Introduction
Lung cancer remains the most frequent cause of
cancer-related death in developed countries (1). Approximately 80% of lung cancers are
classified histopathologically as non-small cell lung cancers
(NSCLC). NSCLCs are subdivided into four major histological
subtypes with distinct pathological characteristics:
adenocarcinoma, squamous cell carcinoma, large cell carcinoma and
neuroendocrine cancer (2).
Patients with NSCLCs in advanced stages rarely survive more than
five years despite aggressive chemotherapy, molecularly-targeted
therapy or chemoradiotherapy (3).
Altered expression of cell surface growth factor
receptors, including the RTK family, has frequently been observed
in many types of human cancer (4,5).
Recently, new targeted therapeutics have been developed to inhibit
oncogenic receptors-mediated signaling, including that in NSCLCs
(3). In NSCLCs, epidermal growth
factor receptor (EGFR) and the RTK for hepatocyte growth factors
(MET) are activated. Signaling by EGFR and MET leads to NSCLC cell
proliferation and promotes survival and invasion (6,7). It
has been shown that MET expression and phosphorylation are
associated with both primary and acquired resistance to tyrosine
kinase inhibitor (TKI) based therapy in patients with NSCLCs, such
as EGFR (8–10). Thus, targeting MET would be an
important approach to overcoming resistance to TKIs in lung
cancer.
The discovery of non-coding RNAs (ncRNAs) in the
human genome was an important conceptual breakthrough in the
post-genome sequencing era (11).
Improved understanding of ncRNAs is necessary for continued
progress in cancer research. microRNAs (miRNAs) repress gene
expression by inhibiting mRNA translation or by promoting mRNA
degradation. Aberrant expression of miRNAs significantly
contributes to cancer development, metastasis and drug resistance
(12–14). Currently, 2,578 human mature miRNAs
are registered at miRBase release 20.0 (http://microrna.sanger.ac.uk/). miRNAs are unique in
their ability to regulate multiple protein-coding genes.
Bioinformatic predictions indicate that miRNAs regulate
approximately 30–60% (or more) of the protein-coding genes in the
human genome (15,16).
Previously, our miRNA expression signature of lung
squamous cell carcinoma (lung-SCC) revealed that
microRNA-206 (miR-206) was significantly reduced in
cancer tissues (17), suggesting
that this miRNA functions as a tumor suppressor in lung-SCC.
Interestingly, MET and EGFR genes have putative
miR-206 binding sites in their 3′-UTRs as determined by
miRNA databases. The aim of this study was to investigate the
functional significance of miR-206 in lung-SCC cells and
whether inhibition of RTKs (MET and EGFR) by miR-206
mediated oncogenic signaling in cancer cells.
Materials and methods
Clinical specimens and RNA
extraction
A total of 32 lung-SCCs and 22 normal lung specimens
were collected from patients who underwent pneumonectomy at
Kagoshima University Hospital from 2010 to 2013. Archival
formalin-fixed paraffin embedded (FFPE) samples were used for
qRT-PCR analysis and immunohistochemistry.
Samples were staged according to the International
Association for the Study of Lung Cancer TNM classification, and
they were histologically graded (18). Our study was approved by the
Institutional Review Board for Clinical Research of Kagoshima
University School of Medicine. Prior written informed consent and
approval were provided by each patient.
FFPE tissues were sectioned to a thickness of 10 μm
and 8 tissue sections were used for RNA extraction. Total RNA
(including miRNA) was extracted using Recover All™ Total Nucleic
Acid Isolation kit (Ambion, Austin, TX, USA) using the
manufacturer’s protocols. The integrity of the RNA was checked with
an RNA 6000 Nano Assay kit and a 2100 Bioanalyzer (Agilent
Technologies, Santa Clara, CA, USA).
Cell culture
We used a human lung-SCC cell line (EBC-1) obtained
from Japanese Cancer Research Resources Bank (JCRB). Cells were
grown in RPMI-1640 medium supplemented with 10% fetal bovine serum
and maintained in a humidified incubator (5% CO2) at
37°C.
Quantitative real-time PCR (qRT-PCR)
The procedure for PCR quantification was as
described previously (19–21). TaqMan probes and primers for
MET (P/N: Hs01565584_m1, Applied Biosystems, Foster City,
CA, USA) and EGFR (P/N: Hs01076078_m1, Applied Biosystems)
were assay-on-demand gene expression products. Stem-loop RT-PCR for
miR-206 (P/N: 000510, Applied Biosystems) was used to
quantify the expression levels of miRNAs according to the
manufacturer’s protocol. To normalize the data for quantification
of MET mRNA and miRNAs, we used human GUSB (P/N:
Hs99999908_m1; Applied Biosystems) and RNU48 (P/N: 001006;
Applied Biosystems), respectively, and the ΔΔCt method was employed
to calculate the fold-change.
Transfections with mature miRNA into
EBC-1 cells
The following mature miRNA species were used in the
present study: Pre-miR™ miRNA precursors (hsa-miR-206; P/N:
AM 17100 and negative control miRNA; P/N: AM 17111). RNAs were
incubated with Opti-MEM (Invitrogen, Carlsbad, CA, USA) and
Lipofectamine RNAiMax reagent (Invitrogen) as described (19–21).
Cell proliferation, migration and
invasion assays
Cells were transfected with 10 nM miRNAs by reverse
transfection and plated in 96-well plates at 3×103 cells
per well. After 72 h, cell proliferation was determined with the
XTT assay using the Cell Proliferation Kit II (Roche Molecular
Biochemicals, Mannheim, Germany) as described (19–21).
Cell migration activity was evaluated with wound
healing assays. Cells were plated in 6-well plates at
8×105 cells per well, and after 48 h of transfection,
the cell monolayer was scraped using a P-20 micropipette tip. The
initial gap length (0 h) and the residual gap length 48 h after
wounding were calculated from photomicrographs as described
(19–21).
Cell invasion assays were performed using modified
Boyden chambers, consisting of Transwell-precoated Matrigel
membrane filter inserts with 8-μm pores in 24-well tissue culture
plates (BD Biosciences, Bedford, MA, USA). After 72 h of
transfection, cells were plated in 24-well plates at
1×105 cells per well. Minimum essential medium
containing 10% fetal bovine serum in the lower chamber served as
the chemoattractant as described previously (19–21).
All experiments were performed in triplicate.
Flow cytometry
EBC-1 cells were transiently transfected with miRNAs
and were harvested 72 h later by trypsinisation. The analysis of
apoptosis was done as previously described (22). Cells for cell cycle analysis were
stained with PI using the CycleTest™ Plus DNA Reagent kit (BD
Biosciences) following their protocol and analyzed with a FACScan
(BD Biosciences). The percentage of the cells in the G0/G1, S and
G2/M phase were counted and compared. Experiments were done in
triplicate.
Western blotting
After a 72-h period of transfection, protein lysates
(1 μg for MET and 20 μg for others) were separated on NuPAGE on
4–12% Bis-Tris gels (Invitrogen) and transferred to polyvinylidene
fluoride membranes. Immunoblotting was done with the following
diluted (1:1,000) antibodies from Cell Signaling, Danvers, MA, USA:
polyclonal anti-EGFR antibody (#4267), anti-p-EGFR (Tyr1045)
antibody (#2237), anti-p-EGFR (Tyr1068) antibody (#3777), anti-MET
antibody (#8198), anti-p-MET (Tyr1234/1235) antibody (#3077),
anti-p-MET (Tyr1003) antibody (#3135), anti-p-MET (Tyr1349)
antibody (#3133), anti-p44/42 MAPK (Erk1/2) antibody (#4965),
anti-p-Erk1/2 antibody (#4370), anti-Akt (pan) antibody (#4691),
anti-p-Akt antibody (#4060). Anti-GAPDH antibody (MAB374) was from
Chemicon, Temecula, CA, USA. The membrane was washed and then
incubated with anti-rabbit-IgG, HRP-linked antibody (#7074; Cell
Signaling). Specific complexes were visualized with an
echochemiluminescence (ECL) detection system (GE Healthcare, Little
Chalfont, UK) as described previously (19–22).
