Introduction
Prostate cancer is common worldwide (1–4).
Because most prostate cancers are initially dependent on androgens
for their growth, patients with prostate cancer receive androgen
depletion therapies including surgical castration and
administration of luteinizing hormone-releasing hormone analogs.
However, in order to achieve total androgen blockade, add-on
androgen receptor (AR) antagonists such as bicalutamide (Casodex)
or flutamide (Eulexin) are required. Although such total androgen
blockade therapy is initially effective (5,6),
most patients acquire resistance and progress to more aggressive
castration-resistant prostate cancer (CRPC) within several years.
Thus, the development of effective therapeutic approaches including
the development of more advanced drugs is eagerly anticipated for
treating patients with CRPC (7,8).
AR is a transcription factor that is known to be
ligand-specifically activated. Unliganded AR, primarily located in
the cytoplasm, translocates into the nucleus following androgen
binding where it binds to specific DNA sequences and subsequently
activates the transcription of its target genes (9). Expression of AR and prostate-specific
antigen (PSA) persists in most CRPC specimens (7,8,10),
suggesting that the AR signal remains obstinately active.
Therefore, AR is still considered to be a potential therapeutic
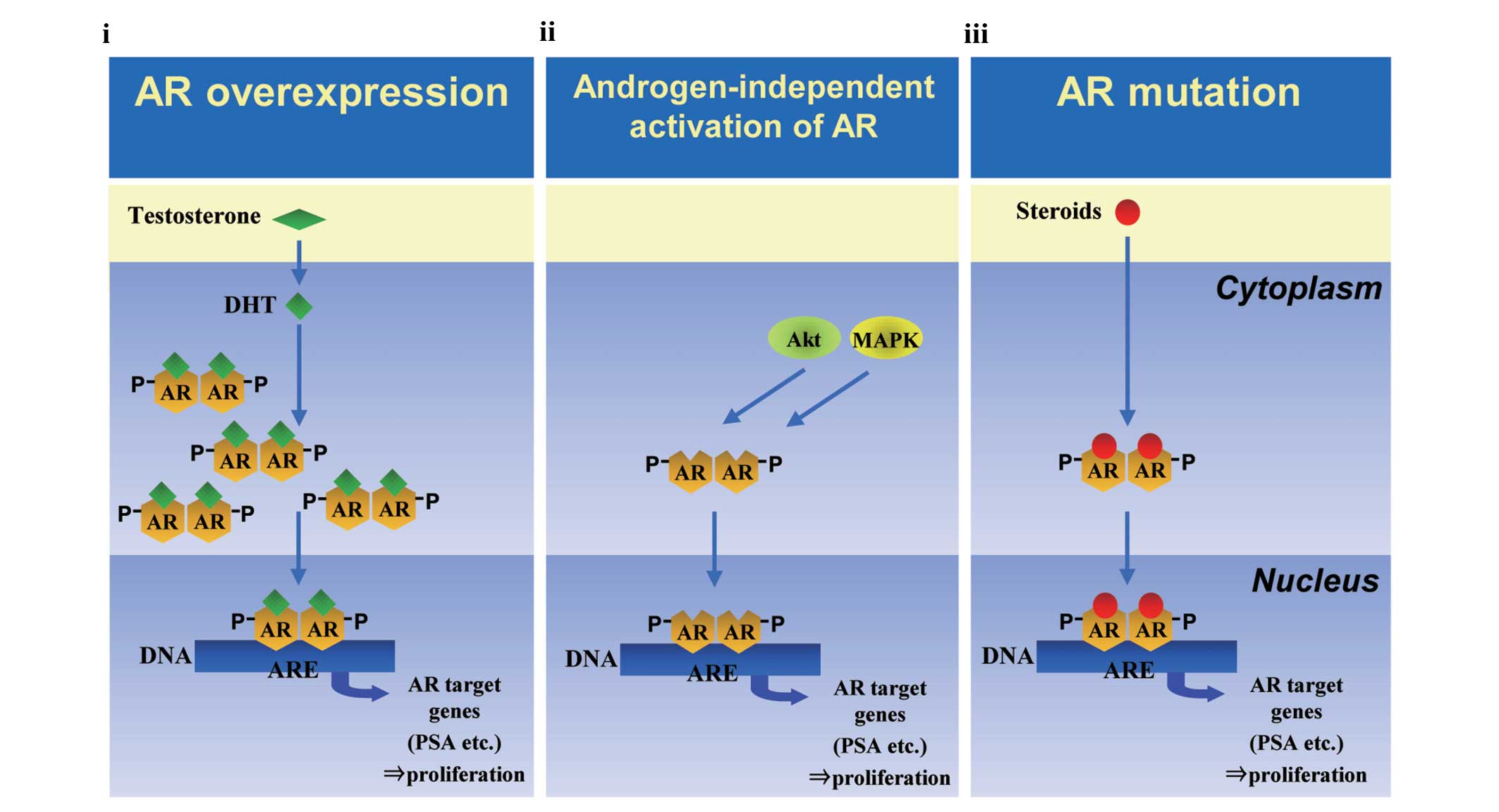
target for CRPC. Pienta and Bradley proposed three major AR-related
mechanisms of castration resistance (Fig. 1) (11): i) hypersensitivity to androgen due
to overexpression of AR (12–14);
ii) androgen-independent activation of AR mediated by deregulated
growth factors and cytokines, termed ‘outlaw’ activation (15–21);
and iii) loss of ligand specificity following an AR mutation
(22–26). We have previously reported that
existing AR antagonists, such as bicalutamide, have major
limitations in that they have a so-called partial agonist profile
(27–29). Specifically, these drugs have
agonistic activity in addition to the intended antagonistic
activity, due to which the drugs show no or at most weak antitumor
effect against prostate cancers with castration resistance acquired
by the three abovementioned mechanisms (28–30).
It is well known that tamoxifen, an estrogen
receptor α (ERα) antagonist widely used against breast cancers,
also has a partial agonist profile and tumors can develop
resistance to tamoxifen (31). To
conquer the problem, ERα pure antagonists, such as fulvestrant
(Faslodex), have been introduced and demonstrated to be efficacious
against tamoxifen-resistant breast cancers (32–34).
In an analogous fashion to ERα, we hypothesized that an AR
antagonist without agonistic activity, namely an AR pure
antagonist, would be efficacious against CRPC.
Tran et al reported a new compound effective
against CRPC with AR-overexpression (35). The compound reduced the efficiency
of AR nuclear translocation. However, the compound was reported to
have a partial agonist activity and could not completely inhibit
the translocation of AR into the nucleus. In light of these
findings, and taking into account the successful experience with
ERα pure antagonists, we screened ~2,000 compounds for their
properties as an AR nuclear translocation pure antagonist, and
discovered a new compound, CH5137291, showing high-quality
properties (36,37).
In the present study, we minutely examined the
characteristics of CH5137291 as well as its antitumor activity in
comparison with that of bicalutamide in several CRPC models with
different mechanisms of resistance.
Materials and methods
Reagents
CH5137291 and fulvestrant were synthesized in our
laboratories. Bicalutamide was purchased as Casodex tablets
(AstraZeneca, London, UK) and purified in our laboratories.
Synthetic androgen R1881 was purchased from NEN Life Science
Products (Boston, MA, USA). Progesterone, mifepristone,
dexamethasone, and β-estradiol were purchased from Sigma-Aldrich
(St. Louis, MO, USA). Aldosterone was purchased from Acros Organics
(Geel, Belgium). Abiraterone acetate was synthesized in our
laboratories.
Cells
LNCaP, VCaP, PC3, HeLa, and COS-7 cells were
obtained from the American Type Culture Collection (ATCC, Manassas,
VA, USA). LNCaP-BC2andLNCaP-CS10cellswere previously established in
our laboratory (28,29). LuCaP35V cells that progressed in
castrated male mice and expressed wild-type AR were a gift from
Professor Robert L. Vessella (University of Washington, Seattle,
WA, USA) (38). LNCaP cells were
maintained in RPMI-1640 medium (Invitrogen, Carlsbad, CA, USA)
supplemented with 10% FBS. VCaP cells were maintained in DMEM (high
glucose) (Invitrogen) supplemented with 10% FBS. LNCaP-BC2 cells
were maintained in phenol red-free RPMI-1640 supplemented with 10%
FBS and 2 μM bicalutamide. LNCaP-CS10 cells were maintained in
phenol red-free RPMI-1640 supplemented with 10% dextran-coated
charcoal-treated FBS (DCC-FBS; Hyclone Laboratories, Logan, UT,
USA) and 10 μM bicalutamide. PC3 cells were maintained in F-12
Kaighn’s medium (Invitrogen) supplemented with 10% FBS. HeLa cells
were maintained in EMEM medium (Sigma-Aldrich) supplemented with
10% FBS, 1 mM sodium pyruvate (Invitrogen), 0.1 mM non-essential
amino acids (Invitrogen), and 1.5 g/l sodium bicarbonate
(Sigma-Aldrich). COS-7 cells were maintained in DMEM (high glucose)
supplemented with 10% FBS, 1.5 mM L-glutamine (Sigma-Aldrich), 4.5
g/l D-glucose (Sigma-Aldrich), and 1.5 g/l sodium bicarbonate. The
characteristics in terms of hormone sensitivity, AR status, and AR
mutation of these cell lines are summarized in Table I.
 | Table ICastration-resistant prostate cancer
models. |
Table I
Castration-resistant prostate cancer
models.
| Model | Hormone-sensitive
or castration-resistant | AR status | AR mutation |
|---|
| VCaP |
Hormone-sensitive | Benchmark | Wild-type |
| LNCaP |
Hormone-sensitive | Benchmark | T877A |
| LNCaP-BC2 |
Castration-resistant | AR
overexpression | T877A |
| LNCaP-CS10 |
Castration-resistant |
Androgen-independent activation of AR | T877A |
| LuCaP35V |
Castration-resistant | Unknown | Wild-type |
Animals
Five-week-old male C.B-17/Icr-scid Jcl severe
combined immune-deficient (SCID) mice were purchased from CLEA
Japan (Tokyo, Japan). Four- to 6-year-old cynomolgus monkeys
(Macaca fascicularis) were bred in the Chugai Research
Institute for Medical Science (Kanagawa, Japan). All animals were
housed in a pathogen-free environment under controlled conditions
(temperature 20–26°C, humidity 35–75%, light/dark cycle 12/12 h).
