Introduction
Despite recent advances in developing therapeutic
strategy for multiple myeloma (MM), MM still remains incurable and
thus a novel therapeutic approach is urgently needed (1). By screening natural compound
libraries, we found that shikonin (SHK), a natural naphthoquinone
derivative isolated from the root of Lithospermum
erythrorhizon, efficiently induced cell death in MM cells.
SHK has been reported to induce cell death in
various tumor cell lines, such as breast cancer, leukemia and
prostate cancer (2–6). Recent report showing efficacy of SHK
to human promyelocytic leukemia cells by interacting with
thioredoxin reductase may promise efficacy of SHK to human
hematopoietic tumors, not only myeloma cells, through multiple
activities.
However, there are no reports showing the efficacy
of SHK in MM cells. Although mechanisms by which SHK regulates cell
death have not been fully analyzed, SHK has been shown to induce
activation of caspases (4,5) by modulating Bcl-2 family proteins,
p27 and p53 (7), which eventually
leads to apoptosis. Involvement of reactive oxygen species (ROS)
(8,9) and the NFκB signaling pathway
(10) in SHK-mediated apoptosis
have also been reported. SHK also functions as a proteasome
inhibitor (11) and topoisomerase
I inhibitor (12). SHK has also
been demonstrated to induce the molecular chaperone heat shock
protein 70 (HSP70) (13,14).
Previous studies have implicated a function for
HSP70 in MM cells. Inhibition of HSP70 reversed drug resistance and
induced apoptosis in MM cells (15–17),
indicating a contribution of HSP70 to the survival of MM cells.
However, the role of HSP70 in MM cells has not been extensively
analyzed.
Interestingly, SHK is also an inducer of necroptosis
in osteosarcoma and glioma cells (18,19).
Necroptosis (programmed necrosis) is a type of cell death distinct
from apoptosis (20). Necroptosis
depends on the kinase activities of receptor-interacting proteins 1
and 3 (RIP1 and RIP3) and is specifically inhibited by the small
molecule necrostatin-1 (Nec-1), which targets RIP1 (21–24)
resulting in caspase-independent cell death. Induction of
necroptosis by SHK was reported in human hematopoietic cells
(25,26). However, no study has examined the
potential for SHK-mediated induction of necroptosis in MM
cells.
In this study, we investigated the mechanisms
underlying SHK regulation of cell death in MM cells, including
apoptosis and necroptosis.
Materials and methods
MM cell lines and patient samples
Human myeloma cell lines KMS-12-PE (27), KMS-12-BM (27), RPMI-8226 (28), KMM1 (29), U266 (30), KMS11 (31), and the bortezomib-resistant myeloma
cell line KMS11/BTZ (32), were
cultured in RPMI-1640 medium containing 10% fetal bovine serum at
37 °C under 5% CO2. KMS11/BTZ cells were kindly provided
by Kyowa Hakko Kirin Co. Ltd. Bone marrow sample was obtained from
an MM patient treated at Kumamoto University Hospital clinically
refractory to both bortezomib and lenalidomide under written
informed consent. After isolation of mononuclear cells from bone
marrow samples using Ficoll-Paque Plus (GE Healthcare, Uppsala,
Sweden), myeloma cells were purified using CD138-immunomagnetic
beads (Miltenyi Biotech, Paris, France) as previously described
(33).
Reagents
Shikonin, Necrostatin-1 and thapsigargin were
purchased from Sigma-Aldrich (St. Louis, MO, USA). VER-155008, an
ATP-derivative inhibitor of HSP70, was purchased from Enzo Life
Sciences (Farmingdale, NY, USA). These reagents were dissolved in
phosphate-buffered saline. Bortezomib was purchased from Janssen
Pharmaceutical (Tokyo, Japan).
Cell viability assay
Cell viability was determined by WST-8 assay using
the Cell Counting Kit-8 (Dojindo, Kumamoto, Japan). Briefly, cells
were seeded in 96-well plates at a concentration of
2×104/100 μl and incubated with reagents SHK, bortezomib
or VER-155008, for 24 h. Following treatment, cells were incubated
with WST-8 reagent for a subsequent 3 h. The light absorbance of
each well was measured at 450 nm using a VMax absorbance microplate
reader (Molecular Devices, Sunnyvale, CA, USA). Data were obtained
from three independent experiments.
Analysis of apoptosis and
necroptosis
Cells were incubated at a concentration of
5×105/ml in the presence of SHK for 7 h, or with
bortezomib or VER-155008 for 24 h. Cell death was evaluated using
the trypan blue exclusion assay (Gibco, Carlsbad, CA, USA).
Inhibitors of pan-caspase, Z-VAD-FMK (MBL, Nagoya, Japan) at a
concentration of 50 μM, and necroptosis, Nec-1 (necrostatin-1) at a
concentration of 60 μM, were employed to distinguish apoptosis and
necroptosis, respectively. In some experiments, MM cells were
pretreated for 20 min with Z-VAD-FMK before analysis of cell death.
Morphological examinations of cells were performed with May-Giemsa
staining and transmission electron microscopy. For transmission
electron microscopy, cells were centrifuged at 2,000 g for 10 min,
fixed in 2.5% glutaraldehyde buffer (pH 7.4, 4°C), and then
post-fixed in 1% osmium tetraoxide and embedded in Epon. Ultrathin
sections were cut with an ultramicrotome, stained with uranyl
acetate and lead citrate, and examined in a Hitachi H-7500 (Tokyo,
Japan).
Western blot analysis
Antibodies against caspase-3, HSP70, HSP90,
ubiquitinated proteins and actin were purchased from Santa Cruz
Biotechnology (Santa Cruz, CA, USA). Antibodies against caspase-8
and RIP-1 were purchased from Cell Signaling Technology (Beverly,
MA, USA). Anti-HSP70 (HSP72) antibody was purchased from Enzo Life
Sciences. Cell lysates were prepared using the M-PER mammalian
protein extraction reagent (Thermo Scientific Inc., Rockford, IL,
USA) after addition of Halt EDTA-free phosphatase inhibitor
cocktail and Halt protease inhibitor cocktail (both from Thermo
Scientific Inc.). The cell lysates were separated in NuPAGE
Bis-Tris precast gels (Invitrogen, Carlsbad, CA, USA) and
transferred to PVDF membranes using an iBlot Dry Blotting system
(Invitrogen). The membranes were blocked with 5% non-fat dry milk
dissolved in Tris-buffered saline (TBS) containing 0.5% Tween-20
(TBS-T) for 1 h at room temperature, followed by incubation with
the primary antibodies at 4°C overnight. After washing with TBS-T,
the membranes were incubated with a horseradish
peroxidase-conjugated secondary antibody (Amersham Biosciences,
Oxford, UK) diluted in TBS-T for 2 h at room temperature. The
antibody-bound proteins were visualized using an ECL plus kit
(Amersham Bioscience).
