Introduction
It is well known that the epithelial-mesenchymal
transition (EMT) can contribute to motile and invasive properties
of cancer cells, which are intimately connected with metastasis
(1). During the process of EMT,
while the protein levels of E-cadherin (epithelial marker) are
decreased, that of N-cadherin and vimentin (mesenchymal markers)
are increased (2). The
alternations of these proteins have been demonstrated to result in
impaired cell-cell adhesion, thereby allowing the spread of cancer
cells from the primary sites. Therefore, EMT is mostly thought to
lead to tumor progression via promoting cancer cell’s ability to
invade into the peripheral microenvironment composed of lymph and
blood vascular systems (3–5). Many reports have suggested that EMT
has a high correlation with the generation of cancer stem cells,
the malignancy of tumor cells and the resistance to anticancer
drugs (5,6). In addition, EMT has been known to be
easily detected at the invasive region of advanced tumors (7,8).
UHRF1 (ubiquitin-like with PHD and RING finger
domains 1) was shown to play a pivotal role in maintaining DNA
methylation by its binding to hemimethylated DNA for recruiting
DNMT1, and correctly imparting the DNA methylation patterns of
mother cells to daughter cells (9). Therefore, UHRF1 is well known as an
epigenetic regulator comprised of multi-domains containing a
N-terminal ubiquitin-like domain, a tandem tudor domain, a PHD
domain, an SRA domain and a RING finger motif domain (10). Particularly, both a PHD and an SRA
domain of UHRF1 recruit and interact with DNMT1 (10). In addition, the SRA domain of UHRF1
recognizes and binds to hemi-methylated DNA and a RING finger motif
accords an E3-ubiquitin-liagase function to UHRF1 (10). Most importantly, UHRF1 is known to
be an E3-ubiquitin-ligase for the degradation of DNMT1 (11,12).
Therefore, overexpression of UHRF1 in cells leads to the global DNA
hypomethylation, a hallmark of cancer cells, by decreasing the
protein stability of DNMT1 (10–12).
Actually, the protein level of UHRF1 is more increased in many
cancer cell types, including colon, lung, bladder, pancreatic,
prostate, cervical and breast cancer cell lines than in normal
human cells (13). Disruption in
forming the PCNA/DNMT1/UHRF1 complex has been also known to cause
the global DNA hypomethylation and high frequency of oncogenic
transformation. Moreover, the global DNA hypomethylation can also
occur via depletion of UHRF1 (14,15).
However, the precise role of UHRF1 depletion in the cancer
development has not been clarified.
In the present study, we investigated the role of
downregulation of UHRF1 in EMT concerned with tumor malignancy, and
found that downregulation of UHRF1 contributes to enhanced EMT of
human cancer cells via increasing the expression level of CXCR4.
Our observation also suggests that UHRF1 depletion may play a
pivotal role in inducing the malignant alteration of cancer
cells.
Materials and methods
Reagents
A mouse monoclonal antibody against UHRF1 (ICBP 90)
was purchased from BD Transduction Laboratories (San Jose, CA,
USA). Mouse monoclonal antibodies against vimentin, E-cadherin,
N-cadherin, Zeb1, Slug, Snail and Twist were purchased from Santa
Cruz Biotechnology (Santa Cruz, CA, USA). Mouse monoclonal
anti-β-actin antibody, horseradish peroxidase-conjugated
anti-rabbit IgG, and anti-mouse IgG antibodies were purchased from
Sigma-Aldrich Co. (St. Louis, MO, USA). APC-conjugated mouse
monoclonal anti-CD44 antibody, PerCP-Cy5.5-conjugated mouse
monoclonal anti-CD117 antibody, PE-Cy7-conjugated mouse monoclonal
anti-CD184 antibody, FITC-conjugated mouse monoclonal antibodies
against CD24, CD47, CD90 and CD340, and phycoerythrin
(PE)-conjugated mouse monoclonal anti-CD126 antibody were purchased
from BD Transduction Laboratories. PE-conjugated mouse monoclonal
antibodies against CD133 and CD326 were purchased from Miltenyi
Biotec Inc. (Auburn, CA, USA).
Cell culture
Human hepatocellular carcinoma HepG2 and Hep3B
cells, human breast cancer MDA-MB-231 and MCF-7 cells, and human
colon cancer HCT116 cells were from the American Type Culture
Collection (ATCC, Manassas, VA, USA). Cells were cultured in a
humidified 5% CO2 atmosphere at 37°C. HepG2 and Hep3B
cells were maintained in Dulbecco’s modified Eagle’s medium
(Sigma-Aldrich) supplemented with 10% (v/v) bovine calf serum,
penicillin (50 units/ml) and streptomycin (50 μg/ml) (all from
Gibco-BRL, Grand Island, NY, USA). MDA-MB-231 and MCF-7 cells were
maintained in DMEM supplemented with 10% (v/v) bovine calf serum,
penicillin (50 units/ml) and streptomycin (50 μg/ml). HCT116 cells
were maintained in McCoy’s 5A medium (Gibco-BRL) with 10% (v/v)
bovine calf serum, penicillin (50 units/ml) and streptomycin (50
μg/ml).
Establishment of inducible UHRF1
shRNA-containing cell lines
The appropriate double-stranded oligonucleotide
(5′-CCG GCC GCA CCA AGG AAT GTA CCA TCT CGA GAT GGT ACA TTC CTT GGT
GCG GTT TTT-3′ and its complement) was cloned into the
pLKO.1-puro-shRNA vector (Sigma-Aldrich). HepG2 cells were
transfected with the plasmid or control empty vector, using
Lipofectamine 2000 (Gibco-Invitrogen Corp., Carlsbad, CA, USA) in
accordance with the manufacturer’s recommendations. UHRF1
shRNA-containing stable clones were selected using 1 μg/ml
puromycin (Sigma-Aldrich). Stable clones were isolated and
endogenous UHRF1 knockdown was determined by western blot analysis
using an anti-UHRF1 antibody.
Small interfering RNA transfection
RNA interference mediated by siRNAs was achieved
using double-stranded RNA molecules. The siRNAs against UHRF1,
Zeb1, Slug, Snail or CXCR4 were purchased from Santa Cruz
Biotechnology. Control siRNA-A (Santa Cruz Biotechnology) was used
as a control. Cells were grown to 30% confluency on 60-mm dishes
and transfected with the siRNA duplexes (100 nM), using
Lipofectamine 2000 (Gibco-Invitrogen) in accordance with the
manufacturer’s instructions. Assays were performed 48 h after
transfection.
Western blot analysis
Cells were treated with lysis buffer [40 mM Tris-HCl
(pH 8.0), 120 mM NaCl and 0.1% (v/v) NP40] supplemented with
protease inhibitor cocktail tablets (1 tablet/50 ml; Boehringer,
Mannheim, Germany) and centrifuged for 15 min at 12,000 × g at 4°C.
Proteins (30 μg) were separated by SDS-PAGE and transferred to
nitrocellulose membranes (Bio-Rad Laboratories, Hercules, CA, USA).
The membranes were blocked with 5% (w/v) non-fat dry milk in
Tris-buffered saline and then incubated for 1 h with primary
antibodies at the dilution of 1:1,000, at room temperature.
Specific reaction bands were detected using peroxidase-conjugated
secondary antibodies at the dilution of 1:500, and proteins were
visualized using an enhanced chemiluminescence system (Amersham
Biosciences, Piscataway, NJ, USA).