Plasmid construction and dualluciferase
reporter assay
Partial wild-type sequence of the MET 3′-UTR
or those with a mutant miR-206 target site (position 499–505
or position 814–820 of the MET 3′-UTR) were inserted between
the XhoI-PmeI restriction sites in the 3′-UTR of the
hRluc gene in the psiCHECK-2 vector (C8021; Promega, Madison, WI,
USA). Similarly, partial wild-type sequences of the EGFR
3′-UTR or those with a mutant miR-206 target site (position
746–752 of the EGFR 3′UTR) were inserted into the
vector.
The synthesized DNA was cloned into the psiCHECK-2
vector. EBC-1 cells were transfected with 20 or 50 ng vector, 10 nM
miRNAs and 1 μl Lipofectamine 2000 (Invitrogen) in 100 μl Opti-MEM
(Invitrogen). The activities of firefly and Renilla luciferases in
cell lysates were determined with a dualluciferase assay system
(E1910; Promega). Normalized data were calculated as the quotient
of Renilla/firefly luciferase activities.
Immunohistochemistry
FFPE tissues were sectioned to a thickness of 5 μm
and 2 tissue sections were used for immunohistochemistry. The
tissues were immunostained following the manufacturer’s protocol
with an UltraVision Detection system (Thermo Scientific). The
primary rabbit polyclonal antibodies against MET (#8198; Cell
Signaling) and EGFR (#4267; Cell Signaling) were diluted 1:300 and
1:200, respectively. The slides were treated with biotinylated goat
anti-rabbit antibodies. Diaminobenzidine hydrogen peroxidase was
the chromogen and counterstaining was done with 0.5% hematoxylin.
For immunohistochemical analyses, we followed a previous report
(23). Briefly, a proportional
cut-off of ≥50% was selected to ensure that a majority of the cells
within a given specimen expressed MET/EGFR at either a weak (+),
moderate (++), or strong (+++) intensity level. Specimens with no
or equivocal staining in tumor cells or <50% of tumor cells
staining at any given intensity were considered negative (−). NSCLC
tumors expressing moderate or strong levels of MET/ EGFR in ≥50% of
cells (++ or +++) were classified as MET/ EGFR-positive. Otherwise,
they were classified as negative. Two observers (Hiroko Mataki and
Takeshi Chiyomaru) evaluated the slides simultaneously, and both
were blinded to clinical data. We recorded the mean of the values
determined by the two observers. Interobserver differences were
<5%.
Identification of putative miR-206 target
genes
To identify putative miR-206-regulated genes,
we used the TargetScan database (http://www.targetscan.org/). Candidate miR-206
target genes were analyzed in Kyoto Encyclopedia of Genes and
Genomics (KEGG) pathway categories using the GeneCodis program.
Finally, we investigated the expression status of putative targets
of miR-206 using lung-SCC clinical expression data from GEO
database (accession no. GSE 11117). Our strategies of
identification of putative tumor-suppressive miRNAs target genes
were described in previous studies (19–22).
Statistical analysis
Relationships between two or three variables and
numerical values were analyzed using the Mann-Whitney U test
or Bonferroni-adjusted Mann-Whitney U test. Spearman’s rank
test was used to evaluate the correlation between the expressions
of miR-206 and miR-133b. Expert StatView version 4
was used in these analyses.
Results
Expression levels of miR-206 in lung-SCC
clinical specimens
To validate our past miRNA signature of lung-SCC, we
evaluated the expression of miR-206 in lung-SCC tissues
(n=32) and normal lung tissues (n=22). The patient backgrounds and
clinicopathological characteristics are summarized in Table I. The typical FFPE specimens that
were used for RNA extraction and expression analysis in this study
are shown in Fig. 1.
 | Table ICharacteristics of the patients. |
Table I
Characteristics of the patients.
| A, Lung cancer |
|---|
|
|---|
| Lung cancer | n | (%) |
|---|
| Total number | 32 | |
| Median age
(range) | 71 (50–88) | |
| Gender |
| Male | 30 | (93.7) |
| Female | 2 | (6.3) |
| Pathological tumor
stage |
| IA | 4 | (12.5) |
| IB | 8 | (25.0) |
|
IIA | 4 | (12.5) |
|
IIB | 5 | (15.6) |
|
IIIA | 8 | (25.0) |
|
IIIB | 1 | (3.1) |
| Unknown | 2 | (6.3) |
|
Differentiation |
| Well | 8 | (25.0) |
| Moderately | 19 | (59.4) |
| Poorly | 3 | (9.4) |
| Unknown | 2 | (6.3) |
| Pleural
invasion |
| (+) | 15 | (46.9) |
| (−) | 17 | (53.1) |
| Venous
invasion |
| (+) | 16 | (50.0) |
| (−) | 16 | (50.0) |
| Lymphatic
invasion |
| (+) | 16 | (50.0) |
| (−) | 16 | (50.0) |
| Recurrence |
| (+) | 9 | (28.1) |
| (−) | 20 | (62.5) |
| Unknown | 3 | (9.4) |
|
Immunohistochemistry |
| MET |
| (+) | 1 | (3.1) |
| (++) | 2 | (6.2) |
| (+++) | 0 | |
| EGFR |
| (+) | 1 | (3.1) |
| (++) | 2 | (6.2) |
| (++++) | 2 | (6.2) |
|
| B, Normal lung |
|
| Normal lung | n | |
|
| Total number | 22 | |
| Median age
(range) | 71 (50–88) | |
| Gender |
| Male | 22 | |
| Female | 0 | |
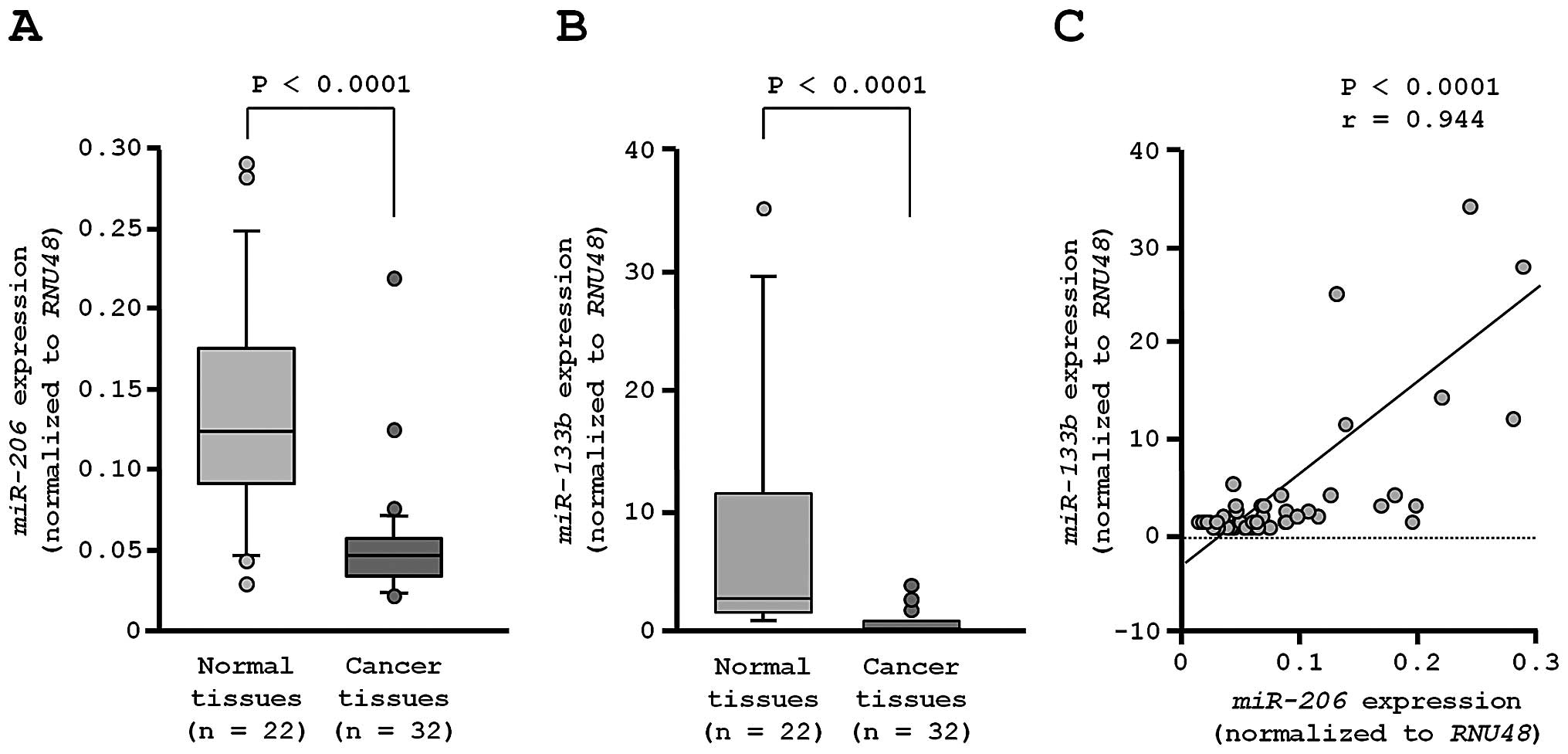
The expression levels of miR-206 were
significantly reduced in tumor tissues compared to corresponding
non-cancer tissues (P<0.0001; Fig.