Proteasome inhibition assay
Proteasome activity was analyzed by 20S proteasome
activity assay kit (Chemicon USA & Canada, cat. no. APT280)
according to the manufacturer’s protocol using Corona
multi-microplate reader MTP-800AFC (Ibaragi, Japan). Data were
obtained from three independent experiments.
Reverse transcription-polymerase chain
reaction (RT-PCR)
RNA was extracted using TRIzol reagent (Invitrogen).
cDNA synthesis was performed using the SuperScript III First-Strand
Synthesis system for RT-PCR (Invitrogen) according to the
manufacturer’s protocol. Thapsigargin, a sarcoplasmic/endoplasmic
reticulum Ca2+ ATPase (SERCA) inhibitor, was used as an
ER stress inducer. ER stress was assessed by detecting activated
XBP-1, which was analyzed by digestion of PCR products with
ApaLI (New England Biolabs Inc., MA, USA), as previously described
(34). Because the ApaLI site in
XBP-1 mRNA is spliced out upon activation, activated
XBP-1 shows one large band after ApaLI digestion, while
inactivated XBP-1 shows two ApaLI-digested bands.
Primers for XBP-1 were 5′-AAA CAG AGT AGC AGC
TCA GAC TGC-3′ (sense) and 5′-CTC CCA GAG GTC TAC CCA GAA GGA -3′
(antisense). GAPDH was used as a normalization control.
Primers for GAPDH were previously described (35).
Statistical analysis and drug combination
analyses
Statistical analyses were examined using Student’s
t-test. P-values <0.05 were considered statistically
significant. The interactions between SHK and VER-155008 was
analyzed by Chou’s combination index (CI) using CalcuSyn software
Version 2.1 (Biosoft, Cambridge, UK) to determine whether the
combination was additive or synergistic (36).
Results
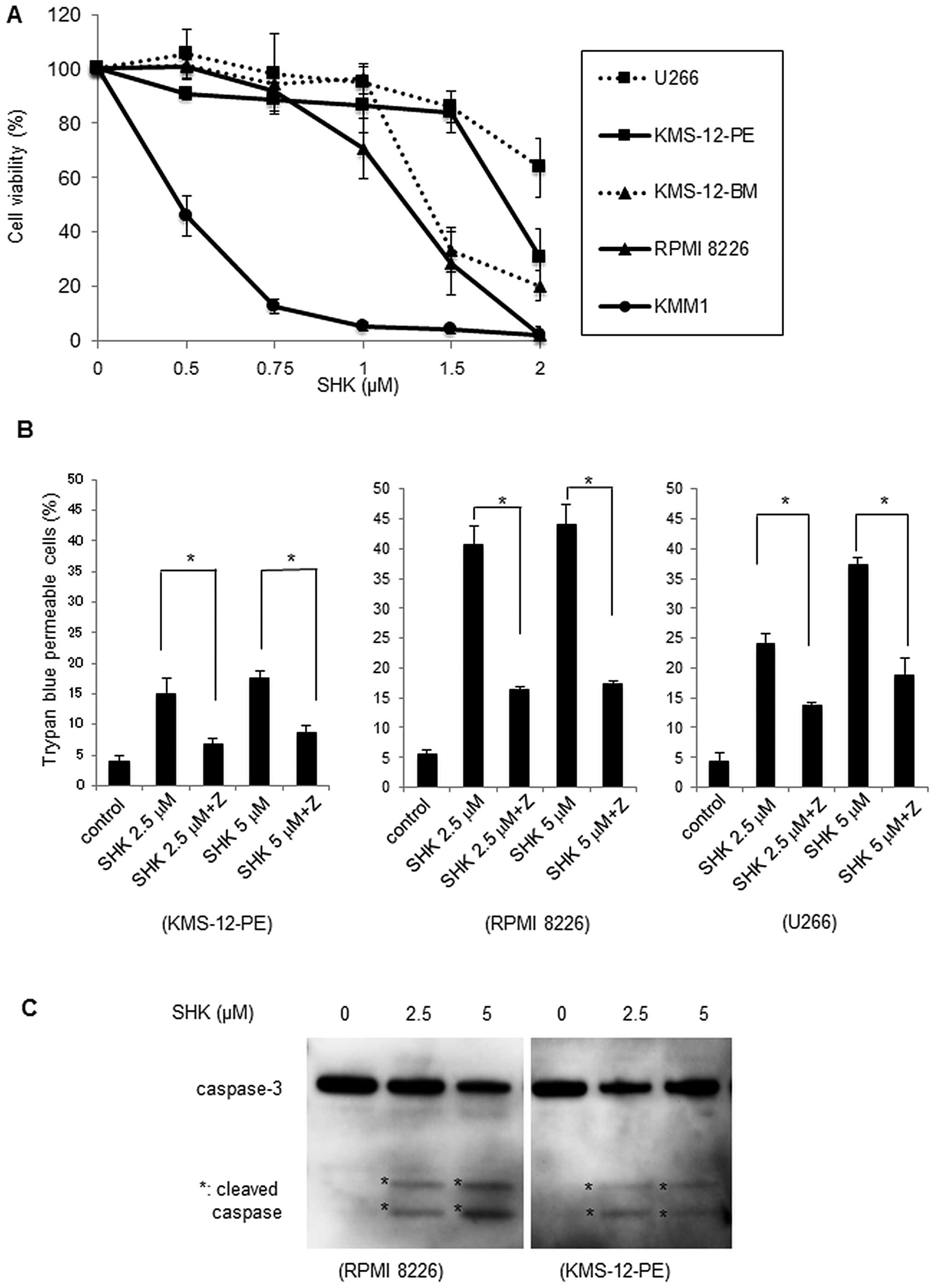
SHK induces cytotoxicity in MM cells
The human MM cell lines were cultured for 24 h in
the presence of various concentrations of SHK and cell viability
was analyzed by WST-8 assay. As shown in Fig. 1A, SHK exerted cytotoxic effects in
all MM cells in a dose-dependent manner, although the effect was
varied. These results clearly indicated that SHK exhibits
cytotoxicity in MM cells at concentrations <2 μM.