RNA preparation and RT-PCR (reverse
transcriptase-polymerase chain reaction)
Total RNA was extracted from the cells using the
TRIzol reagent (Invitrogen), and 1 μg of the isolated total RNAs
was reverse transcribed to complementary DNA (cDNA) using the
iScript™ cDNA synthesis kit (Bio-Rad Laboratories) according to the
manufacturer’s instructions. The reaction was performed at 25°C for
5 min and then at 42°C for 30 min, and was terminated at 85°C for 5
min. Next, the cDNA was diluted to 100 μl, and then 2 μl of it was
used as a template for RT-PCR. Total reaction volume (25 μl)
containing 12.5 μl of Premix Taq (Takara Bio, Shiga, Japan), 2 μl
of a template, and 10 pM of both forward and reverse primers was
used for PCR amplification which was performed on a C1000 thermal
cycler (Bio-Rad Laboratories) using a thermal profile of beginning
at 94°C for 5 min, followed by 30 cycles at 94°C for 10 sec, 58°C
for 15 sec and 72°C for 30 sec, 1 cycle of 72°C for 10 min and
extention at 12°C. All primers were designed using the Primer3
program (http://bioinfo.ut.ee/primer3/). The primer used for
RT-PCR was as follows: CXCR4 forward (5′-TGC TTG CTG AAT TGG
AAG TG-3′) and reverse (5′-AGT CAT AGT CCC CTG AGC CC-3′);
G3PDH forward (5′-GAA ATC CCA TCA CCA TCT TCC AGG-3′) and
reverse (5′-GAG CCC CAG CCT TCT CCA TG-3′). Amplified products were
separated on 1% (w/v) ethidium bromide-stained agarose gels and
expression levels were measured by luminescent image analysis.
Quantitative real-time PCR (qRT-PCR)
Quantitative reverse transcription-PCR was performed
using the SYBR-Green reporter. Total RNA (1 μg) isolated using the
RNeasy Mini kit (Qiagen, Valencia, CA, USA) was subsequently
reverse transcribed to cDNA with the SuperScript First-Strand
Synthesis system (Gibco-Invitrogen). cDNA (3 μl) were added to 17
μl of the reaction mixture containing 5 μl of ddH2O, 10
μl of 2X SYBR-Green Master Mix (Thermo Fisher Scientific, Waltham,
MA, USA) and 1 μl of each primer. PCR amplification was carried out
using a thermal profile of beginning at 94°C for 5 min, followed by
40 cycles at 94°C for 10 sec, 58°C for 15 sec and 72°C for 15 sec,
and last extension at 72°C for 5 min. The amplification specificity
of each qRT-PCR analysis was confirmed by the melting curve
analysis. The transcripts of target genes were analyzed using a
CFX96™ Real-Time PCR detection system (Bio-Rad Laboratories). The
primers used for qRT-PCR were as follows: Zeb1 forward
(5′-GCA CCT GAA GAG GAC CAG AG-3′) and reverse (5′-TGC ATC TGG TGT
TCC ATT TT-3′); Slug forward (5′-AGA TGC ATA TTC GGA CCC
AC-3′) and reverse (5′-CCT CAT GTT TGT GCA GGA GA-3′); Snail
forward (5′-GAA AGG CCT TCA ACT GCA AA-3′) and reverse (5′-TGA CAT
CTG AGT GGG TCT GG-3′); CXCR4 forward (5′-AAA TGG GCT CAG
GGG ACT AT-3′) and reverse (5′-TCC CAA AGT ACC AGT TTG CC-3′);
G3PDH forward (5′-ACC ACA GTC CAT GCC CAT CAC-3′) and
reverse (5′-TCC ACC ACC CTG TTG CTG TA-3′). G3PDH gene was
used as a control to normalize the expression value of Zeb1,
Slug, Snail or CXCR4. The relative expression
level of the genes was calculated using the 2−ΔΔCt
method (16).
Migration and invasion assays
Cell migration and invasion assay were performed
using the Transwell chamber (8-μm pore size; Corning Life Sciences,
Lowell, MA, USA). Cells (2×104) were resuspended in 0.2
ml of serum-free growth medium for both cell migration and invasion
assay. For migration assay, the cells were added to the interior of
the inserts. Growth medium (0.8 ml) supplemented with 10% (v/v)
bovine calf serum was added to the lower chamber. After incubation
for 48 h at 37 °C, the cells attached to the upper surface of the
filter were removed with a cotton swab, and migrated cells on the
lower surface of the filter were fixed and stained for 15 min with
0.25% crystal violet (Sigma-Aldrich), 10% formaldehyde, and 80%
methanol, and then the inserts were washed five times with
ddH2O and photographed (magnification, ×20). Migrated
cells were determined by counting cells in five microscopic fields
per well, and the extent of migration was expressed as an average
number of cells per microscopic field. Cells were imaged by phase
contrast microscopy (Nikon Eclipse 80i; Nikon, Tokyo, Japan). For
invasion assay, the cells were added to the interior of the inserts
precoated with 10 mg/ml growth factor-reduced Matrigel (BD
Biosciences, Bedford, MA, USA). Growth medium (0.8 ml) supplemented
with 10% (v/v) bovine calf serum was added to the lower chamber.
After incubation for 48 h at 37°C, the inserts were processed as
described above for the migration assay.
Flow cytometric analysis
Single cells were detached from the culture dishes
with trypsin-EDTA, counted, ~1×105 cells were
centrifuged at 2,000 rpm for 5 min and then resuspended in PBS
containing 5% FBS and 0.1% NaN3 (Sigma-Aldrich). The
cells were incubated at 4°C with APC-conjugated anti-CD44,
PerCP-Cy5.5-conjugated anti-CD117, PE-Cy7-conjugated anti-CD184,
FITC-conjugated anti-CD24, anti-CD47, anti-CD90, anti-CD340,
phycoerythrin (PE)-conjugated anti-CD126, anti-CD133 or anti-CD326
antibody for 1 h. The labeled cells were washed twice with PBS,
resuspended in PBS, and then analyzed by flow cytometry, FACSAria™
(Special order system, BD Biosciences).
Statistical analysis
The data presented are representative of at least
three independent experiments. Differences between groups were
analyzed using the Student’s t-test (SPSS Statistics version 17.0;
SPSS, Inc., Chicago, IL, USA). P-values <0.05 were considered
significant.
Results
Downregulation of UHRF1 promotes EMT of
human cancer cells
Invasion of cancer cells arises from the loss of
cell-cell interaction leading to their migratory properties.
Importantly, this event is closely related with EMT (3–6). The
high expression level of UHRF1 was shown to be correlated with
tumor progression (10–13). Recently, however, disruption of
DNMT1/PCNA/UHRF1 complex has been suggested to lead to
tumorigenesis, implying that a deficiency in UHRF1 may also be
involved in cancer development (14,15).