2A). In the human genome, miR-206 and miR-133b
are located close together on chromosome 6p12.1 and constitute
clustered miRNAs. Thus, we also investigated the expression levels
of miR-133b in lung-SCC tissues. The miR-133b
expression levels were significantly reduced in cancer tissues
(Fig. 2B). Spearman’s rank test
showed a positive correlation between the expression of
miR-206 and that of miR-133b (r=0.944 and
P<0.0001, Fig. 2C).
There was no significant relationship between the
expression of miRNAs and other clinicopathological parameters
(stage, grade, infiltration).
Effects of miR-206 restoration on the
proliferation, induction of apoptosis and cell cycle arrest of
EBC-1 cells
To examine the functional roles of miR-206,
we performed gain-of-function studies using miRNA transfection into
EBC-1 cells.
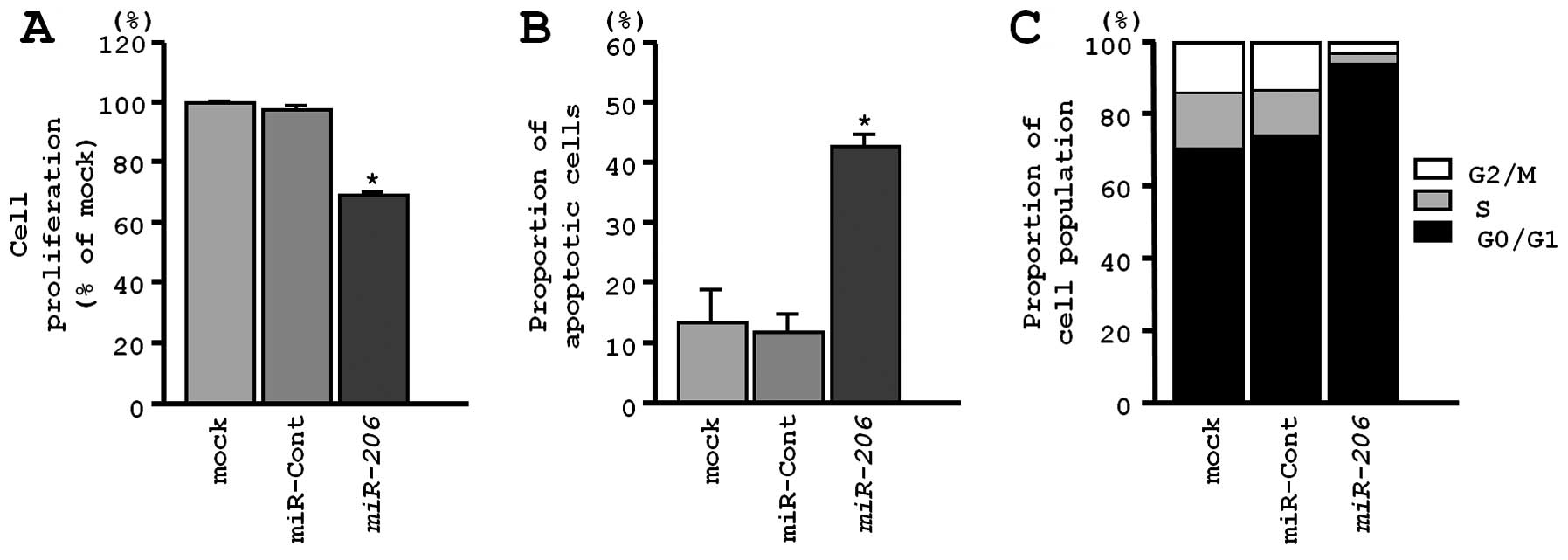
XTT assays revealed significant inhibition of cell
proliferation in EBC-1 cells transfected with miR-206 in
comparison with mock-transfected cells and control transfectants
(P<0.0001, Fig. 3A). Because
miR-206 restoration significantly inhibited cell
proliferation in a lung-SCC cell line, we hypothesized that
miR-206 expression may induce apoptosis or cell cycle
arrest. Using flow cytometry, we investigated the number of
apoptotic cells following restoration of miR-206 expression.
The apoptotic and early apoptotic fractions were greater in
miR-206 transfectants than in the mock transfectants or the
control (Fig. 3B). In terms of the
cell cycle distribution, the number of cells in the G0/1 phase was
significantly greater in miR-206 transfectants than in mock
or miR-control transfectants (Fig.
3C).
