Introduction
Arsenic compounds have been used in traditional
medicine as anti-tumor and anti-inflammatory agents over 2,400
years, however, their clinical use is strikingly decreased because
of the carcinogenic and toxic effects (1). Since successful use of arsenic
trioxide (ATO; As2O3) in patients with acute
promyelocytic leukemia (APL) in China in the 1970s, ATO was
approved for clinical use for patients with relapsed APL (2,3).
Also, in vitro studies have shown that ATO exerts
anti-proliferative effect on solid tumor cells including prostate,
ovarian, stomach, breast, lung and cervical cancer (4–6). The
anti-tumoral effect of ATO is accompanied by tubulin
polymerization, induction of differentiation and apoptosis and
inhibition of angiogenesis (4,6–8).
Arsenic compounds have also been reported to target telomere which
is a region of repetitive nucleotide sequences at each end of a
chromatid and protects the end of the chromosome from deterioration
or from fusion with neighbouring chromosomes (9). Arsenic-induced inhibition of human
telomerase reverse transcriptase (hTERT) is associated with
transcriptional factors, c-Myc and Sp1 (10). However, the anti-tumor effect of
ATO in solid tumors and other hematologic disorders has not been
fully elucidated (11).
Especially, a drawback of ATO is to be administered intravenously,
and not effective in the treatment of acute myeloid leukemia (AML)
except one type called APL.
KML001, sodium metaarsenite, is a water-soluble,
therefore, orally bioavailable, trivalent arsenical compound
(12). KML001 has shown potent
anti-tumoral effect in vitro as well as in human solid tumor
cell line xenografts in vivo, and has entered phase 1/2
clinical trials for the treatment of human prostate cancer
(12,13). It has been suggested that one of
the mechanisms of action of KML001 might be to target the telomeres
of chromosomes in cancer cells (12). Although KML001 has shown cytotoxic
effect on a variety of cancer cells, anti-leukemic effect of KML001
has not been well studied yet.
In the present study, we investigated the effect of
KML001 on acute myeloid leukemia (AML) cell lines. KML001 inhibited
the proliferation of all AML cell lines including Ara-C (cytosine
arabinoside)-resistant HL-60 (HL-60R) cells by cell cycle arrest
and induction of apoptosis. In addition, KML001 shortened the
telomere length without affecting telomerase activity. All
together, KML001 seems to be a candidate agent for clinical
investigation in the treatment of refractory acute myeloid
leukemia.
Materials and methods
Cells and cell culture
Human myeloid leukemia cell lines examined in the
present study were HL-60, BV173, HEL, K562, KCL22, KG1, KU812,
MegO1, ML1, NB4 and U937, and they were kindly presented by Dr H.P.
Koeffler (Cedars-Sinai Medical Center/University of California, Los
Angeles, CA, USA). The cells were cultured in tissue flasks in
RPMI-1640 medium (Gibco-BRL, Gaithersburg, MD, USA) supplemented
with 10% (vol/vol) fetal bovine serum (FBS; Gibco-BRL), 100 U/ml
penicillin and 100 μg/ml streptomycin (Sigma Chemical, St. Louis,
MO, USA) and maintained in a humidified atmosphere, 5%
CO2 at 37°C. The culture medium was changed every 3–4
days. The Ara-C-resistant cell line HL-60R was established by
stepwise increase of concentrations of cytosine arabidoside in
medium. Leukemic blasts were harvested from bone marrow aspirates
of 4 AML patients (2 AML M1 and 2 AML M2), who were initially
diagnosed when the blasts counts were high (>85%) at
presentation and gave written informed consent. Mononuclear cells
(MNCs) from bone marrow were collected by separation on
Ficoll-Paque (Amersham Biosciences AB, Uppsala, Sweden) gradients
at a density of 1.077, washed twice in phosphate-buffered saline
(PBS), and suspended in Iscove’s modified Dulbecco’s medium (IMDM;
Gibco-BRL) containing 10% heat-inactivated FBS (Gibco-BRL).
Reagents
KML001 (sodium metaarsenite) was obtained from
Komipharm International, 10−3 M drug stocks were
prepared in distilled water and aliquots were stored at 4°C. The
stock solution was stable for >1 year. Working concentrations
were freshly prepared daily by diluting the stock with RPMI-1640.
As2O3 was purchased from Sigma-Aldrich,
5×10−2 M stock solution was prepared in 5 M NaOH, and
aliquots were stored at 4°C. SP600125, a specific JNK inhibitor,
and SB202190, specific inhibitor of p38 protein were purchased from
Calbiochem (San Diego, CA, USA) (14). These inhibitors were dissolved in
dimethyl sulfoxide (DMSO) at 1 mM as stock solutions and these
stock solutions were stored at −20°C.
Growth inhibition assay
Cellular growth inhibition effect of KML001 was
determined by measuring the MTT [3-
[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide dye
absorbance of living cells as previously described (15). Briefly, cells (1×104
cells/well) were seeded in 96-well microtiter plates (Nunc,
Roskilde, Denmark) and incubated at 37°C for 72 h. MTT solution (50
μl) (2 μg/ml in PBS) from Sigma was added to each well, and the
plates were incubated for an additional 4 h at 37°C. The MTT
solution in the medium was aspirated. To achieve solubilization of
the formazan crystal formed in viable cells, 200 μl of DMSO was
added to each well. The plates were shaken for 30 min at room
temperature, and absorbance was read immediately at a wavelength of
540 nm on a scanning multiwell spectrophotometer (Titert Multiscan
MC; Flow Laboratory, CA).
Cell cycle analysis
Cells were fixed with methanol for 1 h and stained
with 50 μg/ml of propidium iodide (PI) containing 50 μg/ml of
RNaseA. The DNA contents of the cells (10,000 cells/experimental
group) were analyzed using a FACStar flow cytometer
(Becton-Dickinson, San Jose, CA, USA) equipped with a ModFit LT
program (Lysis II, CellFit). The percentage of cell population in
each cell cycle phase (G1, S or G2/M) was calculated from DNA
content histograms, excluding the population in the sub-G1
phase.
Evaluation of apoptosis
Apoptosis was determined by staining cells with
Annexin V-FITC (BD Biosciences, San Diego, CA, USA) and PI
(16). To quantify the apoptosis
of cells, the cells were washed twice with cold PBS and were
resuspended in binding buffer (10 mM HEPES/NaOH, pH 7.4, 140 mM
NaCl, 2.5 mM CaCl2) at a density of 1×106
cells/ml. This cell solution (100 μl) (total 1×105
cells) was transferred to a 5-ml culture tube with 5 μl of Annexin
V-FITC and 10 μl of PI (20 μg/ml) and was analyzed with the FACStar
flow cytometer.
Immunoblot analysis
Cells were suspended in lysis buffer containing 50
mM Tris (pH 7.5), 1% NP-40, 2 mM EDTA, 10 mM NaCl, 20 μg/ml
aprotinin, 20 μg/ml leupeptin and 1 mM phenylmethylsulphonyl
fluoride and were placed on ice for 20 min. Samples containing
20–100 μg of total protein were resolved in SDS-polyacrylamide
denaturing gel, transferred to nitrocellulose membranes, and probed
with antibodies. The blots were developed using the ECL kit (Intron
Biotechnology, Gyeonggi-do, Korea).