Therefore, we investigated whether downregulation of UHRF1 affects
EMT of cancer cells. To fulfill this purpose, we introduced shRNAs
targeting UHRF1 into HepG2 or MDA-MB-231 cells by transfection and
generated stable cell lines. We first used western blot analysis to
measure epithelial and mesenchymal cell markers in these stable
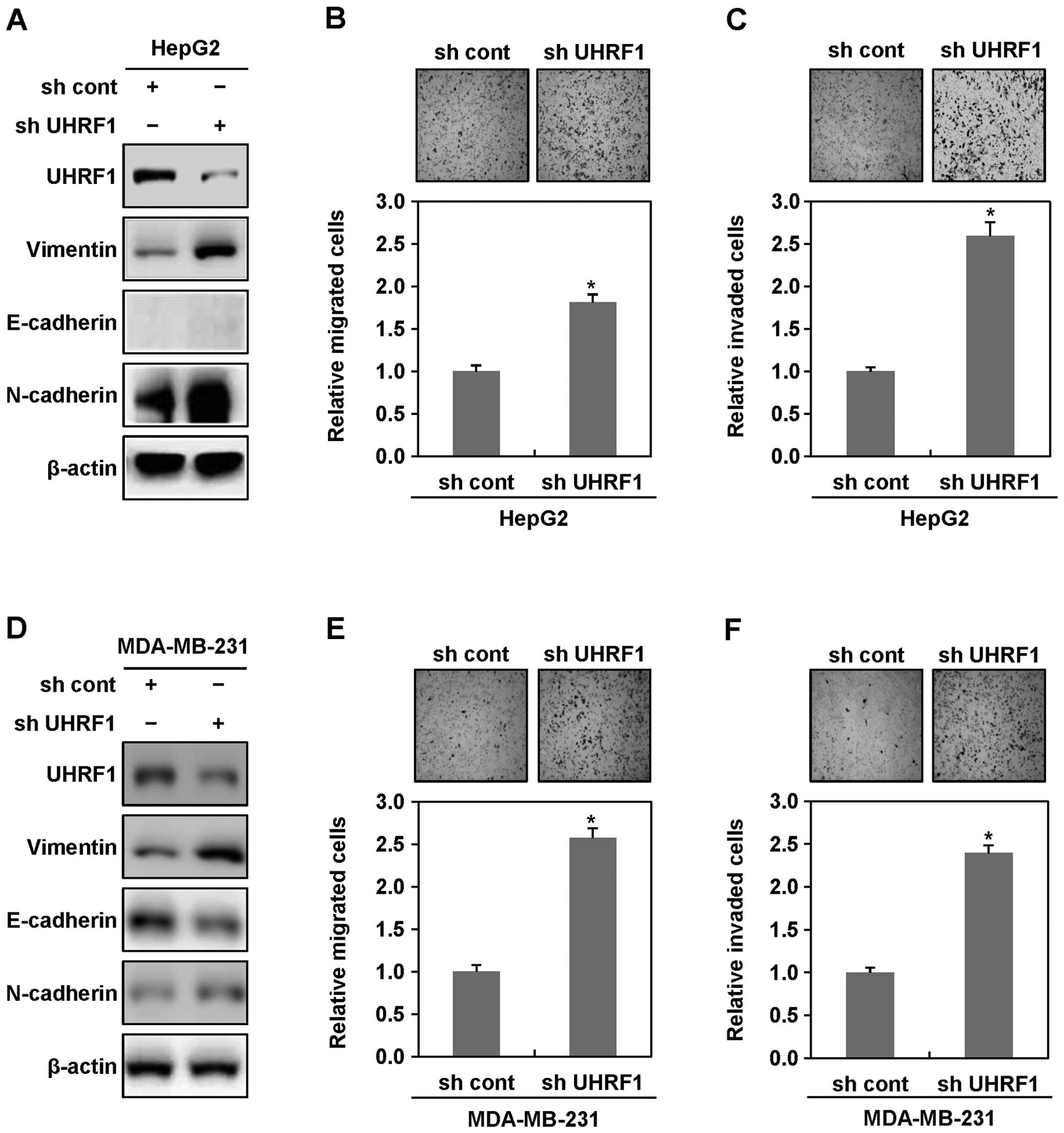
cell lines. As shown in Fig. 1A,
western blot analyses showed an increase in the expression levels
of the mesenchymal cell markers N-cadherin and vimentin in UHRF1
shRNA-expressing HepG2 cells, compared with parental HepG2 cells,
although the expression of the epithelial cell marker E-cadherin,
did not be change. Consistent with these results, UHRF1
shRNA-expressing HepG2 cells displayed more migratory and invasive
properties, compared with parental HepG2 cells (Fig. 1B and C). Moreover, we found that
UHRF1 shRNA-expressing MDA-MB-231 cells also shows an increase in
the expression levels of N-cadherin and vimentin, a decrease in the
expression level of E-cadherin, and displays substantial migratory
and invasive properties, compared with parental MDA-MB-231 cells
(Fig. 1D–F). Collectively, these
results indicate that downregulation of UHRF1 may play a key role
in initiating EMT of cancer cells.
To further confirm a possible causal connection
between downregulation of UHRF1 and EMT in cancer cells, we
transiently transfected HepG2 and MDA-MB-231 cells with siRNAs
targeting UHRF1, and then performed western blot analysis,
migration and invasion assays. In both HepG2 and MDA-MB-231 cells
transiently transfected with siRNAs targeting UHRF1, we obtained
similar results to those observed in stable cell lines (Fig. 2A–F). In addition, we also found
that the expression levels of vimentin increase in Hep3B, MCF-7 and
HCT116 transiently transfected with siRNAs targeting UHRF1
(Fig. 2G). These results
collectively indicated that downregulation of UHRF1 clearly
contributes to initiating EMT of human cancer cells.
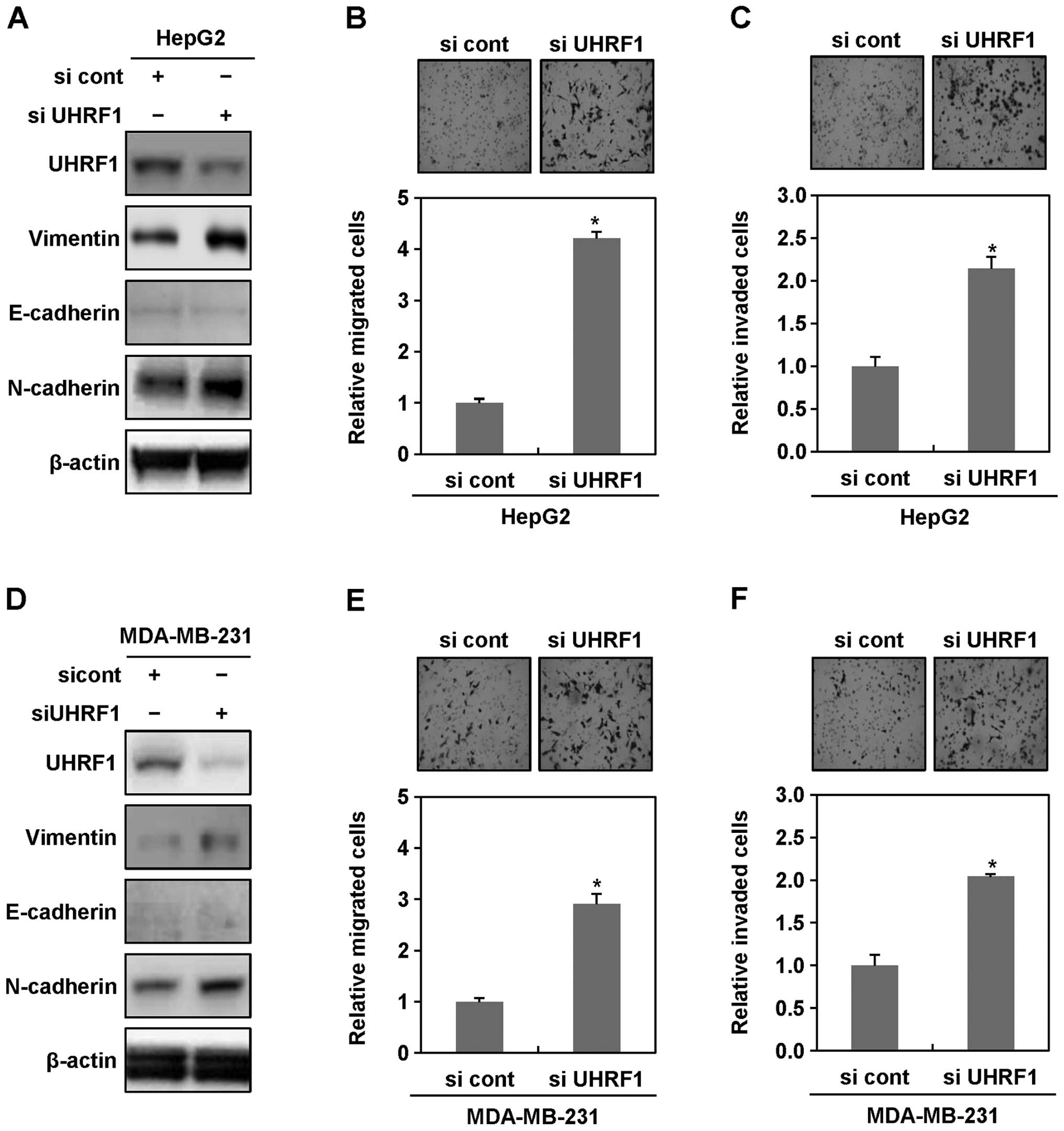 | Figure 2Ectopic downregulation of UHRF1
increases migratory and invasive properties of human cancer cells.