Chlorinated water and irradiated food were provided ad
libitum. The health of the animals was monitored by daily
observation. The protocols of the animal studies were reviewed and
approved by the Institutional Animal Care and Use Committee of
Chugai Pharmaceutical Co., Ltd., and all animal experiments were
performed in accordance with the Guidelines for the Accommodation
and Care of Laboratory Animals promulgated in Chugai Pharmaceutical
Co., Ltd.
Fluorescence imaging analysis
LNCaP cells in 10% DCC-FBS medium were plated 24 h
before transfection. To analyze intracellular translocation of AR,
we used HaloTag system. The cells were transfected with HaloTag
pHT2-hAR-WT, HaloTag pHT2-W741C, or HaloTag pHT2-T877A using FuGENE
HD transfection reagent (Roche, Basel, Switzerland). Forty-eight
hours after the transfection, HaloTag TMR ligand (Promega,
Fitchburg, WI, USA) was added, and the cells were incubated for 0.5
h. CH5137291 or bicalutamide (10 μM) was added in the presence or
absence of R1881 (0.5 nM). After 2-h incubation, fluorescence
imaging of the living cells was performed with a BioZero
fluorescence microscope (Keyence, Osaka, Japan).
Protein fractionation and western
blotting
Cells were plated onto poly-D-lysine coated plates
(Becton-Dickinson, Heidelberg, Germany) in 10% DCC-FBS medium.
After 3-day incubation, 10 μM of CH5137291 or bicalutamide was
added and the plates were incubated for another 24 h. Then, nuclear
and cytoplasmic fractions were prepared by using NE-PER Nuclear and
Cytoplasmic Extraction Reagents (Pierce Biotechnology, Rockford,
IL, USA), and a total fraction was prepared using cell lysis buffer
(Cell Signaling Technology, Danvers, MA, USA) supplemented with
Complete Protease Inhibitor Cocktail Tablets (Roche). AR-specific
antibody N-20, IκBα-specific antibody C-15, and c-Jun-specific
antibody H-79 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) were
used for immunoblotting.
Cell proliferation assay
VCaP, LNCaP, LNCaP-BC2, and LNCaP-CS10 cells were
pre-incubated in phenol red-free RPMI-1640 medium supplemented with
10% DCC-FBS for 2–4 days and were then plated onto
poly-D-lysine-coated plates (Becton-Dickinson) (VCaP,
1×104 cells/well; LNCaP, LNCaP-BC2, and LNCaP-CS10,
5×103 cells/well) in 10% DCC-FBS medium. After overnight
incubation, CH5137291 or bicalutamide at concentrations of 4.6–30
μM was added in the presence of R1881 (VCaP, 0.1 nM; LNCaP, 0.1 nM;
LNCaP-BC2, 0.01 nM) or absence of R1881 (LNCaP-CS10). After an
additional 10-day incubation, the number of remaining cells were
estimated using a DNA quantity assay (FluoReporter Blue
Fluorometric dsDNA Quantitation kit; Invitrogen).
Reporter gene assay of AR
The following plasmids were used: GMLUC, an
MMTV-luciferase reporter plasmid created by replacing the reporter
of GMCAT (ATCC) with luciferase; phRL-CMV (Promega), a
Renilla luciferase plasmid; and human AR expression plasmids
corresponding to mutations (pcDNA3.1-hAR-WT, pcDNA3.1-hAR-W741C and
pcDNA3.1-hAR-T877A).
Twenty-four hours before transfection, the PC3 cells
in 10% DCC-FBS medium were plated onto 96-well plates at
1×104 cells/well. The cells were co-transfected with
GMLUC (50 ng/well), phRL-CMV (0.5 ng/well), and human AR expression
plasmid (10 ng/well) using a TransIT-Prostate Transfection kit
(Mirus, Madison, WI, USA). Six hours after the transfection,
various doses of CH5137291 or bicalutamide were added in the
presence of R1881 (WT, 0.05 nM; W741C, 0.5 nM; T877A, 0.5 nM) or
absence of R1881. Cell lysates were collected 48 h after the
treatment with the compounds, and the luciferase activity of each
sample was measured using a Dual-Luciferase Reporter Assay system
(Promega).
Reporter gene assay of progesterone
receptor (PR), glucocorticoid receptor (GR), mineralocorticoid
receptor (MR), and estrogen receptor α (ERα)
Twenty-four hours before transfection, HeLa cells in
10% DCC-FBS medium were plated onto 96-well plates at
1×104 cells/well for the assays of PR and ERα. COS-7
cells were plated at 0.75×103 cells/well for the assays
of GR and MR. The cells were co-transfected with GMLUC (50
ng/well), phRL-CMV (0.5 ng/well), and expression plasmids of the
nuclear receptors (10 ng/well of pSG5-hPR, pRShGR, or pRShMR) using
FuGENE 6 transfection reagent (Roche). For ERα, the cells were
co-transfected with ERE-reporter vector (50 ng/well), phRL-CMV (0.5
ng/well), and ERα expression vector HEG0 (1 ng/well) using FuGENE 6
transfection reagent. Six hours after transfection, CH5137291 or
bicalutamide (10 μM) were added in the presence or absence of the
corresponding agonist (10 nM progesterone for PR, 10 nM
dexamethasone for GR, 1 nM aldosterone for MR, and 1 nM estradiol
for ERα). Cell lysates were collected 48 h after treatment with the
compounds. Mifepristone (1 nM), mifepristone (10 nM), progesterone
(10 nM), and fulvestrant (100 nM) were used as positive antagonists
for PR, GR, MR and ERα, respectively.
Docking model analysis
Models of CH5137291 docking with wild-type and
W741C-mutant ARs were built based on the X-ray crystal structure of
the W741L-mutant AR in complex with bicalutamide (Protein Data Bank
accession code: 1z95). The three-dimensional structure of CH5137291
was modeled using Sybyl software (Tripos Inc., St. Louis, USA) with
a Tripos force field.
In the docking model of CH5137291 for W741C-mutant
AR, CH5137291 was manually docked to the W741C-mutant AR such that:
i) the crystallographic structure of the W741C-mutant AR was fixed,
ii) helix 12 of the AR was removed from the AR model because of the
intensive collision between CH5137291 and helix 12, iii) the
crystallographic structure of the trifluoromethyl benzonitrile
moiety of bicalutamide and its binding mode were used for CH5137291
and kept fixed during the docking, iv) the binding mode of the
portion other than the trifluoromethyl benzonitrile moiety of
CH5137291 was manually determined so as to avoid a steric collision
between CH5137291 and the AR without helix 12, and v) energy
minimization of the ‘compound/AR without helix 12’ complex was
performed using a molecular mechanics method with the Tripos force
field, on condition that the coordinates of AR without helix 12
were fixed. After the energy minimization, the crystallographic
coordinates of helix 12 were re-built into the AR structure.
The docking models of CH5137291 or bicalutamide for
wild-type AR were built based on the docking model of CH5137291 or
bicalutamide for W741C-mutant AR, respectively. C741 was changed to
W741 such that the conformation of W741 of the model would be
identical to that of the crystallographic structure of W741 of
wild-type AR (PDB ID: 1e3g).
Efficacy experiments in mouse xenograft
models
Xenograft models were prepared and plasma PSA was
quantified as described previously (29). Briefly, LNCaP (2×106
cells), LNCaP-BC2 (2×106 cells), LNCaP-CS10
(2×106 cells), or LuCaP35V (blocks of xenografted tumor)
was subcutaneously inoculated into non-castrated or castrated 6- to
8-week-old male SCID mice. When the tumor size reached 90–400
mm3, the animals were randomized into control and
treatment groups. The agents or vehicle (5% gum arabic;
Sigma-Aldrich) were orally administered at 10 or 100 mg/kg once a
day for 2–17 cycles of 5 days on/2 days off starting from the day
of randomization. The antitumor efficacy was evaluated by tumor
volume (TV) and the percentage of tumor growth inhibition (TGI%).
TV was estimated by using the equation TV =
ab2/2, where a and b are
the length and width of the tumor, respectively. TGI% was
calculated as follows: TGI% = [1 − (mean change in TV in each group
treated with antitumor drugs/mean change in TV in control group)] ×
100. The plasma PSA levels were measured by ELISA (Eiken Chemical,
Tokyo, Japan). Relative PSA concentration (%) = (PSA
concentration/PSA concentration on day 0 of administration) × 100.
Serum testosterone concentrations were measured using LC/MS/MS
(ASKA Pharma Medical, Tokyo, Japan) at the end of the
experiment.