Immunoprecipitation and kinase assay
Cells were suspended in an extraction buffer [50 mM
Tris-Cl (pH 7.5), 250 mM NaCl, 0.1% NP-40, 5 mM EDTA, 50 mM NaF,
0.1 mM NaVO4, 100 mM phenylmethylsulfonyl fluoride, 0.2
mM leupeptin, 10 μg/ml aprotinin, 0.1 mM pepstatin A) and incubated
on ice for 15 min. After centrifugation at 13,000 rpm for 20 min,
the supernatant was collected and protein concentration was
determined using a Bio-Rad assay kit. Two micrograms of each
antibody (CDK2, CDK4 and CDK6) were added to 200 μg of each cell
extract in 500 μl of extraction buffer and incubated for 4 h at 4°C
with continuous agitation. To collect immune complexes, 30 μl of
protein A/G-agarose was added to the mixture, which was then
incubated for 2 h. Immune complexes were centrifuged at 1,200 rpm
for 2 min and the precipitates were washed three times with
extraction buffer and twice with kinase reaction buffer [50 mM
Tris-Cl (pH 7.5), 10 mM MgCl2 and 1 mM DTT]. CDK 2
kinase assays on histone H1 was performed by mixing the respective
immune complexes with 5 μg of histone H1 and 1 μCi of
[γ-32P]-ATP in 35 μl of kinase reaction buffer. CDK 4
and CDK 6 kinase assays on Rb-c residue were performed in the same
way. Kinase reactions were performed at 37°C for 30 min and were
terminated with 2X SDS-PAGE loading buffer. The reaction mixtures
were resolved by SDS-PAGE. The extent of phosphorylation was
determined by autoradiography.
Mean telomere restriction fragment
length
For the measurement of telomere length, Roche
Diagnostics TeloTAGG telomere restriction fragment (TRF) length kit
was used according to manufacturer’s instructions (Roche
Diagnostics GmbH, Mannheim, Germany).
Quantitative hTERT real-time PCR
Total RNA was isolated with TRI reagent (Molecular
Research Center, Inc., Cincinnati, OH, USA) and cDNA was
synthesized from 1 μg of total RNA using ImProm-II Reverse
Transcriptase (Promega Corp., Madison, WI, USA) and random
hexamers. Quantitative PCR was performed using SYBR-Green I as a
double-strand DNA-specific binding dye on iCycler IQ detection
system (Bio-Rad Laboratories). Thermocycling was performed in a
final volume of 20 μl containing; 4 μl cDNA sample, 10 pM of each
primer, 0.125 mM dNTP mixture, 0.25 mg/ml BSA, 0.05% Tween-20, 1X
rTaq reaction buffer containing 1.5 mM MgCl2 (Takara,
Shiga, Japan), 1 unit rTaq DNA polymerase (Takara), and 1X
SYBR-Green I (Molecular Probes, Sunnyvale, CA, USA). After an
initial denaturation at 95°C for 10 min, 35 cycles of 94°C for 30
sec, 53°C for 30 sec, and 72°C for 30 sec were carried out.
All cDNA samples were synthesized in parallel and
PCR reactions were run in triplicate. The mRNA levels were derived
from standard curves and are expressed as relative changes after
normalization vs. β-actin mRNA levels.
Chromatin immunoprecipitation (ChIP)
HL-60 and HL-60R cells were treated with KML001 at a
concentration of 10−7 M for 24, 48 and 72 h. Chromatin
immunoprecipitation (ChIP) assays for studying the specific
association of γ-H2AX with telomeric repeat
sequences were performed as described by d’Adda di Fagagna et
al (17). Briefly, cells were
fixed in 1% formaldehyde in PBS for 10 min at 37°C and lysed with
cell lysis buffer [1% SDS, 50 mM/l Tris-HCl (pH 8.0) and 10 mM/l
EDTA] at a density of 107 cells/ml. Lysates were
sonicated to shear the DNA and centrifuged for 10 min at 4°C.
Lysates (200 μl) were diluted with a buffer [1.8 ml of 0.01% SDS,
1.1% Triton X-100, 1.2 mmol/l EDTA, 16.7 mmol/l Tris-HCl (pH 8.0)
and 150 mmol/l NaCl] and precleaned with a protein G plus/protein A
agarose suspension (Calbiochem, San Diego, CA, USA)/salmon sperm
DNA (Invitrogen, Carlsbad, CA, USA). Control IgG (mouse; Santa Cruz
Biotechnology, Santa Cruz, CA, USA), mouse monoclonal
anti-phospho-H2AX (EMD Millipore, Darmstadt,
Germany; clone JBW301), or anti-telomere repeat biding factor 1
antibodies (clone C-19; Santa Cruz Biotechnology) were added to the
lysates and incubated overnight at 4°C. Immunoprecipitated pellets
were washed with 0.1% SDS, 1% Triton X-100, 2 mmol/l EDTA (pH 8.0)
and 20 mmol/l Tris-HCl (pH 8.0) containing 150 mmol/l NaCl in the
first wash and 500 mmol/l NaCl in the second wash. Further washes
were with 0.25 mol/l LiCl, 1% NP-40, 1% sodium deoxycholate, 1
mmol/l EDTA (pH 8.0) and 10 mmol/l Tris-HCl (pH 8.0) and with 10
mmol/l Tris-HCl (pH 8.0) and 1 mmol/l EDTA. Chromatin was eluted
from the beads with 500 μl of 1% SDS and 0.1 mol/l
NaHCO3. After addition of 20 μl of 5 mol/l NaCl,
cross-links were reversed for 4 h at 65°C. Samples were then
treated with 20 μl of 1 mol/l Tris-HCl (pH 6.5), 10 μl of 0.5 mol/l
EDTA and 20 μg Proteinase K (Invitrogen) and incubated at 45°C for
1 h. Phenol-chloroform extractions were done and the DNA was
precipitated overnight at −80°C. The precipitate was dissolved in
20 μl water just prior to setting up qPCR runs (18).
Statistical analysis
The data represent mean values ± SEM (error bars) of
experiments repeated at least 3 times. Statistical significance was
determined using the Student’s t-tests. P-values <0.05 were
considered statistically significant.
Results
Antileukemic effect of KML001 on human
leukemic cells
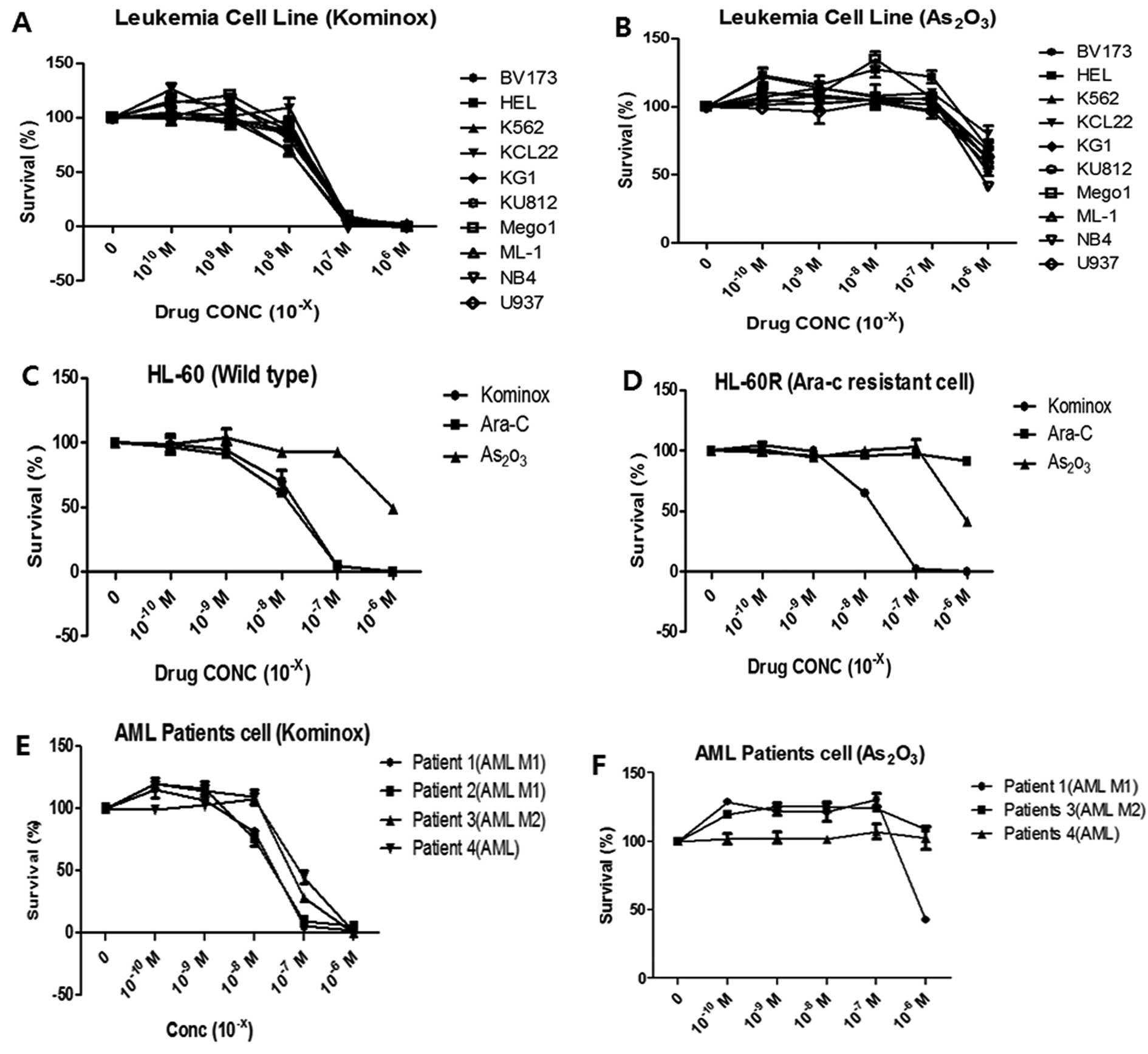
KML001 inhibited the cellular proliferation in all
AML cell lines in a dose-dependent manner with IC50 of
5×10−8 M, while arsenic trioxide
(As2O3) did not (Fig. 1A and B). KML001 effectively
inhibited cellular proliferation of HL-60 cells (IC50;
5×10−8 M) as well as HL-60R cells (IC50;
1×10−8 M), and its anti-leukemic effect was almost the
same as Ara-C (IC50; 5×10−8 M). Ara-C did not
inhibit cellular proliferation in HL-60R cells as expected
(Fig. 1C and D). In addition,
growth of primary leukemic blasts from AML patients was inhibited
in a dose-dependent manner by KML001 with IC50 of
5×10−7 M, however, arsenic trioxide did not inhibit the
primary leukemic blast cells (Fig. 1E
and F). These data indicated that KML001 might be a potent
anti-leukemic agent, and have biological effects different from
arsenic trioxide on AML cells including Ara-C-resistant HL-60
cells.