(A) After HepG2 cells were grown in presence or absence of siRNA
targeting UHRF1 for 48 h, cell lysates were subjected to western
blot analysis using anti-UHRF1, anti-vimentin, anti-E-cadherin,
anti-N-cadherin and anti-β-actin antibodies. Experiments were
conducted in triplicate, and the data shown are representative of a
typical experiment. (B and C) Increased migratory and invasive
properties of HepG2 cells transiently transfected with siRNA
targeting UHRF1. Migration and invasion assay were performed using
the Transwell chamber. Results from three independent experiments
are expressed as means ± SEMs. (*P<0.05). (D) After
MDA-MB-231 cells were grown in presence or absence of siRNA
targeting UHRF1 for 48 h, cell lysates were subjected to western
blot analysis using anti-UHRF1, anti-vimentin, anti-E-cadherin,
anti-N-cadherin and anti-β-actin antibodies. Experiments were
conducted in triplicate, and the data shown are representative of a
typical experiment. (E and F) Increased migratory and invasive
properties of MDA-MB-231 cells transiently transfected with siRNA
targeting UHRF1. Migration and invasion assay were performed using
the Transwell chamber. Results from three independent experiments
are expressed as means ± SEMs. (*P<0.05). (G) After
Hep3B, MCF-7 and HCT116 cells were grown in presence or absence of
siRNA targeting UHRF1 for 48 h, cell lysates were subjected to
western blot analysis using anti-UHRF1, anti-vimentin,
anti-E-cadherin, anti-N-cadherin and anti-β-actin antibodies.
Experiments were conducted in triplicate, and the data shown are
representative of a typical experiment. |
Downregulation of UHRF1 triggers EMT
through the induction of Zeb1 and Snail in human cancer cells
Next, we examined whether downregulation of UHRF1
promotes EMT via EMT-regulating transcription factors, Zeb1, Slug,
Snail and Twist in cancer cells. As shown in Fig. 3A and B, we found that Zeb1, Slug
and Snail are generally upregulated at the protein level by both
shRNAs and siRNAs targeting UHRF1 in HepG2 and MDA-MB-231 cells,
whereas there was no change in the protein level of Twist in both
cell lines (Fig. 3A and B).
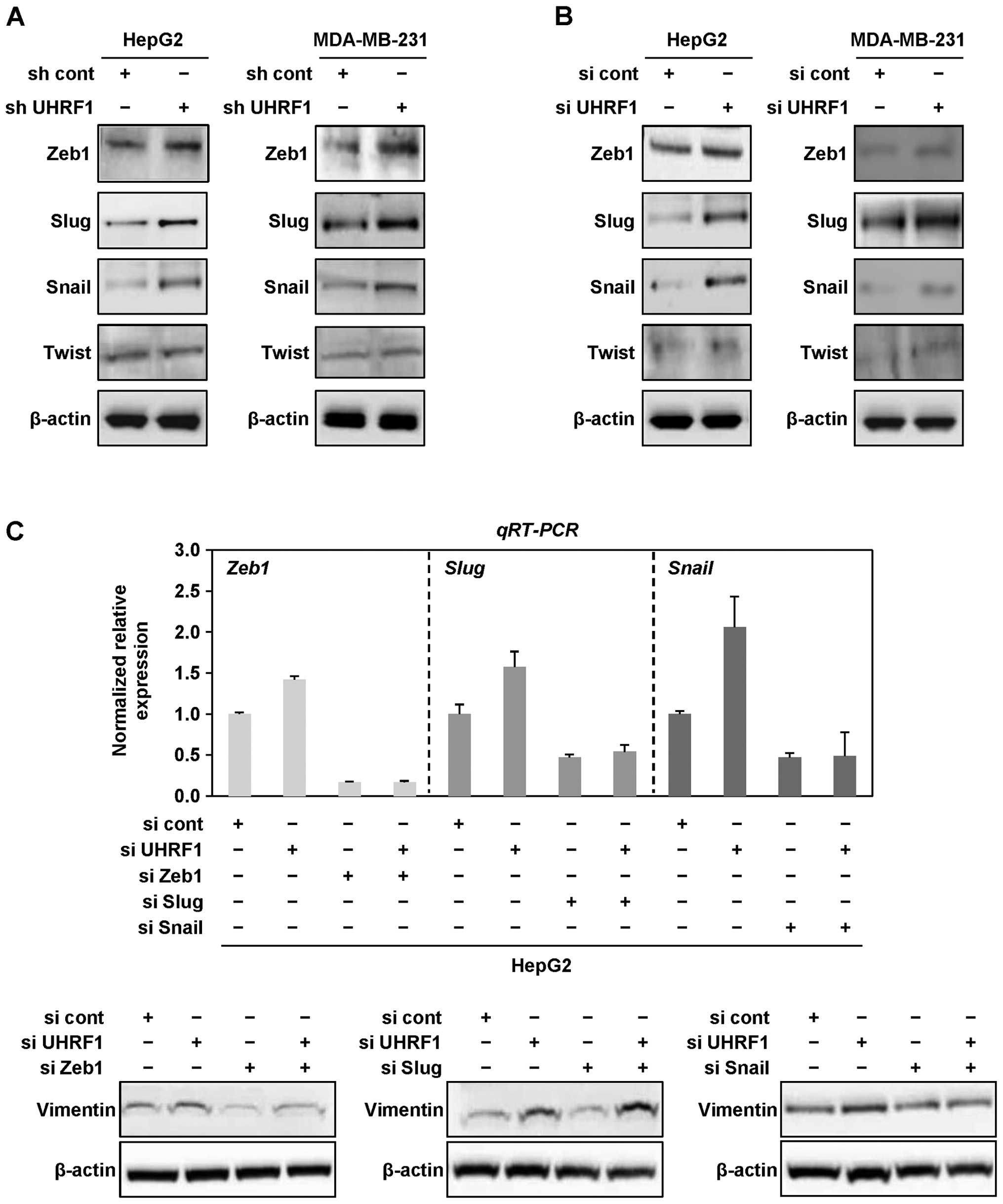 | Figure 3Downregulation of UHRF1 leads to EMT
via EMT transcription factors Zeb1 and Snail in human cancer cells.