Effects of miR-206 restoration on
migration and invasion activities of EBC-1 cells
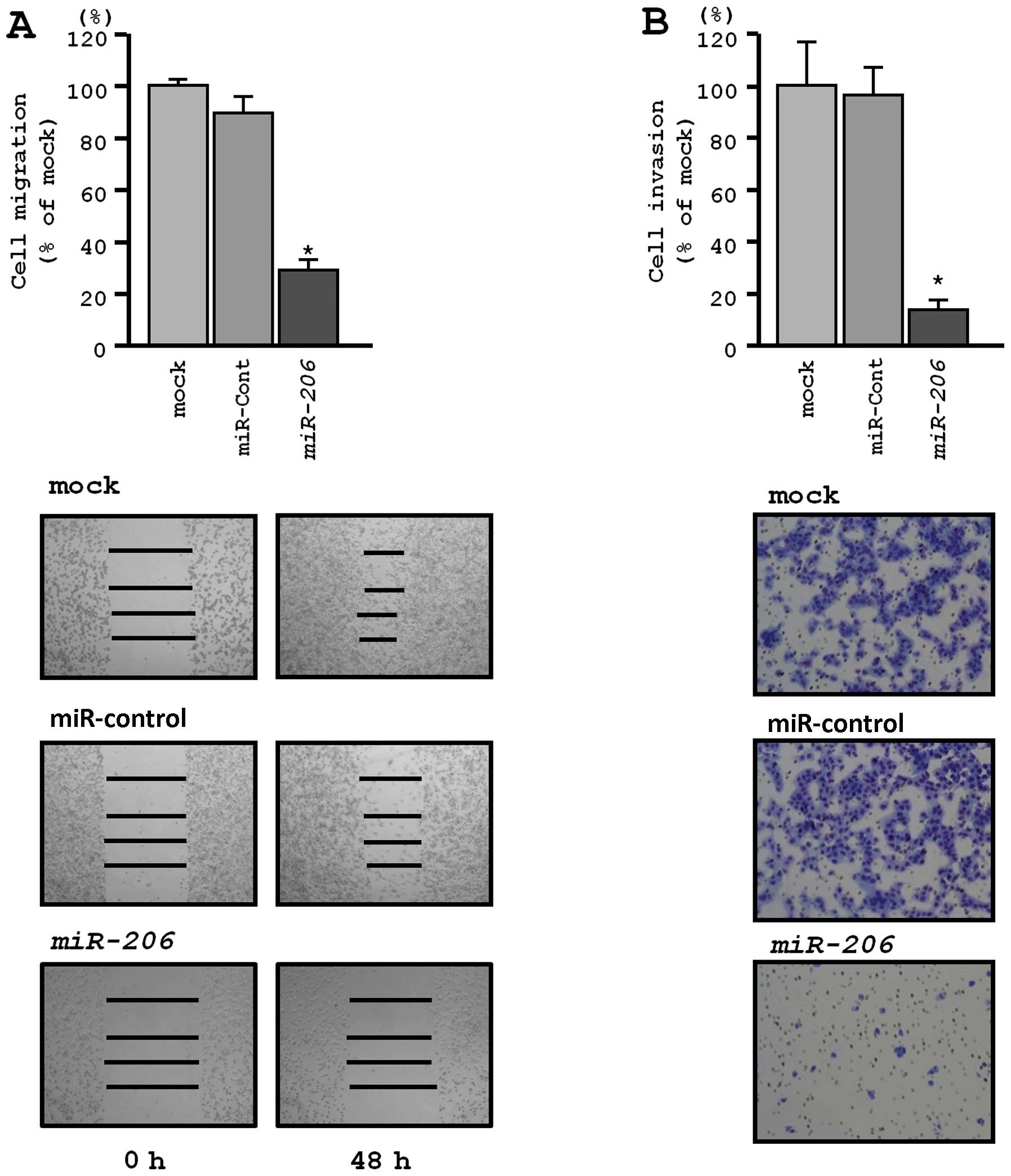
Wound healing assays revealed significant inhibition
of EBC-1 cell migration after transfection with miR-206
(P<0.0001, respectively; Fig.
4A). Similarly, Matrigel invasion assays revealed that
transfection with miR-206 reduced cell invasion. Indeed, the
number of invading cells was significantly decreased in EBC-1 cells
transfected with miR-206 (P<0.0001) (Fig. 4B).
Identification of candidate genes
targeted by miR-206 in lung-SCC
To identify molecular targets of miR-206, we
used combination of in silico analysis and lung-SCC gene
expression data from GEO (accession no. GSE 11117) as described in
our previous studies (19–22). A total of 3,117 genes were putative
targets of miR-206 according to the TargetScan database.
Among those 3,117 genes, 836 were upregulated in lung-SCC clinical
specimens according to GEO database. The 836 genes were categorized
to known pathways according to KEGG and top 10 pathways and
involved genes are shown in Table
II.
| Table IISignificantly enriched annotations
regulated by miR-206. |
Table II
Significantly enriched annotations
regulated by miR-206.
| No. of genes | KEGG entry no. | Annotations | P-value | Genes |
|---|
| 33 | 4110 | Cell cycle | 3.33679E-26 | PTTG2, CCNE2,
ESPL1, STAG1, MCM4, CCNB2, YWHAZ, CDC20, CCNA2, PTTG1, YWHAQ, MCM7,
ATR, CHEK1, CDK4, E2F1, CCNB1, CDC27, RAD21, MAD2L1, PRKDC, MCM3,
CDK2, MYC, MCM5, STAG2, MCM6, BUB1B, CDC25A, BUB1, RB1, PCNA,
MCM2 |
| 20 | 3030 | DNA
replication | 7.75961E-24 | RNASEH2A, MCM4,
MCM7, RPA3, RFC2, RFC1, POLE2, POLD1, RFC5, RFC3, MCM3, MCM5, MCM6,
PRIM1, FEN1, RPA1, PCNA, MCM2, POLE, RFC4 |
| 12 | 3430 | Mismatch
repair | 1.53163E-13 | MSH2, RPA3, RFC2,
RFC1, MSH6, POLD1, EXO1, RFC5, RFC3, RPA1, PCNA, RFC4 |
| 31 | 5200 | Pathways in
cancer | 1.29618E-11 | MET, EGFR, MSH2,
CCNE2, CDC42, LAMA3, CUL2, HIF1A, ITGA6, FZD6, BIRC5, PIK3CA,
PIK3CB, CDK4, RAC2, E2F1, ITGA2, RAD51, TPM3, IL8, MSH6, TPR, CDK2,
MYC, RAC3, CKS1B, ITGAV, RB1, LAMB1, PPARG, CBL |
| 13 | 3420 | Nucleotide excision
repair | 1.67092E-11 | DDB2, RPA3, RFC2,
RFC1, GTF2H1, POLE2, POLD1, RFC5, RFC3, RPA1, PCNA, POLE, RFC4 |
| 17 | 240 | Pyrimidine
metabolism | 7.152E-11 | TYMS, DCK, CAD,
CTPS, POLR3K, NME2, RRM1, POLE2, POLD1, DTYMK, NT5C3, POLR2K,
PRIM1, NT5E, RRM2, TK1, POLE |
| 21 | 230 | Purine
metabolism | 9.08289E-11 | DCK, PRPS1, HPRT1,
POLR3K, NME2, PDE8A, IMPDH1, PDE4D, PPAT, RRM1, ADCY3, POLE2,
POLD1, GMPS, NT5C3, POLR2K, PRIM1, NT5E, PAICS, RRM2, POLE |
| 18 | 4114 | Oocyte meiosis | 9.94037E-11 | PTTG2, CCNE2,
ESPL1, CCNB2, YWHAZ, CDC20, PTTG1, FBXO5, YWHAQ, CCNB1, CDC27,
MAD2L1, ADCY3, PPP2R5C, CDK2, BUB1, RPS6KA3, PPP1CB |
| 22 | 4810 | Regulation of actin
cytoskeleton | 2.64705E-09 | DIAPH3, CDC42,
ITGA6, PIK3CA, PIK3CB, WASF1, PPP1R12A, ROCK2, RAC2, ITGA2, NCKAP1,
GNA13, TMSB4X, ARPC1A, PAK1, ARHGEF4, ITGB4, PPP1CB, EGFR, RAC3,
ITGAV, ROCK1, ARHGEF4, ITGB4, PPP1CB |
| 21 | 4510 | Focal adhesion | 4.95501E-09 | MET, EGFR, CAV2,
CDC42, LAMA3, ITGA6, PIK3CA, PIK3CB, PPP1R12A, ROCK2, RAC2, ITGA2,
CAV1, ARHGAP5, PAK1, RAC3, ITGAV, ROCK1, ITGB4, LAMB1, PPP1CB |
Because RTKs contribute to cancer progression and
metastasis, we focused on RTKs that contained miR-206
binding sites in their 3′-UTRs and are upregulated in lung-SCC
clinical specimens. We found that two RTK genes (MET and EGFR) were
involved in ‘pathways in cancer’ and ‘focal adhesion’ pathways.
Therefore, we focused on these two genes for further studies.