SHK induces apoptosis in MM cells
To further investigate the mechanisms of SHK in
regulating cell death, we utilized the pan-caspase inhibitor
Z-VAD-FMK. After pretreatment of three MM cell lines, KMS-12-PE,
RPMI-8226 and U266, with Z-VAD-FMK for 20 min, cells were incubated
with SHK at 2.5 or 5 μM for 7 h and then analyzed by trypan blue
assay. As shown in Fig. 1B, the
percentages of trypan blue permeable cells under treatment with SHK
were partly inhibited by treatment with Z-VAD-FMK (P<0.01),
indicating that SHK induced cell death in MM cells via activated
caspases.
Western blot analysis revealed that caspase-3 was
activated by SHK (Fig. 1C).
Morphological analysis of SHK-treated MM cells showed typical
apoptotic changes, such as decreased cellular volume, chromatin
condensation, and nuclear fragmentation, and these apoptotic
morphological changes were inhibited by Z-VAD-FMK treatment
(Fig. 1D). Together these
observations suggest that SHK induces apoptosis in MM cells.
In combination with bortezomib, SHK increased
cytotoxic effects for KMS-12-PE cells at a concentration of 0.5 μM
(Fig. 1E), a concentration at
which SHK alone was unable to induce cytotoxic effects (Fig. 1A). This indicates that low dose SHK
sensitizes MM cells to bortezomib.
SHK induces apoptosis in
bortezomib-resistant cells and freshly isolated MM cells
We further investigated whether SHK can overcome
resistance to bortezomib. We used the bortezomib resistant MM cell
line KMS-11/BTZ, which has a 9.9-fold higher IC50 value
to bortezomib than that of its parental cell line KMS11 (Fig. 2A). As shown in Fig. 2B, viability of both KMS11 and
KMS-11/BTZ cells was inhibited by SHK in a dose-dependent manner.
Interestingly, the IC50 value of SHK for KMS11/BTZ cells
was even lower than that for KMS11 cells (1.1 vs. 1.56 μM,
respectively). These results suggest that SHK may overcome
refractoriness of MM cells to bortezomib. We then examined the
effects of a combination of SHK and bortezomib on KMS11/BTZ cells.
As shown in Fig. 2C, treatment of
KMS11/BTZ cells with very low concentration of SHK (0.5 μM)
significantly increased the sensitivity to bortezomib, suggesting
re-sensitization of bortezomib-resistant MM cells to bortezomib by
SHK.
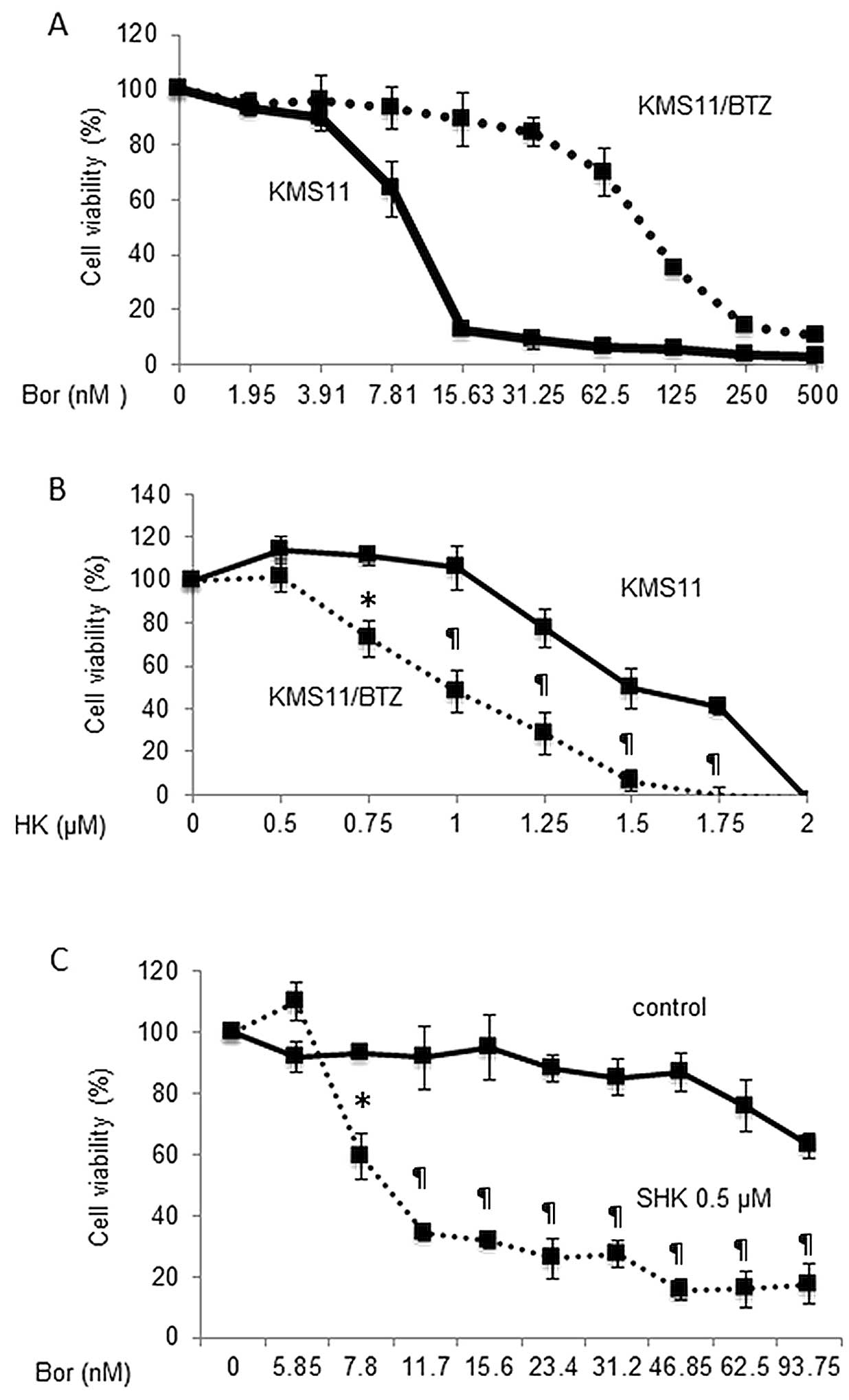 | Figure 2Antitumor effect of SHK on bortezomib
resistant cells, primary MM cells and peripheral blood mononuclear
cells. The bortezomib resistant MM cell line KMS-11/BTZ, and the
parental cell line KMS11, were cultured with various concentrations
of bortezomib (A), SHK (B) or in combination (C) for 24 h and cell
viabilities were analyzed by WST-8 assay. (A) The bortezomib
resistant MM cell line KMS-11/BTZ (dotted line), and the parental
cell line, KMS11 (solid line), were treated with bortezomib for 24
h and subsequently analyzed by WST8 assay. The IC50
values of the KMS11 and KMS11/BTZ cells were 9.9 and 98.5 nM,
respectively. (B) The IC50 value of SHK for KMS11/BTZ
cells (dotted line) was even lower than that of KMS11 cells (solid
line) (1.1 vs. 1.56 μM, respectively). *P<0.05,
¶P<0.01. (C) Treatment of KMS11/BTZ with (dotted
line) or without (solid line) low concentration of SHK (0.5 μM),
which alone does not show cytotoxic effects, increased the
sensitivity to bortezomib. *P<0.005,
¶P<0.0001. (D and E) MM cells from primary bone
marrow sample were incubated with 0.5 μM SHK for 16 h and then
either evaluated by cytospin analysis (D) or WST-8 assay (E). Both
analyses revealed marked increase of dead cells in response to
treatment with SHK and inhibition by Z-VAD-FMK. (F) Lack of
cytotoxic effect of SHK in normal PBMCs. PBMCs were cultured with
0.5 μM SHK for 24 h and evaluated by trypan blue dye exclusion
assay. There was no increase of dead cells by SHK. |
We further investigated whether SHK could induce
cell death in primary MM cells from a patient clinically refractory
to both bortezomib and lenalidomide. We observed marked cell death
in response to treatment with SHK, and the proportion of dead cells
was clearly inhibited by Z-VAD-FMK (Fig. 2D and E) while SHK alone did not
influence viability of peripheral blood mononuclear cells (PBMCs)
from a healthy donor (Fig. 2F).
