Introduction
Rhabdomyosarcoma (RMS) is a tumor that derives from
early skeletal muscle cells and displays characteristics of muscle
differentiation (1–6). There are two major histologic
subtypes of RMS: alveolar (A)RMS and embryonal (E)RMS (7). Clinical evidence indicates that ARMS
is more aggressive and has a significantly worse outcome than ERMS
(8). It is well known that RMS
cells, particularly ARMS cells, can infiltrate the bone marrow (BM)
and, because they can resemble hematologic blasts, are sometimes
misdiagnosed as acute leukemia blasts.
Genetic characterization of RMS has identified
markers that show a correlation with histologic subtype.
Specifically, ARMS is characterized by the translocation
t(2;13)(q35;q14) in 70% of cases or the variant t(1;13)(p36;q14) in
a smaller percentage of cases (9).
These translocations disrupt the paired box 3 (PAX3) and 7
(PAX7) genes on chromosome 2 and 1, respectively, and the
forkhead (FKHR) gene on chromosome 13, which leads to
generation of PAX3-FKHR and PAX7-FKHR fusion genes in
RMS (10–13). These fusion genes encode the fusion
proteins PAX3-FKHR and PAX7-FKHR, which are involved in cell
survival and dysregulation of the cell cycle in ARMS cells
(14).
Moreover, both ARMS and ERMS are characterized by
important epigenetic mutations and an epigenetic imbalance that may
affect some important genes. One of the most common epigenetic
changes is loss of imprinting (LOI) at chromosome locus 11p15.5,
which alters the expression of the tandem gene IGF2-H19
(15–18). Encoded by the insulin-like growth
factor 2 (IGF2) gene, IGF2 is a growth factor essential for
normal embryonic and neonatal growth and development, while
H19 gives rise to a non-coding mRNA that has the opposite
effect on cell proliferation.
IGF2 signals through the tyrosine kinase
insulin-like growth factor 1 receptor (IGF1R), which is frequently
overexpressed in human RMS (19–25)
and a number of other cancers (25). The function of H19-derived
non-coding RNA (ncRNA) is largely unknown and still under debate.
H19 is maternally expressed and may act either as a tumor
suppressor or as an oncogene, depending on cancer type (26,27).
It spans >2 kb and encodes short non-coding microRNAs, including
miR-675, which was recently shown to be a negative regulator of
IGF1R expression in mice (28).
LOI in RMS cells due to hypermethylation of a differentially
methylated region (DMR) that controls IGF2-H19 expression leads to
formation of a potent autocrine loop (IGF2-IGF1R) and, at the same
time, decreases the level of growth-suppressing H19-derived
miRNAs.
The methylation of genomic DNA in cells is catalyzed
by DNA methyltransferases (DNMTs). Recent studies have shown that
application of methyltransferase inhibitors, such as 5-azacitidine
(AzaC), may become a clinically relevant strategy for tumor growth
inhibition (29,30). DNMT inhibitors, which are usually
analogues of the cytidine nucleoside, bind to DNA methyltransferase
1, which results in DNA hypomethylation. As a result of DNA
hypomethylation, some silenced tumor-suppressor genes like
H19 may become activated.
In this study we evaluated the effect of the DNMT
inhibitor AzaC on the tumorigenic potential of RMS cells. We found
that DNA hypomethylation led to inhibition of RMS growth. This
inhibition correlated with reactivation of H19 and
upregulation of miR-675, which, as expected, negatively affected
the expression of not only IGF1R (28) but also the insulin receptor (INSR),
which we show for the first time. Thus, the downregulation of both
receptors inhibits signaling of IGF2, insulin-like growth factor 1
(IGF1), and insulin (INS), which are all potent growth factors in
RMS cells.
Materials and methods
Cell lines
We used human RMS cell lines (a gift of Dr Peter
Houghton, World Children’s Research Hospital, Columbus, OH, USA),
including the RH30 ARMS cell line and the RD ERMS cell line. RMS
cells used for experiments were cultured in Roswell Park Memorial
Institute medium (RPMI)-1640 (Sigma, St. Louis, MO, USA),
supplemented with 100 IU/ml penicillin, 10 μg/ml streptomycin, and
50 μg/ml neomycin (Life Technologies, Grand Island, NY, USA) in the
presence of 10% heat-inactivated fetal bovine serum (FBS) (Life
Technologies). The cells were cultured in a humidified atmosphere
at 37°C in 5% CO2 at an initial cell density of
2.5×104 cells/flask (Corning, Cambridge, MA, USA), and
the medium was changed every 48 h.
Flow cytometry analysis of receptor
expression
The expression of IGF1R, and INSR in RMS cell lines
was evaluated by flow cytometry analysis as previously described
(31). The antigens were detected
with anti-human/mouse INS R/CD220 conjugated with APC (R&D
Systems, Minneapolis, MN, USA); and phycoerythrin (PE)-anti-IGF1R
monoclonal antibody, clone no. 33255 (R&D Systems). Briefly,
the cells were stained in phosphate-buffered saline (PBS)
(Ca2+- and Mg2+-free) supplemented with 2%
bovine calf serum (BCS) (HyClone Laboratories, Inc., Logan, UT,
USA). After the final wash, cells were resuspended in PBS and
analyzed by FACS using the Navios Flow Cytometer (Beckman Coulter,
Inc., Brea, CA, USA).
Quantitative reverse transcription-PCR
(RT-qPCR)
Total RNA was isolated from cells treated with
5-azacitidine (AzaC), 2′-deoxy-5-azacytidine (DAC), or trichostatin
A (TsA) (all from Sigma) and from cell controls with the RNeasy kit
(Qiagen, Valencia, CA, USA). The RNA was reverse-transcribed with
MultiScribe Reverse Transcriptase and oligo-dT primers (Applied
Biosystems, Foster City, CA, USA). Quantitative assessment of mRNA
levels was performed by RT-qPCR on an ABI 7500 instrument with
Power SYBR-Green PCR Master Mix reagent. Real-time conditions were
as follows: 95°C (15 sec), 40 cycles at 95°C (15 sec), and 60°C (1
min). According to melting point analysis, only one PCR product was
amplified under these conditions. The relative quantity of a
target, normalized to the endogenous control β-2 microglobulin gene
and relative to a calibrator, is expressed as 2−ΔΔCt
(fold difference), where Ct is the threshold cycle, ΔCt = (Ct of
target genes) - (Ct of endogenous control gene, β-2 microglobulin),
and ΔΔCt = (ΔCt of samples for target gene) - (ΔCt of calibrator
for the target gene). The following primer pairs were used: IGF2
forward, CCATGTCCTCCTCGCATCTC and reverse, CGTGGCAGAGCTGGTGAAG; H19
forward, GGCTCTGGAAGGTGAAGCTAGA and reverse, GCGGGCGCTGCTGTT.