To evaluate the efficacy of CH5137291 on tumors that
had become resistant to bicalutamide, LNCaP-xenografted
non-castrated mice were orally administered bicalutamide (100
mg/kg) until resistant tumors appeared. The mice bearing resistant
tumors were selected, re-randomized into two groups, and moved on
to secondary treatment. Bicalutamide (100 mg/kg) was administered
in one group and CH5137291 (100 mg/kg) in the other group. The
tumors with acquired resistance were defined as those that
fulfilled both of the following two criteria: i) (PSA concentration
on day 10 of administration/PSA concentration on day 0 of
administration) × 100 < 85%, and ii) the tumor volume on the day
of re-randomization/minimum tumor volume during primary treatment
> 1.4.
Exposure and serum PSA in cynomolgus
monkeys
For the exposure assay, the monkeys received a
single oral administration of CH5137291 at doses of 0.1, 1, 10 or
100 mg/kg. The vehicle was 1% hydroxypropylcellulose (Nippon Soda,
Tokyo, Japan). The serum concentration of CH5137291 was measured by
using LC/MS/MS (API3200; Applied Biosystems, Foster City, CA, USA).
To assess the effect of CH5137291 on serum PSA, we selected 4- to
6-year-old monkeys with initial serum PSA concentrations of
>0.25 ng/ml, as measured by using chemiluminescent immunoassay
at BML, Inc. (Tokyo, Japan). The animals were randomized, and
CH5137291 was orally administered daily for 7 days at doses of 0.1,
1, 10 or 100 mg/kg. Twenty-four hours after the last
administration, serum was harvested and PSA concentration was
measured.
Results
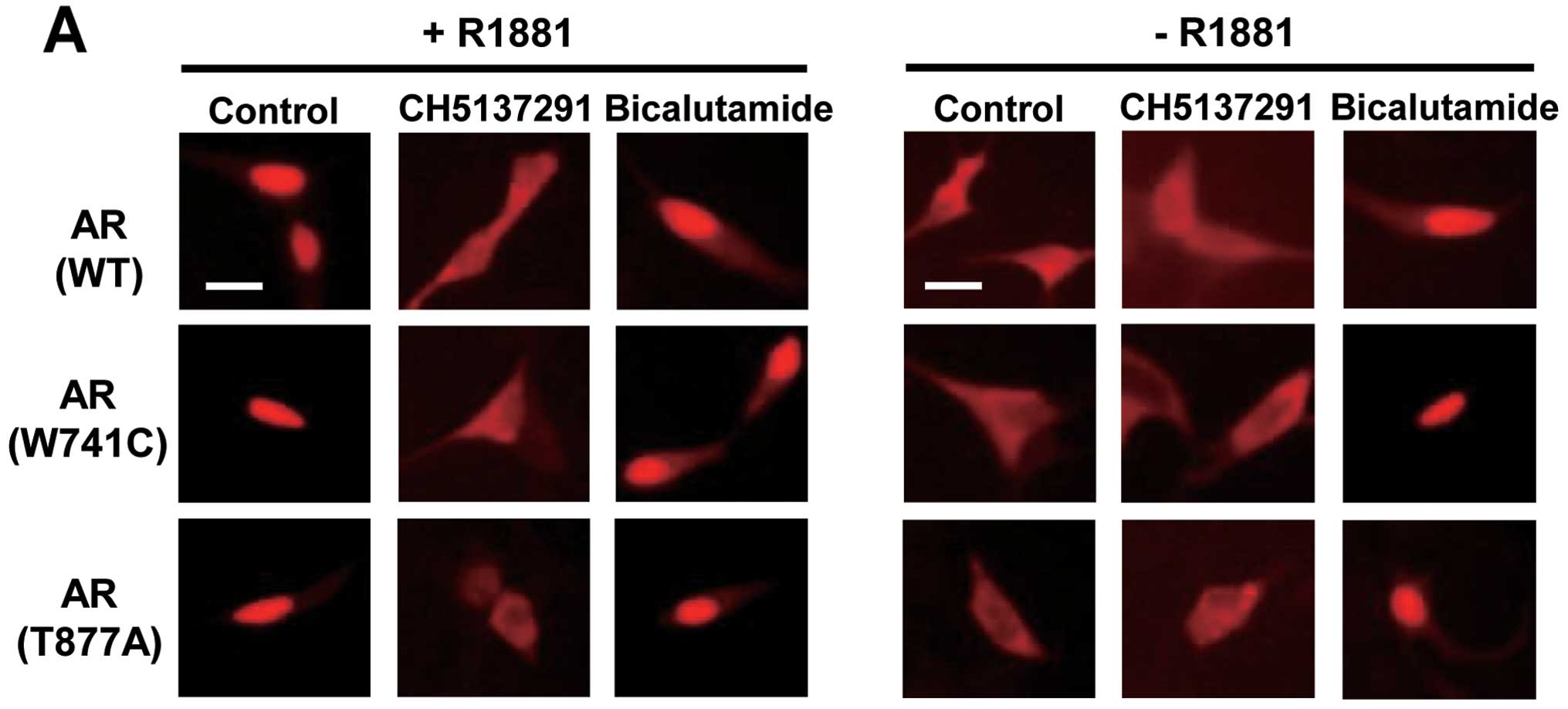
Inhibition of AR nuclear translocation by
CH5137291
Kawata et al reported the inhibitory effect
of CH5137291 on AR nuclear translocation by using COS-7 cells
transiently transfected with human wild-type AR (37). In the present study, we examined
the effect of CH5137291 on subcellular localization in prostate
cancer cell lines with wild-type or mutated AR. For this purpose,
we used LNCaP cells transfected with HaloTag-fused wild-type AR,
W741C-mutant AR, or T877A-mutant AR. In each of these cells with
three different types of AR, CH5137291 clearly inhibited AR nuclear
translocation in the presence of synthetic androgen R1881.
CH5137291 in the absence of R1881 did not induce AR nuclear
translocation suggesting that CH5137291 does not have an agonistic
effect on AR (Fig. 2A). In
contrast, bicalutamide did not inhibit AR nuclear translocation in
the presence of R1881 and induced AR nuclear translocation in the
absence of R1881.
Next, we examined the effect of CH5137291, in
comparison with bicalutamide, on the intracellular distribution of
AR protein. For this purpose, LNCaP-CS10 cells were used, because
AR in LNCaP-CS10 cells is androgen-independently activated.
Bicalutamide reportedly acts as a full agonist on LNCaP-CS10 cells
(29). We found that CH5137291
decreased the nuclear AR level and increased the cytoplasmic AR
level without affecting the total AR level (Fig. 2B). On the other hand, as was
expected, bicalutamide increased the nuclear AR level but did not
affect the total AR level.
These results indicate that CH5137291 acts as an AR
nuclear translocation inhibitor, not only in androgen-dependent
prostate cancer cells with wild-type or mutant AR but also in
androgen-independent prostate cancer cells having outlaw
pathways.
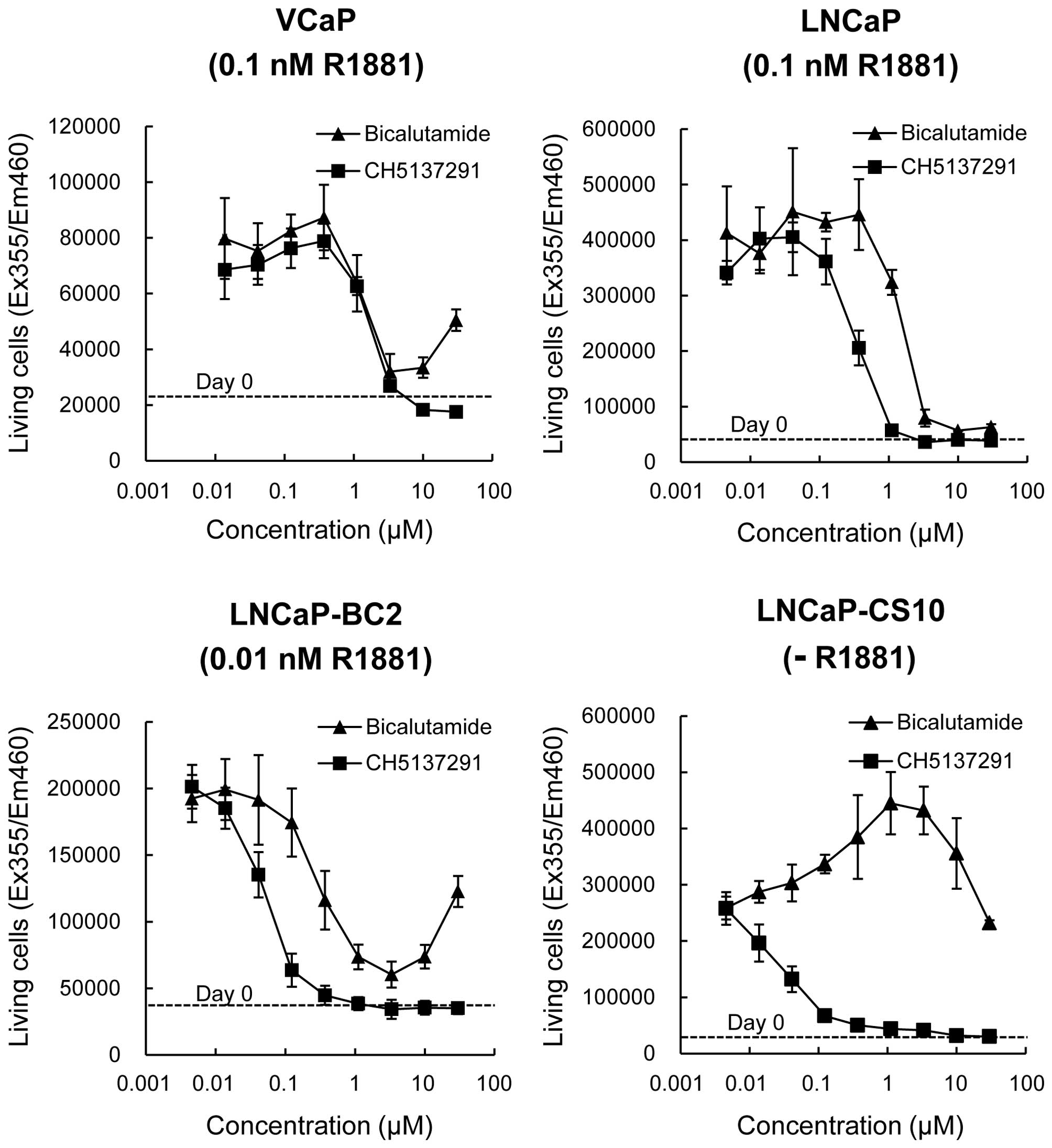
Cell growth inhibition by CH5137291
against hormone-sensitive prostate cancers and CRPCs in vitro
The proliferation inhibitory activity of CH5137291,
compared with bicalutamide, was examined in vitro using four
prostate cancer cell lines (VCaP, LNCaP, LNCaP-BC2, and LNCaP-CS10)
each with different hormone sensitivity and AR status (Table I and Fig. 3). The assays were all performed in
the presence of optimal concentrations of synthetic androgen R1881
except for LNCaP-CS10, because LNCaP-CS10 does not require androgen
for its proliferation (29).