Together this suggests that SHK exerts anti-tumor effects in
primary MM cells refractory to bortezomib while sparing toxicity to
normal PBMCs.
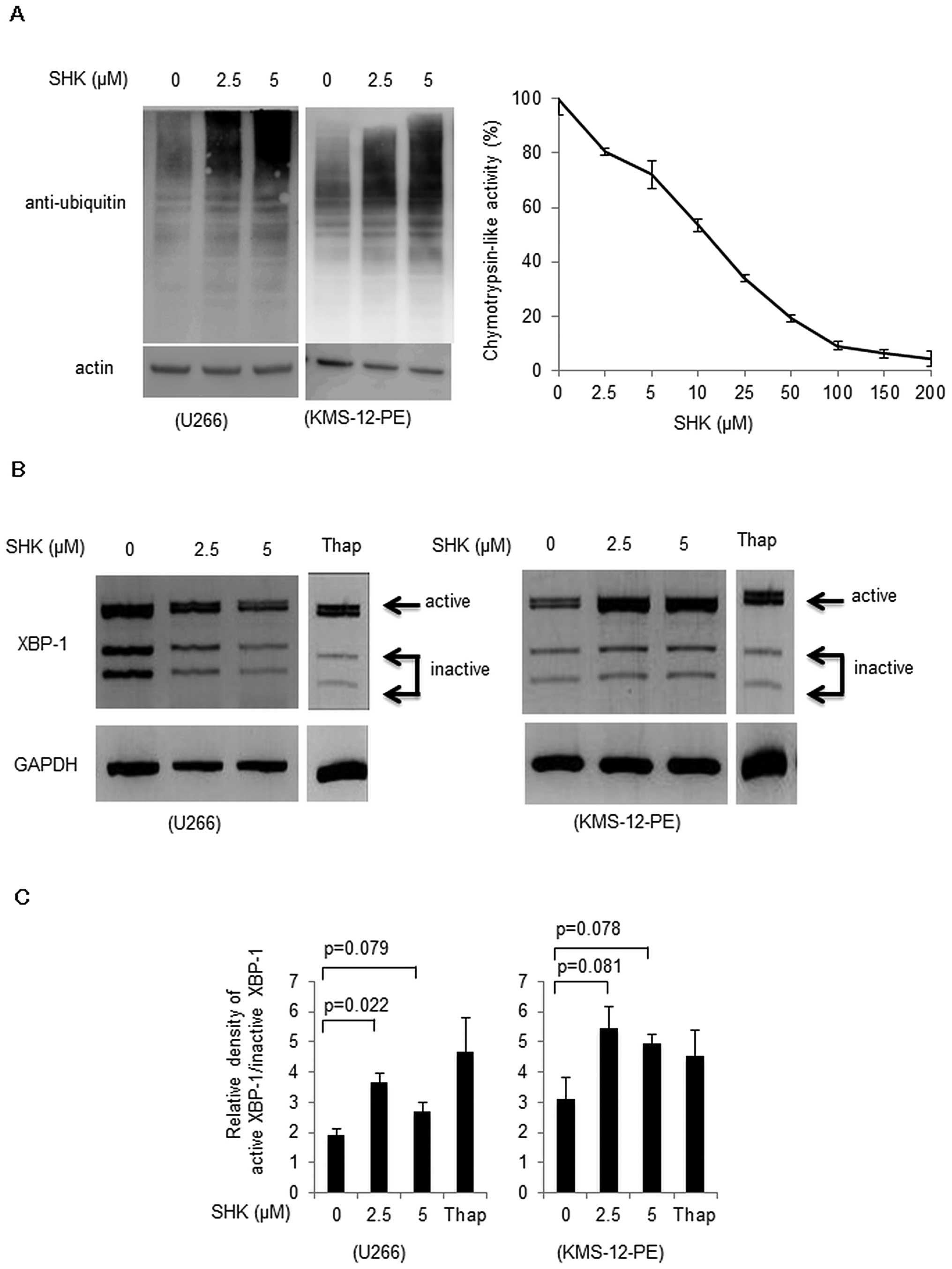
SHK inhibits proteasome functions and
induces ER stress response
We further analyzed in detail the mechanisms
underlying SHK-induced apoptosis. A previous report (11) suggested possible proteasome
inhibitor function of SHK, and we also found that SHK induced an
accumulation of ubiquitinated proteins in a dose-dependent manner
(Fig. 3A, left panel). To further
confirm proteasome inhibitory mechanisms by SHK, direct interaction
between SHK and 20S proteasome was analyzed. We found that SHK
inhibited proteasome enzymatic activity at a dose-dependent manner
(Fig. 3A, right panel).
As shown in Fig. 3B and
C, SHK induced spliced form of XBP-1 in both U266 cells and
KMS-12-PE cells. These results suggest that SHK may function as a
proteasome inhibitor, which eventually evokes the ER stress
response.