Methylation studies using a combined
bisulfite restriction analysis (COBRA) assay and bisulfite
sequencing
DNA was isolated with the DNeasy Blood and Tissue
kit (Qiagen), and exactly 100 ng of genomic DNA prepared from the
indicated cells were used for bisulfite modification with the
EpiTect Bisulfite kit (Qiagen) according to the manufacturer’s
instructions. Bisulfite-treated DNA was subjected to nested PCR
with methylation-specific primers for the human H19-IGF2 DMR
(32): 1st outer primer hH19O
forward, AGG TGT TTT AGT TTT ATG GAT GAT GG and reverse, TCC TAT
AAA TAT CCT ATT CCC AAA TAA CC; 2nd inner primer hH19I forward, TGT
ATA GTA TAT GGG TAT TTT TGG AGG TTT and hH19-DMR-I reverse, same as
hH19-DMR-O reverse. Next, the PCR products were cut with BstUI
restriction enzyme and visualized on an agarose gel. In parallel
after agarose gel electrophoresis, the PCR products were eluted
using the QIAquick Gel Extraction kit (Qiagen). The eluted
amplicons were subsequently ligated into the pCR2.1-TOPO vector and
transformed into TOP10 bacteria using the TOPO TA Cloning kit
(Invitrogen Life Technologies, Carlsbad, CA, USA). The plasmids
were prepared using the QIAprep Spin Miniprep kit (Qiagen),
sequenced with M13 forward and reverse primers, and the methylation
pattern visualized using CpGviewer software. All experiments were
conducted with three independent isolations of all cell
populations.
miR-675 transfection and H19 gene
cloning
Full-length H19 and Exon1 complementary DNA
(GenBank: AF087017.1) was PCR-amplified and cloned into the
pcDNA3.1 expression vector, and the vector was introduced into RH30
cells using Lipofectamine 2000 (both from Invitrogen Life
Technologies). Cells were positively selected with G418 (200 μg/ml)
for 2 weeks. The level of H19 overexpression was assessed by
RT-qPCR, and the cells were subjected to proliferation assays.
Enhanced expression of miR-675 was performed by transfecting RD and
RH30 cells with miR-675-3p, -5p, or both (Sigma). Cells were seeded
into 24-well plates (3×104) and transfected with
Lipofectamine 2000 (Invitrogen Life Technologies) and 40 μM
microRNA for 24 h. In parallel, cells were transfected with MISSION
miRNA Negative Control (Sigma). Next, cells were replated for
proliferation assays or receptor expression analysis by flow
cytometry.
Cell cycle analysis
The cells were incubated with or without 0.5, 1, 2,
5, 10, 20, or 50 μM AzaC (Sigma). After 96 h, the cells were
collected, washed with PBS, centrifuged at 1,200 rpm for 8 min, and
resuspended in 1 ml RPMI-1640 medium supplemented with 10% FBS at a
concentration of 106 cells/ml. Then, 2 μl of Vybrant
DyeCycle Orange Stain (Invitrogen Life Technologies) was added to
the cells, which were gently vortexed. Samples were kept at 37°C
for 30 min in the dark and were analyzed using a flow cytometer
(Navios, Beckman Coulter, Inc.).
Annexin V/propidium iodide (PI) assays
for apoptosis
For Annexin V/PI assays, cells were stained with
Annexin V-FITC and PI and evaluated for apoptosis by flow cytometry
according to the manufacturer’s instructions (BD Biosciences, San
Diego, CA, USA). Briefly, 1×106 cells were washed twice
with PBS and stained with 5 μl of Annexin V-FITC and 10 μl of PI (5
μg/ml) in 1X binding buffer (10 mM HEPES, pH 7.4, 140 mM NaOH, 2.5
mM CaCl2) for 15 min at room temperature in the dark.
The apoptotic cells were determined using a Navios flow cytometer
(Beckman Coulter, Inc.). Annexin V+ PI− cells
represented the early apoptotic populations, while Annexin
V+ PI+ cells represented either late
apoptotic or secondary necrotic populations.
Chemotaxis assay
Polycarbonate membranes (8-μm) were covered with 50
μl of 0.5% gelatin. The cells were detached with 0.5 mmol/l
ethylenediaminetetraacetic acid (EDTA), washed in RPMI-1640,
resuspended in RPMI-1640 with 0.5% bovine serum albumin (BSA), and
seeded at a density of 3×104 in 120 μl into the upper
chambers of Transwell inserts (Corning). The lower chambers were
filled with IGF1 (100 ng/ml), IGF2 (100 ng/ml), INS (10 ng/ml), or
0.5% BSA in RPMI-1640 (control). After 24 h, the inserts were
removed from the Transwell supports. Cells remaining in the upper
chambers were scraped off with cotton wool, and cells that had
transmigrated were stained by HEMA 3 according to the
manufacturer’s instructions (Thermo Fisher Scientific, Pittsburgh,
PA, USA) and counted either on the lower side of the membrane or on
the bottom of the Transwell chamber.
Colony-formation assay
Cells were incubated with or without 1 or 5 μM AzaC
for 72 h, collected, counted, mixed in 0.35% top agar (in RPMI-1640
medium supplemented with 10% FBS) and plated at 1,250 cells/well
onto 24-well plates containing a solidified bottom layer (0.5% base
agar in the same growth medium). Every 3 days, colonies were fed
with 250 μl/well culture medium with 10% FBS. After 21 days,
unstained colonies were counted.
Cell proliferation
Cells were plated in culture flasks at an initial
density of 104 cells/cm2 in the presence or
absence of AzaC and miR-675-3p or -5p or both. The cell number was
calculated at 24, 48, 72, and 96 h after culture initiation. At the
indicated time points, the cells were harvested from the culture
flasks by trypsinization, and the number of cells was determined
using a Navios cytometer.
Phosphorylation of intracellular pathway
proteins and western blotting
Western blotting was performed on extracts prepared
from RMS cell lines (2×106 cells) that were kept in RPMI
medium containing low levels of BSA (0.5%) to render the cells
quiescent, as previously described (33). The cells were divided and
stimulated with optimal doses of IGF1 (100 ng/ml), IGF2 (100
ng/ml), INS (10 ng/ml) for 5 min at 37°C and then lysed (for 10
min) on ice in RIPA buffer (Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA), containing protease and phosphatase inhibitors
(Roche Diagnostics Corp., Indianapolis, IN, USA). Subsequently, the
extracted proteins were separated by either 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and the
fractionated proteins were transferred to a PVDF membrane (Bio-Rad,
Hercules, CA, USA). Phosphorylation of the intracellular kinases,
p42/44 mitogen-activated protein kinase (MAPK) (Thr202/Tyr204) was
detected using commercial mouse phospho-specific mAb (p42/44)
polyclonal antibody (Cell Signaling Technology, Inc., Danvers, MA,
USA) with horseradish peroxidase (HRP)-conjugated goat anti-mouse
IgG secondary antibody (Santa Cruz Biotechnology, Inc.). Equal
loading in the lanes was evaluated with appropriate mAb: p42/44
anti-MAPK clone no. 9102 (Cell Signaling Technology, Inc.). The
membranes were developed with an enhanced chemiluminescence (ECL)
reagent (Amersham Pharmacia Biotech, Little Chalfont, UK).
Statistical analysis
All results are presented as mean ± standard error
of the mean (SEM). Statistical analysis of the data was performed
using the non-parametric Mann-Whitney test or Student’s t-test,
with p<0.05 considered significant.
Results
Hypermethylation at the DMR for the
IGF2-H19 tandem gene is erased after exposure of RMS cells to
AzaC
Hypermethylation of the DMR at the IGF2-H19 locus
(known as LOI) is an important epigenetic change occurring in ARMS
and ERMS cells and leads to overexpression of IGF2 and
downregulation of H19. As mentioned above, while IGF2 is an
autocrine factor for RMS cells that stimulates proliferation of
these cells after binding to the IGF1R and INSR, H19-derived
ncRNA gives rise to several miRNAs that negatively regulate cell
proliferation. One of these miRNAs, miR-675, downregulates IGF1R,
which in turn binds IGF2 and IGF1 in murine placental cells
(28).