In VCaP cells, growth was completely inhibited by
CH5137291 at concentrations of 3–30 μM. In contrast, bicalutamide
almost completely inhibited the cell growth at 3 μM, however at 30
μM, it induced cell growth. In LNCaP cells, CH5137291 inhibited
cell growth more strongly than bicalutamide, and completely
inhibited cell growth at concentrations of 1–30 μM. In LNCaP-BC2
cells, CH5137291 also showed stronger inhibition of cell growth
than bicalutamide, and complete inhibition was observed at
CH5137291 concentrations as low as 0.3 μM. Although bicalutamide
showed strong inhibition of cell growth at 3 μM, cell growth was
revived at higher concentrations. In LNCaP-CS10 cells, CH5137291
showed strong inhibition of cell growth, whereas bicalutamide
enhanced cell growth.
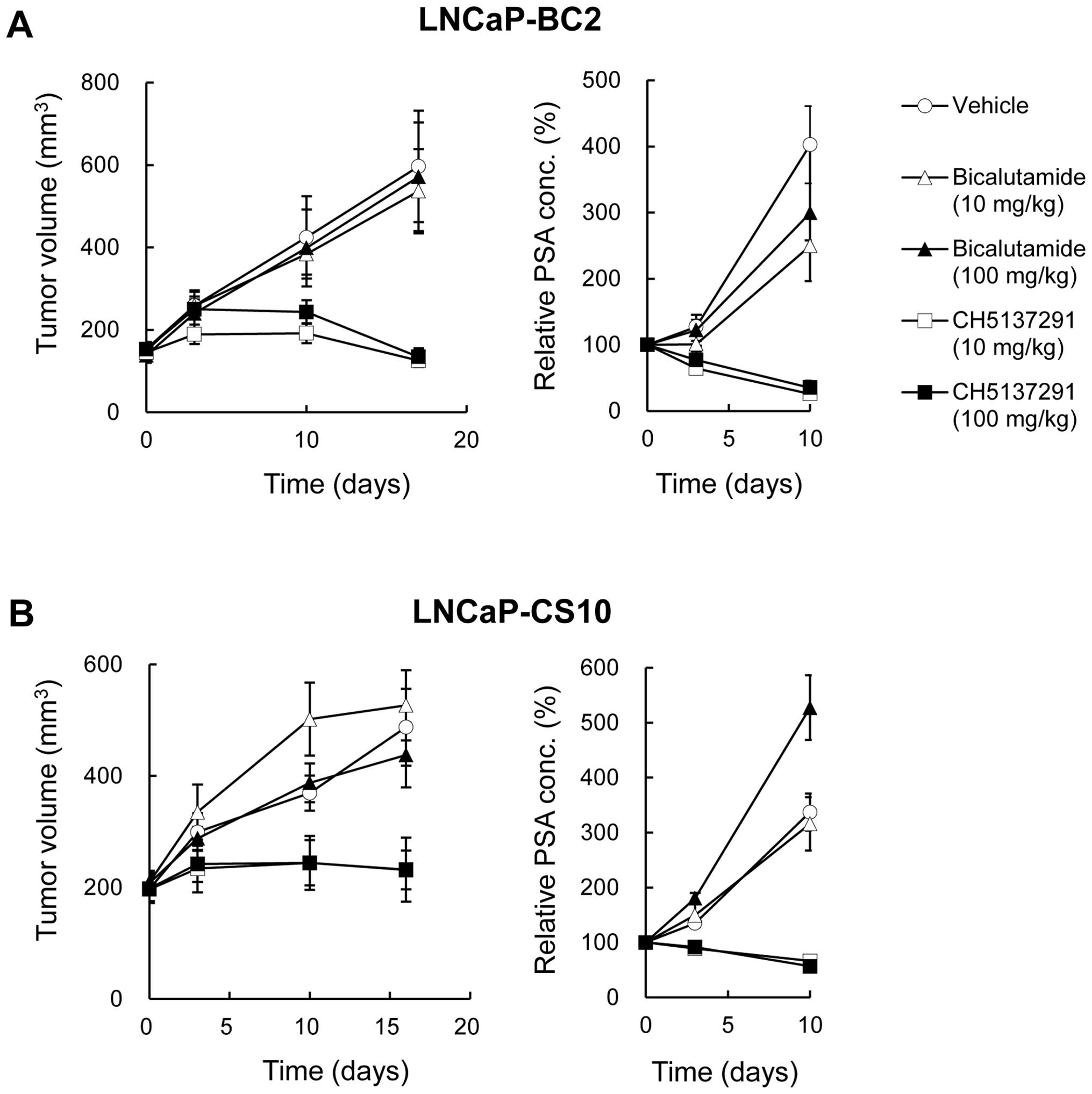
Antitumor activity of CH5137291 against
CRPC xenografts
The in vivo efficacy of CH5137291 on CRPCs
was examined by using xenograft models of castrated SCID mice
bearing LNCaP-BC2 and LNCaP-CS10. CH5137291 treatment (10 and 100
mg/kg) potently inhibited tumor growth in both xenograft models.
The TGI% in each model was as follows: LNCaP-BC2, 104% and 104% on
day 17 (at 10 and 100 mg/kg CH5137291, respectively); LNCaP-CS10,
88% and 88% on day 16 (at 10 and 100 mg/kg CH5137291, respectively)
(Fig. 4). The plasma PSA level was
also measured as a pharmacodynamic biomarker of AR antagonist
activity. In both of the xenograft models tested, CH5137291
completely inhibited the increase in plasma PSA level, even at the
dose of 10 mg/kg, without agonistic activity (Fig. 4). In contrast, bicalutamide
treatment (10 and 100 mg/kg) showed almost no or little effect on
either TV or PSA level even at the dose of 100 mg/kg.
Pure antagonistic effect of CH5137291 on
the transcriptional activity of wild-type and mutant AR
The antagonistic effect of CH5137291 on AR
transcriptional activity was examined by reporter gene assay. For
this purpose, PC3 cells transiently co-transfected with AR
(wild-type, W741C, or T877A) expression vectors and GMLUC reporter
vectors including an MMTV promoter, a well-characterized
AR-targeting promoter, were used. The W741C and T877A mutations are
known to cause resistance to bicalutamide and flutamide,
respectively (23,26,30).
In the three transfectants, CH5137291 inhibited
transcriptional activity of AR in a dose-dependent manner in the
presence of R1881 (Fig. 5).