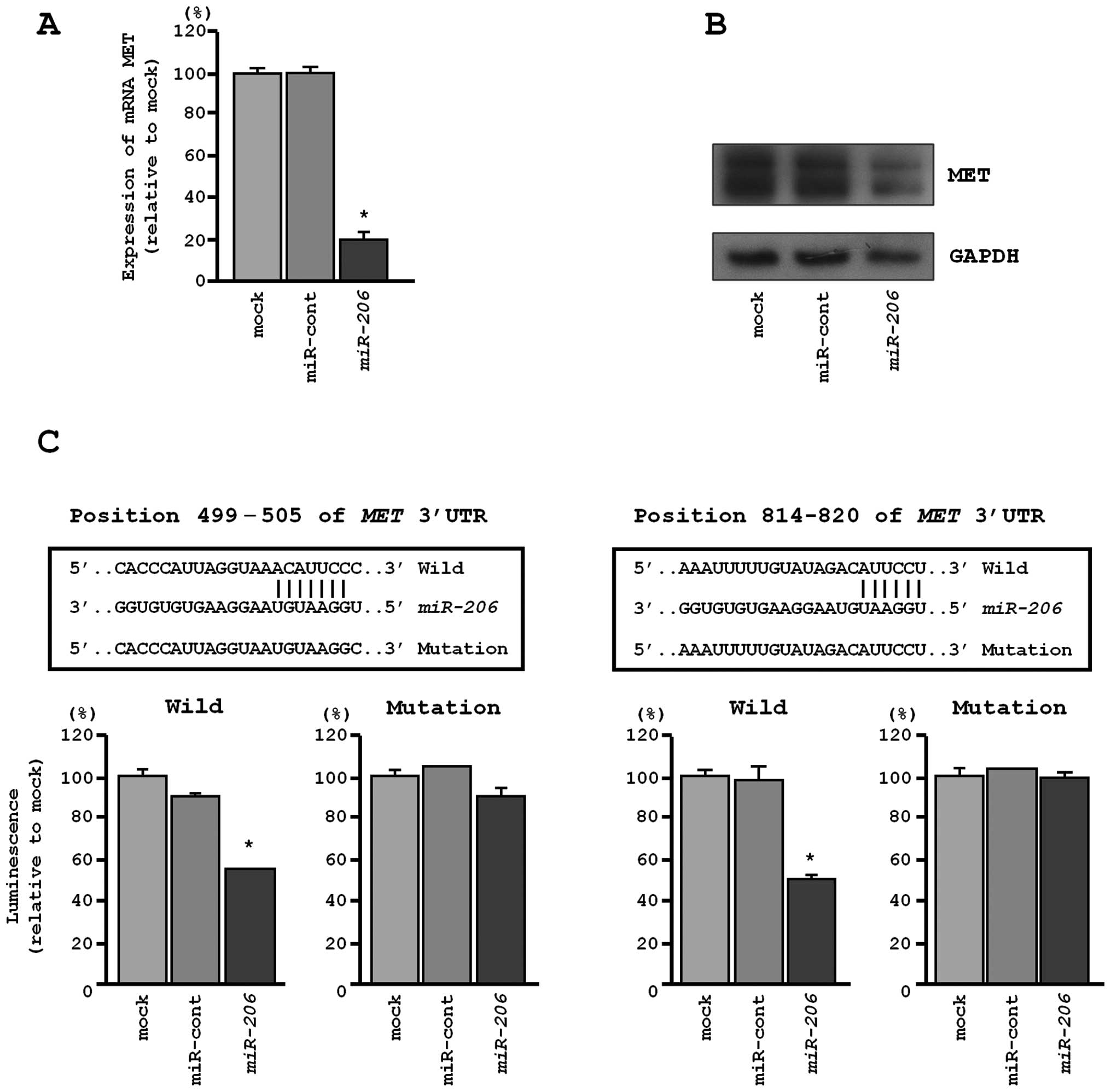
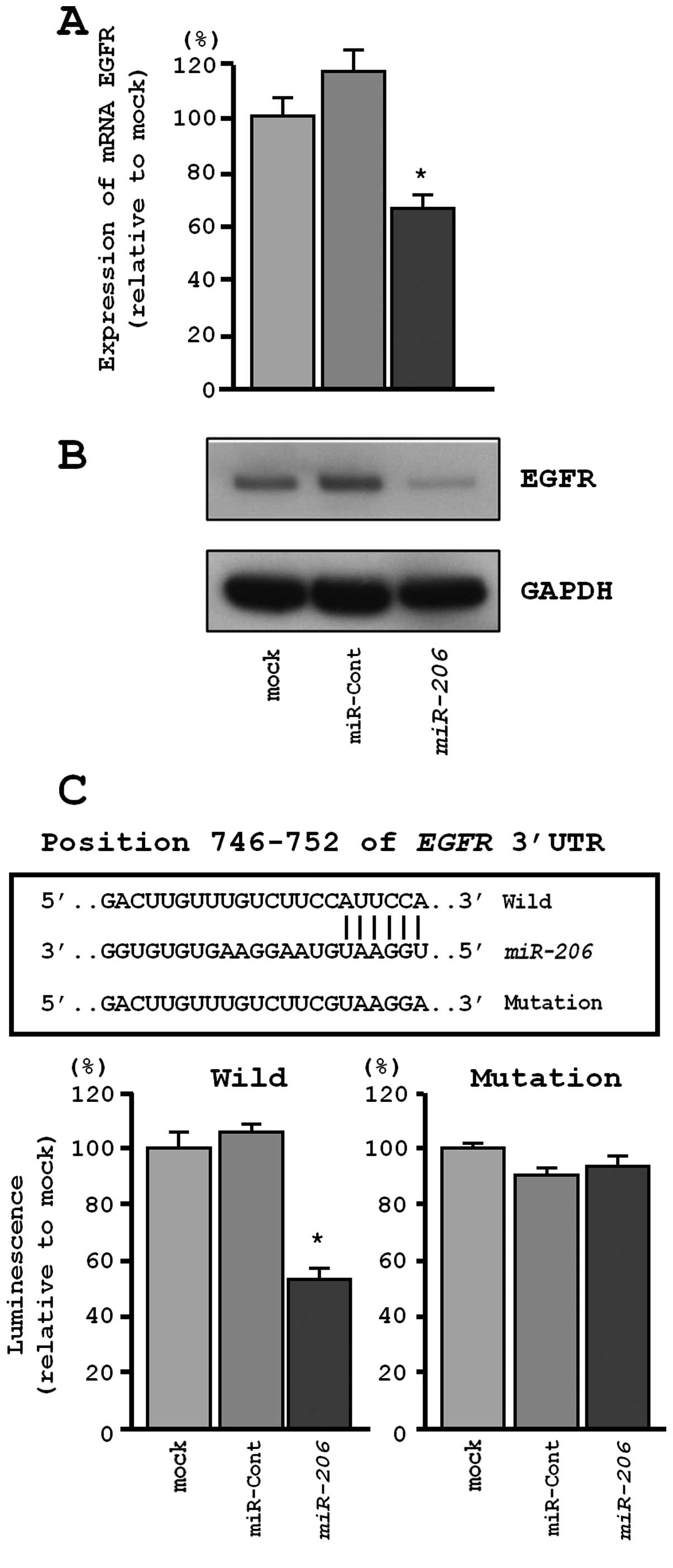
MET and EGFR were directly regulated by
miR-206 in EBC-1 cells
We performed qRT-PCR and western blotting to confirm
MET downregulation following restoration of miR-206
expression in EBC-1. The mRNA and protein expression levels of MET
and EGFR were significantly repressed in miR-206
transfectants in comparison with mock or miR-control transfectants
(P<0.001, Figs. 5A and B, and
6A and B).
The TargetScan database identified two putative
target sites in the 3′-UTR of MET (Fig. 5C, upper). A luciferase reporter
assay confirmed that the 3′-UTR of MET was indeed an actual target
of miR-206. Luciferase activity was significantly decreased
in two miR-206 target sites (positions 499–505 and 814–820
in the 3′-UTR of MET) (Fig.
5C, lower).
