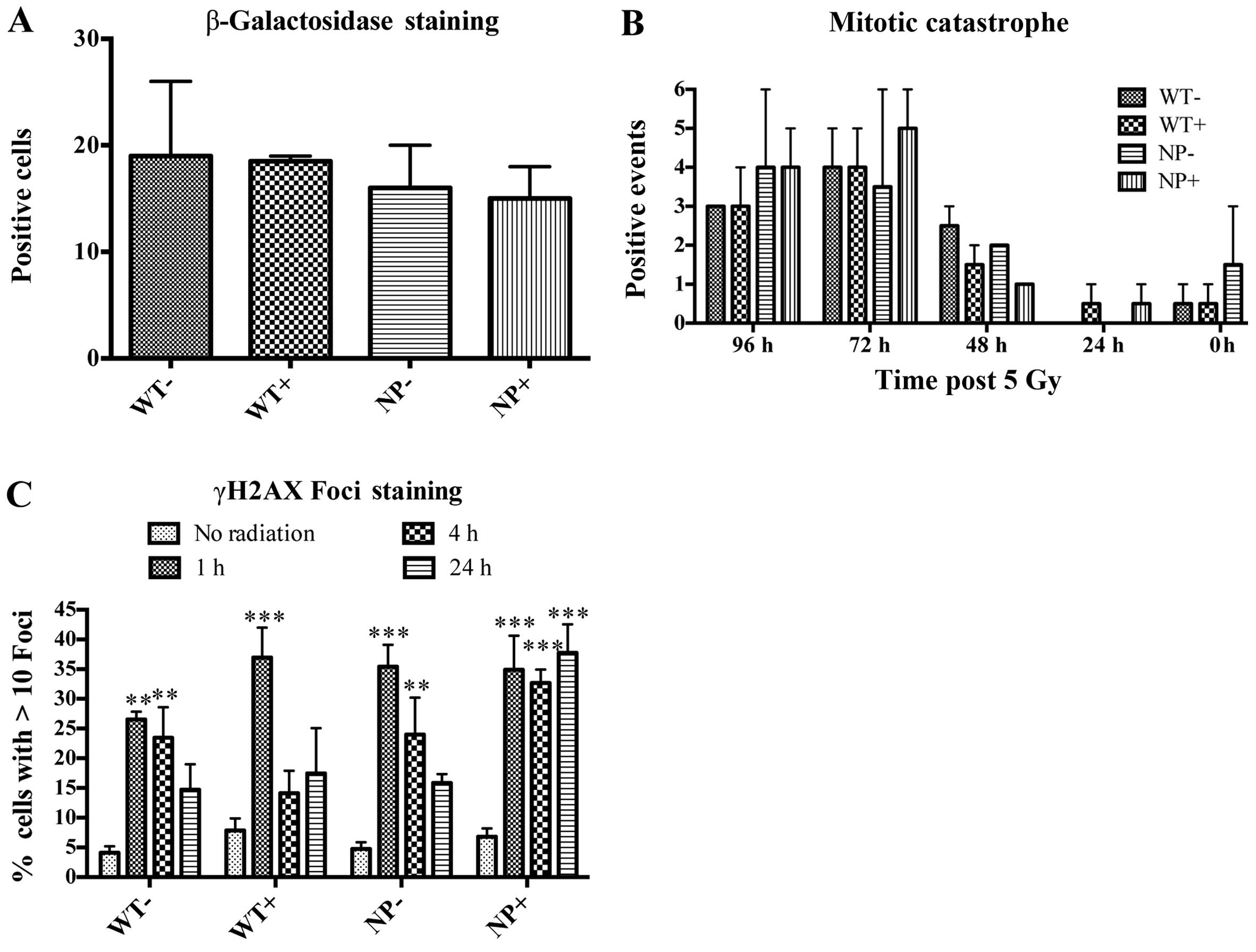
Previously, we have observed that overexpression of
wild-type MARCKS in GBM cells led to a state of cellular senescence
(3). To rule out senescence being
the cause of lower clonogenicity seen with NP-MARCKS expression, we
measured β-galactosidase, an enzyme upregulated during senescence.
Comparing WT-MARCKS with (+) and without (−) doxycycline along with
NP-MARCKS with (+) and without (−) doxycycline there was no
significant differences in β-galactosidase positive cells 8 days
post the 5 Gy irradiation (Fig.