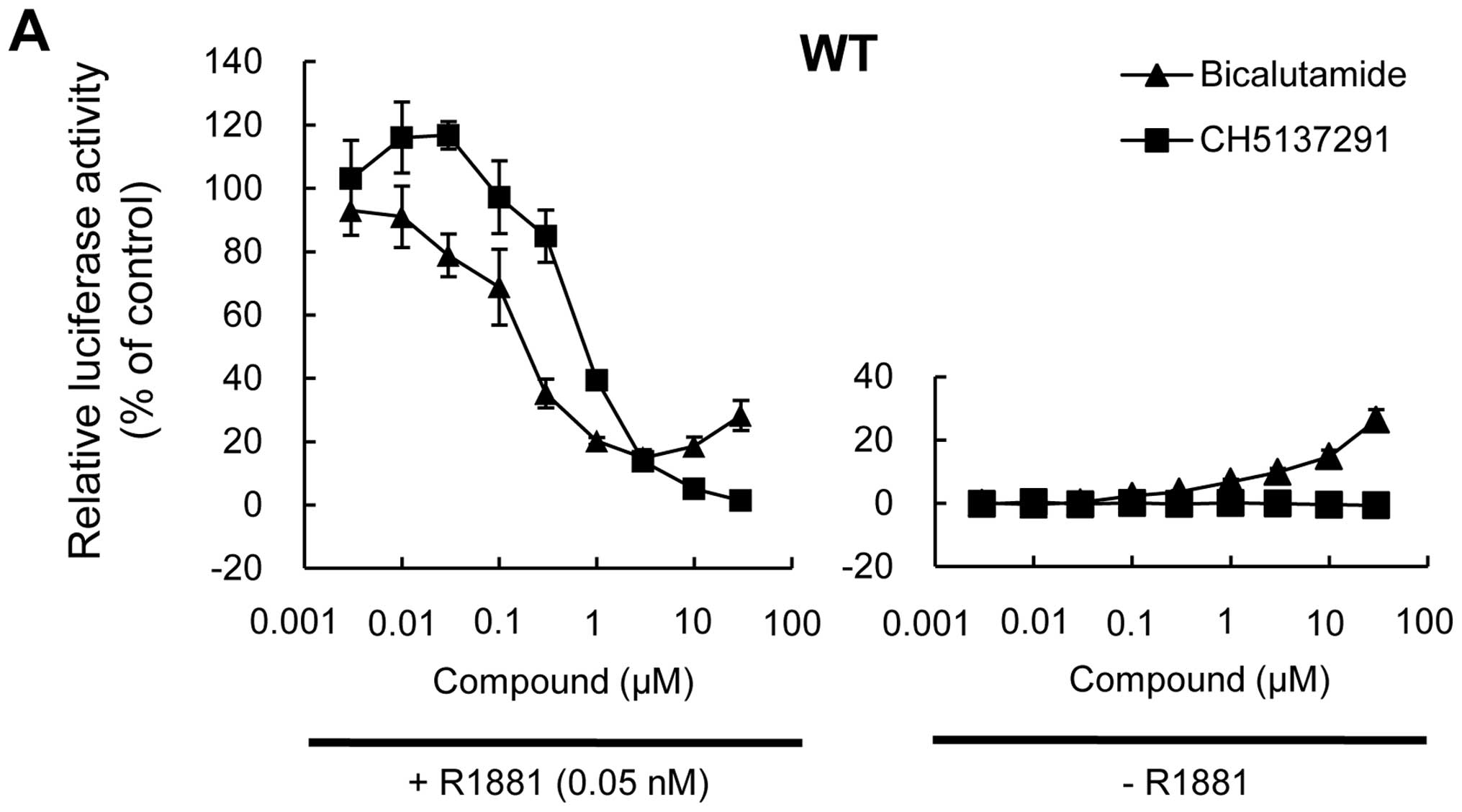
Bicalutamide showed different behaviors in the three transfectants.
With wild-type AR, bicalutamide showed dose-dependent inhibition in
transcriptional activity ≤3 μM; however, resurgence was observed at
higher bicalutamide concentrations (Fig. 5A). With W741C-mutant AR,
bicalutamide had no inhibitory effect throughout the concentrations
used (Fig. 5B). With T877A-mutant
AR, bicalutamide showed a weaker inhibitory effect compared with
CH5137291. In the absence of R1881, CH5137291 showed no agonistic
effect on the transcriptional activity of AR in any of the three
transfectants (Fig. 5). In
contrast, bicalutamide strongly increased the transcriptional
activity of W741C-mutant AR in a dose-dependent manner (Fig. 5B). With wild-type and T877A-mutant
AR, bicalutamide increased the transcriptional activity at
relatively high concentrations (Fig.
5A and C).
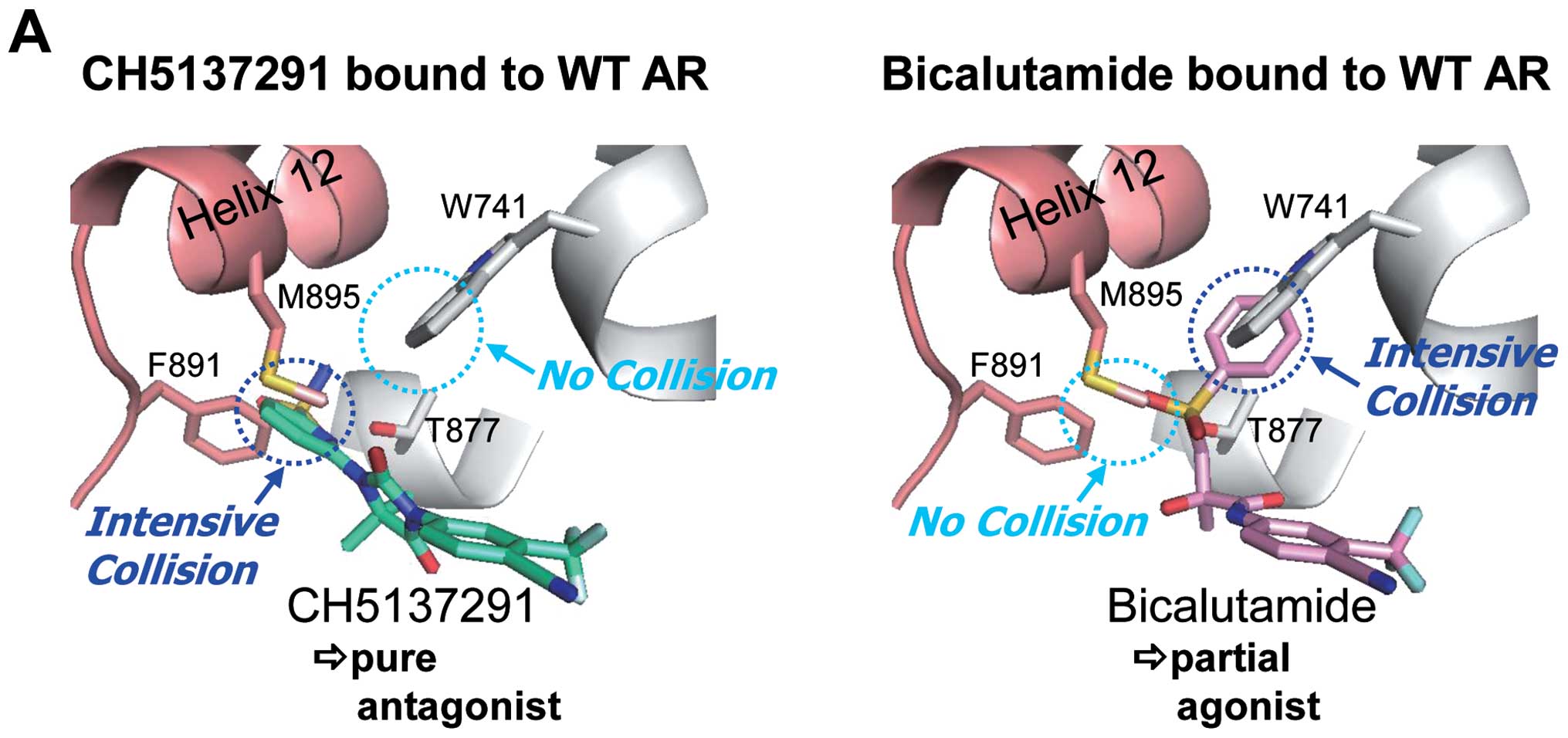
Complete inhibition of helix 12 folding
by CH5137291 in wild-type and W741C-mutant AR
The folding of helix 12, present in the
ligand-binding domain of AR, is reported to be necessary for a
ligand-induced agonist effect on AR transcriptional activity
(39,40). The crystal structure of the
bicalutamide/W741C-mutant AR complex revealed that bicalutamide
does not collide with the C741 of the AR as it does with W741 in
the wild-type AR; therefore, helix 12 of the
bicalutamide/W741C-mutant AR complex is folded in the same manner
as when a ligand, such as R1881, binds to it (Fig. 6) (41). Reportedly, this is a reason for
bicalutamide exhibiting an agonist effect in W741C-mutant AR. To
investigate the differences in the mechanisms of CH5137291 and
bicalutamide with respect to agonist/antagonist effects against
wild-type or W741C-mutant ARs, we examined a docking model of
CH5137291 against wild-type and W741C-mutant ARs. The docking model
indicated that the terminal sulfonamide group of CH5137291
intensively collided with the M895 residue of helix 12 in both the
wild-type AR and the W741C-mutant AR. The collision would cause a
complete inhibition of helix 12 folding and would result in a pure
antagonist effect in both wild-type and W741C-mutant ARs. The
varying interactions between the compound and AR may lead to the
differing agonist and antagonist characteristics of CH5137291 and
bicalutamide, respectively, in wild-type and W741C-mutant ARs.