Similarly, the TargetScan database identified one
putative target site in the 3′-UTR of EGFR (Fig. 6C, upper). A luciferase reporter
assay confirmed that the 3′-UTR of EGFR was the actual target of
miR-206. Specifically, the luciferase activity was
significantly decreased at the miR-206 target site (position
746–752 in the 3′-UTR of EGFR) (Fig.
6C, lower).
Restoration of miR-206 inhibited ERK and
AKT signaling in EBC-1 cells
To investigate the effect of miR-206 on
pathway signaling, we checked the state of the phosphorylation of
MET, EGFR and the downstream proteins of these RTKs (ERK and AKT)
following miR-206 expression.
As shown in Fig. 7,
restoration of miR-206 inhibited phosphorylation of MET
(Tyr1003, Tyr1234/1239 and Tyr1349) and EGFR (Tyr1068 and Tyr1045)
in EBC-1 cells. We also confirmed miR-206-mediated
inhibition of phosphorylation of ERK1/2 and AKT that are downstream
from MET and EGFR (Fig. 7).
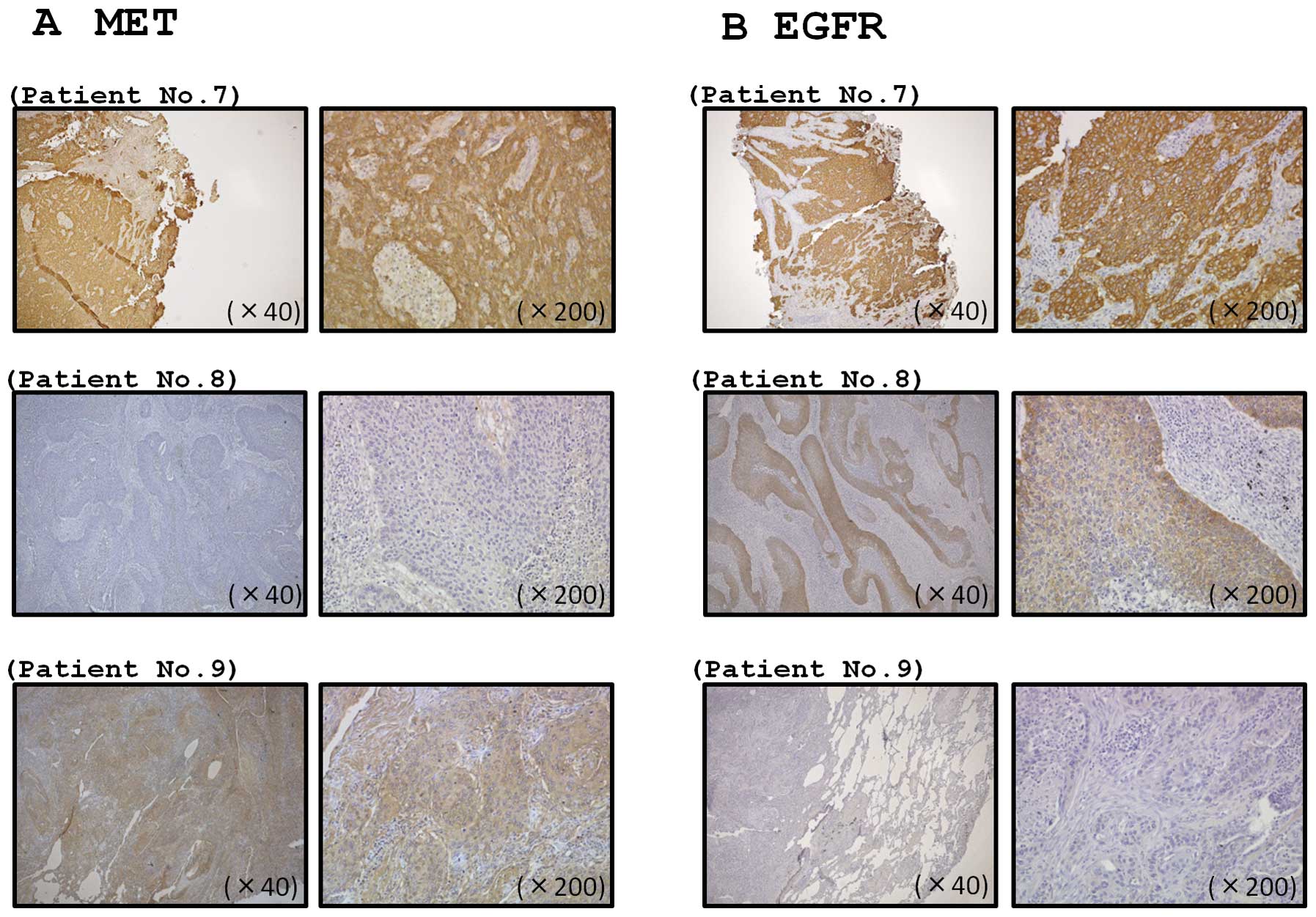
Expression of MET and EGFR in lung-SCC
clinical specimens
We confirmed the expression status of MET and EGFR
in lung-SCC clinical specimens using immunohistochemical staining.
A total of 32 specimens were checked in this study, and two and
four samples stained positively (≥50% of positive cells with
moderate or strong staining) for MET and EGFR, respectively
(Fig. 8). One sample stained
positively for both MET and EGFR (Fig.
8). Clinicopathological characteristics are summarized in
Table III.
| Table IIIImmunohistochemistry status and
characteristics of the patients. |
Table III
Immunohistochemistry status and
characteristics of the patients.
| | | | | TNM
classification | |
Immunohistochemistry |
|---|
| | | | |
| |
|
|---|
| Patient no. | Pleural
invasion | Venous
invasion | Lymphatic
invasion |
Differentiation | T | N | M | Pathological tumor
stage | MET | EGFR |
|---|
| 1 | (+++) | (+) | (+) | Moderately | 3 | 0 | 0 | IIB | (−) | (−) |
| 2 | (−) | (−) | (−) | Well | 3 | 2 | 0 |
IIIA | (−) | (−) |
| 3 | (−) | (+) | (+) | Moderately | 2b | 2 | 0 |
IIIA | (−) | (−) |
| 4 | (−) | (+) | (+) | Unknown | 1a | 1 | 0 | IIA | (−) | (−) |
| 5 | (−) | (+) | (+) | Well | 2a | 1 | 0 | IIA | (−) | (−) |
| 6 | (+++) | (−) | (−) | Unknown | 3 | 0 | 0 | IIB | (−) | (−) |
| 7 | (+++) | (+) | (+) | Poorly | 3 | X | 0 | Unknown | (++) | (+++) |
| 8 | (−) | (+) | (−) | Moderately | 1b | 0 | 0 | IA | (−) | (+++) |
| 9 | (−) | (−) | (−) | Well | 1a | X | 0 | Unknown | (++) | (−) |
| 10 | (−) | (+) | (−) | Well | 2a | 1 | 0 | IIA | (−) | (−) |
| 11 | (−) | (−) | (+) | Moderately | 4 | 2 | 0 |
IIIB | (−) | (++) |
| 12 | (+) | (+) | (−) | Poorly | 3 | 0 | 0 | IIB | (−) | (−) |
| 13 | (−) | (−) | (−) | Well | 1b | 0 | 0 | IA | (−) | (−) |
| 14 | (−) | (−) | (−) | Moderately | 2a | 0 | 0 | IB | (−) | (−) |
| 15 | (+) | (−) | (−) | Moderately | 3 | 0 | 0 | IIB | (−) | (−) |
| 16 | (−) | (−) | (−) | Moderately | 2a | 0 | 0 | IB | (+) | (++) |
| 17 | (−) | (+) | (−) | Moderately | 1a | 0 | 0 | IA | (−) | (−) |
| 18 | (−) | (−) | (+) | Moderately | 1b | 0 | 0 | IA | (−) | (−) |
| 19 | (−) | (−) | (+) | Moderately | 2a | 2 | 0 |
IIIA | (−) | (−) |
| 20 | (+++) | (+) | (+) | Moderately | 3 | 1 | 0 |
IIIA | (−) | (−) |
| 21 | (+) | (+) | (−) | Moderately | 2a | 0 | 0 | IB | (−) | (−) |
| 22 | (−) | (−) | (+) | Moderately | 2b | 2 | 0 |
IIIA | (−) | (−) |
| 23 | (+) | (−) | (+) | Moderately | 2a | 0 | 0 | IB | (−) | (+) |
| 24 | (++) | (+) | (+) | Moderately | 2a | 0 | 0 | IB | (−) | (−) |
| 25 | (++) | (−) | (+) | Moderately | 2a | 0 | 0 | IB | (−) | (−) |
| 26 | (−) | (+) | (+) | Moderately | 1a | 2 | X |
IIIA | (−) | (−) |
| 27 | (−) | (+) | (+) | Moderately | 1b | 2 | 0 |
IIIA | (−) | (−) |
| 28 | (+) | (+) | (−) | Well | 2a | 0 | 0 | IB | (−) | (−) |
| 29 | (+) | (+) | (−) | Well | 2a | 0 | 0 | IB | (−) | (−) |
| 30 | (+++) | (−−) | (−) | Moderately | 3a | 0 | 0 | IIB | (−) | (−) |
| 31 | (+) | (−) | (+) | Poorly | 2a | 1 | 0 | IIA | (−) | (−) |
| 32 | (+++) | (−) | (−) | Well | 3 | 1 | 0 |
IIIA | (−) | (−) |
Discussion
Aberrant expression of miRNAs can disrupt tightly
regulated RNA networks in normal cells, thereby promoting the
development and progression of human cancers. The first step in
defining the contribution of miRNAs to human cancers is to identify
the miRNAs that are differentially expressed in cancer cells.