4A). In addition, we examined for mitotic catastrophe following
radiation exposure of these cells. Again, comparing WT-MARCKS with
(+) and without (−) doxycycline along with NP-MARCKS with (+) and
without (−) doxycycline there was no significant mitotic
catastrophe difference over the course of 96 h post the 5 Gy
irradiation (Fig. 4B). As we
observed no difference in senescence or mitotic catastrophe, we
anticipated that NP-MARCKS was influencing DNA damage compared to
WT-MARCKS. As shown in Fig. 4C,
DNA damage was studied by probing for DNA double-strand breaks
(DSB) by measuring γH2AX foci (26). As expected, irradiation of A549
cells increased γH2AX foci formation within 1-h post irradiation.
At 24 h, all groups approached basal level γH2AX foci formation
except for NP-MARCKS (+) overexpressing cells that demonstrated
persistent γH2AX foci staining, suggesting that NP-MARCKS promotes
prolonged DNA damage, leading to a decrease in cell survival
following radiation exposure.
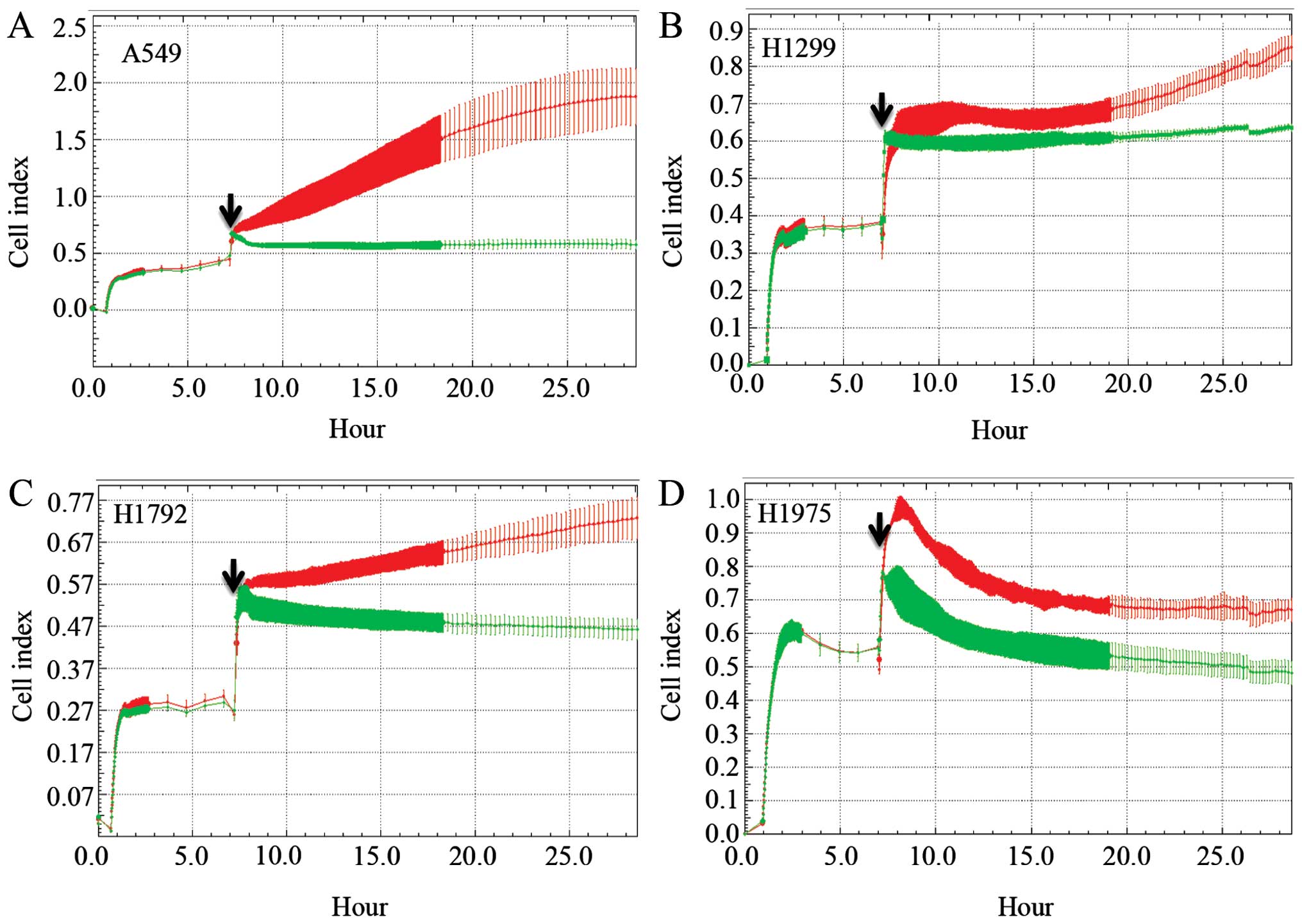
Using a MARCKS ED-targeting peptide
mimetic in lung cancer cells
Because MARCKS has no enzymatic activity, we
utilized a peptide mimetic to modulate MARCKS behavior by targeting
the ED of MARCKS. We engineered a cell permeable peptide mimetic
composed of the cell permeable TAT peptide conjugated to the 25
amino acid sequence (KKKKKRFSFKKSFKLSGFSFKKNKK) of the ED region of
MARCKS (19). A control peptide
was generated based on the Expasy random peptide generator using
the average amino acid composition computed from Swiss-Prot
(CEIEEHAWNTV EMFSSFPGTQLYNDA). This sequence was also conjugated to
TAT. A549, H1299, H1792 and H1975 lung cancer cell lines were
plated in the iCELLigence instrument for a proliferation experiment
with the MARCKS-ED or control peptide. In all four cell lines, the
control peptide group had an increased Cell index over the duration
of the assay (Fig. 5). However,
the MARCKS-ED TAT peptide treated cells showed a marked decrease in
their Cell index, suggesting that targeting the MARCKS ED is a
potential strategy against lung cancer.
Discussion
The role of MARCKS in the development and
progression of cancer is quite controversial in that there is
evidence for both tumor suppression (1–5) and
tumor promotion (6,7). As we have previously postulated
(3), the cellular environment and
co-existent signaling pathways almost certainly contribute to the
importance and role of MARCKS. In the present study, we identified
a key role of the ED of MARCKS in terms of cellular response,
particularly to radiation. The ED of MARCKS is a very important
domain as it determines MARCKS subcellular localization, binds