Cell cycle analysis in HL-60 and HL-60R
cells
The effect of KML001 on the cell cycle was
determined in HL-60 and HL-60R cells by FACS analysis. Three
independently performed DNA flow cytometric analyses indicated that
KML001 induced a G1 arrest in HL-60 and HL-60R cells at a
concentration of 1×10−7 M during 72 h of exposure;
population of HL-60 cells in G1 phase was 46.21% at 0 h and 64.56%
at 72 h (Table I). Population of
HL-60R cells was 28.82% at 0 h and 59.67% at 72 h (Table II). In contrast, no cell cycle
arrest was observed in HL-60 and HL-60R cells with
As2O3 treatment (data not shown).
 | Table ICell cycle analysis in KML001-treated
HL-60. |
Table I
Cell cycle analysis in KML001-treated
HL-60.
| 0 h | 12 h | 18 h | 24 h | 48 h | 72 h |
|---|
| G0–G1 | 46.21 | 48.02 | 41.61 | 42.33 | N.D | 64.56 |
| S | 40.48 | 38.76 | 29.05 | 34.52 | N.D | 23.77 |
| G2/M | 12.31 | 13.22 | 29.33 | 21.15 | N.D | 11.67 |
| SubG1 | 2.10 | 11.22 | 70.79 | 51.30 | 74.58 | 48.04 |
| Debris | 2.04 | 11.33 | 90.77 | 66.07 | N.D | 50.36 |
| Table IICell cycle analysis in KML001-treated
HL-60R. |
Table II
Cell cycle analysis in KML001-treated
HL-60R.
| 0 h | 12 h | 18 h | 24 h | 48 h | 72 h |
|---|
| G0–G1 | 28.28 | 36.34 | 50.90 | 35.13 | N.D | 59.67 |
| S | 56.11 | 46.13 | 38.48 | 18.96 | N.D | 36.95 |
| G2/M | 15.07 | 17.53 | 10.62 | 15.90 | N.D | 3.37 |
| SubG1 | 3.99 | 21.81 | 11.13 | 3.08 | 63.71 | 57.86 |
| Debris | 6.27 | 21.59 | 11.89 | 4.37 | N.D | 66.34 |
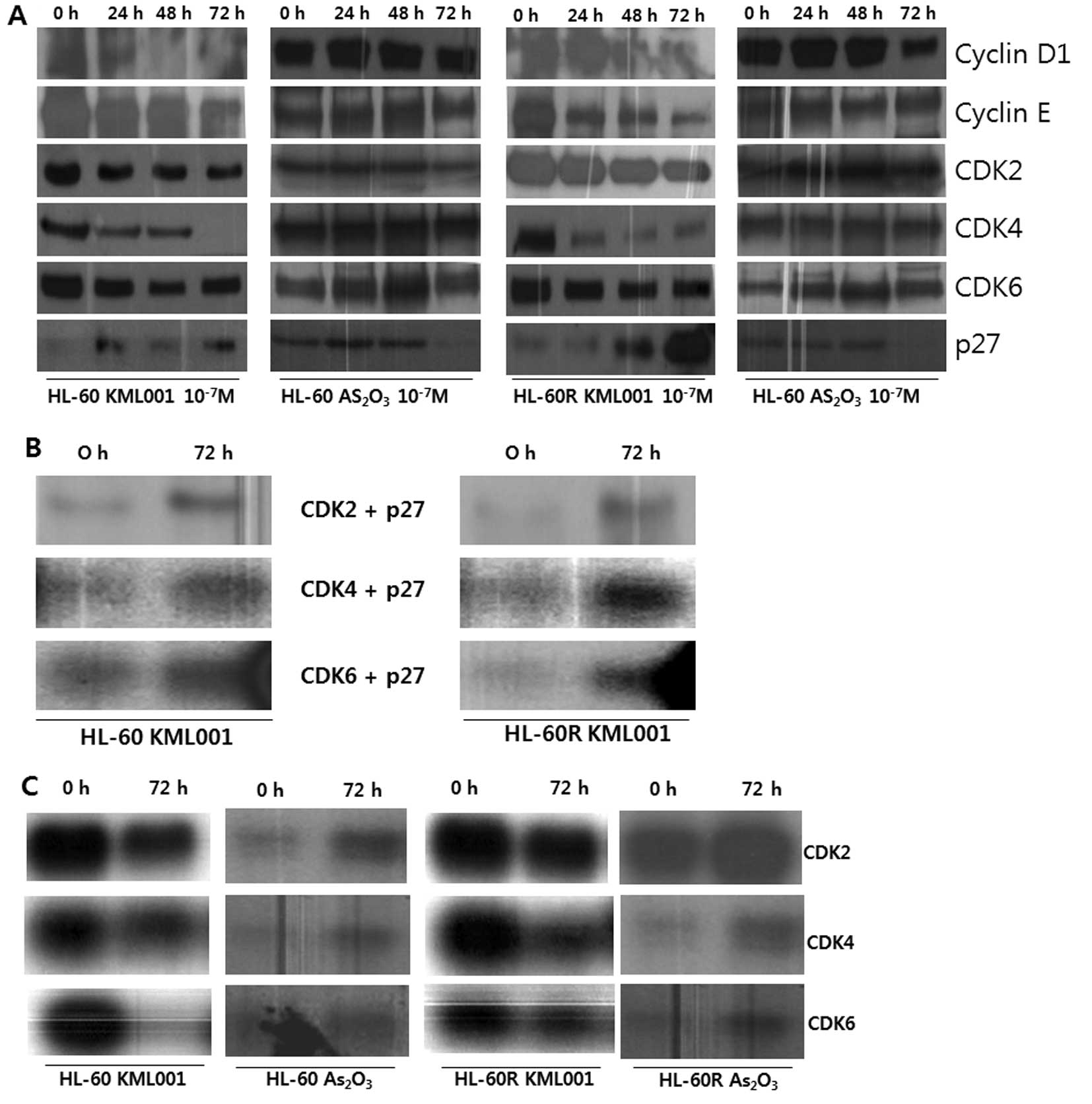
Cell cycle arrest was associated with downregulation
of CDKs of CDK4, CDK6, as well as cyclins of cyclin D1 and cyclin
E. In addition, the expression of p21 and p27 proteins, and
cyclin-dependent kinase inhibitors (CDKIs), was increased in HL-60
and HL-60R cells. In contrast, no significant change of CDKs and
cyclins was observed and the expression of CDKIs was decreased in
arsenic trioxide-treated HL-60 and HL-60R cells (Fig. 2A).
