Introduction
Angiosarcoma (AS) is a rare and aggressive
malignancy of the endothelium (reviewed in refs. 1,2).
Angiosarcoma has no single identified cause. Rather, mutations in
several different genes have been reported. Most recently, Bejhati
et al reported that mutations in PTPRB and PLCG1 were
detected in 10/39 and 3/34 tumors, respectively (3). In addition, constitutive activation
of KRAS-2 (4–6) and VEGF receptor 2 (7) have been documented. Both of these
signal through the mitogen-activated
protein/extracellular-regulated kinase (MAPK/ERK) signaling
pathway. Consistent with this, we have reported that AS shows focal
to widespread ERK activity and expresses ERK-responsive genes
(8). Furthermore, canine
angiosarcoma tumorgrafts are sensitive to inhibitors that target
MAPK/ERK kinase (MEK), the upstream activator of ERK (8). These data indicate the MEK/ERK
pathway plays a central role in AS tumor growth.
MEK 1 and 2 are kinases that drive diverse basic
biological processes such as cellular proliferation and cellular
survival. Aberrant activation of these kinases has been linked with
developmental syndromes and to as many as one-third of all cancers
(reviewed in refs. 9,10). While MEK activation is
predominately associated with melanoma (11), MEK dependency has been documented
in a variety of other cancers, including osteosarcoma (12), Ewing sarcoma (13), fibrosarcoma (10,14),
and Kaposi sarcoma (15). Thus,
the MEK/ERK pathway is a therapeutic target with a broad spectrum
of applications.
Despite the well-documented role of MEK signaling in
cancer, MEK inhibitors historically have had limited utility in the
clinic. The MEK1/2 inhibitor CI-1040 showed poor efficacy in Phase
II study (16). PD0325901, a
CI-1040 derivative, also showed poor tumor response in Phase II
clinical study (17), and dose
increases were limited by neurological and ocular toxicities
(18). Currently, trametinib is
the only FDA-approved MEK inhibitor for advanced melanoma. Even
with this success, trametinib has failed to show additional benefit
in patients who had been treated with BRAF inhibitors (19). Additional therapeutic strategies
are needed to overcome dose-response and resistance mechanisms.
Combinations of multiple drugs having different
mechanisms of action have been used effectively to treat diseases
such as HIV, cancer, and bacterial infections (20–22),
but the combined effects of drugs are not easily predicted. The
combination often acts like a third drug with effects that are
distinct from those of the original drugs (23). Moreover, the interaction of the
combined drugs can be influenced by the cellular or genetic context
in which they meet. Such interactions between drugs can promote
greater selectivity, efficacy, lower toxicity, and delayed
resistance, but they can also be antagonistic or promote greater
toxicity. We and others have observed that one ratio of combined
drugs may have a synergic effect but a different ratio of the same
drugs may act in an antagonistic fashion (23). Thus, designing a combinatorial
therapy first requires a rigorous in vitro evaluation to
determine the optimal ratios and doses to elicit the greatest
response. Since their interaction can be influenced by the cellular
or genetic context, an in vitro evaluation must be performed
for each tumor type tested. Finally, because strategies that are
additive or synergic for tumor response may instead be more toxic,
any new combination therapy requires an equally rigorous in
vivo evaluation of toxicity and efficacy.
Herein we report our efforts to identify drugs that
synergize with the MEK1/2 inhibitor PD0325901 in order to design a
more effective therapy for angiosarcoma. Drugs were selected based
on their ability to inhibit 11 of the conserved cancer pathways
(24). The goal of these tests was
to identify the optimal drug combination, i.e., the combination
showing the greatest additive or synergic interaction with
effective inhibition of cell viability at the lowest concentration.
Using a systematic approach, we have discovered that angiosarcomas
are insensitive to mTOR inhibition. However, treatment with
nanomolar levels of an mTOR inhibitor renders these cells as
sensitive to MEK inhibition as melanoma with mutant BRAF. Similar
results were observed in B-Raf wild-type melanoma and in
vivo, where treatment of canine AS (cAS) tumorgrafts with MEK
and mTOR inhibitors is more effective than monotherapy. Our results
show that a low dose of an mTOR inhibitor can dramatically enhance
the response to MEK inhibition and thus may widen the field of
applications for MEK-targeted therapy.