PIP2 and F-actin allowing MARCKS to influence both
proliferation and migration of cancer cells. Intrinsic modulators
of the ED of MARCKS include serine kinases, including PKCs and
ROCKs, as well as calcium-calmodulin (Ca2+-CAM), which
bind in a mutually exclusive manner. Indeed, when we induced
expression of a non-phosphorylatable MARCKS (that should not bind
Ca2+-CAM either), we noted radiation sensitization
concomitant with a prolongation of DNA double strand breaks.
Treatment with a peptide mimetic of the ED was effective in
blocking tumor cell growth as well. These findings complement our
previous work in GBM cell lines (3) showing that loss of MARCKS promotes
radiation protection and increased cell growth, while
overexpression of MARCKS suppressed growth.
Recent studies of MARCKS in lung cancer have also
suggested that interfering with the N-terminal region of MARCKS can
manipulate cellular behavior. An N-terminal peptide mimetic
corresponding to the first 24 amino acids of MARCKS reduced levels
of phospho-MARCKS and inhibited cellular migration (18). A proposed mechanism for a peptide
mimetic interfering with MARCKS function is that the peptide
therapy overwhelms the cell and prevents MARCKS from cycling to and
from the plasma membrane. This proposed mechanism is consistent
with the decreased levels of phospho-MARCKS observed in our
studies. Several studies have shown that phosphorylated MARCKS
(cytoplasmic localization) promotes cancer cell migration (7,31).
The role of MARCKS expression and subcellular localization upon
cell migration is reported to be influenced by the cycling of
MARCKS from the plasma membrane to the cytoplasm and back to the
plasma membrane (32), which is
consistent with our observations. Additionally, MARCKS contains two
actin binding sites in the ED, that are disrupted by ED
phosphorylation, thus preventing cross-linking (33). MARCKS may have additional indirect
effects on this process since actin-binding proteins, such as
Arp2/3 and N-WASP, bind to PIP2, once again, pointing to
the importance of the ability of MARCKS to sequester the
phospholipid (34).
Additionally, we describe here the expression
pattern of the MARCKS protein in a variety of human lung tumor
histologies. To the best of our knowledge, the present study is the
first to report such findings. MARCKS expression was predominately
expressed in the adenocarcinoma and squamous cell carcinoma tumor
samples. Other cases, including small cell carcinoma and atypical
carcinoid had very few samples that stained positive for MARCKS.
Currently, we cannot comment on clinical outcomes relative to
MARCKS protein expression since the TMA lacked outcome data.