Therefore, we have constructed miRNA expression signatures in
various cancers, allowing us to identify tumor-suppressive miRNAs
and their regulated cancer pathways (22,24,25).
Our lung-SCC signature revealed that miR-206 was
significantly reduced in cancer tissues (17).
The chromosomal location of miR-206 in the
human genome is of significant interest. miR-1-1/miR-133a-2,
miR-1-2/miR-133a-1 and miR-206/miR-133b form
clusters in three different chromosomal regions, 20q13.33, 18q11.2
and 6p12.1, respectively. Our previous studies demonstrated that
miR-1/133a clustered miRNAs function as tumor suppressors in
various types of human cancers, targeting several oncogenes
(26). The mature sequence of
miR-206 is similar to that of miR-1 in terms of
expression and function, but its sequence differs from that of
miR-1 by four nucleotides (26).
Our present data showed that restoration of
miR-206 significantly inhibited proliferation, migration and
invasion in EBC-1 cells, suggesting that miR-206 functions
as a tumor suppressor in lung-SCC. A tumor-suppressive function of
miR-206 has been reported in other types of cancers
(27–31). These findings indicate that
miR-206 is closely involved in human cancer. We also found
that expression of miR-133b was reduced in lung-SCC tissues
(Fig. 2B), and Spearman’s rank
test showed a positive correlation between the expression of
miR-206 and that of miR-133b (Fig. 2C). It is likely that the
miR-206/miR-133b cluster is frequently reduced in
cancer tissues and they function as tumor suppressors in lung-SCC.
For patients with advanced lung-SCC, the standard therapeutic
approach remains chemotherapy. Therefore, additional options to
treat lung-SCC are needed. Elucidation of the molecular targets and
pathways regulated by tumor suppressive miR-206 or
miR-133b in lung-SCC enhances our understanding of the
disease and suggests more effective strategies for future
therapeutic interventions.
The next problem we pursued was the identification
of the pathways/targets that were regulated by tumor-suppressive
miR-206 in lung-SCC cells. We used a combination of
expression data and in silico database analysis to identify
tumor-suppressive miR-206 regulated targets. In this
screening, several putative pathways and targets were annotated to
be subject to miR-206 regulation. Among them, we focused on
the RTKs because their overexpression is often observed in cancer,
and it is known that they contribute to anti-apoptotic signaling,
cell proliferation, angiogenesis and invasion, metastasis and drug
resistance (4,5). These findings have led to the
development of several therapeutic agents targeting RTKs. These
agents are now available for lung cancer, including gefitinib and
erlotinib for mutations of EGFR and crizotinib for the EML4-ALK
fusion gene (32–35).
In this study, we focused on MET and
EGFR as putative targets of tumor-suppressive
miR-206. We demonstrated that these RTKs were directly
regulated by miR-206. Overexpression of MET protein in tumor
tissue (relative to adjacent normal tissues) occurs in 27–77% of
NSCLC and is associated with a poor prognosis (35). Also, upregulation of EGFR was
reported in 40–80% of patients (36). MET signaling pathways are tightly
regulated in normal cells. However, in cancer cells, activating MET
signals promote cell proliferation, invasion, metastasis and
angiogenesis (37–39). Activation of MET signals causes
transcriptional deregulation, genetic abnormalities and crosstalk
between MET and other RTKs (37–39).
Although patients with NSCLC initially benefit from EGFR targeted
therapies, some patients ultimately acquire resistance to agents,
leading to disease progression (8). Importantly, in patients who have
acquired resistance to EGFR TKI, the MET amplification rate is
approximately 20% (9,10). Therefore, inhibition of MET
signaling must be targeted in this disease. Such therapeutics is in
fact now available (35,37–39).
In the present study, we found one patient with overexpression of
both MET and EGFR in lung-SCC lesions. In this situation, dual
inhibition treatment of MET and EGFR is necessary.
Several studies reported that MET or EGFR were
directly regulated by several miRNAs, such as miR-1/206,
miR-7 and miR-146a in several cancer cell types
(40–43). A recent study demonstrated that
miR-27a regulated both EGFR and MET in NSCLC (44). Our present data demonstrated that
miR-206 clearly inhibited both MET and EGFR
expression and their associated signaling in cancer cells. Dual
inhibition of tyrosine kinases by tumor-suppressive miR-27a
and miR-206 is a very attractive treatment option for the
treatment of lung-SCC lesions.
In conclusion, miR-206 was significantly
downregulated in lung-SCC clinical specimens. It appeared to
function as a tumor suppressor through regulation of oncogenic RTKs
(MET and EGFR) and their associated downstream signaling.
Elucidation of the cancer pathways and target genes regulated by
tumor-suppressive miR-206 should provide new approaches and
potential therapeutic targets in the treatment of lung-SCC.
Acknowledgements
We thank Ms. Mutsumi Miyazaki for her excellent
laboratory assistance.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Travis WD: Pathology of lung cancer. Clin
Chest Med. 32:669–692. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Reck M, Heigener DF, Mok T, Soria JC and
Rabe KF: Management of non-small-cell lung cancer: recent
developments. Lancet. 382:709–719. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gschwind A, Fischer OM and Ullrich A: The
discovery of receptor tyrosine kinases: targets for cancer therapy.
Nat Rev Cancer. 4:361–370. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cadena DL and Gill GN: Receptor tyrosine
kinases. FASEB J. 6:2332–2337. 1992.PubMed/NCBI
|
|
6
|
Engelman JA and Cantley LC: The role of
the ErbB family members in non-small cell lung cancers sensitive to
epidermal growth factor receptor kinase inhibitors. Clin Cancer
Res. 12:S4372–S4376. 2006. View Article : Google Scholar
|
|
7
|
Gherardi E, Birchmeier W, Birchmeier C and
Vande Woude G: Targeting MET in cancer: rationale and progress. Nat
Rev Cancer. 12:89–103. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sacher AG, Janne PA and Oxnard GR:
Management of acquired resistance to epidermal growth factor
receptor kinase inhibitors in patients with advanced non-small cell
lung cancer. Cancer. 120:2289–2298. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Engelman JA, Zejnullahu K, Mitsudomi T, et
al: MET amplification leads to gefitinib resistance in lung cancer
by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bean J, Brennan C, Shih JY, et al: MET
amplification occurs with or without T790M mutations in EGFR mutant
lung tumors with acquired resistance to gefitinib or erlotinib.