Association of p27 with cell
cycle-regulatory proteins in KML001-treated HL-60 and HL-60R
cells
Since western blot analysis showed that KML001
induced a marked accumulation of p27 protein, we investigated
whether the KML001-induced p27 protein existed in complexes with
CDKs active in the G1 phase of the cell cycle in HL-60 and HL-60R
cells. As shown in Fig. 2B, the
complexes immunoprecipitated with anti-CDK2, anti-CDK4 and
anti-CDK6 antibodies exhibited higher levels of immunodetectable
p27 protein in HL-60 and HL-60R cells at 72 h.
Effect of KML001 on CDK-associated kinase
activity
To determine whether the increased CDKIs and the
changed cell cycle-regulatory proteins result in the inhibition of
CDK activity in KML001-treated HL-60 and HL-60R cells, in
vitro CDK activity assay on Rb-c substrate and histone H1 was
performed using immunoprecipitation with anti-CDK2, anti-CDK4 and
anti-CDK6 antibodies. HL-60 and HL-60R cells treated with KML001 at
a concentration of 1×10−7 M showed a decrease in
CDK2-associated kinase activity on histone H1, and CDK4- and
CDK6-associated kinase activity on Rb-c substrate at 72 h (Fig. 2C).
Taken together, these results suggest that p27
protein could play a critical role in a G1 arrest via its increased
binding to CDK2, CDK4 and CDK6 proteins and subsequent inhibition
of CDK2-, CDK4-, CDK6-associated kinase activities in
KML001-treated HL-60 and HL-60R cells.
In contrast, arsenic trioxide did not reduce the
activity of CDK2-, CDK4- and CDK6-associated kinase in HL-60 and
HL-60R cells (Fig. 2C).
Induction of apoptosis by KML001 in HL-60
and HL-60R cells
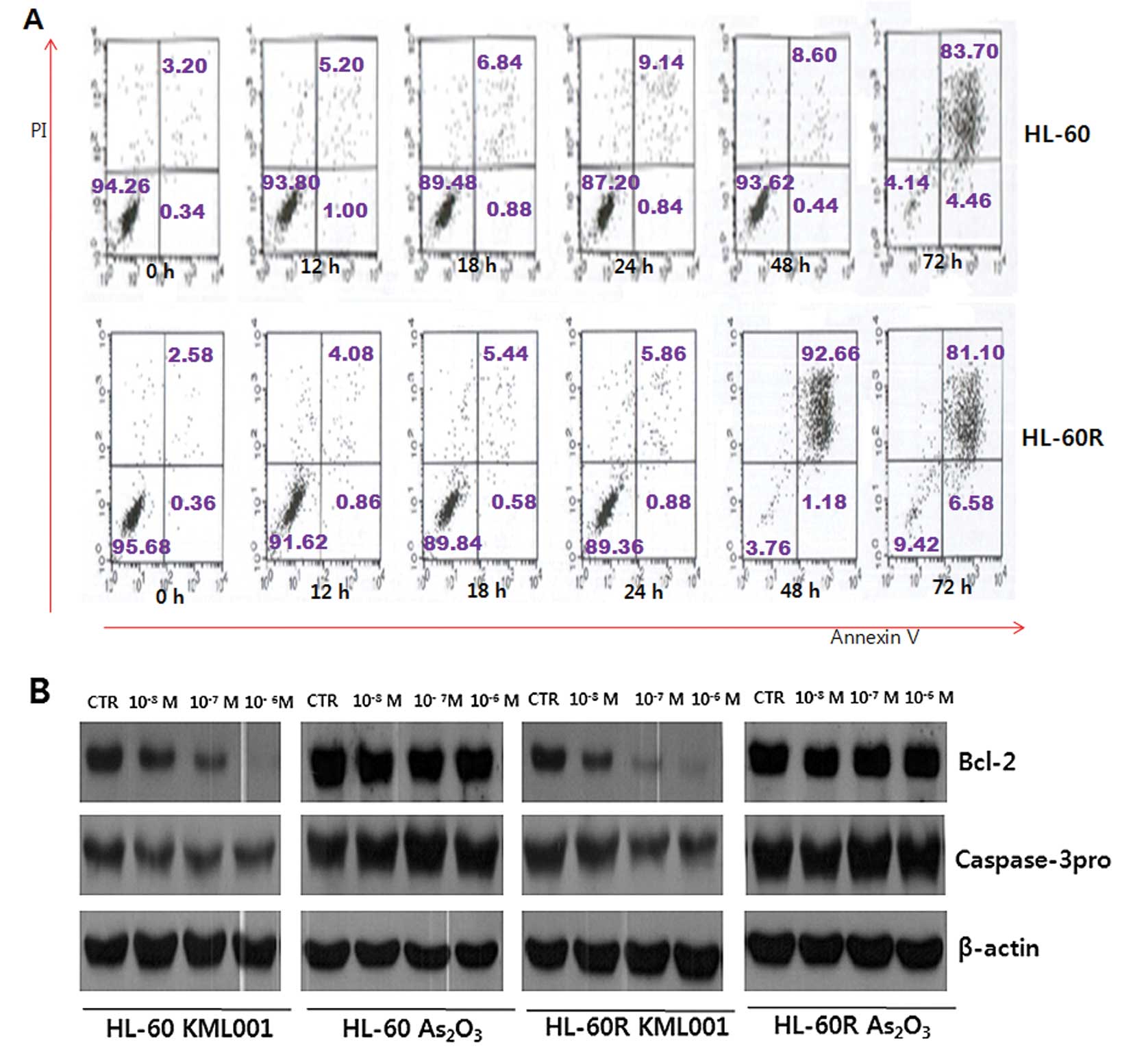
In order to determine whether KML001 treatment could
induce apoptosis in HL-60 and HL-60R cells, in vitro
apoptosis detection assay was performed. Cells were stained with
Annexin V-FITC and propidium iodide (PI) and analyzed by FACS. The
proportion of Annexin V-FITC-stained cells was dramatically
increased in a time-dependent manner following treatment of HL-60
and HL-60R cells with KML001 at a concentration of
1×10−7 M (Fig. 3A). In
addition, Bcl-2 protein was downregulated in a dose-dependent
manner in HL-60 and HL-60R cells (Fig.
3B). Next, it was evaluated whether caspase might be activated
during the induction of apoptosis by KML001, since cell death can
be completed through caspase activation after external stimuli. The
expression of effector caspase-3 was downregulated in a
dose-dependent manner (Fig. 3B).
Collectively, these results indicate that induction of apoptosis
could be another mechanism of the antiproliferative effect of
KML001 besides cell cycle arrest in HL-60 and HL-60R cells.