However, as discussed above, the cellular response to MARCKS
expression will depend on its phosphorylation status and cellular
localization, which has not been consistently examined in clinical
specimens. For example Hanada et al (17) described that squamous cell
carcinomas with higher MARCKS expression had poorer prognosis, yet,
neither phosphorylation status or subcellular location was
described. Future work on MARCKS, particularly in lung cancer, will
require a more in depth investigation beyond gene expression or
protein expression levels to include the status of MARCKS
modulators and ED phosphorylation.
Acknowledgements
We would like to thank Shawn Williams for his
assistance with the confocal microscopy. Funding sources included:
UAB Radiation Oncology Intramural Pilot Grant (awarded to T.D.R.
and C.D.W.); UAB Brain SPORE Pilot Project (awarded to C.D.W.); and
ASTRO Junior Faculty Training Research Award (awarded to
C.D.W.).
References
|
1
|
Bickeboller M, Tagscherer KE, Kloor M, et
al: Functional characterization of the tumor-suppressor MARCKS in
colorectal cancer and its association with survival. Oncogene.
24–March;2014.(Epub ahead of print). View Article : Google Scholar
|
|
2
|
Brooks G, Brooks SF and Goss MW: MARCKS
functions as a novel growth suppressor in cells of melanocyte
origin. Carcinogenesis. 17:683–689. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jarboe JS, Anderson JC, Duarte CW, et al:
MARCKS regulates growth and radiation sensitivity and is a novel
prognostic factor for glioma. Clin Cancer Res. 18:3030–3041. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li T, Li D, Sha J, Sun P and Huang Y:
MicroRNA-21 directly targets MARCKS and promotes apoptosis
resistance and invasion in prostate cancer cells. Biochem Biophys
Res Commun. 383:280–285. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Masaki T, Tokuda M, Yoshida S, et al:
Comparison study of the expressions of myristoylated alanine-rich C
kinase substrate in hepatocellular carcinoma, liver cirrhosis,
chronic hepatitis, and normal liver. Int J Oncol. 26:661–671.
2005.PubMed/NCBI
|
|
6
|
Browne BC, Hochgräfe F, Wu J, et al:
Global characterization of signalling networks associated with
tamoxifen resistance in breast cancer. FEBS J. 280:5237–5257. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Techasen A, Loilome W, Namwat N, et al:
Myristoylated alanine-rich C kinase substrate phosphorylation
promotes cholangiocarcinoma cell migration and metastasis via the
protein kinase C-dependent pathway. Cancer Sci. 101:658–665. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Janku F, Garrido-Laguna I, Petruzelka LB,
Stewart DJ and Kurzrock R: Novel therapeutic targets in non-small
cell lung cancer. J Thorac Oncol. 6:1601–1612. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Newton AC: Lipid activation of protein
kinases. J Lipid Res. 50(Suppl): S266–S271. 2009. View Article : Google Scholar :
|
|
10
|
Engelman JA: Targeting PI3K signalling in
cancer: opportunities, challenges and limitations. Nat Rev Cancer.
9:550–562. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ellena JF, Burnitz MC and Cafiso DS:
Location of the myristoylated alanine-rich C-kinase substrate
(MARCKS) effector domain in negatively charged phospholipid
bicelles. Biophys J. 85:2442–2448. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang J, Gambhir A, Hangyas-Mihalyne G,
Murray D, Golebiewska U and McLaughlin S: Lateral sequestration of
phosphatidylinositol 4,5-bisphosphate by the basic effector domain
of myristoylated alanine-rich C kinase substrate is due to
nonspecific electrostatic interactions. J Biol Chem.
277:34401–34412. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tanabe A, Kamisuki Y, Hidaka H, Suzuki M,
Negishi M and Takuwa Y: PKC phosphorylates MARCKS Ser159 not only
directly but also through RhoA/ROCK. Biochem Biophys Res Commun.
345:156–161. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Thelen M, Rosen A, Nairn AC and Aderem A:
Regulation by phosphorylation of reversible association of a
myristoylated protein kinase C substrate with the plasma membrane.
Nature. 351:320–322. 1991. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Clarke PR, Siddhanti SR, Cohen P and
Blackshear PJ: Okadaic acid-sensitive protein phosphatases
dephosphorylate MARCKS, a major protein kinase C substrate. FEBS
Lett. 336:37–42. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Stumpo DJ, Graff JM, Albert KA, Greengard
P and Blackshear PJ: Molecular cloning, characterization, and
expression of a cDNA encoding the ‘80- to 87-kDa’ myristoylated
alanine-rich C kinase substrate: a major cellular substrate for
protein kinase C. Proc Natl Acad Sci USA. 86:4012–4016. 1989.