Proc Natl Acad Sci USA. 104:20932–20937. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hobert O: Gene regulation by transcription
factors andmi-croRNAs. Science. 319:1785–1786. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Iorio MV and Croce CM: MicroRNAs in
cancer: small molecules with a huge impact. J Clin Oncol.
27:5848–5856. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rolfo C, Fanale D, Hong DS, et al: Impact
of microRNAs in resistance to chemotherapy and novel targeted
agents in non-small cell lung cancer. Curr Pharm Biotechnol.
15:475–485. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Filipowicz W, Bhattacharyya SN and
Sonenberg N: Mechanisms of post-transcriptional regulation by
microRNAs: are the answers in sight? Nat Rev Genet. 9:102–114.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Friedman JM and Jones PA: MicroRNAs:
critical mediators of differentiation, development and disease.
Swiss Med Wkly. 139:466–472. 2009.PubMed/NCBI
|
|
17
|
Moriya Y, Nohata N, Kinoshita T, et al:
Tumor suppressive microRNA-133a regulates novel molecular networks
in lung squamous cell carcinoma. J Hum Genet. 57:38–45. 2012.
View Article : Google Scholar
|
|
18
|
Goldstraw P, Crowley J, Chansky K, et al:
The IASLC Lung Cancer Staging Project: proposals for the revision
of the TNM stage groupings in the forthcoming (seventh) edition of
the TNM Classification of malignant tumours. J Thorac Oncol.
2:706–714. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kojima S, Chiyomaru T, Kawakami K, et al:
Tumour suppressors miR-1 and miR-133a target the oncogenic function
of purine nucleoside phosphorylase (PNP) in prostate cancer. Br J
Cancer. 106:405–413. 2012. View Article : Google Scholar :
|
|
20
|
Nohata N, Hanazawa T, Kinoshita T, et al:
Tumour-suppressive microRNA-874 contributes to cell proliferation
through targeting of histone deacetylase 1 in head and neck
squamous cell carcinoma. Br J Cancer. 108:1648–1658. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kinoshita T, Nohata N, Hnasawa T, et al:
Tumour-suppressive microRNA-29s inhibit cancer cell migration and
invasion by targeting laminin-integrin signalling in head and neck
squamous cell carcinoma. Br J Cancer. 109:2636–2645. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yoshino H, Chiyomaru T, Enokida H, et al:
The tumour-suppressive function of miR-1 and miR-133a targeting
TAGLN2 in bladder cancer. Br J Cancer. 104:808–818. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Koeppen H, Yu W, Zha J, et al: Biomarker
analyses from a placebo-controlled phase II study evaluating
erlotinib ± onartuzumab in advanced non-small cell lung cancer: MET
expression levels are predictive of patient benefit. Clin Cancer
Res. 20:4488–4498. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Itesako T, Seki N, Yoshino H, et al: The
microRNA expression signature of bladder cancer by deep sequencing:
the functional significance of the miR-195/497 cluster. PLoS One.
9:e843112014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hidaka H, Seki N, Yoshino H, et al: Tumor
suppressive microRNA-1285 regulates novel molecular targets:
aberrant expression and functional significance in renal cell
carcinoma. Oncotarget. 3:44–57. 2012.PubMed/NCBI
|
|
26
|
Nohata N, Hanazawa T, Enokida H and Seki
N: microRNA-1/133a and microRNA-206/133b clusters: dysregulation
and functional roles in human cancers. Oncotarget. 3:9–21.
2012.PubMed/NCBI
|
|
27
|
Georgantas R, Streicher K, Greenless L, et
al: MicroRNA-206 induces G1 arrest in melanoma by inhibition of
CDK4 and Cyclin D. Pigment Cell Melanoma Res. 27:275–286. 2014.
View Article : Google Scholar
|
|
28
|
Missiaglia E, Shepherd CJ, Patel S, et al:
MicroRNA-206 expression levels correlate with clinical behaviour of
rhabdomyosarcomas. Br J Cancer. 102:1769–1777. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yan D, da Dong XE, Chen X, et al:
MicroRNA-1/206 targets c-Met and inhibits rhabdomyosarcoma
development. J Biol Chem. 284:29596–29604. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen X, Yan Q, Li S, et al: Expression of
the tumor suppressor miR-206 is associated with cellular
proliferative inhibition and impairs invasion in ERalpha-positive
endometrioid adenocarcinoma. Cancer Lett. 314:41–53. 2012.
View Article : Google Scholar
|
|
31
|
Kondo N, Toyama T, Sugiura H, et al:
miR-206 expression is down-regulated in estrogen receptor
alpha-positive human breast cancer. Cancer Res. 68:5004–5008. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Maemondo M, Inoue A, Kobayashi K, et al:
Gefitinib or chemotherapy for non-small-cell lung cancer with
mutated EGFR. N Engl J Med. 362:2380–2388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhou C, Wu YL, Chen G, et al: Erlotinib
versus chemotherapy as first-line treatment for patients with
advanced EGFR mutation-positive non-small-cell lung cancer
(OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase
3 study. Lancet Oncol. 12:735–742. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shaw AT, Yeap BY, Solomon BJ, et al:
Effect of crizotinib on overall survival in patients with advanced
non-small-cell lung cancer harbouring ALK gene rearrangement: a
retrospective analysis. Lancet Oncol. 12:1004–1012. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Scagliotti GV, Novello S and von Pawel J:
The emerging role of MET/HGF inhibitors in oncology. Cancer Treat
Rev. 39:793–801. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gately K, Forde L, Cuffe S, et al: High
coexpression of both EGFR and IGF1R correlates with poor patient
prognosis in resected non-small-cell lung cancer. Clin Lung Cancer.
15:58–66. 2014. View Article : Google Scholar
|
|
37
|
Maroun CR and Rowlands T: The Met receptor
tyrosine kinase: a key player in oncogenesis and drug resistance.
Pharmacol Ther. 142:316–338. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sadiq AA and Salgia R: MET as a possible
target for non-small-cell lung cancer. J Clin Oncol. 31:1089–1096.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cipriani NA, Abidoye OO, Vokes E and
Salgia R: MET as a target for treatment of chest tumors. Lung
Cancer. 63:169–179. 2009. View Article : Google Scholar :
|
|
40
|
Nasser MW, Datta J, Nuovo G, et al:
Down-regulation of micro-RNA-1 (miR-1)in lung cancer. Suppression
of tumorigenic property of lung cancer cells and their
sensitization to doxorubicin-induced apoptosis by miR-1. J Biol
Chem. 283:33394–33405. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Taulli R, Bersani F, Foglizzo V, et al:
The muscle-specific microRNA miR-206 blocks human rhabdomyosarcoma
growth in xenotransplanted mice by promoting myogenic
differentiation. J Clin Invest. 119:2366–2378. 2009.PubMed/NCBI
|
|
42
|
Kefas B, Godlewski J, Comeau L, et al:
microRNA-7 inhibits the epidermal growth factor receptor and the
Akt pathway and is down-regulated in glioblastoma. Cancer Res.
68:3566–3572. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li Y, Vandenboom TG II, Wang Z, et al:
miR-146a suppresses invasion of pancreatic cancer cells. Cancer
Res. 70:1486–1495. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Acunzo M, Romano G, Palmieri D, et al:
Cross-talk between MET and EGFR in non-small cell lung cancer
involves miR-27a and Sprouty2. Proc Natl Acad Sci USA.
110:8573–8578. 2013. View Article : Google Scholar : PubMed/NCBI
|