Effect of KML001 on cell signaling in
HL-60 and HL-60R cells
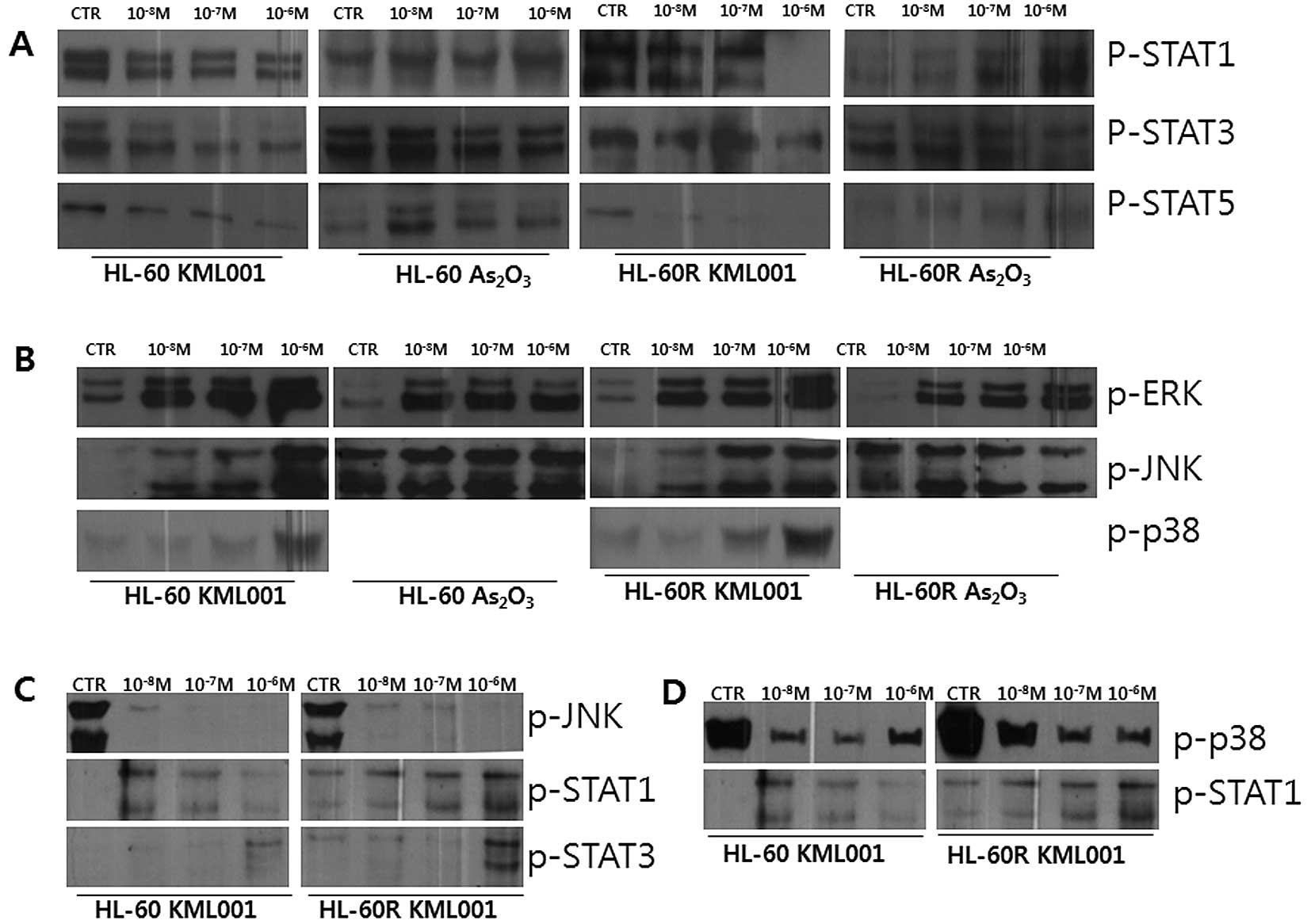
STATs are cytoplasmic transcription factors and key
mediators of cytokine and growth factor signaling pathways. Many
malignant cells have shown to consistently express STAT molecules.
In order to investigate the role of STAT molecules in anti-leukemic
effect of KML001, HL-60 and HL-60R cells were treated with KML001
for 2 h at indicated concentrations.
The expression of p-STAT1, p-STAT3 and p-STAT5 was
downregulated in HL-60 and HL-60R cells following treatment with
KML001, while no significant expressional change was noted in
As2O3-treated HL-60 and HL-60R cells
(Fig. 4A). Regarding the MAPK
signaling, the phosphorylated forms of p42/p44 ERK, JNK and p38
were increased following treatment of HL-60 and HL60-R cells with
KML001 for 2 h in a dose-dependent manner, while no dramatic
expressional change was shown in
As2O3-treated cells, suggesting that the
KML001-induced activation of MAPK pathways might be related to the
growth inhibition of HL-60 and HL-60R cells (Fig. 4B). Furthermore, pre-treatment of
cells with 10 μM of SP600125, a specific JNK inhibitor, for 12 h
blocked KML001-induced phosphorylation of JNK, and subsequently
abrogated the effect of KML001 on STAT1 and STAT3 (Fig. 4C).
Additionally, pre-treatment of HL-60 and HL-60R
cells for 12 h with 20 μM of SB202190, specific inhibitor of p38
protein, blocked KML001-induced p-p38, and subsequently abrogated
the effect of KML001 on STAT1 (Fig.
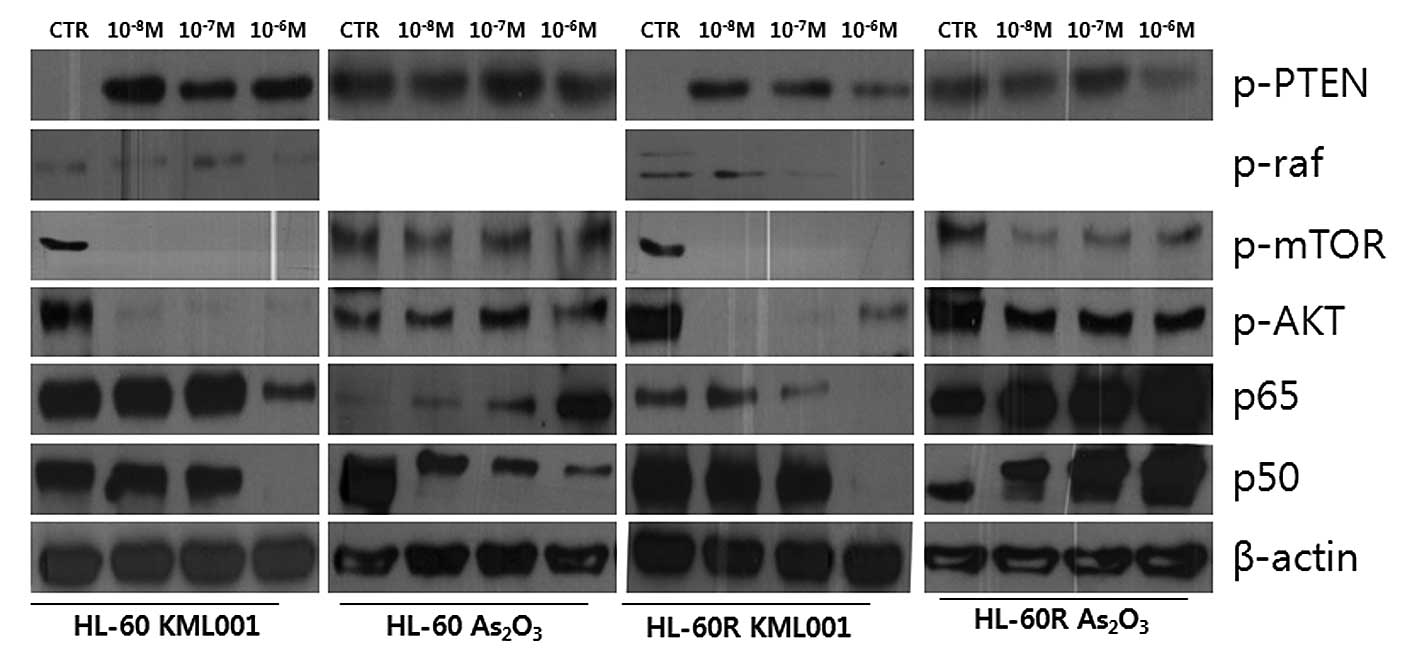
4D). The examination of the effect of KML001 on PI3K/Akt
signaling showed downregulation of p-mTOR, p-Akt and p-Raf
proteins, while p-PTEN was upregulated in HL-60 and HL-60R cells
(Fig. 5). In contrast, no
alteration in expressions of the proteins was observed in
As2O3-treated HL-60 and HL-60R cells.