Materials and methods
Cell culture
cAS primary isolate VCT115 was grown as previously
described (8). VCT220, VCT345, and
VCT511 were isolated from cAS tumor samples as previously described
(8) and were grown in DMEM
containing 10% heat-inactivated fetal bovine serum (FBS; Life
Technologies, Carlsbad, CA, USA) and 1% penicillin/1% streptomycin
(Life Technologies). VCT261e was isolated from cAS tumor as
previously described (8) and grown
in EGM (Lonza, Basel, Switzerland) supplemented with EGM2
SingleQuots (Lonza). The melanoma-derived SK-MEL28 cells were grown
as described, and WM-3211 (Coriell Institute, Camden, NJ, USA)
cells were grown as previously described (25,26).
In vitro combination index studies
PD0325901, sorafenib tosylate, dasatinib (LC
Laboratories, Woburn, MA, USA), rapamycin doxorubicin, Nutlin-3a,
SGX-523 (Selleck, Houston, TX, USA), and KT5270 (Santa Cruz
Biotechnology, Dallas, TX, USA) were prepared in DMSO
(Sigma-Aldrich, St. Louis, MO, USA). Cells were seeded into a
96-well plate using an epMotion 5075 pipetting system (Eppendorf,
Hamburg, Germany). Treatments began when cells reached 30%
confluency. Cells were treated for 72 h. Cell viability was
measured using the CellTiter 96 Aqueous Non-Radioactive Cell
Proliferation Assay (Promega, Madison, WI, USA) according to the
manufacturer’s instructions. Assays were measured using a Benchmark
Plus microplate spectrophotometer (Bio-Rad, Hercules, CA, USA) at
490 and 700 nm reference wavelengths. Cell viability was performed
twice in duplicate wells. Treated wells were normalized to
non-treated wells and then were normalized to DMSO-treated plates.
The concentration of compound required to cause 50% inhibition of
cell viability (IC50) was calculated (23). A combinatorial index was calculated
following Chou and Talalay (23).
If a drug combination resulting in synergy (CI<1) that
combination was repeated for a total of three separate experiments
in duplicate wells. The IC50 and CI were then calculated
as previously described (23).
Western blot analysis
The cAS primary SK-MEL28 and WM-3211 cells were
seeded in the appropriate medium and were incubated overnight.
Cells were grown to 30% confluency, then treated with a drug
combination near the IC60 of the optimal drug molar
ratio for 72 h. Total cell lysates were collected in RIPA buffer
[50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 2 mM
Na3VO4, 20 mM sodium pyrophosphate, 1% Triton
X-100, 1% sodium deoxycholate, and 0.1% SDS, with Complete
EDTA-free Protease Tablets (Roche Corp., Palo Alto, CA, USA)] and
were sonicated three times using a Misonix Sonicator 3000
(Farmingdale, NY, USA). Protein concentrations were determined
using the BCA Protein Assay kit (Pierce, Rockford, IL, USA).
Cellular lysates were resolved in Novex Pre-Cast Tris-glycine gels
(Life Technologies) and then transferred onto polyvinylidene
fluoride (PVDF) membranes (Millipore, Billerica, CA, USA).
Membranes were blocked with 10% non-fat milk and then incubated
with antibodies against phospho-ERK (Thr202/Tyr204) (E10; Cell
Signaling, Danvers, MA, USA), ERK (Cell Signaling), α-tubulin
(Sigma-Aldrich), phospho-S6 (S235/236) (2F9; Cell Signaling)
phospho-4E-BP1 (S65) (Cell Signaling), 4E-BP1 (53H11, Cell
Signaling), and Bim (C34C5, Cell Signaling). The membranes were
washed three times with TBST (50 mM Tris, 150 mM NaCl, and 0.1%
Tween-20) and then incubated with the appropriate HRP-conjugated
secondary antibodies (KPL, Gaithersburg, MD, USA) overnight at 4°C.
Washed blots were incubated with Super Signal West Pico
Chemiluminescent Substrate (Fisher Scientific, Pittsburgh, PA, USA)
and were exposed to Hyblot CL film (Denville Scientific, South
Plainfield, NJ, USA). The film was processed in an X-OMAT 2000A
Processor (Kodak, Rochester, NY, USA).
In vivo combination studies
Mice were bred and maintained according to
established guidelines and a protocol approved by VARI’s
Institutional Animal Care and Use Committee. For the toxicity
studies, athymic nudes were treated at a 4:1 molar ratio
PD0325901:temsirolimus (Selleck) (with a range of 4.38/2.35 to
3.5/1.87 mg/kg) for two weeks. PD0325901 was administered daily by
oral gavage in 100 μl of 0.5% hydroxylpropyl methylcellulose plus
0.2% Tween-80 (27). Temsirolimus
was administered i.p. in 200 μl using a 5 days on, two days off
schedule. A 50 mg/ml stock of temsirolimus was prepared in 100%
ethanol. On day of injection, it was diluted with 5% Tween-80, 5%
polyethyleneglycol-400 for a final concentration of 0.4% ethanol as
previously described (28).