(A) Cell lysates from HepG2 and MDA-MB-231 cells stably transfected
with UHRF1 shRNA were subjected to western blot analysis using
anti-Zeb1, anti-Slug, anti-Snail, anti-Twist and anti-β-actin
antibodies. Experiments were conducted in triplicate, and the data
shown are representative of a typical experiment. (B) After HepG2
and MDA-MB-231 cells were grown in presence or absence of siRNA
targeting UHRF1 for 48 h, cell lysates were subjected to western
blot analysis using anti-Zeb1, anti-Slug, anti-Snail, anti-Twist
and anti-β-actin antibodies. Experiments were conducted in
triplicate, and the data shown are representative of a typical
experiment. (C) After HepG2 cells were transfected with siRNA
targeting Zeb1, Slug or Snail in presence or absence of UHRF1 siRNA
for 48 h, qRT-PCR was conducted using primers designed for Zeb1,
Slug or Snail, and cell lysates were subjected to western blot
analysis using anti-vimentin and anti-β-actin antibodies.
Experiments were conducted in triplicate, and the data shown are
representative of a typical experiment. (D) After HepG2 cells were
transfected with siRNA targeting Zeb1 in presence or absence of
UHRF1 siRNA for 48 h, qRT-PCR was conducted using primers designed
for Zeb1. (E and F) Migration and invasion assay were performed
using the Transwell chamber. Results from three independent
experiments are expressed as means ± SEMs. (*P<0.05).
(G) After HepG2 cells were transfected with siRNA targeting Snail
in presence or absence of UHRF1 siRNA for 48 h, cell lysates were
subjected to western blot analysis using anti-UHRF1, anti-Snail and
anti-β-actin antibodies. (H and I) Migration and invasion assay
were performed using the Transwell chamber. Results from three
independent experiments are expressed as means ± SEMs
(*P<0.05). |
To further determine which of these transcription
factors is dominantly involved in EMT caused by downregulation of
UHRF1, we transfected HepG2 cells with siRNAs targeting Zeb1, Slug
or Snail in presence or absence of UHRF1 siRNAs. Notably, siRNAs
targeting Zeb1 or Snail effectively attenuated the expression level
of vimentin increased by downregulation of UHRF1, whereas siRNAs
targeting Slug did not (Fig. 3C).
Furthermore, we found that treatment with siRNAs targeting Zeb1 or
Snail efficiently blocks the migratory and invasive properties of
HepG2 cells increased by downregulation of UHRF1 (Fig. 3D–I). Taken together, these results
suggest that downregulation of UHRF1 promotes EMT through
upregulation of Zeb1 and Snail in human cancer cells.
Downregulation of UHRF1 induces the
expressions of cancer stem cell-related cell surface markers
As EMT has been suggested to lead to an increase in
the population of cancer stem cells (17,18),
we next determined the expression of cancer stem cell-related cell
surface markers in presence or absence of shRNAs targeting UHRF1 in
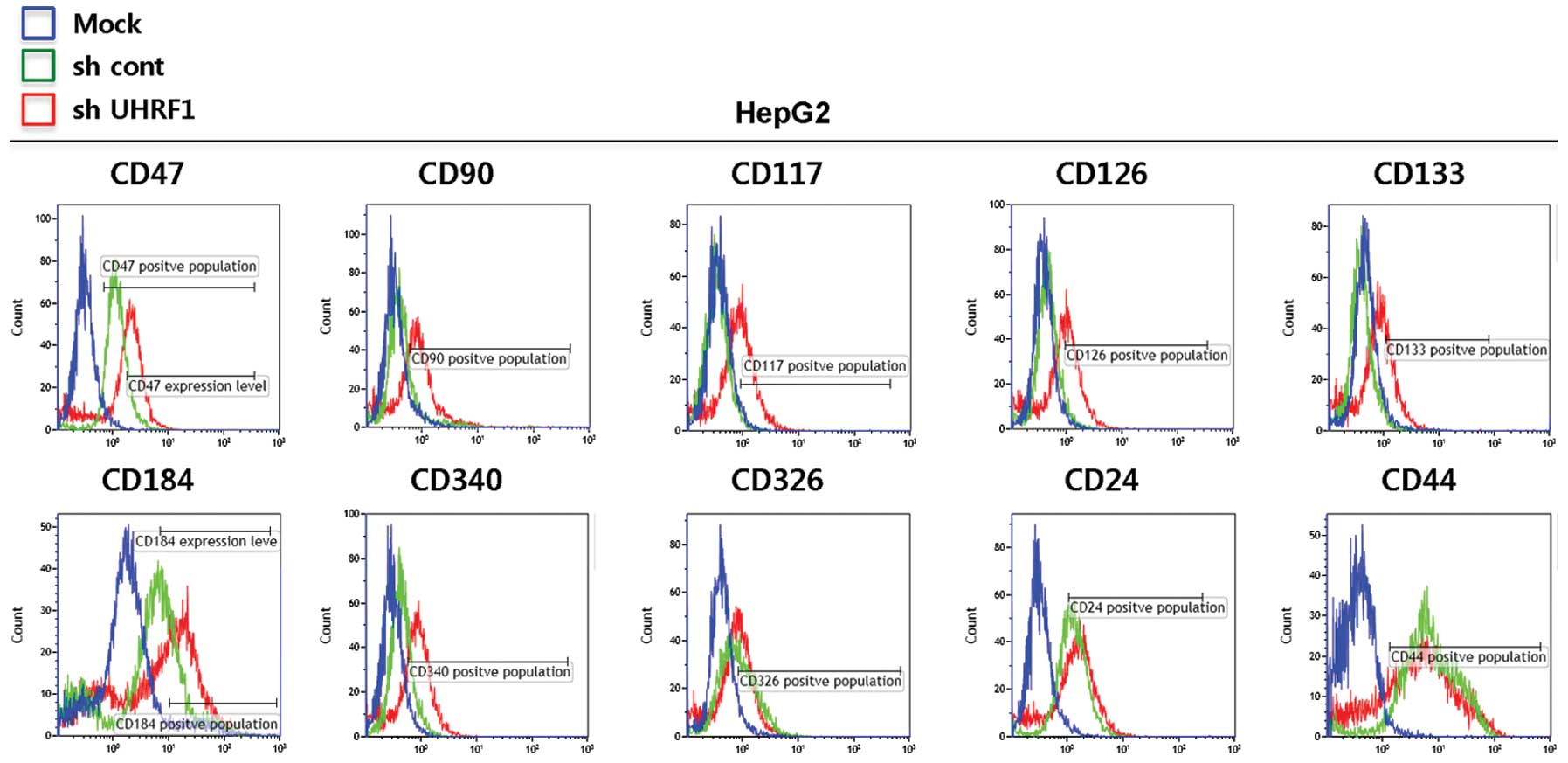
HepG2 cells. As shown in Fig. 4,
flow cytometric data revealed that the cells expressing UHRF shRNAs
show an increase in the expressions of CD47, CD90, CD117, CD126,
CD133 and CD340, but no significant change in those of CD326, CD24
and CD44. These results indicated that downregulation of UHRF1 can
increase the expression of cancer stem cell-related cell surface
markers, which are thought to be closely associated with the
generation of cancer stem cells.
CXCR4 contributes to EMT caused by
downregulation of UHRF1
Especially, CD90 (Thy-1), CD117 (c-Kit), CD340
(HER2) and CD184 (CXCR4) increased in HepG2 cells expressing UHRF1
shRNAs are well known to be receptor-tyrosine kinases and a G
protein-coupled receptor possessing a great ability in a variety of
cellular events (19–22). Moreover, among them, CXCR4 was
demonstrated to induce an aggressively malignant phenotype of
cancer cells via driving EMT (23,24).