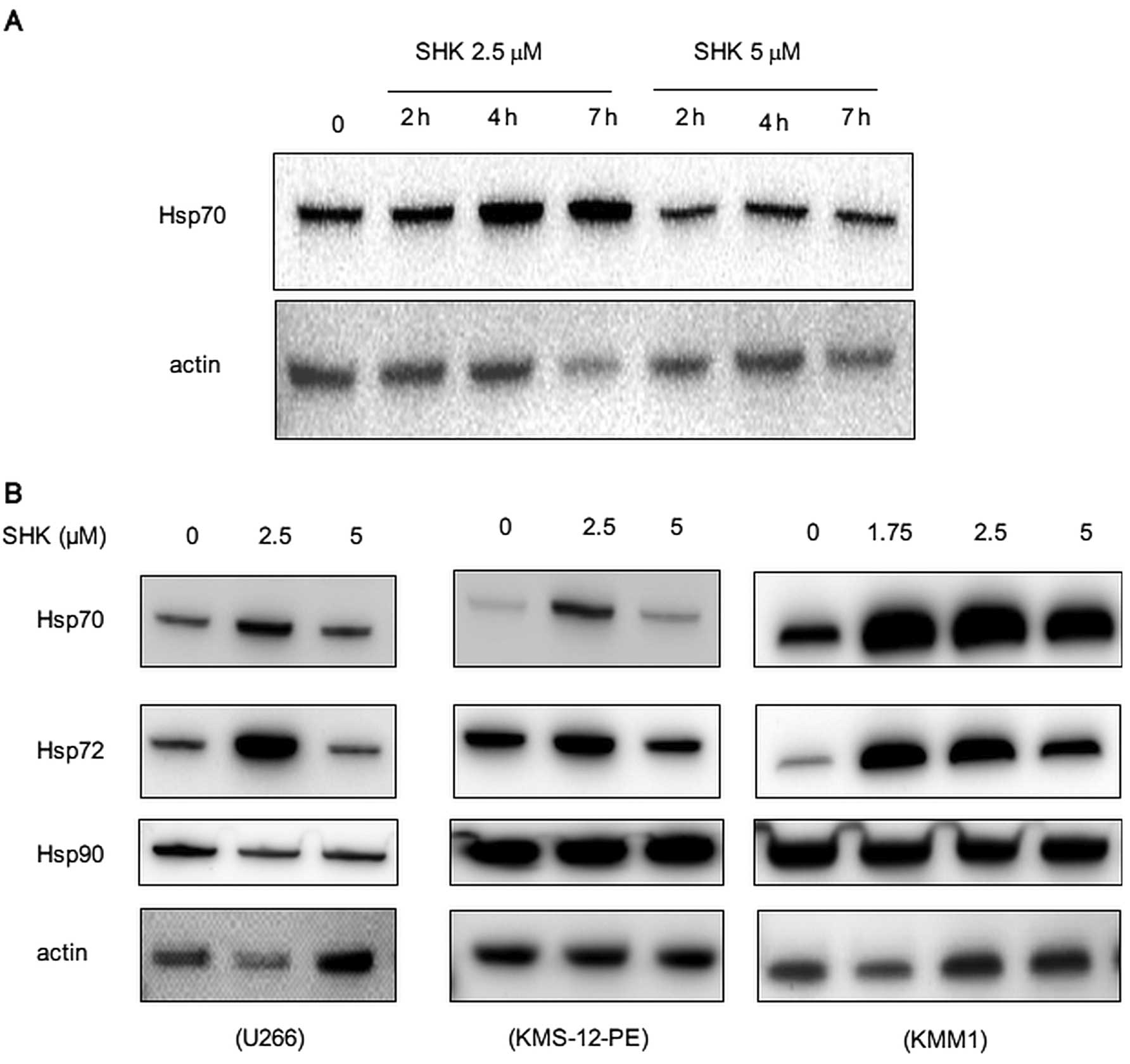
SHK increases HSP 70/72 and exerts cytotoxic effect
in combination with HSP70/72-inhibitor, VER-155008. Our preliminary
observations using mass-spectrometry analysis suggested induction
of heat shock proteins (HSPs) by SHK (data not shown), which was
consistent with previous reports in monocytes and leukemia cells
(13,14). We next analyzed if SHK induced HSPs
in MM cells. As expected, SHK transiently increased amounts of
HSP70 after 2–7 h of treatment (Fig.
4A). This was more prominent at a concentration of 2.5 μM than
5 μM, suggesting that induction of HSP70 is dose-sensitive. We also
detected a similar induction of HSP72, an inducible form of HSP70,
while there was no significant change in expression of HSP90
(Fig. 4B).
Because HSPs are known to support cell survival
under various stresses, we utilized the HSP70/72 inhibitor
VER-155008, to analyze the influence of HSPs in cytotoxic effects
delivered by SHK. VER-155008 alone increased cytotoxicity of MM
cells in a dose-dependent manner (Fig.
4C), indicating that inhibition of constitutive HSPs is capable
of induction of cell death. Interestingly, addition of VER-155008
to cells treated with low dose SHK prepared at concentrations
<0.5 μM enhanced the proportion of dead cells, whereas SHK alone
did not influence cell viability (Fig.
4D). The combination index analysis (CI=0.72) according to the
method of Chou (36) revealed a
synergistic effect of SHK and VER-155008. The proportion of dead
cells induced by the combination of SHK and VER-155008 was partly
reduced by treatment with Z-VAD-FMK, indicating that the
combination of SHK and VER-155008 induced caspase-mediated
apoptosis (Fig. 4E). Analysis
using PBMCs from a normal donor showed no cytotoxic effect by this
combination treatment (Fig.
4F).