To better address the LOI phenomenon at the DMR for
the IGF2-H19 locus and the role of IGF2 and H19-derived
miR-675 in the proliferation of RMS cells, we employed the DNMT
inhibitor AzaC to reverse hypermethylation of the DMR at the
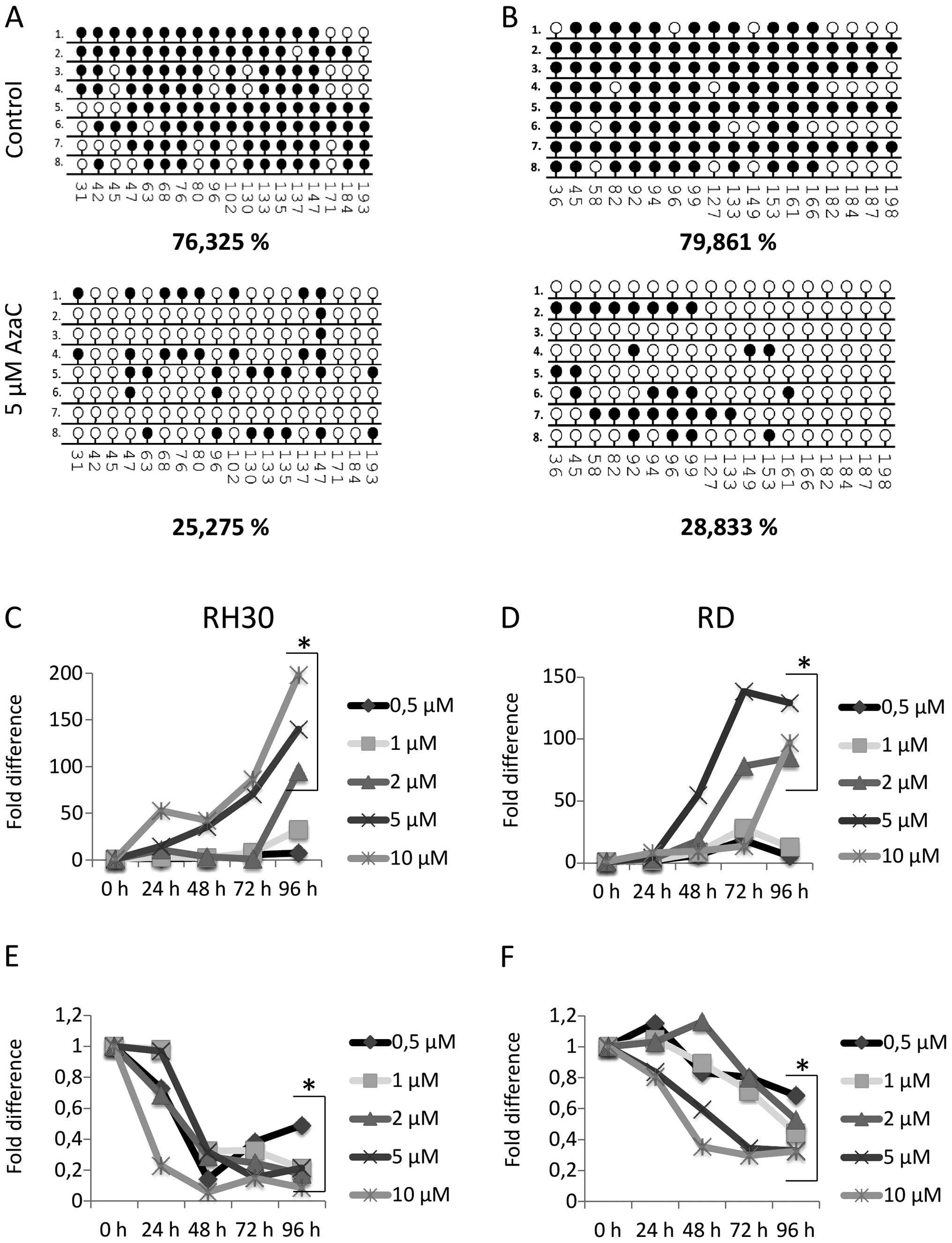
IGF2-H19 locus (Fig. 1A and B). We
found that exposure of the RH30 ARMS cell line and the RD ERMS cell
line to 5 μM AzaC resulted in demethylation of the hypermethylated
DMR for this tandem gene from ~76 to 25% and from ~80 to 29%,
respectively.
The erasure of methylation at the DMR for the
IGF2-H19 locus in response to increasing doses of AzaC resulted, as
predicted, in upregulation of H19 and downregulation of IGF2
expression at the mRNA level (Fig.
1C–F). Similar results were obtained by employing another DNMT
inhibitor, decitabine (data not shown). At the same time, TsA,
which is a potent deacetylating agent, did not change the IGF2/H19
mRNA ratio in RMS cells (data not shown).
Downregulation of IGF2 after AzaC treatment was
subsequently confirmed at the protein level in RH30- and RD-derived
conditioned media by employing a sensitive ELISA (data not
shown).
Exposure to AzaC inhibits proliferation
of RMS cells