Treatment was initiated when tumors reached 50–100 mm3.
Tumors were randomly selected into five different treatment arms:
PD0325901 (3.5 mg/kg) and temsirolimus (1.87 mg/kg), PD0325901
single therapy (3.5 mg/kg), temsirolimus single therapy (1.87
mg/kg), vehicle, and non-treated. Monotherapy animals were given
the corresponding vehicle on the appropriate schedule. Vehicle
animals were given both vehicles on the appropriate schedule. Mice
were weighed and monitored three times a week. Mice were sacrificed
when tumors reached 1000 mm3 or treatment day 38, which
ever came first, and terminal bleeds were collected for biochemical
serum analysis (Abaxis VetScan Blood Chemistry Analyzer, Abaxis,
Union City, CA, USA). cAS cardiac-derived tumorgrafts were
characterized and implanted into athymic nude mice as previously
described (8).
Immunohistochemistry
Formalin-fixed, paraffin-embedded tumors were
sectioned and immunostained with optimized standard protocols using
a Ventana Discovery XL instrument (Ventana Medical Systems, Tucson,
AZ, USA) and antibodies against phospho-ERK (Thr202/Tyr204) (20G11;
Cell Signaling), CD31/PECAM-1 (Lab Vision, Kalamazoo, MI, USA),
phospho-S6 (S235/236) (2F9; Cell Signaling), and Ki67 (ab833;
ABCAM, Cambridge, MA, USA). Slides were incubated with
HRP-conjugated anti-rabbit IgG secondary antibody (Ventana Medical
Systems) and developed with 3-3′-diaminobenzidine (DAB) chromagen
substrate. Images were acquired using a Nikon E800 Epifluorescent
microscope equipped with a Spot RT3 CCD camera (Diagnostic
Instruments, Sterling Heights, MI, USA) and Spot Advanced
software.
Results
Synergism in vitro
We have published data showing that human and canine
angiosarcomas express focal to widespread active ERK and are
sensitive to MEK inhibition (8).
In this follow-up study, we wanted to identify drugs or compounds
that synergize with the MEK inhibitor PD0325901. We treated five
canine angiosarcoma cell isolates with different ratios of the