Therefore, we next examined whether CXCR4 is critically involved in
EMT caused by downregulation of UHRF1. To better assess the
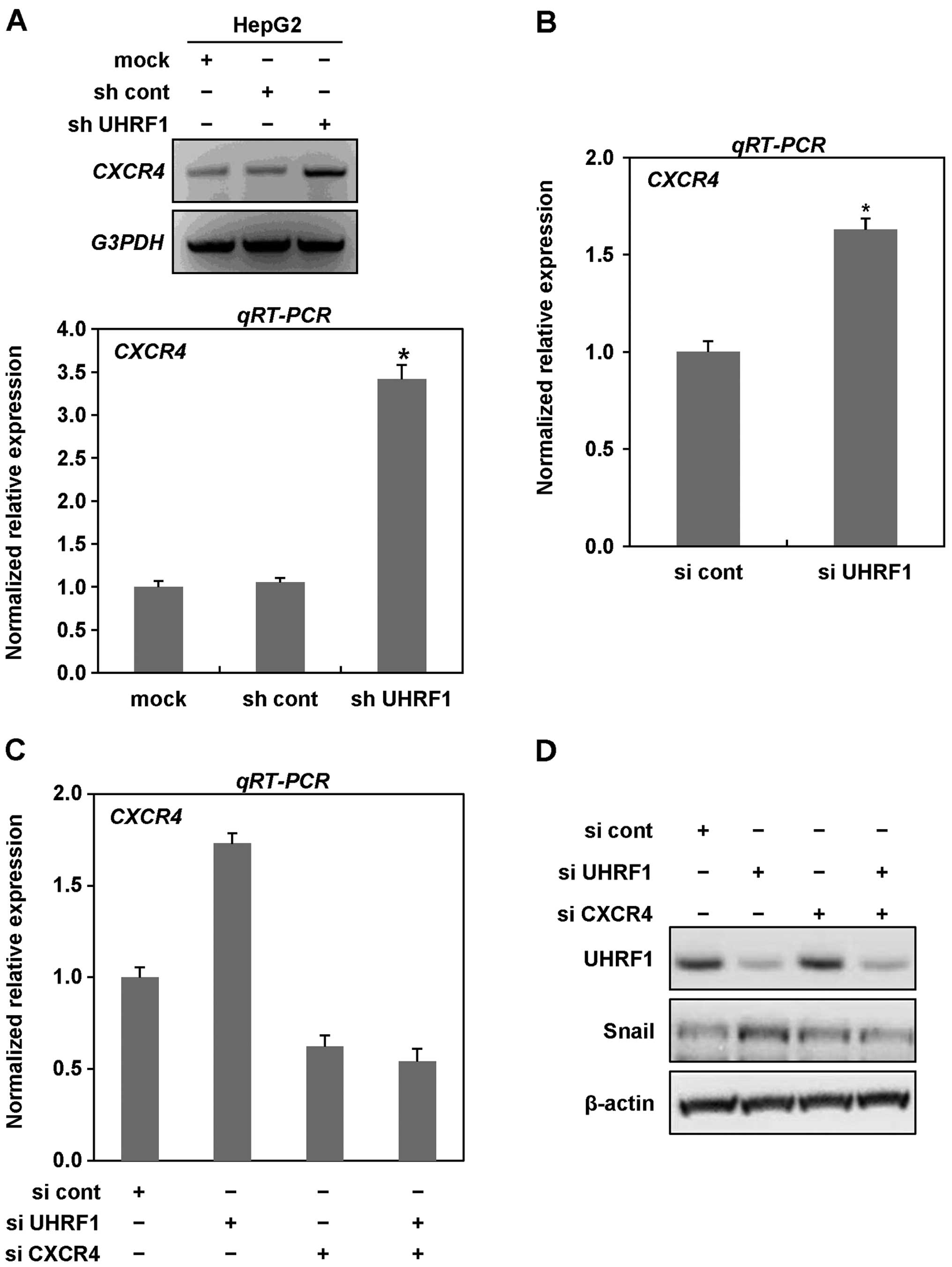
expression level of CXCR4 in the presence or absence of shRNAs
targeting UHRF1 in HepG2 cells, we performed RT and qRT-PCR
analysis. As shown in Fig. 5A,
mRNA expression of CXCR4 was markedly increased in the cells
expressing UHRF1 shRNAs. To further confirm the increase in mRNA
expression of CXCR4, we transiently transfected HepG2 cells with
siRNAs targeting UHRF1 and performed qRT-PCR analysis. In HepG2
cells transiently transfected with siRNAs targeting UHRF1, we
obtained similar results to those observed in stable cell lines,
indicating that downregulation of UHRF1 efficiently increases the
expression of CXCR4 (Fig. 5B).
We next examined whether CXCR4 contributes to EMT
caused by downregulation of UHRF1. As shown in Fig. 5C–E, we found that siRNA-mediated
CXCR4 knockdown greatly inhibited the expression of both Snail and
Zeb1 increased by downregulation of UHRF1. Moreover, siRNA-mediated
CXCR4 knockdown effectively modulated both migratory and invasive
properties of HepG2 cells increased by downregulation of UHRF1
(Fig. 5F and G). Collectively,
these results clearly indicate that CXCR4 plays key roles in EMT
induced by downregulation of UHRF1 in human cancer cells.
Discussion
Recently, EMT defining that epithelial cells acquire
mesenchymal characteristics has been implicated to be highly
relevant to the acquisition of malignant properties by cancer cells
(1,3–6).
Especially, EMT confers the early steps of metastasis including
local invasion and subsequent spreading of cancer cells to distant
locations (25,26). Although, in this context, the
epigenetic pathways activating EMT are extensively investigated
(26), the epigenetic mechanism
for initiating the malignancy of cancer cells is well not
understood. In the present study, we found that downregulation of
UHRF1, an epigenetic regulator, contributes to the increase in EMT
of human cancer cells via inducing the expression of CXCR4.
UHRF1 has been reported to be involved in a variety
of physiological and pathological events, from development to
cancer progression (13,27). The expression levels of UHRF1 are
highly upregulated in diverse cancer types (13). Indeed, overexpression of UHRF1 has
been demonstrated to play pivotal roles in malignant cellular
transformation via repressing either transcriptional or protein
level of tumor suppressors, and inducing the global DNA
hypomethylation which leads to genomic instability, a hallmark of
cancer cells (10–12). However, the precise mechanism by
which UHRF1 inhibits tumor-suppressors has not yet been
clarified.
Furthermore, it has been reported that cells
overexpressing UHRF1 show increased growth and migration with
morphological features resembling those occurring in EMT in
colorectal cancer, suggesting that high expression level of UHRF1
may lead to the phenotypic changes of cancer cells by inducing EMT
(28). In addition, several
reports showed that overexpression of UHRF1 promotes metastasis in
colorectal, gastric and breast cancer (13,29–31).
Thus, UHRF1 is considered as an oncogene driving tumorigenesis
(32).
Since UHRF1 does not only recognize hemimethylated
DNA to recruit DNMT1 for methylation on newly synthesized DNA
strands, but also has a RING domain with ubiquitin E3 ligase
activity, UHRF1 can readily reduce the protein stability of DNMT1
via ubiquitin-proteasome pathway (10–12).