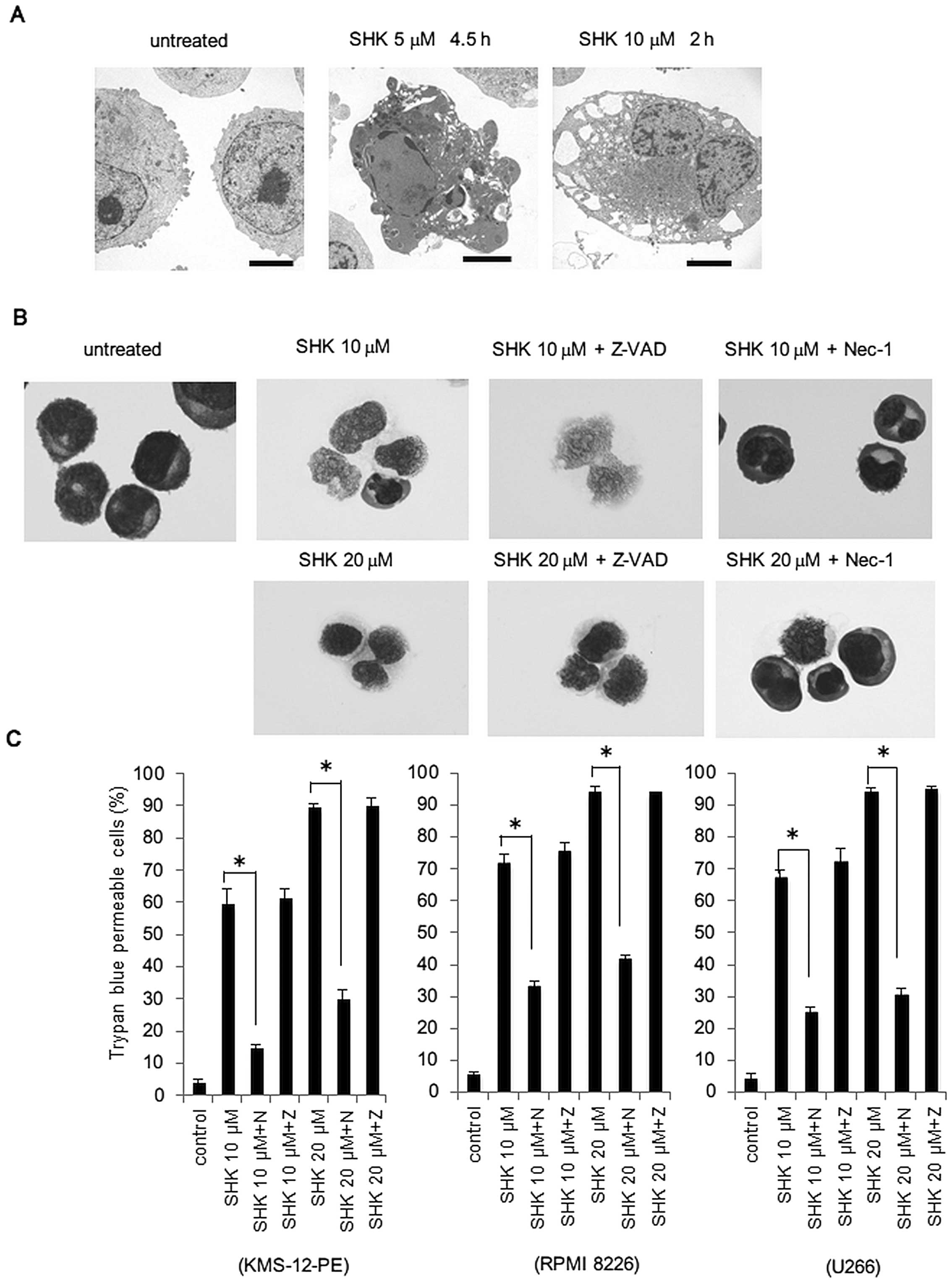
SHK induces necroptosis in MM cells at
high concentrations
Because SHK has been reported to induce necroptosis
in various tumor cell lines, we examined whether SHK could induce
necroptosis in MM cells. As shown in Fig. 5A, electron microscopic examination
revealed typical necrotic changes, such as translucent cytoplasm
and swelling of cell membranes by treatment with SHK at a
concentration of 10 μM for 2 h, while apoptotic changes were
observed with treatment with SHK at lower concentrations. To
further confirm the induction of necroptosis, cells were either
treated with Z-VAD-FMK or the necroptosis inhibitor, necrostatin-1
(Nec-1), in combination with SHK at 10 and 20 μM. As shown in
Fig. 5B, cell death was completely
inhibited by (Nec-1), and unaffected by Z-VAD-FMK, indicating that
cell death delivered by SHK at high concentration was necroptosis.
Trypan blue dye exclusion assay showed that SHK greatly increased
the proportion of dead cells at 10 or 20 μM, and this was again
inhibited by Nec-1 and not affected by Z-VAD-FMK (Fig. 5C), confirming the induction of
necroptosis by high-dose SHK. Western blot analysis revealed
activation of caspase-8 and -3 by SHK at concentrations starting
from 2.5 μM and maximizing at 5 μM, and decreasing at higher
concentrations (Fig. 5D, upper
panels), indicating that activation of caspases are specifically
dependent on concentration of SHK. Because necroptosis is dependent
on RIP1 function, we further evaluated the status of RIP1 in
response to SHK. We detected cleavage of RIP1 by treatment with SHK
at lower concentrations (~10 μM), while it was not cleaved and
remained active at higher concentrations (Fig. 5D, lower panel), indicating that SHK
dynamically regulates the cleavage of RIP1 in a
concentration-dependent manner.
Discussion
Our above results demonstrate that SHK exerted
antitumor effects in MM cells. No previously examination of the
potential cytotoxic effects of SHK in MM cells, has been reported
and to the best of our knowledge, ours is the first report
providing possible therapeutic efficacy of SHK in MM. We also
showed that SHK exerted cytotoxicity to both the
bortezomib-resistant cell line and freshly isolated MM cells from a
patient clinically refractory in both bortezomib and lenalidomide.
The IC50 value of SHK to a bortezomib-resistant cell
line was even lower than that of the parental cell line. The
mechanisms regulating cell death in bortezomib resistant cell line
is unknown. Western blot analysis of KMS11/BTZ treated with SHK
revealed slight accumulation of ubiquitinated proteins but the
amount was rather less than what found in parental cells (data not
shown), suggesting SHK should possess other mechanisms than
proteasome inhibition. Collectively, these findings suggested that
monotherapy using SHK may overcome refractoriness to bortezomib.
Alternatively, since treatment of the bortezomib resistant MM cell
line KMS-11/BTZ, with very low concentration of SHK (0.5 μM)
sensitized cells to bortezomib; this suggests that SHK may be
utilized as a combinational reagent with bortezomib. These results
indicate that SHK may overcome refractoriness of MM cells to
bortezomib by either monotherapy or in combination with bortezomib.
Future studies in other refractory cases should be performed to
examine this possibility.
Next, we attempted to elucidate the biological
mechanisms of SHK in inducing cell death. Because previous reports
showed induction of HSP70 (13,14)
or inhibition of proteasome activity by SHK (11), and our preliminary observations
using mass-spectrometry analysis suggested induction of HSPs by SHK
(data not shown), we investigated whether SHK had an effect on
HSP70 or ubiquitinated proteins. Accumulation of ubiquitinated
proteins and activation of XBP-1 were found in response to
treatment with SHK at low concentration (<5 μM), suggesting that
SHK may function as a proteasome inhibitor eventually leading to
accumulation of ER stress. This ER stress inducing property of SHK
may explain the coordinate action of SHK and bortezomib in respect
to augmented inhibition of proteasome function.