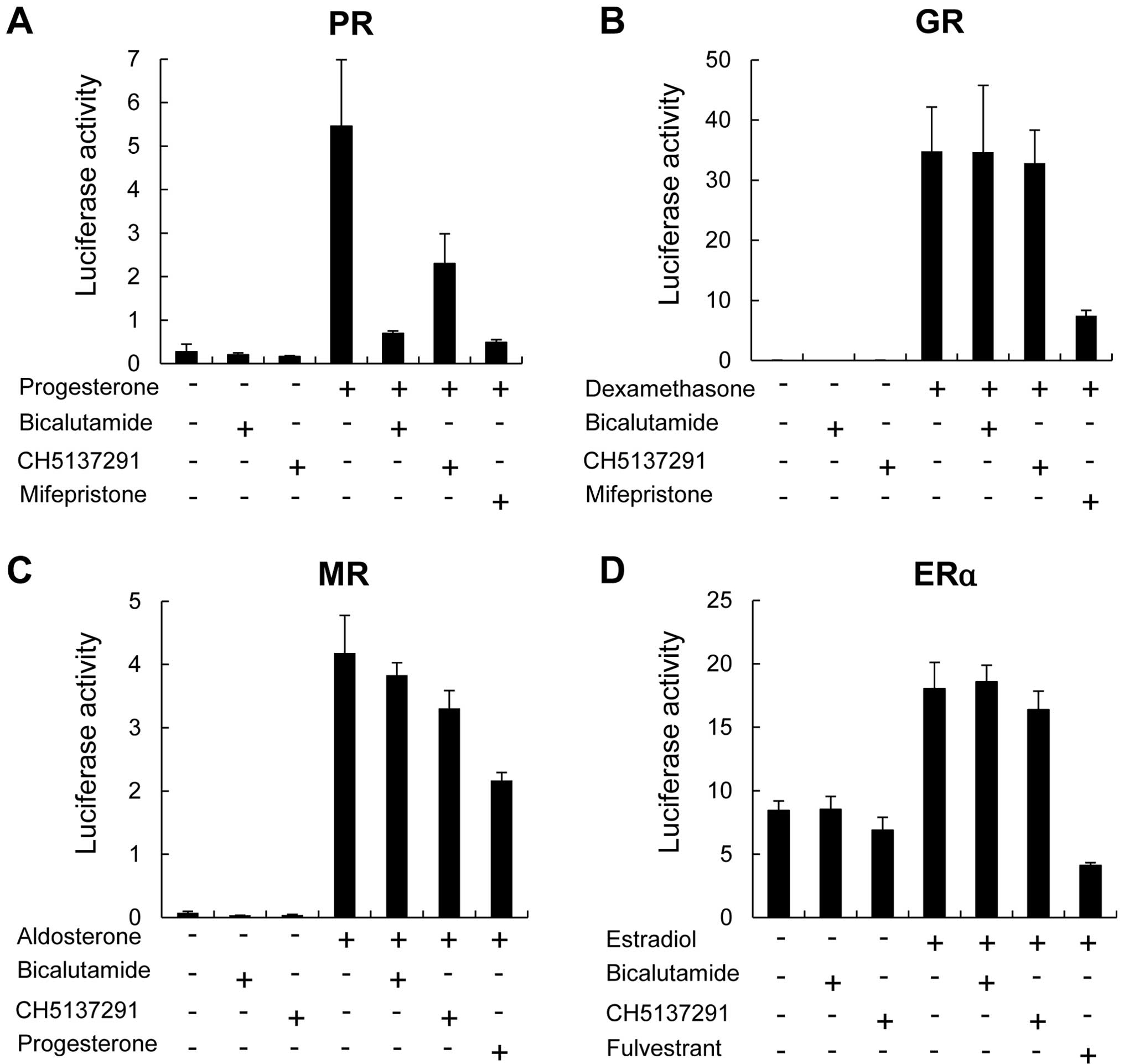
Nuclear receptor specificity of
CH5137291
Because of the highly similar structures within the
nuclear receptor superfamily, we used a reporter gene assay to
profile the effect of CH5137291 on the transcriptional activity of
other nuclear receptors, including PR, GR, MR and ERα, in the
presence or absence of ligands. CH5137291 exhibited weaker
antagonistic effects on the progesterone receptor as compared to
bicalutamide. Moreover, it exhibited neither agonistic nor
antagonistic effects on glucocorticoid receptor, mineralocorticoid
receptor, or estrogen receptor α (Fig.
7).
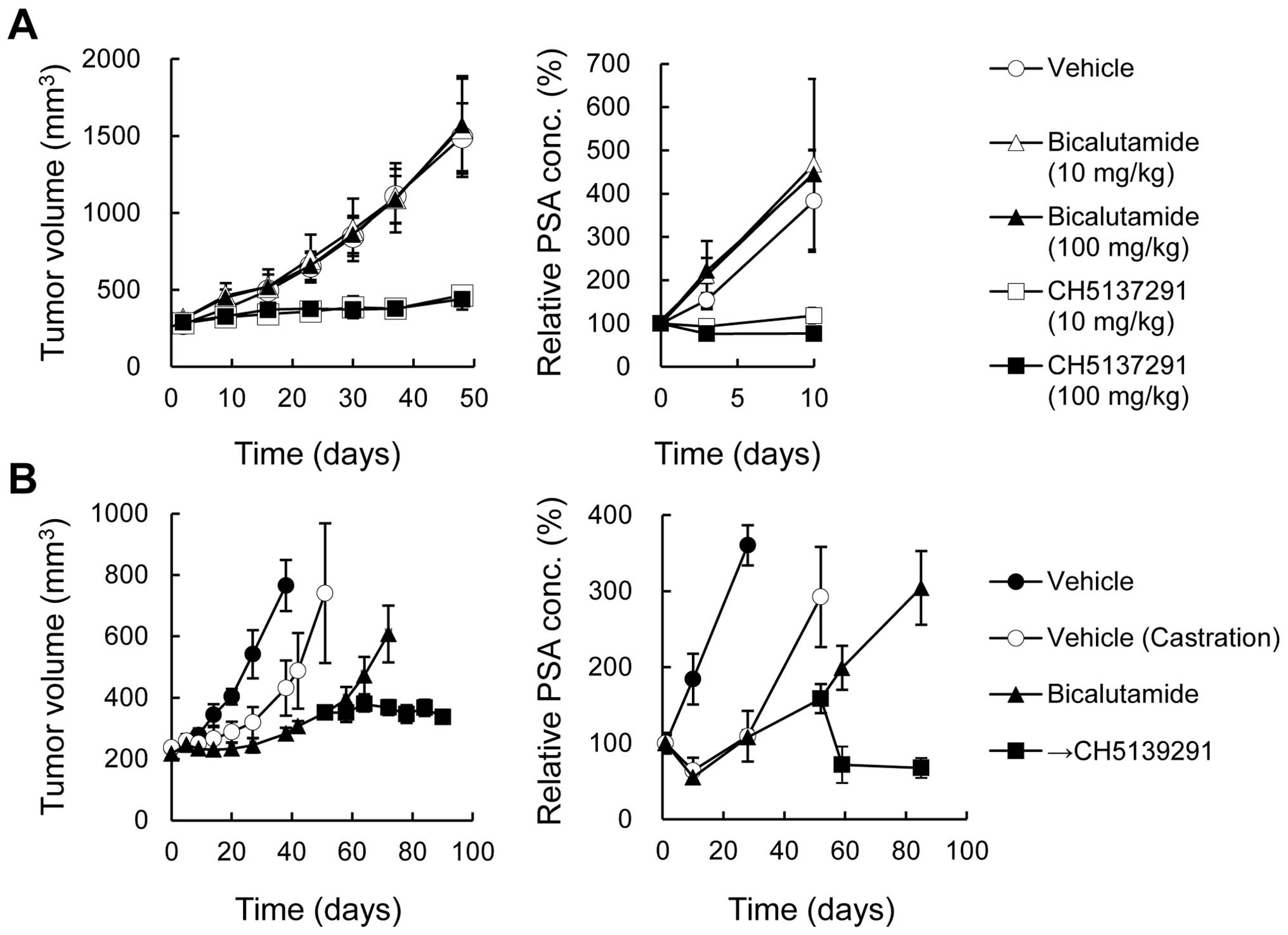
Antitumor activity of CH5137291 in
LuCaP35V xenografts that progressed after castration and in LNCaP
xenografts that progressed during bicalutamide treatment
The effects of CH5137291 was examined against
LuCaP35V xenografted tumors that progressed in castrated mice and
LNCaP xenografted tumors that had failed to respond to initial
bicalutamide treatment. In the LuCaP35V xenograft model, CH5137291
treatment (10 and 100 mg/kg) potently inhibited the tumor growth
and plasma PSA level. The TGI% was 82% and 85% on day 48 (at 10 and
100 mg/kg, respectively) (Fig.
8A). In contrast, bicalutamide treatment (10 and 100 mg/kg)
showed almost no effect on TV or PSA level. In the LNCaP xenograft
model, 50 days after initiation of bicalutamide treatment, animals
with tumors that showed resistance to bicalutamide were selected
and randomized into two groups. One group continued receiving
bicalutamide and the other group was switched to CH5137291
treatment. Only in the group switched to CH5137291 treatment did
the tumor growth become static and the plasma PSA level decrease
(Fig. 8B).
Long-term tumor growth inhibition by
CH5137291 in the hormone-sensitive LNCaP xenograft model
Long-term tumor growth inhibition against
hormone-sensitive prostate cancer was examined by using an LNCaP
xenograft in non-castrated SCID mice (Fig. 8C). The average time taken for the
tumor volume to exceed double the initial volume was 113 days in
the CH5137291 group and 58 days in the bicalutamide group. In
addition, CH5137291 suppressed the plasma PSA concentration and the
reduction in body weight during the observation period. These
results indicate that CH5137291 can control tumor growth of
hormone-sensitive prostate cancer for nearly twice as long as the
period of control with bicalutamide.
Exposure of CH5137291 and serum PSA
levels in cynomolgus monkeys
Finally, to estimate the clinical therapeutic
potential of CH5137291, cynomolgus monkeys were treated with
CH5137291. Serum concentration of the drug and PSA as a
pharmacodynamic biomarker was measured. The CH5137291 concentration
dose-dependently increased. PSA concentration decreased in inverse
proportion to the dosage of CH5137291 with a maximum 80% inhibition
at 100 mg/kg (Fig. 9).