Next, we performed in vitro assays to see
whether AzaC, by down-regulating IGF2 and upregulating H19, affects
proliferation of RMS cells. First, to exclude a non-specific toxic
effect of AzaC treatment on RMS cells, we evaluated the effect of
increasing doses of AzaC on Annexin V binding in RH30 and RD cells
(data not shown). By employing PI staining and Annexin V binding,
we found that AzaC does not significantly affect cell survival at
concentrations <10 μM.
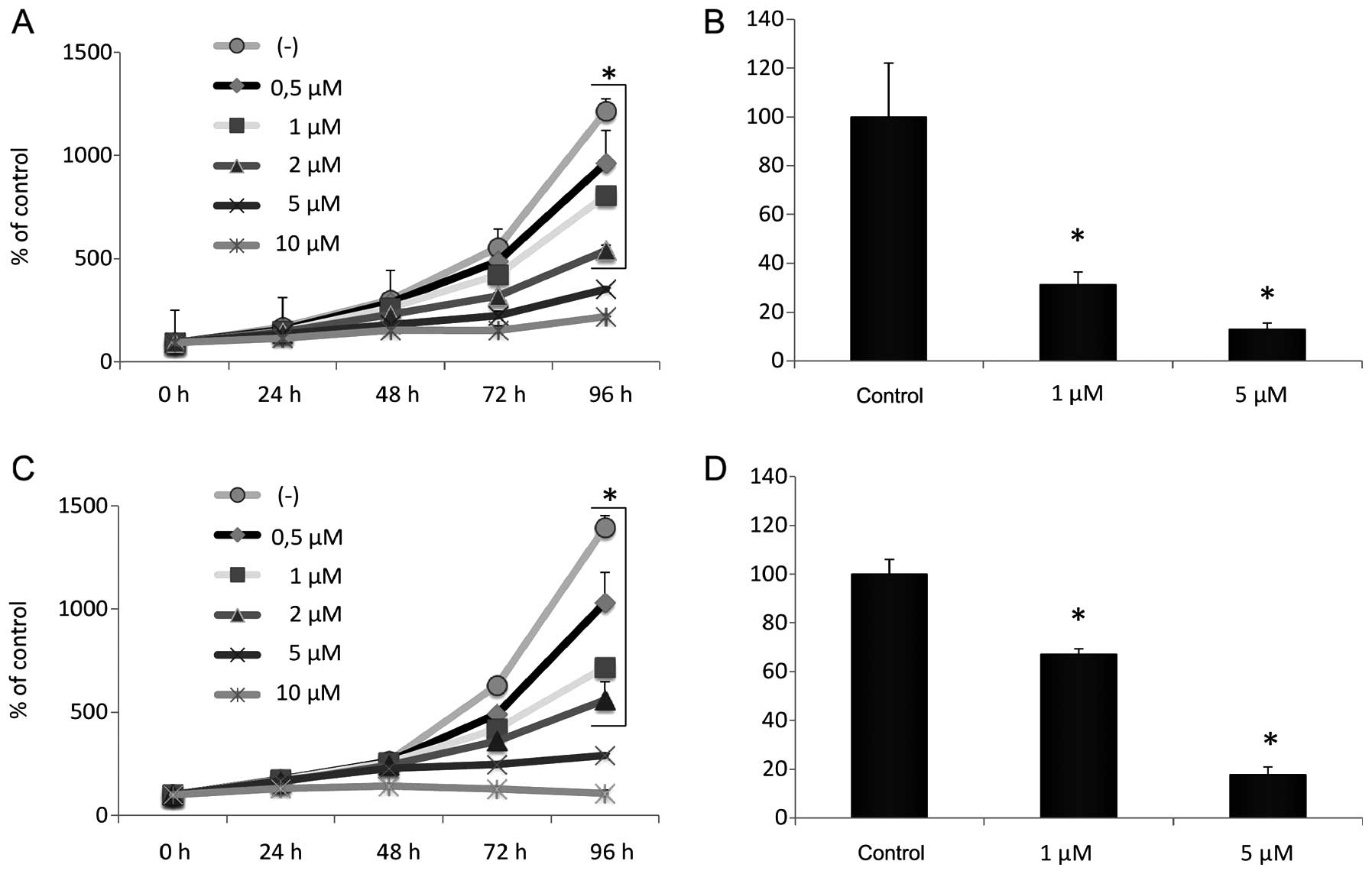
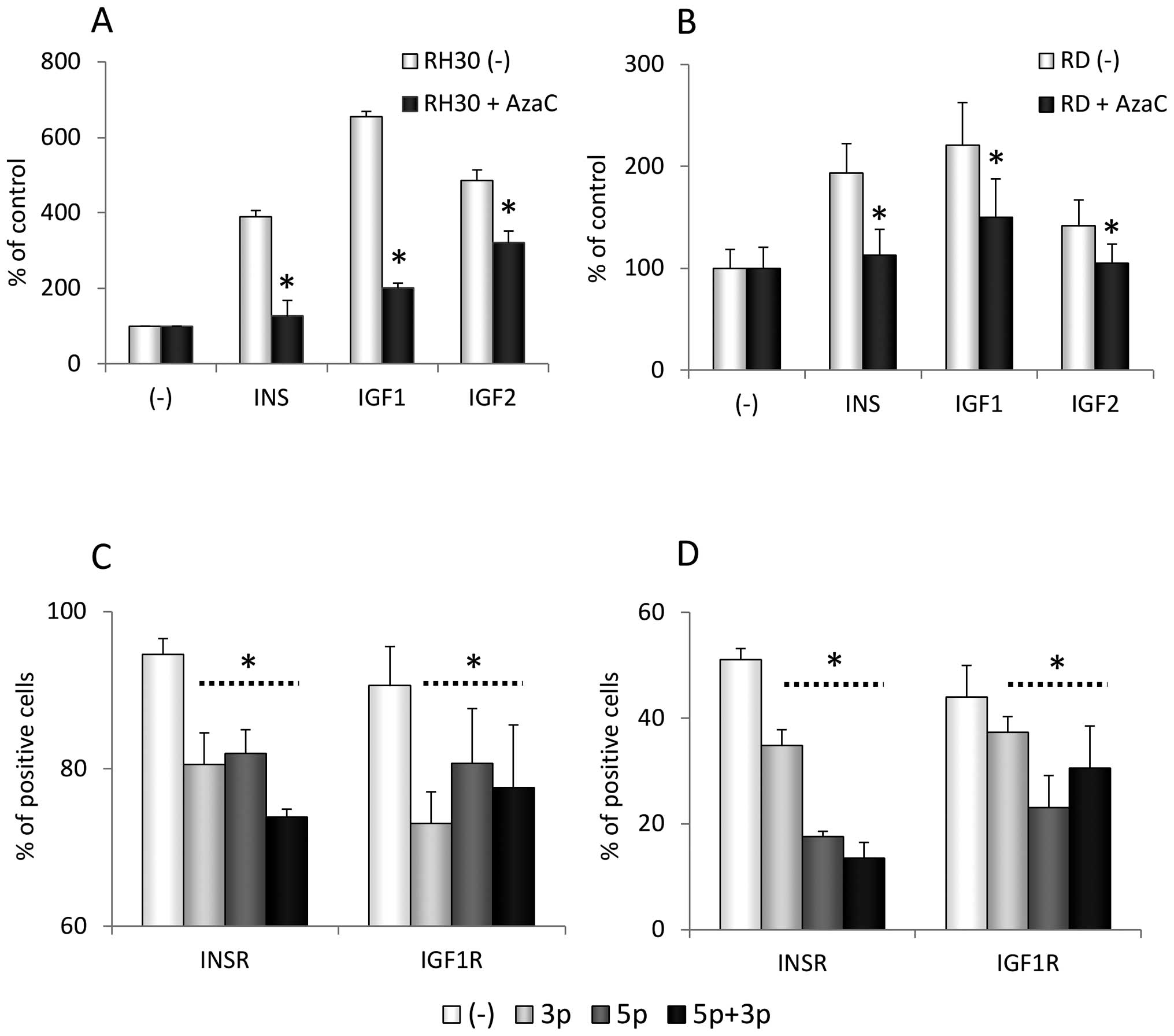
Next, by employing an anchorage-dependent
proliferation assay in plastic dishes (Fig. 2A and C) and an
anchorage-independent colony-forming assay in soft agar (Fig. 2B and D), we observed an AzaC
dose-dependent inhibition of RH30 (Fig. 2A and B) and RD (Fig. 2C and D) cell proliferation. The
effect of AzaC on proliferation of RMS cells was subsequently
evaluated by employing FACS-based time-lapse monitoring of the cell
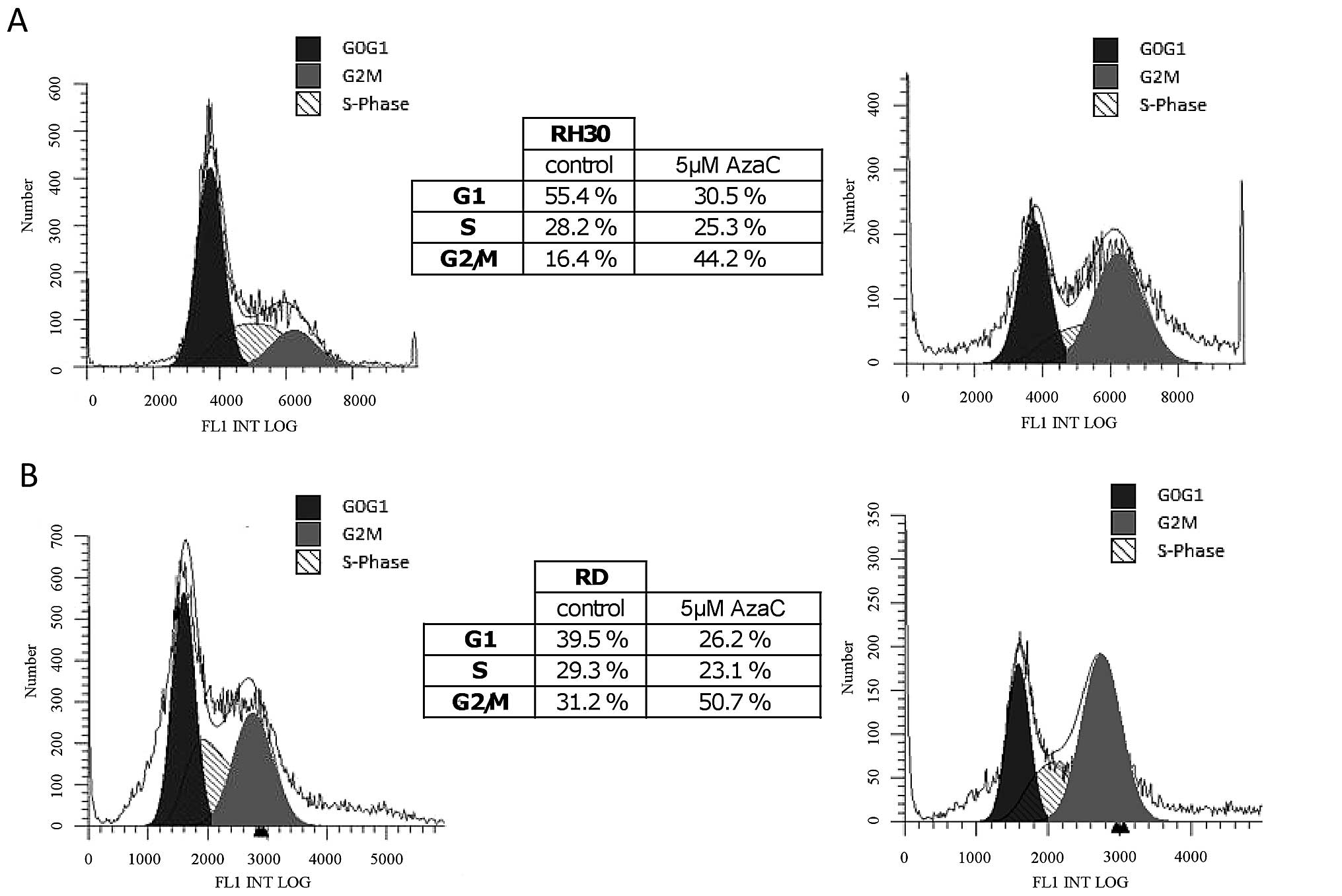
cycle (Fig. 3). We found that AzaC
at a dose of 5 μM inhibits cell proliferation in G2/M phase and
reduces the number of cells in G1 phase in both RH30 (Fig. 3A) and RD (Fig. 3B) cells.