That HSP70 induction is detected by SHK treatment
may seem conflicting, as SHK alone causes cytotoxic effects and
HSP70 is considered to support cell survival. HSP70 is
overexpressed in MM cells and HSP70 inhibition is reported to
reverse drug resistance (15–17).
Our observation that transient increase of both HSP70 and its
inducible form of HSP70 (HSP72) by SHK may be explained by
accumulation of ER stress by SHK, although the exact mechanisms
underlying SHK regulation of HSP70 is not known. Although SHK
eventually induces apoptosis, cells may try to overcome these
stress by inducing molecules such as heat shock proteins. In that
situation, imbalance of stress and stress-relief should be a key
for survival of tumor cells.
To evaluate if antitumor effects delivered by SHK
could be enhanced by HSP70 inhibition, a combination of SHK and the
HSP70 inhibitor VER-155008 was used. As expected, VER-155008 alone
exerted cytotoxicity to MM cells in a dose-dependent manner. This
was not surprising because HSP inhibitors are reported to be
therapeutic candidates for MM (37,38).
However, combination of VER-155008 at 3 μM, which exerts ~40%
reduction of cell viability, with SHK at very low concentration
(<0.5 μM) significantly increased the proportion of dead cells.
These results show, for the first time, that besides SHK alone
potentially serving as a treatment approach in MM, it significantly
shows efficacy in inhibiting growth of MM cells in combination with
the HSP70 inhibitor. Although efficacy of SHK may be limited,
combining HSP70 inhibitor with SHK may promise efficient induction
of cell death. We found that the concentrations of SHK required for
combined treatment with HSP70 inhibitor is quite low, allowing us
to minimize the toxic effect of SHK to normal tissues. Indeed, both
SHK alone at low concentrations and in combination with VER-155008
did not show toxicity to normal PBMCs, suggesting the safety of the
combination. However, extensive analysis regarding toxicity of SHK
to normal tissues should be performed before clinical
utilization.
Furthermore, we showed that SHK at higher
concentrations induced necroptosis in MM cells. Necroptosis
(programmed necrosis) is regulated by RIP1 and RIP3 through complex
formation and activation (20,21,39).
Necroptosis, which is distinct from apoptosis, exhibits specific
characteristics. First, after the death receptor family is
stimulated, necroptosis is induced under activation of RIP by
inhibition of caspase-8, resulting in caspase-independent cell
death (24). By contrast, under
activation of caspase-8, it cleaves RIP1 and RIP3, resulting in
inhibition of necroptosis and induction of apoptosis. Second,
Nec-1, an inhibitor of RIP-kinase-1, specifically inhibits
necroptosis (22). Third,
translucent cytoplasm and swelling of cell membranes are
morphological characteristics of necroptosis, and distinct from
those in apoptosis, such as condensed chromatin and fragmented
nucleus. In the present report, because activation of caspase-8 and
-3 followed by cleavage of RIP1 were found by treatment with SHK at
lower concentrations, this suggests that mechanisms required for
induction of necroptosis is disrupted by SHK at its low
concentration. On the other hand, SHK did not activate caspase-3
and caspase-8 at high concentrations, allowing RIP-1 to remain in
an intact form, which leads to induction of necroptosis. Thus, our
findings indicate apoptosis and necroptosis are strictly regulated,
depending on the amount of SHK.
Taken together, our results show that SHK at low
concentrations may have a potential role as an inducer of apoptosis
in MM cells, including drug resistant clones, through affecting
caspases. SHK shows potential to be a new therapeutic agent for
treating MM in combination with HSP70 inhibitors. Moreover, SHK at
high concentrations may have a potential role as an inducer of
necroptosis in MM cells. Since necroptosis has not been considered
as a therapeutic strategy in treating MM, this approach might serve
as a new modality leading to better control of MM cells.
Elucidating the mechanisms underlying the multiple effects of SHK
such as reversal of bortezomib resistance and induction of
necroptosis by determination of molecules targeted by SHK is
currently underway.
Acknowledgements
We are grateful to the Institute of Natural
Medicine, Toyama University, Japan for providing natural herbal
compounds. This study was supported in part by a grant from the
Amyloidosis Research Committee from the Ministry of Health, Labour,
and Welfare, Japan. We are also grateful to Ms. Y. Otake for
technical assistance. We thank Gene Technology Center, Institute of
Resource Development and Analysis, Kumamoto University for
technical assistances regarding proteasome activity analysis.
References
|
1
|
Palumbo A and Anderson K: Multiple
myeloma. N Engl J Med. 364:1046–1060. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
He S, Liao TT, Chen YT, Kuo HM and Lin YL:
Glutathione-S-transferase enhances proliferation-migration and
protects against shikonin-induced cell death in breast cancer
cells. Kaohsiung J Med Sci. 27:477–484. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Duan D, Zhang B, Yao J, Liu Y and Fang J:
Shikonin targets cytosolic thioredoxin reductase to induce
ROS-mediated apoptosis in human promyelocytic leukemia HL-60 cells.
Free Radic Biol Med. 70:182–193. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yoon Y, Kim YO, Lim NY, Jeon WK and Sung
HJ: Shikonin, an ingredient of Lithospermum erythrorhizon induced
apoptosis in HL60 human premyelocytic leukemia cell line. Planta
Med. 65:532–535. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mao X, Yu CR, Li WH and Li WX: Induction
of apoptosis by shikonin through a ROS/JNK-mediated process in
Bcr/Abl-positive chronic myelogenous leukemia (CML) cells. Cell
Res. 18:879–888. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jang SY, Jang EH, Jeong SY and Kim JH:
Shikonin inhibits the growth of human prostate cancer cells via
modulation of the androgen receptor. Int J Oncol. 44:1455–1460.
2014.PubMed/NCBI
|
|
7
|
Hsu PC, Huang YT, Tsai ML, Wang YJ, Lin JK
and Pan MH: Induction of apoptosis by shikonin through coordinative
modulation of the Bcl-2 family, p27, and p53, release of cytochrome
c, and sequential activation of caspases in human colorectal
carcinoma cells. J Agric Food Chem. 52:6330–6337. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen CH, Lin ML, Ong PL and Yang JT: Novel
multiple apoptotic mechanism of shikonin in human glioma cells. Ann
Surg Oncol. 19:3097–3106. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ahn J, Won M, Choi JH, et al: Reactive
oxygen species-mediated activation of the Akt/ASK1/p38 signaling
cascade and p21(Cip1) downregulation are required for
shikonin-induced apoptosis. Apoptosis. 18:870–881. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Min R, Tong J, Wenjun Y, et al: Growth
inhibition and induction of apoptosis in human oral squamous cell
carcinoma Tca-8113 cell lines by Shikonin was partly through the
inactivation of NF-kappaB pathway. Phytother Res. 22:407–415. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang H, Zhou P, Huang H, et al: Shikonin
exerts antitumor activity via proteasome inhibition and cell death
induction in vitro and in vivo. Int J Cancer. 124:2450–2459. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang FL, Wang P, Liu YH, et al:
Topoisomerase I inhibitors, shikonin and topotecan, inhibit growth
and induce apoptosis of glioma cells and glioma stem cells. PLoS
One. 8:e818152013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ahmed K, Furusawa Y, Tabuchi Y, et al:
Chemical inducers of heat shock proteins derived from medicinal
plants and cytoprotective genes response. Int J Hyperthermia.