Additional study showed that the expression of P65
and P50 subunits of NF-κB was decreased in KML001-treated HL-60 and
HL-60R cells (Fig. 5). These
results suggest that the anti-proliferative effect of KML001 might
be mediated via various cell signaling including JAK/STAT, MAPK/ERK
and PI3K/Akt pathways in HL-60 and HL-60R cells.
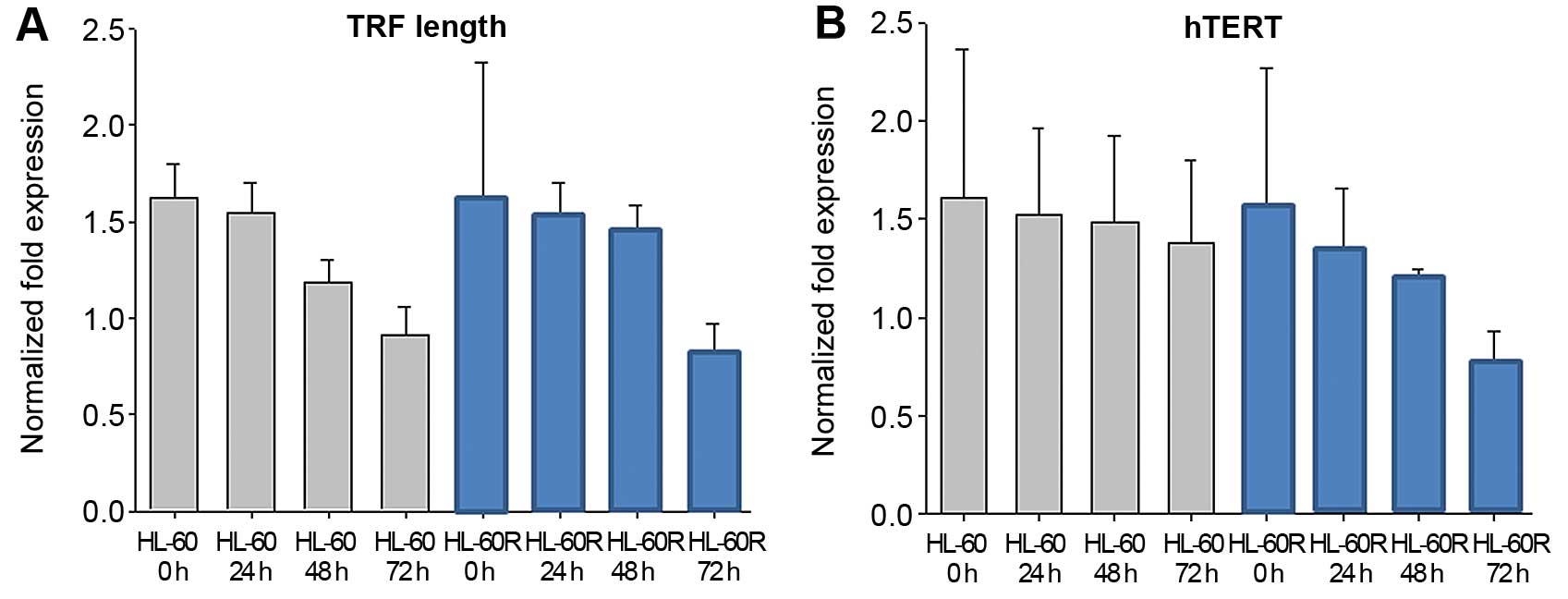
The effect of telomere length and hTERT
in HL-60 and HL-60R
The maintenance of telomere lengths is critical for
chromosomal integrity in cells. It has been shown that KML001
targets the telomere (12). In
this experiment, KML001 (1×10−7 M) induced TRF length
shortening in a time-dependent manner in HL-60 and HL-60R cells
(Fig. 6A). In order to examine
whether KML001 has an effect on the catalytic subunit of
telomerase, hTERT, real-time PCR analysis with RNA extracted from
KML001-treated HL-60 and HL-60R cells was done, showing that hTERT
mRNA expression was inhibited in KML001-treated HL-60 and HL-60R
cells in a time-dependent manner (Fig.
6B).
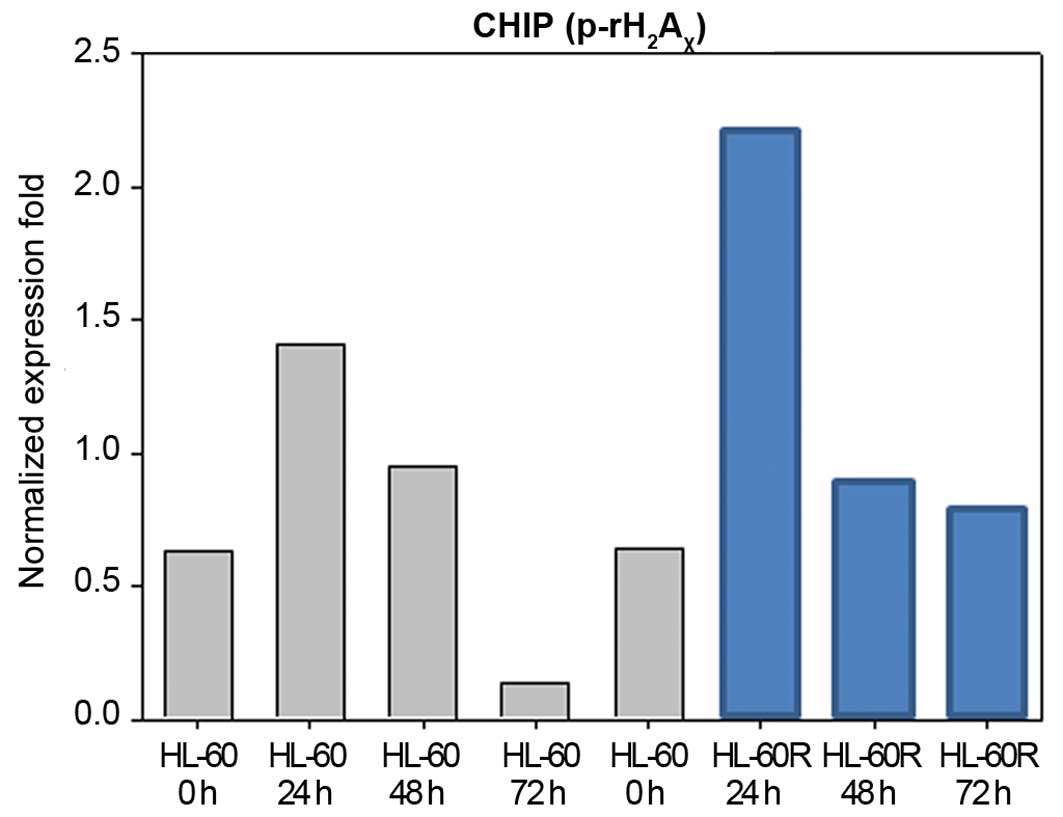
Induction of telomere-associated DNA
damage by KML001
Uncapping the telomere signal can induce cellular
growth arrest/apoptosis via telomere length shortening. It
accelerates the double-strand break-mediated DNA damage signaling
pathways, including phosphorylation of γ-H2AX
at Ser139, which represents an early event of DNA damage signaling
(12,19,20).
Therefore, p-γ-H2AX has been known to be a
marker for DNA damage checkpoint response in telomere-initiated
senescence (20,21). Accordingly, we examined whether
KML001 treatment would induce the γ-H2AX
phosphorylation in HL-60 and HL-60R cells. Our ChIP assay
demonstrated that p-γ-H2AX was upregulated in
HL-60 and HL-60R cells at 24 h after treatment with
1×10−7 M of KML001, and their phosphorylated form of
γ-H2AX was maintained up to 48 h (Fig. 7).
Discussion
In the present study, we demonstrated for the first
time that KML001 potently inhibited the proliferation of HL-60 and
HL-60R cells by inducing cell cycle arrest and triggering
apoptosis. KML001 induced a dose-dependent inhibition of cellular
proliferation of all AML cell lines including HL-60R cells as well
as primary leukemic blasts from AML patients. The cell cycle
analysis revealed that KML001 was able to prominently induce a G1
growth arrest of HL-60 and HL-60R cells at a concentration of
10−7 M during 72-h exposure, in contrast, cell cycle
arrest was not observed in As2O3-treated
HL-60 and HL-60R cells. Among CDKs that regulate the cell cycle,
CDK4 and CDK6 are activated in association with cyclin D during G1
progression, whereas CDK2 is activated primarily in association
with cyclin E in the late G1 phase (22,23).