We are aware that this effect is most likely the
result of a combined increase in H19 expression as well as
epigenetic changes in other genes that regulate the cell cycle. At
this point, however, we observed that AzaC treatment did not
reactivate other epigenetically regulated suppressor genes, such as
p16INK4A and p14, and did not
increase the expression of p21waf1 and
p53 (data not shown). Furthermore, despite the fact that
IGF2 is downregulated in RMS cells after AzaC treatment, addition
of this growth factor to RMS cells did not abrogate AzaC-mediated
G2/M inhibition, which again indicates major involvement of
H19-derived miR-675 in expression of the IGF2 signaling
receptor, IGF1R.
The negative effect of H19 expression on
RMS cell proliferation may be partially explained by
miR-675-mediated downregulation of IGF1R expression
As mentioned above, H19-derived miR-675
downregulates the expression of IGF1R in murine placental cells
(24). To address whether miR-675
is involved in regulation of IGF1R expression in human RMS cells,
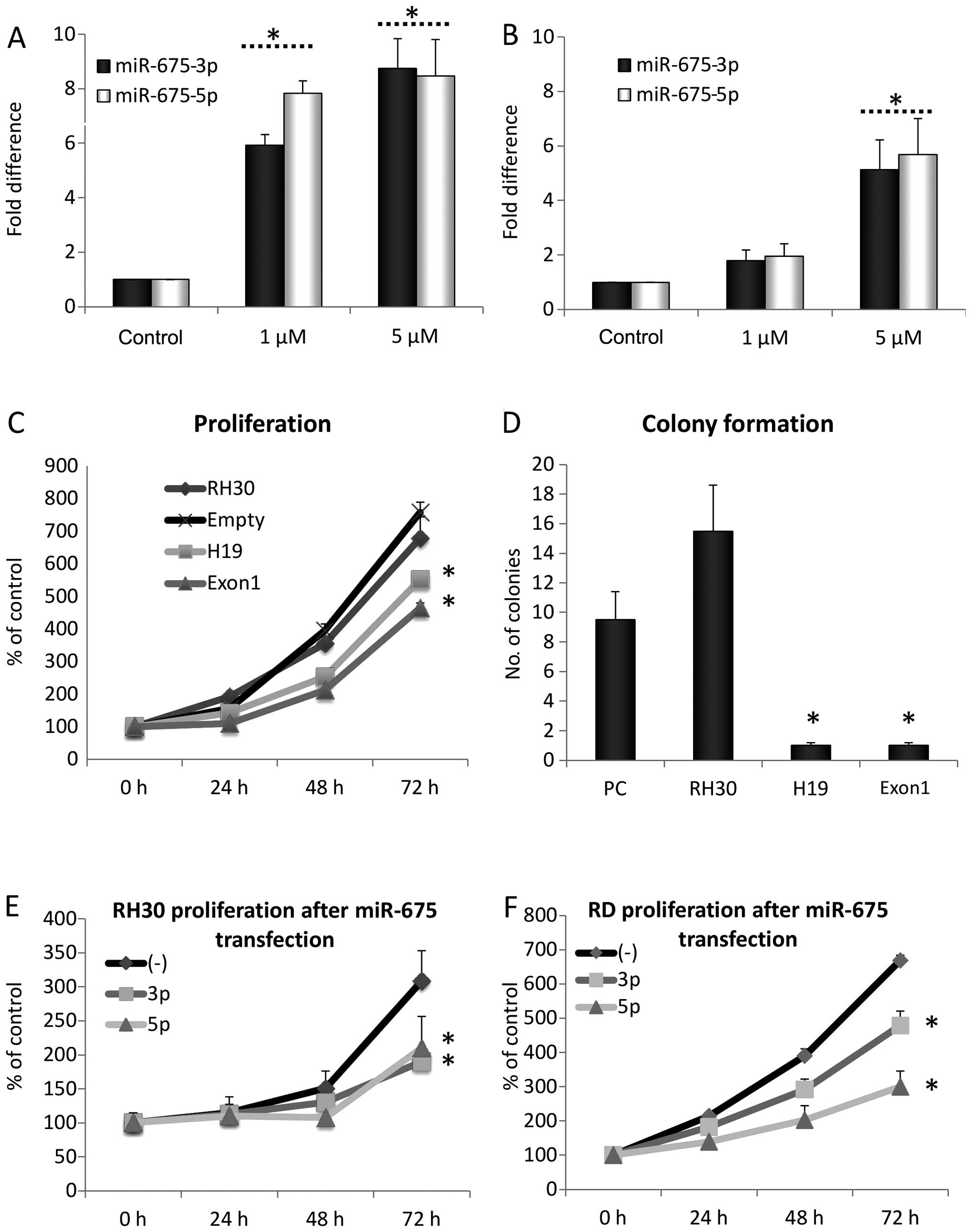
we measured the expression of two isoforms of this miRNA
(miR-675-3p and -5p) in RH30 and RD cells, unexposed and exposed to
AzaC (Fig. 4A and B). We observed
an AzaC dose-dependent increase in both miR-675 isoforms in these
cells.
Next, to shed more light on the role of H19
and miR-675 on proliferation of RMS cells we cloned the entire
H19 gene and H19 Exon1 (E1.1) into the pcDNA3.1
expression vector and transfected them into RH30 cells. We observed
an inhibitory effect of H19 and H19 Exon1 (E1.1)
ncRNA on proliferation of RMS cells, both in plastic dishes
(Fig. 4C) and in a soft agar
colony-forming assay (Fig. 4D). To
better address the potential involvement of H19-encoded
miR-675, we transfected RH30 and RD cells with miR-675-3p or -5p
and observed the expected decrease in proliferation of RH30 and RD
cells (Fig. 4E and F).
Effect of AzaC on the expression of the
IGF1R and INSR
Our results described above demonstrate an
inhibitory effect of AzaC on the proliferation of RMS cells due to
upregulation of miR-675 and downregulation of IGF1R expression.
However, it is known that, in addition to IGF2, IGF1 and INS also
affect the proliferation of RMS cells (19–25).
While IGF2 binds to IGF1R, both IGF1 and INS interact also with the
INSR. Since we did not see a positive effect of IGF1 or INS as a
replacement for IGF2 in AzaC-treated cells, we asked whether
miR-675 also downregulates the INSR.