28:1–8. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Eremenko EM, Antimonova OI, Shekalova OG,
Polonik SG, Margulis BA and Guzhova IV: Novel compounds increasing
chaperone Hsp70 expression and their biological activity.
Tsitologiia. 52:235–241. 2010.(In Russian).
|
|
15
|
Munshi NC, Hideshima T, Carrasco D, et al:
Identification of genes modulated in multiple myeloma using
genetically identical twin samples. Blood. 103:1799–1806. 2004.
View Article : Google Scholar
|
|
16
|
Nimmanapalli R, Gerbino E, Dalton WS,
Gandhi V and Alsina M: HSP70 inhibition reverses cell adhesion
mediated and acquired drug resistance in multiple myeloma. Br J
Haematol. 142:551–561. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chatterjee M, Andrulis M, Stuhmer T, et
al: The PI3K/Akt signaling pathway regulates the expression of
Hsp70, which critically contributes to Hsp90-chaperone function and
tumor cell survival in multiple myeloma. Haematologica.
98:1132–1141. 2013. View Article : Google Scholar :
|
|
18
|
Fu Z, Deng B, Liao Y, et al: The
anti-tumor effect of shikonin on osteosarcoma by inducing RIP1 and
RIP3 dependent necroptosis. BMC Cancer. 13:5802013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Huang C, Luo Y, Zhao J, et al: Shikonin
kills glioma cells through necroptosis mediated by RIP-1. PLoS One.
8:e663262013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Degterev A, Huang Z, Boyce M, et al:
Chemical inhibitor of nonapoptotic cell death with therapeutic
potential for ischemic brain injury. Nat Chem Biol. 1:112–119.
2005. View Article : Google Scholar
|
|
21
|
Hitomi J, Christofferson DE, Ng A, et al:
Identification of a molecular signaling network that regulates a
cellular necrotic cell death pathway. Cell. 135:1311–1323. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Degterev A, Hitomi J, Germscheid M, et al:
Identification of RIP1 kinase as a specific cellular target of
necrostatins. Nat Chem Biol. 4:313–321. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cai Z, Jitkaew S, Zhao J, et al: Plasma
membrane translocation of trimerized MLKL protein is required for
TNF-induced necroptosis. Nat Cell Biol. 16:55–65. 2014. View Article : Google Scholar
|
|
24
|
Gunther C, Martini E, Wittkopf N, et al:
Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and
terminal ileitis. Nature. 477:335–339. 2011. View Article : Google Scholar
|
|
25
|
Sawai H and Domae N: Discrimination
between primary necrosis and apoptosis by necrostatin-1 in Annexin
V-positive/propidium iodide-negative cells. Biochem Biophys Res
Commun. 411:569–573. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chromik J, Safferthal C, Serve H and Fulda
S: Smac mimetic primes apoptosis-resistant acute myeloid leukaemia
cells for cytarabine-induced cell death by triggering necroptosis.
Cancer Lett. 344:101–109. 2014. View Article : Google Scholar
|
|
27
|
Ohtsuki T, Yawata Y, Wada H, Sugihara T,
Mori M and Namba M: Two human myeloma cell lines, amylase-producing
KMS-12-PE and amylase-non-producing KMS-12-BM, were established
from a patient, having the same chromosome marker,
t(11;14)(q13;q32). Br J Haematol. 73:199–204. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Matsuoka Y, Moore GE, Yagi Y and Pressman
D: Production of free light chains of immunoglobulin by a
hematopoietic cell line derived from a patient with multiple
myeloma. Proc Soc Exp Biol Med. 125:1246–1250. 1967. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Togawa A, Inoue N, Miyamoto K, Hyodo H and
Namba M: Establishment and characterization of a human myeloma cell
line (KMM-1). Int J Cancer. 29:495–500. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ikeyama S, Nakagawa S, Arakawa M, Sugino H
and Kakinuma A: Purification and characterization of IgE produced
by human myeloma cell line, U266. Mol Immunol. 23:159–167. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Namba M, Ohtsuki T, Mori M, et al:
Establishment of five human myeloma cell lines. In Vitro Cell Dev
Biol. 25:723–729. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ri M, Iida S, Nakashima T, et al:
Bortezomib-resistant myeloma cell lines: a role for mutated PSMB5
in preventing the accumulation of unfolded proteins and fatal ER
stress. Leukemia. 24:1506–1512. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Uneda S, Hata H, Matsuno F, et al:
Macrophage inflammatory protein-1 alpha is produced by human
multiple myeloma (MM) cells and its expression correlates with bone
lesions in patients with MM. Br J Haematol. 120:53–55. 2003.
View Article : Google Scholar
|
|
34
|
Nakamura M, Gotoh T, Okuno Y, et al:
Activation of the endoplasmic reticulum stress pathway is
associated with survival of myeloma cells. Leuk Lymphoma.
47:531–539. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tatetsu H, Ueno S, Hata H, et al:
Down-regulation of PU.1 by methylation of distal regulatory
elements and the promoter is required for myeloma cell growth.
Cancer Res. 67:5328–5336. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chou TC: Drug combination studies and
their synergy quantification using the Chou-Talalay method. Cancer
Res. 70:440–446. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang L, Fok JJ, Mirabella F, et al: Hsp70
inhibition induces myeloma cell death via the intracellular
accumulation of immunoglobulin and the generation of proteotoxic
stress. Cancer Lett. 339:49–59. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Garcia-Carbonero R, Carnero A and Paz-Ares
L: Inhibition of HSP90 molecular chaperones: moving into the
clinic. Lancet Oncol. 14:e358–369. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Christofferson DE and Yuan J: Necroptosis
as an alternative form of programmed cell death. Curr Opin Cell
Biol. 22:263–268. 2010. View Article : Google Scholar : PubMed/NCBI
|