We found that the expression of CDK2, CDK4, and CDK6 was decreased
in a time-dependent manner following treatment of KML001, and the
expression of cyclin D1 and cyclin E was also decreased in HL-60
and HL-60R cells. These results suggested that KML001 affected the
cell cycle arrest in HL-60 and HL-60R cells through changes in the
cell cycle-regulatory proteins. It is known that a family of
cyclin-dependent kinase inhibitors (CDKIs) plays a major role in
the negative regulation of CDKs and is involved in the arrest of G1
cell cycle resulting in antiproliferative signals. In the present
study, KML001 treatment showed a marked upregulation of CDKI, p27
protein, in HL-60 and HL-60R cells. In addition, the accumulation
of p27 protein in association with G1 arrest was detected largely
in complexes with CDK2, CDK4 and CDK6. These increased formations
of p27-CDK2, p27-CDK4 and p27-CDK6 complexes support the notion
that KML001 may decrease CDK4-, CDK6- and CDK2-associated kinase
activities in HL-60 and HL-60R cells. In fact, our kinase assay
showed that KML001 reduced CDK2-, CDK4- and CDK6-associated kinase
activities in HL-60 and HL-60R cells, in contrast,
As2O3 did not. Taken together, the G1 arrest
by KML001 might be mediated by downregulation of CDK2-, CDK4- and
CDK6 kinase activities in association with induction of p27 in
HL-60 and HL-60R cells.
Our data demonstrated that KML001 markedly induced
the apoptosis in HL-60 and HL-60R cells, showing the downregulation
of Bcl-2 and effector procaspase-3 in a dose-dependent manner in
HL-60 and HL-60R cells, in contrast, As2O3
did not. We confirmed apoptosis using Annexin/PI double staining in
which apoptotic cells were increased in a time-dependent manner in
HL-60 and HL-60R cells.
Signal transduction pathways regulate cellular
differentiation, division and cell death. Therefore, we
investigated the effect of KML001 on the signal transduction
pathways. STATs are latent cytoplasmic transcription factors
activated by JAK which are constitutively associated with cytokines
and growth factor receptors, and frequently activated in many types
of human malignancies including leukemia (24,25).
Activated/phosphorylated STATs by external stimuli are dimerized
and translocated to the nucleus where they bind to response
elements on DNA, and activate the transcription of target genes,
resulting in cellular survival, proliferation, differentiation and
apoptosis (26–28). The present study showed that KML001
decreased p-STAT1, p-STAT3 and p-STAT5 in HL-60 and HL-60R cells in
a dose-dependent manner. Thus, JAK/STAT pathways might be involved
in KML001-induced apoptosis. Mitogen-activated protein kinase
(MAPK) pathways contribute to control and determine the cell fate
in response to many environmental stimuli. It has been suggested
that ERK activation exerts a cytoprotective effect, whereas JNKs
and p38-MAPKs seems to be involved in apoptosis (29,30).
However, the involvement of subfamilies of MAPK regarding the
apoptosis depends on the cell type and stimulating agents (30). In the present study, KML001
increased levels of p-JNK as well as p-p38 protein. Furthermore,
SP600125, a specific JNK inhibitor, blocked KML001-induced
phosphorylation of JNK, and subsequently abrogated the effect of
KML001 on STAT1 and STAT3 in HL-60 and HL-60R. Also, pre-treatment
of HL-60 and HL-60R cells with SB202190, specific inhibitor of p38
protein, blocked KML001-induced p-p38, and subsequently abrogated
the effect of KML001 on STAT1. The amount of phosphorylated ERK was
increased by treatment of KML001. These results suggest that
KML001-induced apoptosis may be dependent upon or tightly regulated
by activation of MAPK pathways.
Phosphatidylinositol 3-kinase (PI3K)/Akt pathway is
crucial in signaling of diverse biological functions, including
cell proliferation, survival and metastasis (31–33).
PTEN, a tumor suppressor gene, downregulates the PI3K signal, thus,
the loss of function of PTEN results in inappropriate signaling to
downstream molecules, including Akt, Raf and mTOR (34–36).
In the present study, the p-PTEN was upregulated, while p-Akt and
p-Raf were downregulated in KML001-treated HL-60 and HL-60R cells.
Taken together, these results suggest that induction of PTEN by
KML001 may downregulate the intermediate molecules of PI3K/Akt
pathway including Raf which deactivates MAPK signaling, or may
directly inhibit MAPK signaling, thereby inhibiting cellular
proliferation of HL-60 and HL-60R cells.
NF-κB is a heterodimer composed of the p65 and p50
subunits that contains the transcriptional activation domain
required for initiation of gene transcription (37). Also, NF-κB protein is known to be a
positive regulator of cell cycle progression, and it activates
target genes, such as cyclin D1, D2, D2 and E (38,39).
In this study, the expression of p65 and p50 was decreased in the
KML001-treated HL-60 and HL-60R cells, suggesting that KML001 may
inhibit the activation of NF-κB.
Arsenic has been found to target telomere by
inhibition of the transcription of the human telomerase reverse
transcriptase (hTERT) and alteration of telomere length, telomerase
activity, and telomere binding proteins (40). We demonstrated that the telomere
length (TRF) in HL-60 and HL-60R cells was shortened after
treatment with KML001. In addition, real-time PCR with RNA
extracted from KML001-treated HL-60 and HL-60R cells showed a
reduction of catalytic subunit of telomerase, hTERT, in a
time-dependent manner.
Uncapping the telomere signal can induce cellular
growth arrest/apoptosis via telomere length shortening. It
accelerates the double-strand break-mediated DNA damage signaling
pathways, including p-γ-H2AX at Ser139, which
represents an early event of DNA damage signaling (20). Therefore,
p-γ-H2AX is known to be a marker for DNA
damage checkpoint response in telomere-initiated senescence
(21). Our telomere associated
p-γ-H2AX ChIP assay showed that KML001
increased γ-H2AX phosphorylaton in HL-60 and
HL-60R cells, suggesting that KML001 might target the telomere.
In summary, KML001 inhibited the cellular
proliferation of human primary leukemic blasts as well as AML cell
lines including HL-60R via induction of the cell cycle arrest,
triggering apoptosis and modulation of STATs, MAPK, PI3K and NF-κB
signaling. In addition, KML001 targeted the telomere. Therefore,
KML001 might be a candidate agent for the treatment of de
novo as well as refractory AML patients.
Acknowledgements
The present study was conducted in the Hanyang
University Hospital Clinical Laboratory, and it was supported in
part by Dong-A Socio Holdings, Inc.
References
|
1
|
Waxman S and Anderson KC: History of the
development of arsenic derivatives in cancer therapy. Oncologist.
2(Suppl 6): 3–10. 2001. View Article : Google Scholar
|
|
2
|
Soignet SL, Maslak P, Wang ZG, Jhanwar S,
Calleja E, Dardashti LJ, Corso D, DeBlasio A, Gabrilove J,
Scheinberg DA, Pandolfi PP and Warrell RP: Complete remission after
treatment of acute promyelocytic leukemia with arsenic trioxide. N
Engl J Med. 339:1341–1348. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Soignet SL, Frankel SR, Douer D, Tallman
MS, Kantarjian H, Calleja E, Stone RM, Kalaycio M, Scheinberg DA,
Steinherz P, Sievers EL, Coutre S, Dahlberg S, Ellison R and
Warrell RP: United States multicenter study of arsenic trioxide in
relapsed acute promyelocytic leukemia. J Clin Oncol. 19:3852–3860.