drugs and evaluated the effect on cell viability after a 72-h
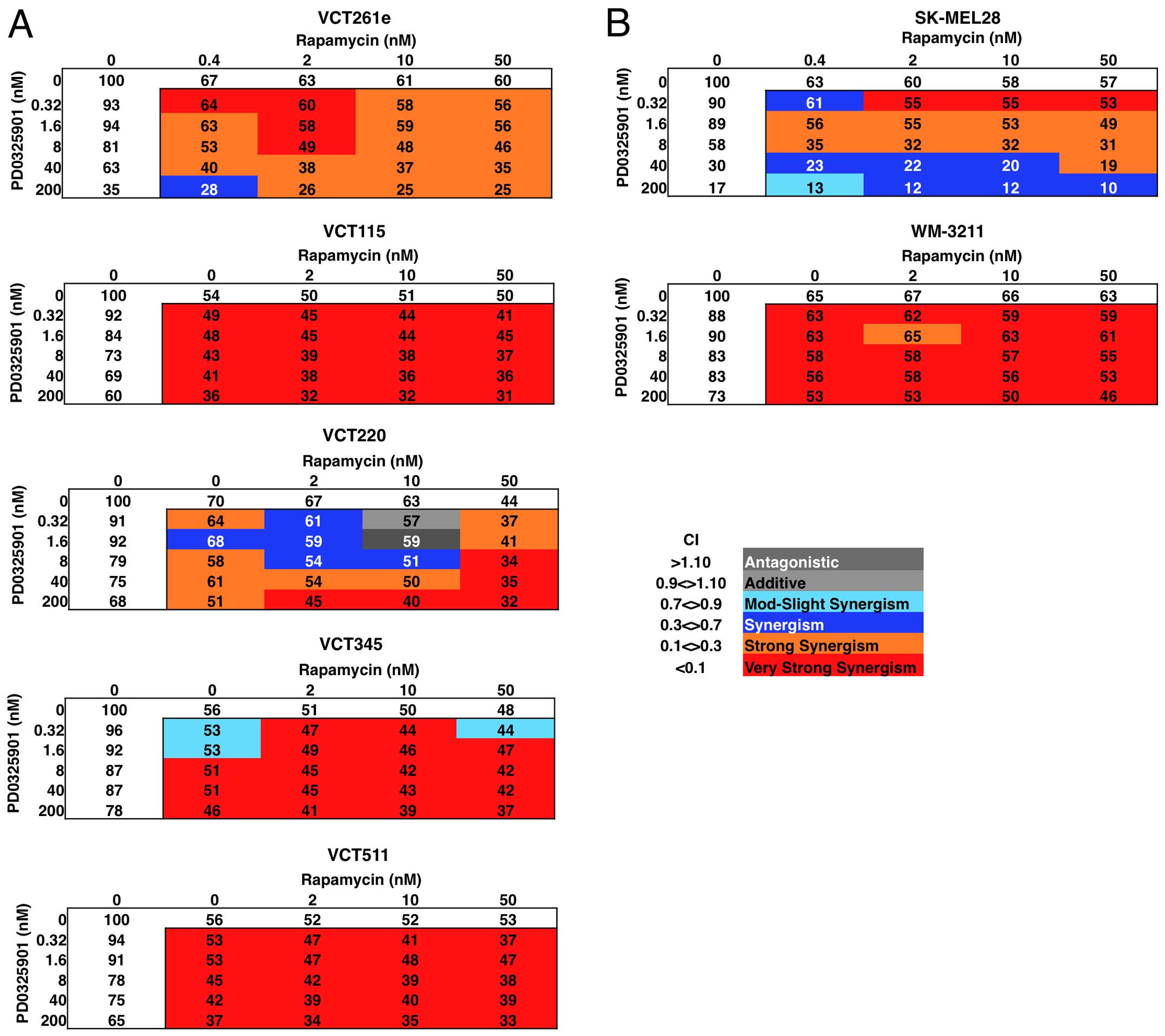
treatment (Fig. 1A). Among the
drugs we tested, mTOR inhibitor rapamycin showed the strongest
synergy with PD0325901 even at subnanomolar concentrations
(Table I, and data not shown).
PD0325901 plus rapamycin had the greatest synergy (CI≤0.08). 4 of
the 5 angiosarcoma primary cell isolates had an optimal 4:1 molar
ratio of PD0325901:rapamycin and the 5th had a 4:5 ratio (Table I, and data not shown).
 | Table ICalculated IC50 for a
single dose of PD0325901, rapamycin, or both at the optimal molar
ratios for cAS primary cell isolate VCT261e and the melanoma cell
lines SK-MEL28 and WM-3211. |
Table I
Calculated IC50 for a
single dose of PD0325901, rapamycin, or both at the optimal molar
ratios for cAS primary cell isolate VCT261e and the melanoma cell
lines SK-MEL28 and WM-3211.
| cAS | Melanoma |
|---|
|
|
|
|---|
| Treatment | VCT261e | SK-MEL28 | WM-3211 |
|---|
| PD0325901 |
| IC50
(nM) | 150±30 | 20±4 | >1,000 |
| Rapamycin |
| IC50
(nM) | >50 | 7±11 | >50 |
| PD0325901 +
Rapamycin |
| IC50
(nM) | 11±6 | 6±8 | 250±250 |
| CI | 0.07 | 0.07 | 0.0003 |
| Molar ratio | 4:1 | 4:5 | 4:1 |
To determine whether this response was unique to
angiosarcoma or was a consequence of their MEK dependency, we
performed the same experiments in melanoma-derived cell lines that
were MEK-dependent (SK-MEL28, which has a BRAF V600E mutation) and
MEK-independent (WM-3211, which is BRAF wild-type but contains a
c-kit L576P mutation) (Fig. 1B).
SK-MEL28 were sensitive to MEK and mTOR inhibition, but showed
greater sensitivity to combined inhibition. In contrast, the
WM-3211 line was insensitive to either MEK or mTOR inhibition but
showed enhanced sensitivity to combination therapy (Table I). Thus, treatment with an mTOR
inhibitor renders angiosarcomas as sensitive to MEK inhibition as
melanomas having mutant BRAF, and it renders MEK
inhibitor-resistant cells sensitive to MEK inhibition.