In order to clarify this issue, we applied a
bioinformatic tool (rna22v1.0) to search for potential miR-675
targets in the INSR mRNA. As with IGF1R, we observed the presence
of miR-675-3p and/or -5p sites in the 3′UTR and coding sequence of
the INSR gene (data available upon request).
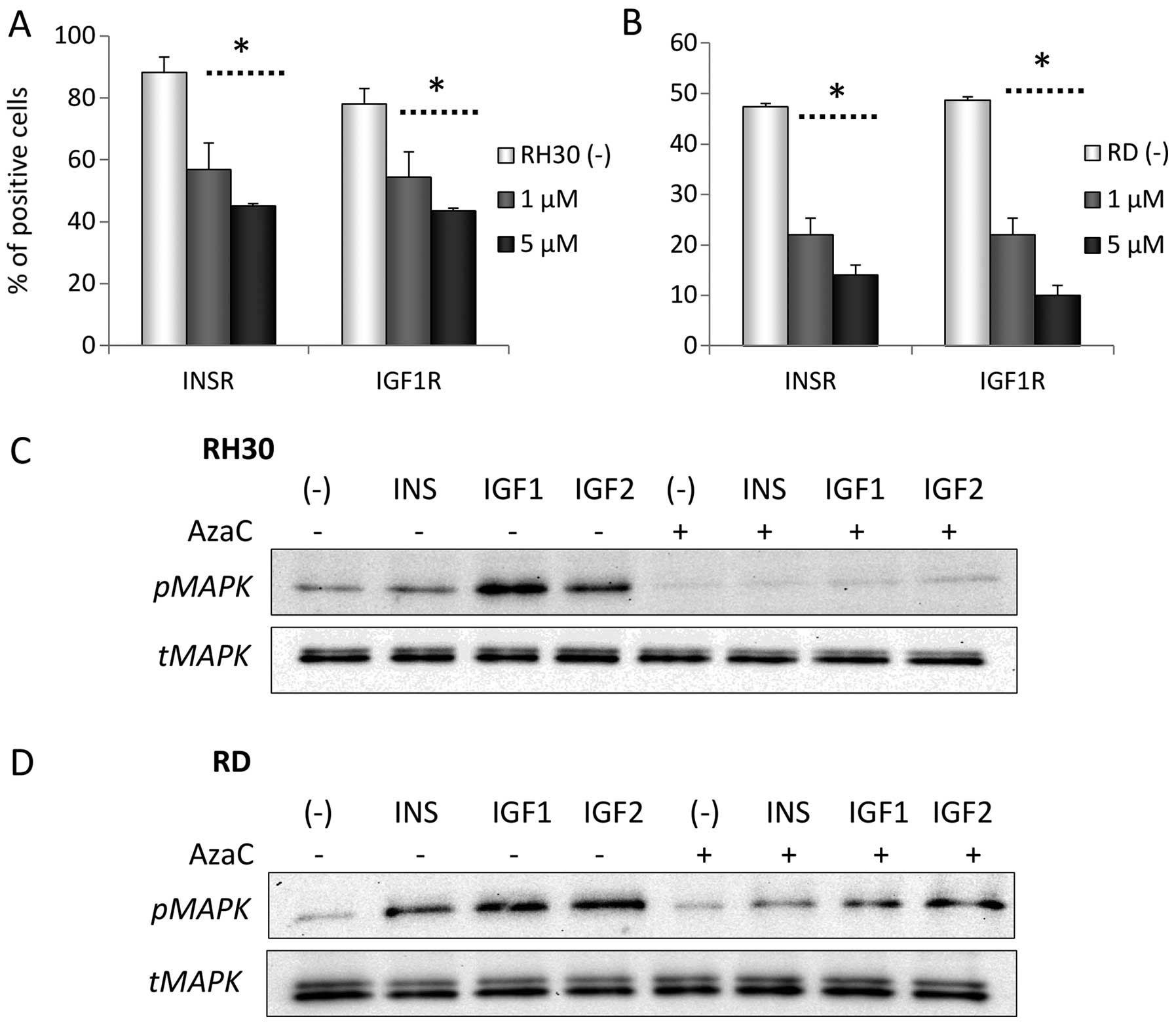
To confirm whether AzaC affects the expression of
the INSR, we first evaluated the expression of the INSR in parallel
with the expression of IGF1R in RH30 and RD cells by FACS analysis
(Fig. 5A and B). We found that
exposure of RMS cells to AzaC decreases the expression of IGF2-,
IGF1-, and INS-binding receptors in a dose-dependent manner and,
moreover, attenuates activation of MAPK p42/44 in RMS cells
stimulated with INS, IGF1 and 2 and exposed to AzaC (Fig. 5C and D).
This effect of AzaC on INSR and IGF1R signaling was
correlated with a decrease in the chemotactic responsiveness of
RH30 and RD cells to INS, IGF1 and 2 gradients (Fig. 6A and B). Importantly, as was the
case for the IGF1R (Fig. 5A and
B), downregulation of the INSR in RH30 and RD cells transfected
with miR-675-3p and -5p isoforms was observed by FACS analysis
(Fig. 6C and D).
Discussion
Changes in DNA methylation are among the most common
molecular alterations in human neoplasia (34–37).
The best characterized are related to hypermethylation of DNA in
the DMRs of genes that regulate cell proliferation, such as the
IGF2-H19 tandem gene (38).
The IGF2-H19 locus is part of a cluster of imprinted
genes on human chromosome 11p15, which also includes KCNQ1
(39). In normal tissues,
methylation of the DMR for this gene on the paternal chromosome
prevents binding of the CTCF insulator protein to the DMR, which
allows the distal enhancer to stimulate the expression of IGF2 from
the paternal chromosome. On the maternal chromosome, the DMR for
IGF2-H19 is demethylated and CTCF binds to this region of DNA, with
the result that the distal enhancer activates transcription of only
the H19 gene (40). Thus,
in normal somatic cells a balance is established between
transcription of IGF2 and H19 from paternally and maternally
derived chromosomes, respectively. This balance is perturbed in
several malignancies in which DMRs on both maternally and
paternally derived chromosomes are hypermethylated, and a distal
enhancer stimulates transcription of the IGF2 gene from both
chromosomes (41). This situation
is known as LOI for this particular DMR.
LOI at the IGF2-H19 locus is observed in patients
suffering from Beckwith-Wiedemann syndrome, in which neonates
suffer from organomegaly and frequently develop pediatric sarcomas,
including RMS (42). However, LOI
at the IGF2-H19 locus that develops independently of
Beckwith-Wiedemann syndrome is also observed in RMS (15,16)
as well as in several other malignancies in mice and humans. In all
these cases, the proliferation of malignant cells is driven by
overexpression of IGF2. These cells also have higher expression of
IGF1R and INSR because of the lack of H19-derived miR-675, which
downregulates the expression of IGF1R (28). What is most important in this
report, we provide for the first time evidence that miR-675 also
downregulates the expression of INSR.
Thus, in toto, LOI at the IGF2-H19 locus
leads to enhanced stimulation of cancer cells by the family of
insulin-like growth factors and INS due to upregulation of
autocrine IGF2 and, as a result of H19 downregulation, to the high
expression of IGF1R and INSR, which bind IGF2 and 1, as well as
INS.
Based on these findings, it was tempting to
postulate that demethylation of the DMR at the IGF2-H19 locus would
have the opposite effect, leading to attenuation of INS and
insulin-like growth factor signaling in RMS cells. To test this
hypothesis, we employed the DNMT inhibitor AzaC to widely
demethylate the DNA in RMS cells. We were aware that this
non-specific approach would also lead to the demethylation of
several other genes. To our surprise, however, exposure of RMS
cells to AzaC resulted in inhibition of cell proliferation that was
paralleled by a decrease in IGF2 expression, both at the mRNA and
protein levels, as well as an increase in H19-derived miR-675,
which led to downregulation of IGF1R and INSR expression. The
inhibitory effect of miR-675 on the expression of IGF1R and also
the INSR (as is shown here) in human RMS cells was also
accomplished by exposure of RMS cells to an H19 expression vector
or by transducing cells with miR-675.