Moreover, DNMT1-deficent cells have been reported to show global
DNA hypomethylation (33,34). Therefore, the global DNA
hypomethylation by upregulation of UHRF1 expression is thought to
due to the reduced protein stability of DNMT1.
Recently, the disruption of DNMT1/PCNA/UHRF1 complex
has been reported to induce tumorigenesis and the global DNA
hypomethylation (14,15). Therefore, it is plausible that
UHRF1 deficiency is also associated with further acquisition of
invasive properties of cancer cells through disrupting the complex
formation. Because both overexpression of and deficiency in UHRF1
affects malignancy of cells, a balanced expression level of UHRF1
is thought to be necessary for the normal behavior of cells. In the
present study, we elucidated the possible role of UHRF1 deficiency
in the induction of EMT in human cancer cells using sh- or
siRNA-mediated knockdown approaches. As expected, we found that
both shRNA and siRNA-mediated knockdown of UHRF1 increase the
migratory and invasive properties of human cancer cells as well as
inducing mesenchymal markers (Figs.
1 and 2), suggesting that
UHRF1 deficiency is also responsible for malignancy of human cancer
cells. In addition, we observed that downregulation of UHRF1
induces EMT through activating the EMT-regulating transcription
factors Zeb1 and Snail, but not Slug and Twist in HepG2 cells
(Fig. 3). These transcription
factors are thought to cooperatively control both genes involved in
epithelial markers, such as E-cadherin, and mesenchymal markers,
such as N-cadherin and vimentin via performing dual functions as an
activator and a repressor (35,36).
Moreover, it has been previously reported that the overexpression
of snail can raise the transcriptional level of Zeb1 via activating
Zeb1 promoter (37). Consistent
with these reports, snail in UHRF1-deficient cancer cells seems to
participate in increasing the expression level of Zeb1 to induce
EMT. Although the EMT-regulating transcription factors are
aberrantly expressed in various cancer types (1), the precise mechanism underlying the
regulation of these factors in cancers are not understood.
Emerging evidence suggested that EMT plays a key
role not only in metastasis, but also in cancer recurrence well
known to be closely associated with generation of cancer stem cells
(17,18). Therefore, we investigated whether
UHRF1 deficiency affects the expression of cancer stem cell-related
surface markers in HepG2 cells. We found that shRNA-mediated
knockdown of UHRF1 increases the expressions of CD47, CD90, CD117,
CD133, CD184 and CD340 in HepG2 cells. These observations
indirectly indicate that UHRF1 deficiency may contribute to the
generation of cancer stem cells. In addition, since CD117 (c-Kit),
CD184 (CXCR4) and CD340 (HER2) are well known to activate
intracellular signaling transduction pathways (19–22),
there is a possibility that they may lead to EMT in UHRF1-deficient
cancer cells.
As one of the best studied chemokine receptors,
CXCR4 plays a key role in survival, growth, vasculogenesis, EMT and
metastasis in a variety of cancer types (38,39),
and is known to interact with its ligand, CXCL12 (also named
stromal derived growth factor-1, SDF-1) to activate intracellular
signaling pathway (40).
Importantly, CXCR4-mediated signaling pathways have been suggested
to be crucial for metastatic malignancy (40). In addition, it has been shown that
inhibition of CXCR4-mediated pathways by treatment with
neutralizing antibodies against, or antisense RNA to CXCR4 greatly
attenuates both invasion and migration of cancer cells, and that
high expression level of CXCR4 is closely related with metastatic
ability of cancer cells in vivo (41). Consistent with these reports, we
found that UHRF1 deficiency efficiently increases the expression
level of CXCR4 in HepG2 cells in vitro (Figs. 4 and 5). Furthermore, siRNA-mediated knockdown
of CXCR4 effectively blocked the expressions of Zeb1 and Snail, and
migratory and invasive properties of UHRF1-deficient HepG2 cells
(Fig. 5). These observations
strongly indicate that CXCR4 contributes to EMT increased in
UHRF1-deficient cancer cells. Recent studies also showed that CXCR4
participates in survival and maintenance of cancer stem cells
during cancer progression (42,43).
In addition, CXCR4 is also deemed a putative cell surface marker
for cancer stem cells (44).
Similarly, because our results indicated that UHRF1 deficiency
effectively increases the cancer stem cell-related cell surface
markers, as well as CXCR4, further studies are required to
elucidate the possible role of UHRF1 deficiency in the generation
of cancer stem cells. Moreover, a recent study indicates that UHRF1
can recruit and cooperate with G9a, a histone lysine
methyltransferase, to suppress the activity of p21 promoter
(45). Similarly, as upregulation
of CXCR4 by UHRF1 deficiency is thought to be due to disruption of
the cooperation between UHRF1 and G9a, additional studies are
needed to clarify the suppressive effect of UHRF1 on the induction
of CXCR4 gene expression.
In conclusion, we demonstrated that shRNA or
siRNA-mediated knockdown of UHRF1 markedly increased EMT in human
cancer cells. Furthermore, we present evidence that downregulation
of UHRF1 is sufficient to induce EMT-regulating transcription
factors Zeb1 and Snail through increasing the expression level of
CXCR4, and thereby promotes EMT in human cancer cells. Our
observations might provide an important clue as to how malignant
tumors develop.
Acknowledgements
The present research was supported by the National
R&D Program through the Dongnam Institute of Radiological and
Medical Sciences (DIRAMS) (50595-2014) and the (Basic Science
Research Program) through the National Research Foundation of Korea
(NRF) (NRF-2014 M2A2A 7043665) funded by the Ministry of Science,
ICT and Future Planning.
References
|
1
|
Tsai JH and Yang J: Epithelial-mesenchymal
plasticity in carcinoma metastasis. Genes Dev. 27:2192–2206. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Larue L and Bellacosa A:
Epithelial-mesenchymal transition in development and cancer: role
of phosphatidylinositol 3′ kinase/AKT pathways. Oncogene.
24:7443–7454. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Thompson EW, Newgreen DF and Tarin D:
Carcinoma invasion and metastasis: a role for
epithelial-mesenchymal transition? Cancer Res. 65:5991–5995. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hugo H, Ackland ML, Blick T, Lawrence MG,
Clements JA, Williams ED and Thompson EW: Epithelial-mesenchymal
and mesenchymal-epithelial transitions in carcinoma progression. J
Cell Physiol. 213:374–383. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Polyak K and Weinberg RA: Transitions
between epithelial and mesenchymal states: acquisition of malignant
and stem cell traits. Nat Rev Cancer. 9:265–273. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Singh A and Settleman J: EMT, cancer stem
cells and drug resistance: an emerging axis of evil in the war on
cancer. Oncogene. 29:4741–4751. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Thiery JP and Sleeman JP: Complex networks
orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell
Biol. 7:131–142. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Christofori G: New signals from the
invasive front. Nature. 441:444–450. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bronner C: Control of DNMT1 abundance in
epigenetic inheritance by acetylation, ubiquitylation, and the
histone code. Sci Signal. 4:pe32011.PubMed/NCBI
|
|
10
|
Hashimoto H, Vertino PM and Cheng X:
Molecular coupling of DNA methylation and histone methylation.
Epigenomics. 2:657–669. 2010. View Article : Google Scholar
|
|
11
|
Du Z, Song J, Wang Y, et al: DNMT1
stability is regulated by proteins coordinating deubiquitination
and acetylation-driven ubiquitination. Sci Signal.
3:ra802010.PubMed/NCBI
|
|
12
|
Hong Q and Shao ZM:
Ubiquitination/deubiquitination and acetylation/deacetylation:
making DNMT1 stability more coordinated. Acta Pharmacol Sin.