View Article : Google Scholar
|
|
17
|
Hanada S, Kakehashi A, Nishiyama N, et al:
Myristoylated alanine-rich C-kinase substrate as a prognostic
biomarker in human primary lung squamous cell carcinoma. Cancer
Biomark. 13:289–298. 2013.PubMed/NCBI
|
|
18
|
Chen CH, Thai P, Yoneda K, Adler KB, Yang
PC and Wu R: A peptide that inhibits function of Myristoylated
Alanine-Rich C Kinase Substrate (MARCKS) reduces lung cancer
metastasis. Oncogene. 33:3696–3706. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Graff JM, Rajan RR, Randall RR, Nairn AC
and Blackshear PJ: Protein kinase C substrate and inhibitor
characteristics of peptides derived from the myristoylated
alanine-rich C kinase substrate (MARCKS) protein phosphorylation
site domain. J Biol Chem. 266:14390–14398. 1991.PubMed/NCBI
|
|
20
|
Solly K, Wang X, Xu X, Strulovici B and
Zheng W: Application of real-time cell electronic sensing (RT-CES)
technology to cell-based assays. Assay Drug Dev Technol. 2:363–372.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nam HY, Han MW, Chang HW, et al:
Radioresistant cancer cells can be conditioned to enter senescence
by mTOR inhibition. Cancer Res. 73:4267–4277. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jarboe JS, Jaboin JJ, Anderson JC, et al:
Kinomic profiling approach identifies Trk as a novel radiation
modulator. Radiother Oncol. 103:380–387. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tu T, Thotala D, Geng L, Hallahan DE and
Willey CD: Bone marrow X kinase-mediated signal transduction in
irradiated vascular endothelium. Cancer Res. 68:2861–2869. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang ES, Wang H, Jiang G, et al:
Lithium-mediated protection of hippocampal cells involves
enhancement of DNA-PK-dependent repair in mice. J Clin Invest.
119:1124–1135. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nowsheen S, Bonner JA and Yang ES: The
poly(ADP-Ribose) polymerase inhibitor ABT-888 reduces
radiation-induced nuclear EGFR and augments head and neck tumor
response to radiotherapy. Radiother Oncol. 99:331–338. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang ES, Nowsheen S, Wang T, Thotala DK
and Xia F: Glycogen synthase kinase 3beta inhibition enhances
repair of DNA double-strand breaks in irradiated hippocampal
neurons. Neuro Oncol. 13:459–470. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gao J, Aksoy BA, Dogrusoz U, et al:
Integrative analysis of complex cancer genomics and clinical
profiles using the cBio-Portal. Sci Signal. 6:pl12013.
|
|
28
|
Cerami E, Gao J, Dogrusoz U, et al: The
cBio cancer genomics portal: an open platform for exploring
multidimensional cancer genomics data. Cancer Discov. 2:401–404.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ohmori S, Sakai N, Shirai Y, et al:
Importance of protein kinase C targeting for the phosphorylation of
its substrate, myristoylated alanine-rich C-kinase substrate. J
Biol Chem. 275:26449–26457. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Franken NA, Rodermond HM, Stap J, Haveman
J and van Bree C: Clonogenic assay of cells in vitro. Nat Protoc.
1:2315–2319. 2006. View Article : Google Scholar
|
|
31
|
Chen X and Rotenberg SA: PhosphoMARCKS
drives motility of mouse melanoma cells. Cell Signal. 22:1097–1103.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Disatnik MH, Boutet SC, Pacio W, et al:
The bi-directional translocation of MARCKS between membrane and
cytosol regulates integrin-mediated muscle cell spreading. J Cell
Sci. 117:4469–4479. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yarmola EG, Edison AS, Lenox RH and Bubb
MR: Actin filament cross-linking by MARCKS: characterization of two
actin-binding sites within the phosphorylation site domain. J Biol
Chem. 276:22351–22358. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kalwa H and Michel T: The MARCKS protein
plays a critical role in phosphatidylinositol 4,5-bisphosphate
metabolism and directed cell movement in vascular endothelial
cells. J Biol Chem. 286:2320–2330. 2011. View Article : Google Scholar :
|