We are aware that AzaC leads to demethylation of
several other genes, but our data tend to confirm a crucial role of
the IGF2-H19 locus in driving the pro-proliferative effect of INS
and insulin-like growth factor signaling in human RMS cells.
Moreover, a similar result was obtained with human ARMS and ERMS
cell lines. In RMS cells, AzaC at non-cytotoxic doses strongly
inhibited anchor-dependent cell proliferation, caused cell cycle
arrest in G2 phase, as well as decreased anchor-independent growth
in a colony-formation assay in soft agar in plastic dishes. Most
importantly, the effect of AzaC on the methylation state of the DMR
at the IGF2-H19 locus was confirmed in our study by DNA bisulfite
exposure and subsequent sequencing.
Our results are in accordance with previously
published study in which AzaC efficiently reactivated the
expression of silenced H19 by demethylation of the DMR within the
IGF2-H19 locus (43). By contrast,
a potent deacetylating agent, TsA, did not influence the expression
from IGF2-H19. This result is in agreement with results obtained
with murine cells in which the epigenetic state of the IGF2-H19
locus was regulated by methylation and not by acetylation (44,45).
AzaC is a chemical analogue of the cytosine
nucleoside, which is present in DNA and RNA. AzaC induces
antineoplastic activity by employing two mechanisms (46,47).
At low doses, it inhibits DNMT activity, causing hypomethylation
and synthesis of DNA. As a ribonucleoside, at high doses AzaC
incorporates into RNA, which leads to the disassembly of
polyribosomes and defective methylation and acceptor function of
transfer RNA and finally results in the inhibition of protein
synthesis. AzaC is employed in some clinical settings, for example,
as an antineoplastic drug to inhibit HIV and HTLV infection in
leukemia or to induce γ-globin synthesis in thalassemia patients
(48–52). Our results indicate that AzaC could
also be employed as a drug for RMS patients. This, however, will
require further study in animal xenotransplant models of RMS.
In conclusion, we have demonstrated for the first
time the in vitro efficacy of AzaC as a novel potential drug
for treatment of RMS. Our data also confirm a pivotal role for LOI
at the IGF2-H19 locus in the proliferation and migration state of
RMS cells. We also show that IGF1R is regulated by miR-675 in human
cells, as has been reported for murine cells, and for the first
time that miR-675 also regulates the expression of human INSR.
Further studies are needed to assess whether changes in the
methylation state of other genes also contribute to the observed
effects of AzaC.
Acknowledgements
Supported by Maestro grant 2011/02/A/NZ4/00035 and
Innovative Economy Operational Programme POIG.01.01.02-00-109/09 to
M.Z.R., and FNP ‘Homing PLUS’ programme co-financed from European
Union, Regional Development Fund to M.T.
References
|
1
|
Barr FG, Galili N, Holick J, Biegel JA,
Rovera G and Emanuel BS: Rearrangement of the PAX3 paired box gene
in the paediatric solid tumour alveolar rhabdomyosarcoma. Nat
Genet. 3:113–117. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Collins MH, Zhao H, Womer RB and Barr FG:
Proliferative and apoptotic differences between alveolar
rhabdomyosarcoma subtypes: a comparative study of tumors containing
PAX3-FKHR or PAX7-FKHR gene fusions. Med Pediatr Oncol. 37:83–89.
2001. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hazelton BJ, Houghton JA, Parham DM,
Douglass EC, Torrance PM, Holt H and Houghton PJ: Characterization
of cell lines derived from xenografts of childhood
rhabdomyosarcoma. Cancer Res. 47:4501–4507. 1987.PubMed/NCBI
|
|
4
|
Kelly KM, Womer RB and Barr FG: PAX3-FKHR
and PAX7-FKHR gene fusions in rhabdomyosarcoma. J Pediatr Hematol
Oncol. 20:517–518. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sandberg AA, Stone JF, Czarnecki L and
Cohen JD: Hematologic masquerade of rhabdomyosarcoma. Am J Hematol.
68:51–57. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sharp R, Recio JA, Jhappan C, et al:
Synergism between INK4a/ARF inactivation and aberrant HGF/SF
signaling in rhabdomyosarcomagenesis. Nat Med. 8:1276–1280. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gordon T, McManus A, Anderson J, Min T,
Swansbury J, Pritchard-Jones K and Shipley J; United kingdom
Children’s Cancer Study Group; United Kingdom Cancer Cytogenetics
Group. Cytogenetic abnormalities in 42 rhabdomyosarcoma: a United
Kingdom Cancer Cytogenetics Group Study. Med Pediatr Oncol.
36:259–267. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gurney JG, Severson RK, Davis S and
Robison LL: Incidence of cancer in children in the United States.
Sex-, race-, and 1-year age-specific rates by histologic type.
Cancer. 75:2186–2195. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Davis RJ, D’Cruz CM, Lovell MA, Biegel JA
and Barr FG: Fusion of PAX7 to FKHR by the variant t(1;13)(p36;q14)
translocation in alveolar rhabdomyosarcoma. Cancer Res.
54:2869–2872. 1994.PubMed/NCBI
|
|
10
|
Galili N, Davis RJ, Fredericks WJ,
Mukhopadhyay S, Rauscher FJ 3rd, Emanuel BS, Rovera G and Barr FG:
Fusion of a fork head domain gene to PAX3 in the solid tumour
alveolar rhabdomyosarcoma. Nat Genet. 5:230–235. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bennicelli JL, Advani S, Schäfer BW and
Barr FG: PAX3 and PAX7 exhibit conserved cis-acting transcription
repression domains and utilize a common gain of function mechanism
in alveolar rhabdomyosarcoma. Oncogene. 18:4348–4356. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Barr FG, Nauta LE, Davis RJ, Schäfer BW,
Nycum LM and Biegel JA: In vivo amplification of the PAX3-FKHR and
PAX7-FKHR fusion genes in alveolar rhabdomyosarcoma. Hum Mol Genet.
5:15–21. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Anderson J, Gordon A, Pritchard-Jones K
and Shipley J: Genes, chromosomes, and rhabdomyosarcoma. Genes
Chromosomes Cancer. 26:275–285. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Barr FG: Gene fusions involving PAX and
FOX family members in alveolar rhabdomyosarcoma. Oncogene.
20:5736–5746. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Anderson J, Gordon A, McManus A, Shipley J
and Pritchard-Jones K: Disruption of imprinted genes at chromosome
region 11p15.5 in paediatric rhabdomyosarcoma. Neoplasia.
1:340–348. 1999. View Article : Google Scholar
|
|
16
|
Casola S, Pedone PV, Cavazzana AO, Basso
G, Luksch R, d’Amore ES, Carli M, Bruni CB and Riccio A: Expression
and parental imprinting of the H19 gene in human rhabdomyosarcoma.
Oncogene. 14:1503–1510. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhan S, Shapiro DN and Helman LJ:
Activation of an imprinted allele of the insulin-like growth factor
II gene implicated in rhabdomyosarcoma. J Clin Invest. 94:445–448.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schneider G, Bowser MJ, Shin DM, Barr FG
and Ratajczak MZ: The paternally imprinted DLK1-GTL2 locus is
differentially methylated in embryonal and alveolar
rhabdomyosarcomas. Int J Oncol. 44:295–300. 2014.
|
|
19
|
El-Badry OM, Minniti C, Kohn EC, Houghton
PJ, Daughaday WH and Helman LJ: Insulin-like growth factor II acts
as an autocrine growth and motility factor in human
rhabdomyosarcoma tumors. Cell Growth Differ. 1:325–331.