Discussion
Three major AR-related mechanisms of castration
resistance in prostate cancer have been reported: i)
hypersensitivity to androgen caused by AR overexpression, ii)
androgen-independent activation of AR mediated by deregulated
growth factors and cytokines; and iii) loss of ligand specificity
due to AR mutation (Fig. 1)
(11).
Following binding with androgen, AR exhibits
transcriptional activity through dimerization, nuclear
translocation, and binding to the androgen response element. Each
of these processes is essential for AR signaling. Among these
processes, we focused on the nuclear translocation, and screened
for compounds that inhibited nuclear translocation of AR without
agonistic activity. We finally found a candidate, the CH5137291. In
the present study, we conducted a detailed investigation into the
inhibitory activity of CH5137291 on nuclear translocation of AR. We
found that CH5137291 inhibited, regardless of the presence of
androgen, the nuclear translocation of wild-type,
bicalutamide-resistant type (W741C), and flutamide-resistant type
(T877A) ARs which were exogenously expressed in LNCaP cells. In
contrast, bicalutamide induced the nuclear translocation of all of
the above AR types, similar to the action of androgen (Fig. 2A). We also confirmed that CH5137291
inhibited the nuclear translocation of the AR that was endogenously
expressed in LNCaP-CS10 (Fig.
2B).
Because the nuclear translocation of AR is required
for all three of the abovementioned mechanisms of castration
resistance, it was considered that the inhibition of AR nuclear
translocation was the most important characteristic of
CH5137291.
To investigate the activities of CH5137291 against
the three mechanisms of castration resistance, we used the
following cells corresponding to each mechanism of resistance: i)
LNCaP-BC2 cells as an AR overexpression model, ii) LNCaP-CS10 cells
as an androgen-independent AR activation model, and iii) PC3 cells
expressing mutant ARs for reporter gene assay as an AR mutation
model. LNCaP-BC2 cells can proliferate at androgen concentrations
as low as 1/10 of those required for parental LNCaP cells in
vitro, and can grow in castrated male mice (28). LNCaP-CS10 cells proliferate
AR-dependently in the absence of androgen in vitro, and can
grow in castrated male mice (29).
In in vitro studies, CH5137291 completely
inhibited the cell growth of both LNCaP-BC2 and LNCaP-CS10 cells;
however, bicalutamide showed a biphasic activity on the
proliferation of LNCaP-BC2 cells, and surprisingly, it stimulated
the proliferation of LNCaP-CS10 cells even at the lower
concentrations tested in vitro (Fig. 3). In xenograft models of castrated
mice with these cell lines, CH5137291 showed significant antitumor
activity along with a reduction in plasma PSA levels (Fig. 4). In contrast, bicalutamide did not
inhibit tumor growth at all in these xenograft models. These
results coincide well with our previous reports (28,29),
and suggest that CH5137291 will be effective on those castration
resistant prostate cancers which have AR overexpression or
androgen-independent activation of AR and do not respond to
bicalutamide.
The reporter gene assays performed using different
types of mutant AR clearly showed that bicalutamide acts as a full
agonist on the transcriptional activity in bicalutamide-resistant
W741C-mutant AR, and as a partial agonist in wild-type AR and
flutamide-resistant T877A-mutant AR (Fig. 5) (23,26,30).
In contrast, CH5137291 acted as a pure antagonist on the
transcriptional activity in each of the ARs (Fig. 5). These results suggest that
CH5137291 is potentially superior to bicalutamide as an AR
antagonist against all of the three mechanisms of castration
resistance.
To elucidate the pure AR antagonistic nature of
CH5137291 in terms of three-dimensional structure, a docking model
analysis was performed. The folding of helix 12 of AR is reported
to be caused by ligand binding and is considered necessary for a
ligand-induced AR agonist effect (39,40).
The docking model revealed that CH5137291 intensively collided with
the M895 residue of helix 12 in both wild-type AR and W741C-mutant
AR (Fig. 6). This collision of
CH5137291 would cause a complete inhibition of helix 12 folding and
would result in a pure antagonist effect in both the wild-type AR
and W741C-mutant AR. On the other hand, bicalutamide collided with
the W741 residue in the wild-type AR, but did not collide with the
C741 residue in the W741C-mutant AR; subsequently, helix 12 of the
bicalutamide/W741C-mutant AR complex was folded in the same manner
as when the ligand binds to wild-type AR (41). This would be one of the reasons for
the partial agonistic effect of bicalutamide in wild-type AR and
its fully agonistic effect in W741C-mutant AR.
Recently, reports regarding the development of the
CYP17 inhibitor abiraterone acetate and the AR antagonist MDV3100
have described their efficacy against CRPC (35,42–44).
Our LNCaP-CS10 xenograft study showed that abiraterone acetate did
not exert an effect on CRPC with an androgen-independent AR
activation mechanism of resistance because the mode of action of
the agent is the inhibition of androgen synthesis, whereas
LNCaP-CS10 tumors grow in an androgen-independent manner (data not
shown) (29). MDV3100 is reported
to have partial agonist activity on AR nuclear translocation
(35); on the other hand,
CH5137291 exhibited pure antagonist activity on AR nuclear
translocation. These data suggest the advantages of CH5137291 over
other agents against CRPCs.
In addition to superior non-clinical efficacy, fewer
adverse effects and higher exposure in humans are indispensable for
the development of a compound for therapeutic use. Because AR
belongs to the nuclear steroid receptor family and has a structure
highly similar to that of other nuclear receptors (e.g.,
progesterone, glucocorticoid, mineralocorticoid and estrogen
receptors), any drug targeting AR holds the possibility for cross
reactivity-related adverse effects (45,46).
Our results indicated that CH5137291 exhibited weaker antagonistic
effects on the progesterone receptor as compared to bicalutamide
and did not exhibit any agonist/antagonist effects on
glucocorticoid receptor, mineralocorticoid receptor, or estrogen
receptor α (Fig. 7). We therefore
consider that CH5137291 would not cause any adverse events through
cross reactivity with other nuclear receptors.
We also examined the effectiveness of CH5137291 in
various therapeutic situations. To mimic second-line hormonal
therapy, we used a castration-resistant LuCaP35V and LNCaP
xenograft models (38). In the
latter model, treatment was changed to CH5137291 when the tumor
became resistant to bicalutamide after initial tumor growth
inhibition by bicalutamide. CH5137291 showed antitumor activity
against both of the two models (Fig.
8A and B). To mimic first-line hormonal therapy, the duration
of tumor stabilization in the LNCaP xenograft model was examined.
CH5137291 showed long-term antitumor efficacy as compared to
bicalutamide (Fig. 8C). These
results suggest that CH5137291 was potentially efficacious not only
against CRPCs that progress despite castration or bicalutamide
treatment but also against hormone therapy-naïve prostate
cancers.
Concerning exposure, other pure AR antagonists such
as CH4892280 have failed to achieve adequate plasma levels due to
their metabolic instability (27,40).
However, based on the present results, sufficient exposure of
CH5137291 is expected in humans by virtue of its remarkable
stability in the liver microsomes of mice, rats, monkeys, and
humans [(37) and data not shown]
as well as the dose-dependent increase in levels in plasma/serum,
which were adequately efficacious not only in mouse xenografts but
also in cynomolgus monkeys (Fig.
9). These results suggest that CH5137291 shows promising
clinical efficacy.
In conclusion, we have described here the superior
characteristics of the novel AR pure antagonist CH5137291 which
inhibited AR signaling through the inhibition of AR nuclear
translocation. The mechanism of action was the inhibition of helix
12 folding of AR by immediate interference with the M895 residue.
Compared with bicalutamide, CH5137291 exhibited not only a stronger
therapeutic efficacy against CRPCs with AR overexpression,
androgen-independent AR activation, and AR mutation as the
mechanisms of resistance but also showed long-term inhibition of
growth of hormone-sensitive prostate cancer. We expect that
CH5137291 will be involved in novel therapeutic approaches for
prostate cancer.
Acknowledgements
The authors thank Dr Kentaro Furumoto for his
technical advice; Dr Yoichiro Moriya for his help in manuscript
preparation; and Mr. Hitoshi Arakawa, Mr. Toshio Hani, Mr. Ko
Saito, Ms. Hiroko Igarashi, Ms. Yuka Sugiura and Ms. Ayako
Takahashi for their excellent technical assistance. Funding
assistance for this study was received from Chugai Pharmaceutical
Co., Ltd., Kanagawa, Japan.
References
|
1
|
Bosetti C, Bertuccio P, Levi F, Lucchini
F, Negri E and La Vecchia C: Cancer mortality in the European
Union, 1970–2003, with a joinpoint analysis. Ann Oncol. 19:631–640.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jemal A, Siegel R, Ward E, Hao Y, Xu J and
Thun MJ: Cancer statistics, 2009. CA Cancer J Clin. 59:225–249.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Matsuda T, Marugame T, Kamo K, Katanoda K,
Ajiki W and Sobue T: Cancer incidence and incidence rates in Japan
in 2002: based on data from 11 population-based cancer registries.
Jpn J Clin Oncol. 38:641–648. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Qiu D, Katanoda K, Marugame T and Sobue T:
A Joinpoint regression analysis of long-term trends in cancer
mortality in Japan (1958–2004). Int J Cancer. 124:443–448. 2009.
View Article : Google Scholar
|
|
5
|
Taplin ME and Balk SP: Androgen receptor:
a key molecule in the progression of prostate cancer to hormone
independence. J Cell Biochem. 91:483–490. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Arai Y, Akaza H, Deguchi T, et al:
Evaluation of quality of life in patients with previously untreated
advanced prostate cancer receiving maximum androgen blockade
therapy or LHRHa monotherapy: a multicenter, randomized,
double-blind, comparative study. J Cancer Res Clin Oncol.