2001.PubMed/NCBI
|
|
4
|
Uslu R, Sanli UA, Sezgin C, Karabulut B,
Terzioglu E, Omay SB and Goker E: Arsenic trioxide-mediated
cytotoxicity and apoptosis in prostate and ovarian carcinoma cell
lines. Clin Cancer Res. 6:4957–4964. 2000.
|
|
5
|
Zhang TC, Cao EH, Li JF, Ma W and Qin JF:
Induction of apoptosis and inhibition of human gastric cancer
MGC-803 cell growth by arsenic trioxide. Eur J Cancer.
35:1258–1263. 1999. View Article : Google Scholar
|
|
6
|
Ling YH, Jiang JD, Holland JF and
Perez-Soler R: Arsenic trioxide produces polymerization of
microtubules and mitotic arrest before apoptosis in human tumor
cell lines. Mol Pharmacol. 62:529–538. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Miller WH, Schipper HM, Lee JS, Singer J
and Waxman S: Mechanisms of action of arsenic trioxide. Cancer Res.
62:3893–3903. 2002.PubMed/NCBI
|
|
8
|
Chen GQ, Zhu J, Shi XG, Ni JH, Zhong HJ,
Si GY, Jin XL, Tang W, Li XS, Xong SM, Shen ZX, Sun GL, Ma J, Zhang
P, Zhang TD, Gazin C, Naoe T, Chen SJ, Wang ZY and Chen Z: In vitro
studies on cellular and molecular mechanisms of arsenic trioxide
(As2O3) in the treatment of acute
promyelocytic leukemia: As2O3 induces NB4
cell apoptosis with downregulation of Bcl-2 expression and
modulation of PML-RAR alpha/PML proteins. Blood. 88:1052–1061.
1996.PubMed/NCBI
|
|
9
|
Hug N and Lingner J: Telomere length
homeostasis. Chromosoma. 115:413–425. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chou WC, Hawkins AL, Barrett JF, Griffin
CA and Dang CV: Arsenic inhibition of telomerase transcription
leads to genetic instability. J Clin Invest. 108:1541–1547. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Litzow MR: Arsenic trioxide. Expert Opin
Pharmacother. 9:1773–1785. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Phatak P, Dai F, Butler M, Nandakumar MP,
Gutierrez PL, Edelman MJ, Hendriks H and Burger AM: KML001
cytotoxic activity is associated with its binding to telomeric
sequences and telomere erosion in prostate cancer cells. Clin
Cancer Res. 14:4593–4602. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Glienke W, Chow KU, Bauer N and Bergmann
L: Down-regulation of wt1 expression in leukemia cell lines as part
of apoptotic effect in arsenic treatment using two compounds. Leuk
Lymphoma. 47:1629–1638. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jin JO, Song MG, Kim YN, Park JI and Kwak
JY: The mechanism of fucoidan-induced apoptosis in leukemic cells:
involvement of ERK1/2, JNK, glutathione, and nitric oxide. Mol
Carcinog. 49:771–782. 2010.PubMed/NCBI
|
|
15
|
Yoon JS, Won YW, Kim SJ, Oh SJ, Kim ES,
Kim BK, Cho CG, Choi JH, Park BB, Lee MH and Lee YY: Anti-leukemic
effect of 2-pyrone derivatives via MAPK and PI3 kinase pathways.
Invest New Drugs. 30:2284–2293. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vermes I, Haanen C, Steffens-Nakken H and
Reutelingsperger C: A novel assay for apoptosis. Flow cytometric
detection of phosphatidylserine expression on early apoptotic cells
using fluorescein labelled Annexin V. J Immunol Methods. 184:39–51.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
d’Adda di Fagagna F, Hande MP, Tong WM,
Roth D, Lansdorp PM, Wang ZQ and Jackson SP: Effects of DNA
nonhomologous end-joining factors on telomere length and
chromosomal stability in mammalian cells. Curr Biol. 11:1192–1196.
2001. View Article : Google Scholar
|
|
18
|
Cawthon RM: Telomere length measurement by
a novel monochrome multiplex quantitative PCR method. Nucleic Acids
Res. 37:e212009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Blackburn EH: Telomere states and cell
fates. Nature. 408:53–56. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
d’Adda di Fagagna F, Reaper PM,
Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G,
Carter NP and Jackson SP: A DNA damage checkpoint response in
telomere-initiated senescence. Nature. 426:194–198. 2003.
View Article : Google Scholar
|
|
21
|
Phatak P, Cookson JC, Dai F, Smith V,
Gartenhaus RB, Stevens MF and Burger AM: Telomere uncapping by the
G-quadruplex ligand RHPS4 inhibits clonogenic tumour cell growth in
vitro and in vivo consistent with a cancer stem cell targeting
mechanism. Br J Cancer. 96:1223–1233. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sherr CJ: Cancer cell cycles. Science.
274:1672–1677. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dynlacht BD: Regulation of transcription
by proteins that control the cell cycle. Nature. 389:149–152. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Darnell JE, Kerr IM and Stark GR: Jak-STAT
pathways and transcriptional activation in response to IFNs and
other extracellular signaling proteins. Science. 264:1415–1421.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bowman T, Garcia R, Turkson J and Jove R:
STATs in oncogenesis. Oncogene. 19:2474–2488. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Akira S: Functional roles of STAT family
proteins: lessons from knockout mice. Stem Cells. 17:138–146. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hirano T, Ishihara K and Hibi M: Roles of
STAT3 in mediating the cell growth, differentiation and survival
signals relayed through the IL-6 family of cytokine receptors.
Oncogene. 19:2548–2556. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Smithgall TE, Briggs SD, Schreiner S,
Lerner EC, Cheng H and Wilson MB: Control of myeloid
differentiation and survival by Stats. Oncogene. 19:2612–2618.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cross TG, Scheel-Toellner D, Henriquez NV,
Deacon E, Salmon M and Lord JM: Serine/threonine protein kinases
and apoptosis. Exp Cell Res. 256:34–41. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wada T and Penninger JM: Mitogen-activated
protein kinases in apoptosis regulation. Oncogene. 23:2838–2849.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Willems L, Tamburini J, Chapuis N, Lacombe
C, Mayeux P and Bouscary D: PI3K and mTOR signaling pathways in
cancer: new data on targeted therapies. Curr Oncol Rep. 14:129–138.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cantley LC: The phosphoinositide 3-kinase
pathway. Science. 296:1655–1657. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hidalgo M and Rowinsky EK: The
rapamycin-sensitive signal transduction pathway as a target for
cancer therapy. Oncogene. 19:6680–6686. 2000. View Article : Google Scholar
|
|
34
|
Ortega-Molina A and Serrano M: PTEN in
cancer, metabolism, and aging. Trends Endocrinol Metab. 24:184–189.
2013. View Article : Google Scholar
|
|
35
|
Sawyers CL: Rational therapeutic
intervention in cancer: kinases as drug targets. Curr Opin Genet
Dev. 12:111–115. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Janus A, Linke A, Cebula B, Robak T and
Smolewski P: Rapamycin, the mTOR kinase inhibitor, sensitizes acute
myeloid leukemia cells, HL-60 cells, to the cytotoxic effect of
arabinozide cytarabine. Anticancer Drugs. 20:693–701. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Baldwin AS: The NF-kappa B and I kappa B
proteins: new discoveries and insights. Annu Rev Immunol.
14:649–683. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Guttridge DC, Albanese C, Reuther JY,
Pestell RG and Baldwin AS: NF-kappaB controls cell growth and
differentiation through transcriptional regulation of cyclin D1.
Mol Cell Biol. 19:5785–5799. 1999.PubMed/NCBI
|
|
39
|
Hinz M, Krappmann D, Eichten A, Heder A,
Scheidereit C and Strauss M: NF-kappaB function in growth control:
regulation of cyclin D1 expression and G0/G1-to-S-phase transition.
Mol Cell Biol. 19:2690–2698. 1999.PubMed/NCBI
|
|
40
|
Zhang Y, Cao EH and Qin JF: Up-regulation
of telomere-binding TRF1, TRF2 related to reactive oxygen species
induced by As2O3 in MGC-803 cells. Eur J
Pharmacol. 516:1–9. 2005. View Article : Google Scholar : PubMed/NCBI
|