32:139–140. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bronner C, Krifa M and Mousli M:
Increasing role of UHRF1 in the reading and inheritance of the
epigenetic code as well as in tumorogenesis. Biochem Pharmacol.
86:1643–1649. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pacaud R, Brocard E, Lalier L, Hervouet E,
Vallette FM and Cartron PF: The DNMT1/PCNA/UHRF1 disruption induces
tumorigenesis characterized by similar genetic and epigenetic
signatures. Sci Rep. 4:42302014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hervouet E, Lalier L, Debien E, et al:
Disruption of Dnmt1/PCNA/UHRF1 interactions promotes tumorigenesis
from human and mice glial cells. PLoS One. 5:e113332010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
17
|
Scheel C and Weinberg RA: Cancer stem
cells and epithelial-mesenchymal transition: concepts and molecular
links. Semin Cancer Biol. 22:396–403. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Morel AP, Lievre M, Thomas C, Hinkal G,
Ansieau S and Puisieux A: Generation of breast cancer stem cells
through epithelial-mesenchymal transition. PLoS One. 3:e28882008.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jung MJ, Rho JK, Kim YM, et al:
Upregulation of CXCR4 is functionally crucial for maintenance of
stemness in drug-resistant non-small cell lung cancer cells.
Oncogene. 32:209–221. 2013. View Article : Google Scholar
|
|
20
|
Korkaya H, Kim GI, Davis A, et al:
Activation of an IL6 inflammatory loop mediates trastuzumab
resistance in HER2+ breast cancer by expanding the
cancer stem cell population. Mol Cell. 47:570–584. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sukowati CH, Rosso N, Croce LS and
Tiribelli C: Hepatic cancer stem cells and drug resistance:
relevance in targeted therapies for hepatocellular carcinoma. World
J Hepatol. 2:114–126. 2010.PubMed/NCBI
|
|
22
|
Yang ZF, Ho DW, Ng MN, et al: Significance
of CD90+ cancer stem cells in human liver cancer. Cancer
Cell. 13:153–166. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li X, Ma Q, Xu Q, et al: SDF-1/CXCR4
signaling induces pancreatic cancer cell invasion and
epithelial-mesenchymal transition in vitro through non-canonical
activation of Hedgehog pathway. Cancer Lett. 322:169–176. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bertran E, Caja L, Navarro E, Sancho P,
Mainez J, Murillo MM, Vinyals A, Fabra A and Fabregat I: Role of
CXCR4/SDF-1 alpha in the migratory phenotype of hepatoma cells that
have undergone epithelial-mesenchymal transition in response to the
transforming growth factor-beta. Cell Signal. 21:1595–1606. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wan L, Pantel K and Kang Y: Tumor
metastasis: moving new biological insights into the clinic. Nat
Med. 19:1450–1464. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tam WL and Weinberg RA: The epigenetics of
epithelial-mesenchymal plasticity in cancer. Nat Med. 19:1438–1449.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chu J, Loughlin EA, Gaur NA, et al: UHRF1
phosphorylation by cyclin A2/cyclin-dependent kinase 2 is required
for zebrafish embryogenesis. Mol Biol Cell. 23:59–70. 2012.
View Article : Google Scholar :
|
|
28
|
Sabatino L, Fucci A, Pancione M, et al:
UHRF1 coordinates peroxisome proliferator activated receptor gamma
(PPARG) epigenetic silencing and mediates colorectal cancer
progression. Oncogene. 31:5061–5072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang F, Yang YZ, Shi CZ, Zhang P, Moyer
MP, Zhang HZ, Zou Y and Qin HL: UHRF1 promotes cell growth and
metastasis through repression of p16ink4a in colorectal
cancer. Ann Surg Oncol. 19:2753–2762. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhou L, Zhao X, Han Y, et al: Regulation
of UHRF1 by miR-146a/b modulates gastric cancer invasion and
metastasis. FASEB J. 27:4929–4939. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li XL, Xu JH, Nie JH and Fan SJ: Exogenous
expression of UHRF1 promotes proliferation and metastasis of breast
cancer cells. Oncol Rep. 28:375–383. 2012.PubMed/NCBI
|
|
32
|
Mudbhary R, Hoshida Y, Chernyavskaya Y, et
al: UHRF1 overexpression drives DNA hypomethylation and
hepatocellular carcinoma. Cancer Cell. 25:196–209. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Karpf AR and Matsui S: Genetic disruption
of cytosine DNA methyltransferase enzymes induces chromosomal
instability in human cancer cells. Cancer Res. 65:8635–8639. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu S, Liu Z, Xie Z, et al: Bortezomib
induces DNA hypomethylation and silenced gene transcription by
interfering with Sp1/NF-kappaB-dependent DNA methyltransferase
activity in acute myeloid leukemia. Blood. 111:2364–2373. 2008.
View Article : Google Scholar
|
|
35
|
Batlle E, Sancho E, Franci C, Dominguez D,
Monfar M, Baulida J and Garcia De Herreros A: The transcription
factor snail is a repressor of E-cadherin gene expression in
epithelial tumour cells. Nat Cell Biol. 2:84–89. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cano A, Perez-Moreno MA, Rodrigo I,
Locascio A, Blanco MJ, del Barrio MG, Portillo F and Nieto MA: The
transcription factor snail controls epithelial-mesenchymal
transitions by repressing E-cadherin expression. Nat Cell Biol.
2:76–83. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Guaita S, Puig I, Franci C, et al: Snail
induction of epithelial to mesenchymal transition in tumor cells is
accompanied by MUC1 repression and ZEB1 expression. J Biol Chem.
277:39209–39216. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Darash-Yahana M, Pikarsky E, Abramovitch
R, et al: Role of high expression levels of CXCR4 in tumor growth,
vascularization, and metastasis. FASEB J. 18:1240–1242.
2004.PubMed/NCBI
|
|
39
|
Scala S, Giuliano P, Ascierto PA, et al:
Human melanoma metastases express functional CXCR4. Clin Cancer
Res. 12:2427–2433. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wald O, Shapira OM and Izhar U:
CXCR4/CXCL12 axis in non small cell lung cancer (NSCLC) pathologic
roles and therapeutic potential. Theranostics. 3:26–33. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Su L, Zhang J, Xu H, Wang Y, Chu Y, Liu R
and Xiong S: Differential expression of CXCR4 is associated with
the metastatic potential of human non-small cell lung cancer cells.
Clin Cancer Res. 11:8273–8280. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dubrovska A, Hartung A, Bouchez LC, Walker
JR, Reddy VA, Cho CY and Schultz PG: CXCR4 activation maintains a
stem cell population in tamoxifen-resistant breast cancer cells
through AhR signalling. Br J Cancer. 107:43–52. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gelmini S, Mangoni M, Serio M, Romagnani P
and Lazzeri E: The critical role of SDF-1/CXCR4 axis in cancer and
cancer stem cells metastasis. J Endocrinol Invest. 31:809–819.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Miki J, Furusato B, Li H, et al:
Identification of putative stem cell markers, CD133 and CXCR4, in
hTERT-immortalized primary nonmalignant and malignant tumor-derived
human prostate epithelial cell lines and in prostate cancer
specimens. Cancer Res. 67:3153–3161. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kim JK, Esteve PO, Jacobsen SE and Pradhan
S: UHRF1 binds G9a and participates in p21 transcriptional
regulation in mammalian cells. Nucleic Acids Res. 37:493–505. 2009.
View Article : Google Scholar :
|