134:1385–1396. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Roudier MP, True LD, Higano CS, et al:
Phenotypic heterogeneity of end-stage prostate carcinoma metastatic
to bone. Hum Pathol. 34:646–653. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shah RB, Mehra R, Chinnaiyan AM, et al:
Androgen-independent prostate cancer is a heterogeneous group of
diseases: lessons from a rapid autopsy program. Cancer Res.
64:9209–9216. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tomura A, Goto K, Morinaga H, et al: The
subnuclear three-dimensional image analysis of androgen receptor
fused to green fluorescence protein. J Biol Chem. 276:28395–28401.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gregory CW, He B, Johnson RT, et al: A
mechanism for androgen receptor-mediated prostate cancer recurrence
after androgen deprivation therapy. Cancer Res. 61:4315–4319.
2001.PubMed/NCBI
|
|
11
|
Pienta KJ and Bradley D: Mechanisms
underlying the development of androgen-independent prostate cancer.
Clin Cancer Res. 12:1665–1671. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Visakorpi T, Hyytinen E, Koivisto P, et
al: In vivo amplification of the androgen receptor gene and
progression of human prostate cancer. Nat Genet. 9:401–406. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Linja MJ, Savinainen KJ, Saramaki OR,
Tammela TL, Vessella RL and Visakorpi T: Amplification and
overexpression of androgen receptor gene in hormone-refractory
prostate cancer. Cancer Res. 61:3550–3555. 2001.PubMed/NCBI
|
|
14
|
Chen CD, Welsbie DS, Tran C, et al:
Molecular determinants of resistance to antiandrogen therapy. Nat
Med. 10:33–39. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Culig Z, Hobisch A, Cronauer MV, et al:
Androgen receptor activation in prostatic tumor cell lines by
insulin-like growth factor-I, keratinocyte growth factor, and
epidermal growth factor. Cancer Res. 54:5474–5478. 1994.PubMed/NCBI
|
|
16
|
Gioeli D, Ficarro SB, Kwiek JJ, et al:
Androgen receptor phosphorylation. Regulation and identification of
the phosphorylation sites. J Biol Chem. 277:29304–29314. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ueda T, Mawji NR, Bruchovsky N and Sadar
MD: Ligand-independent activation of the androgen receptor by
interleukin-6 and the role of steroid receptor coactivator-1 in
prostate cancer cells. J Biol Chem. 277:38087–38094. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Debes JD, Schmidt LJ, Huang H and Tindall
DJ: p300 mediates androgen-independent transactivation of the
androgen receptor by interleukin 6. Cancer Res. 62:5632–5636.
2002.PubMed/NCBI
|
|
19
|
Gregory CW, Whang YE, McCall W, et al:
Heregulin-induced activation of HER2 and HER3 increases androgen
receptor transactivation and CWR-R1 human recurrent prostate cancer
cell growth. Clin Cancer Res. 11:1704–1712. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Culig Z, Steiner H, Bartsch G and Hobisch
A: Interleukin-6 regulation of prostate cancer cell growth. J Cell
Biochem. 95:497–505. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ponguta LA, Gregory CW, French FS and
Wilson EM: Site-specific androgen receptor serine phosphorylation
linked to epidermal growth factor-dependent growth of
castration-recurrent prostate cancer. J Biol Chem. 283:20989–21001.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhao XY, Malloy PJ, Krishnan AV, et al:
Glucocorticoids can promote androgen-independent growth of prostate
cancer cells through a mutated androgen receptor. Nat Med.
6:703–706. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shi XB, Ma AH, Xia L, Kung HJ and de Vere
White RW: Functional analysis of 44 mutant androgen receptors from
human prostate cancer. Cancer Res. 62:1496–1502. 2002.PubMed/NCBI
|
|
24
|
Taplin ME, Rajeshkumar B, Halabi S, et al:
Androgen receptor mutations in androgen-independent prostate
cancer: Cancer and Leukemia Group B Study 9663. J Clin Oncol.
21:2673–2678. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hara T, Nakamura K, Araki H, Kusaka M and
Yamaoka M: Enhanced androgen receptor signaling correlates with the
androgen-refractory growth in a newly established MDA PCa 2b-hr
human prostate cancer cell subline. Cancer Res. 63:5622–5628.
2003.PubMed/NCBI
|
|
26
|
Yoshida T, Kinoshita H, Segawa T, et al:
Antiandrogen bicalutamide promotes tumor growth in a novel
androgen-dependent prostate cancer xenograft model derived from a
bicalutamide-treated patient. Cancer Res. 65:9611–9616. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tachibana K, Imaoka I, Yoshino H, et al:
Discovery of 7alpha-substituted dihydrotestosterones as androgen
receptor pure antagonists and their structure-activity
relationships. Bioorg Med Chem. 15:174–185. 2007. View Article : Google Scholar
|
|
28
|
Kawata H, Ishikura N, Watanabe M,
Nishimoto A, Tsunenari T and Aoki Y: Prolonged treatment with
bicalutamide induces androgen receptor overexpression and androgen
hypersensitivity. Prostate. 70:745–754. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ishikura N, Kawata H, Nishimoto A,
Nakamura R, Ishii N and Aoki Y: Establishment and characterization
of an androgen receptor-dependent, androgen-independent human
prostate cancer cell line, LNCaP-CS10. Prostate. 70:457–466.
2010.
|
|
30
|
Hara T, Miyazaki J, Araki H, et al: Novel
mutations of androgen receptor: a possible mechanism of
bicalutamide withdrawal syndrome. Cancer Res. 63:149–153.
2003.PubMed/NCBI
|
|
31
|
Osborne CK, Coronado-Heinsohn EB,
Hilsenbeck SG, et al: Comparison of the effects of a pure steroidal
antiestrogen with those of tamoxifen in a model of human breast
cancer. J Natl Cancer Inst. 87:746–750. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hoffmann J, Bohlmann R, Heinrich N, et al:
Characterization of new estrogen receptor destabilizing compounds:
effects on estrogen-sensitive and tamoxifen-resistant breast
cancer. J Natl Cancer Inst. 96:210–218. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Osborne CK, Pippen J, Jones SE, et al:
Double-blind, randomized trial comparing the efficacy and
tolerability of fulvestrant versus anastrozole in postmenopausal
women with advanced breast cancer progressing on prior endocrine
therapy: results of a North American trial. J Clin Oncol.
20:3386–3395. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Howell A, Robertson JF, Quaresma Albano J,
et al: Fulvestrant, formerly ICI 182,780, is as effective as
anastrozole in postmenopausal women with advanced breast cancer
progressing after prior endocrine treatment. J Clin Oncol.
20:3396–3403. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tran C, Ouk S, Clegg NJ, et al:
Development of a second-generation antiandrogen for treatment of
advanced prostate cancer. Science. 324:787–790. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yoshino H, Sato H, Shiraishi T, et al:
Design and synthesis of an androgen receptor pure antagonist
(CH5137291) for the treatment of castration-resistant prostate
cancer. Bioorg Med Chem. 18:8150–8157. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kawata H, Arai S, Nakagawa T, et al:
Biological properties of androgen receptor pure antagonist for
treatment of castration-resistant prostate cancer: optimization
from lead compound to CH5137291. Prostate. 71:1344–1356.
2011.PubMed/NCBI
|
|
38
|
Corey E, Quinn JE, Buhler KR, et al: LuCaP
35: a new model of prostate cancer progression to androgen
independence. Prostate. 55:239–246. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tachibana K, Imaoka I, Shiraishi T, et al:
Discovery of an orally-active nonsteroidal androgen receptor pure
antagonist and the structure-activity relationships of its
derivatives. Chem Pharm Bull. 56:1555–1561. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tachibana K, Imaoka I, Yoshino H, et al:
Discovery and structure-activity relationships of new steroidal
compounds bearing a carboxy-terminal side chain as androgen
receptor pure antagonists. Bioorg Med Chem Lett. 17:5573–5576.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bohl CE, Gao W, Miller DD, Bell CE and
Dalton JT: Structural basis for antagonism and resistance of
bicalutamide in prostate cancer. Proc Natl Acad Sci USA.
102:6201–6206. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
O’Donnell A, Judson I, Dowsett M, et al:
Hormonal impact of the 17alpha-hydroxylase/C(17,20)-lyase inhibitor
abiraterone acetate (CB7630) in patients with prostate cancer. Br J
Cancer. 90:2317–2325. 2004.
|
|
43
|
Fizazi K, Scher HI, Molina A, et al:
Abiraterone acetate for treatment of metastatic
castration-resistant prostate cancer: final overall survival
analysis of the COU-AA-301 randomised, double-blind,
placebo-controlled phase 3 study. Lancet Oncol. 13:983–992. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Beer TM, Armstrong AJ, Rathkopf DE, et al:
Enzalutamide in metastatic prostate cancer before chemotherapy. N
Engl J Med. 371:424–433. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Longui CA: Glucocorticoid therapy:
minimizing side effects. J Pediatr (Rio J). 83:S163–S177. 2007.
View Article : Google Scholar
|
|
46
|
Joffe HV and Adler GK: Effect of
aldosterone and mineralocorticoid receptor blockade on vascular
inflammation. Heart Fail Rev. 10:31–37. 2005. View Article : Google Scholar : PubMed/NCBI
|