1990.PubMed/NCBI
|
|
20
|
Wang W, Kumar P, Wang W, Epstein J, Helman
L, Moore JV and Kumar S: Insulin-like growth factor II and
PAX3-FKHR cooperate in the oncogenesis of rhabdomyosarcoma. Cancer
Res. 58:4426–4433. 1998.PubMed/NCBI
|
|
21
|
Hahn H, Wojnowski L, Specht K, et al:
Patched target Igf2 is indispensable for the formation of
medulloblastoma and rhabdomyosarcoma. J Biol Chem. 275:28341–28344.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Makawita S, Ho M, Durbin AD, Thorner PS,
Malkin D and Somers GR: Expression of insulin-like growth factor
pathway proteins in rhabdomyosarcoma: IGF-2 expression is
associated with translocation-negative tumors. Pediatr Dev Pathol.
12:127–135. 2009. View Article : Google Scholar
|
|
23
|
Rikhof B, de Jong S, Suurmeijer AJ, Meijer
C and van der Graaf WT: The insulin-like growth factor system and
sarcomas. J Pathol. 217:469–482. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Martins AS, Olmos D, Missiaglia E and
Shipley J: Targeting the insulin-like growth factor pathway in
rhabdomyosarcomas: rationale and future perspectives. Sarcoma.
2011:2097362011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gallagher EJ and LeRoith D: The
proliferating role of insulin and insulin-like growth factors in
cancer. Trends Endocrinol Metab. 21:610–618. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yoshimizu T, Miroglio A, Ripoche MA, et
al: The H19 locus acts in vivo as a tumor suppressor. Proc Natl
Acad Sci USA. 105:12417–12422. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lottin S, Adriaenssens E, Dupressoir T,
Berteaux N, Montpellier C, Coll J, Dugimont T and Curgy JJ:
Overexpression of an ectopic H19 gene enhances the tumorigenic
properties of breast cancer cells. Carcinogenesis. 23:1885–1895.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Keniry A, Oxley D, Monnier P, Kyba M,
Dandolo L, Smits G and Reik W: The H19 lincRNA is a developmental
reservoir of miR-675 that suppresses growth and Igf1r. Nat Cell
Biol. 14:659–665. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kimura S, Kuramoto K, Homan J, Naruoka H,
Ego T, Nogawa M, Sugahara S and Naito H: Antiproliferative and
antitumor effects of azacitidine against the human myelodysplastic
syndrome cell line SKM-1. Anticancer Res. 32:795–798.
2012.PubMed/NCBI
|
|
30
|
Thakur S, Feng X, Qiao Shi Z, Ganapathy A,
Kumar Mishra M, Atadja P, Morris D and Riabowol K: ING1 and
5-azacytidine act synergistically to block breast cancer cell
growth. PLoS One. 7:e436712012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Grymula K, Tarnowski M, Wysoczynski M,
Drukala J, Barr FG, Ratajczak J, Kucia M and Ratajczak MZ:
Overlapping and distinct role of CXCR7-SDF-1/ITAC and CXCR4-SDF-1
axes in regulating metastatic behavior of human rhabdomyosarcomas.
Int J Cancer. 127:2554–2568. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kerjean A, Dupont JM, Vasseur C, Le
Tessier D, Cuisset L, Pàldi A, Jouannet P and Jeanpierre M:
Establishment of the paternal methylation imprint of the human H19
and MEST/PEG1 genes during spermatogenesis. Hum Mol Genet.
9:2183–2187. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tarnowski M, Schneider G, Amann G, Clark
G, Houghton P, Barr FG, Kenner L, Ratajczak MZ and Kucia M: RasGRF1
regulates proliferation and metastatic behavior of human alveolar
rhabdomyosarcomas. Int J Oncol. 41:995–1004. 2012.PubMed/NCBI
|
|
34
|
Baylin SB, Herman JG, Graff JR, Vertino PM
and Issa JP: Alterations in DNA methylation: a fundamental aspect
of neoplasia. Adv Cancer Res. 72:141–196. 1998. View Article : Google Scholar
|
|
35
|
Bird A: The essentials of DNA methylation.
Cell. 70:5–8. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Leonhardt H and Cardoso MC: DNA
methylation, nuclear structure, gene expression and cancer. J Cell
Biochem. (Suppl 35): 78–83. 2000. View Article : Google Scholar
|
|
37
|
Esteller M: Cancer epigenetics: DNA
methylation and chromatin alterations in human cancer. Adv Exp Med
Biol. 532:39–49. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dammann RH, Kirsch S, Schagdarsurengin U,
Dansranjavin T, Gradhand E, Schmitt WD and Hauptmann S: Frequent
aberrant methylation of the imprinted IGF2/H19 locus and LINE1
hypomethylation in ovarian carcinoma. Int J Oncol. 36:171–179.
2010.
|
|
39
|
Smilinich NJ, Day CD, Fitzpatrick GV, et
al: A maternally methylated CpG island in KvLQT1 is associated with
an antisense paternal transcript and loss of imprinting in
Beckwith-Wiedemann syndrome. Proc Natl Acad Sci USA. 96:8064–8069.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bell AC and Felsenfeld G: Methylation of a
CTCF-dependent boundary controls imprinted expression of the Igf2
gene. Nature. 405:482–485. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Feinberg AP: Phenotypic plasticity and the
epigenetics of human disease. Nature. 447:433–440. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Feinberg AP: The two-domain hypothesis in
Beckwith-Wiedemann syndrome. J Clin Invest. 106:739–740. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lynch CA, Tycko B, Bestor TH and Walsh CP:
Reactivation of a silenced H19 gene in human rhabdomyosarcoma by
demethylation of DNA but not by histone hyperacetylation. Mol
Cancer. 1:22002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Grandjean V, O’Neill L, Sado T, Turner B
and Ferguson-Smith A: Relationship between DNA methylation, histone
H4 acetylation and gene expression in the mouse imprinted Igf2-H19
domain. FEBS Lett. 488:165–169. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Svensson K, Mattsson R, James TC, Wentzel
P, Pilartz M, MacLaughlin J, Miller SJ, Olsson T, Eriksson UJ and
Ohlsson R: The paternal allele of the H19 gene is progressively
silenced during early mouse development: the acetylation status of
histones may be involved in the generation of variegated expression
patterns. Development. 125:61–69. 1998.
|
|
46
|
Jones PA, Taylor SM and Wilson VL:
Inhibition of DNA methylation by 5-azacytidine. Recent Results
Cancer Res. 84:202–211. 1983.PubMed/NCBI
|
|
47
|
Taylor SM and Jones PA: Mechanism of
action of eukaryotic DNA methyltransferase. Use of
5-azacytosine-containing DNA. J Mol Biol. 162:679–692. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hollenbach PW, Nguyen AN, Brady H,
Williams M, Ning Y, Richard N, Krushel L, Aukerman SL, Heise C and
MacBeth KJ: A comparison of azacitidine and decitabine activities
in acute myeloid leukemia cell lines. PLoS One. 5:e90012010.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Greggs WM 3rd, Clouser CL, Patterson SE
and Mansky LM: Discovery of drugs that possess activity against
feline leukemia virus. J Gen Virol. 93:900–905. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Dapp MJ, Clouser CL, Patterson S and
Mansky LM: 5-Azacytidine can induce lethal mutagenesis in human
immunodeficiency virus type 1. J Virol. 83:11950–11958. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Clouser CL, Patterson SE and Mansky LM:
Exploiting drug repositioning for discovery of a novel HIV
combination therapy. J Virol. 84:9301–9309. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ley TJ, DeSimone J, Anagnou NP, Keller GH,
Humphries RK, Turner PH, Young NS, Keller P and Nienhuis AW:
5-Azacytidine selectively increases gamma-globin synthesis in a
patient with beta+ thalassemia. N Engl J Med. 307:1469–1475. 1982.
View Article : Google Scholar : PubMed/NCBI
|