Increased inhibition of canine AS
tumorgrafts using combined MEK and mTOR inhibitors
In vitro combination matrices detailed the
PD0325901 and rapamycin dual treatment at 4:1 molar ratio was the
most efficacious. This dual treatment regimen was then examined
in vivo on patient derived xenografts. Before the drug study
was initiated, in vivo toxicity testing was performed to
determine whether the combined therapy was safe in mice. For these
studies we used the mTOR inhibitor temsirolimus, which is a
pro-drug that is metabolized to yield rapamycin in vivo
(29). Temsirolimus was used
because, compared with rapamycin, it has a more favorable
pharmacokinetic profile and greater solubility in water (30). Using 4:1 combinations of PD0325901
plus temsirolimus, over a two-week period we observed significant
(>10%) weight loss, elevated serum phosphorus, and dry skin when
daily doses of PD0325901 exceeded 4 mg/kg. In contrast, when the
dose of PD0325901 was 3.5 mg/kg, we found no adverse effects over
two weeks.
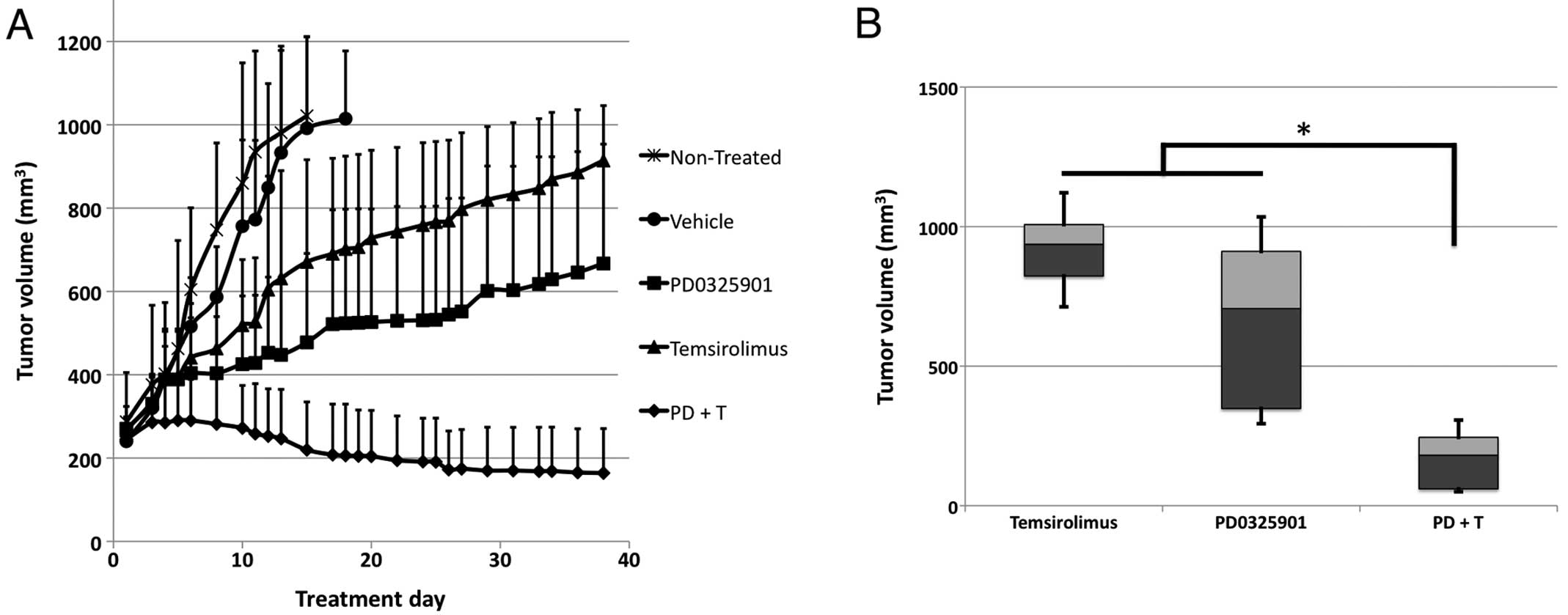
Consequently, a 4:1 molar ratio of PD0325901 (at 3.5
mg/kg) and temsirolimus (at 1.9 mg/kg) was used to treat mice
bearing canine cardiac angiosarcoma tumorgrafts. After only two
weeks, the PD0325901/temsirolimus combination decreased the tumor
volume. By three weeks, all vehicle control tumorgrafts had grown
to 1000 mm3, while tumorgrafts treated with
PD0325901/temsirolimus had virtually no growth. On day 38 of
treatment, the tumors were significantly smaller than those treated
with either PD0325901 or temsirolimus alone (Fig. 2A and B). No weight loss was found
in mice treated with the combination over the treatment period.
Thus, the combination of MEK and mTOR inhibition produced an
efficacious response with no observable toxicities.
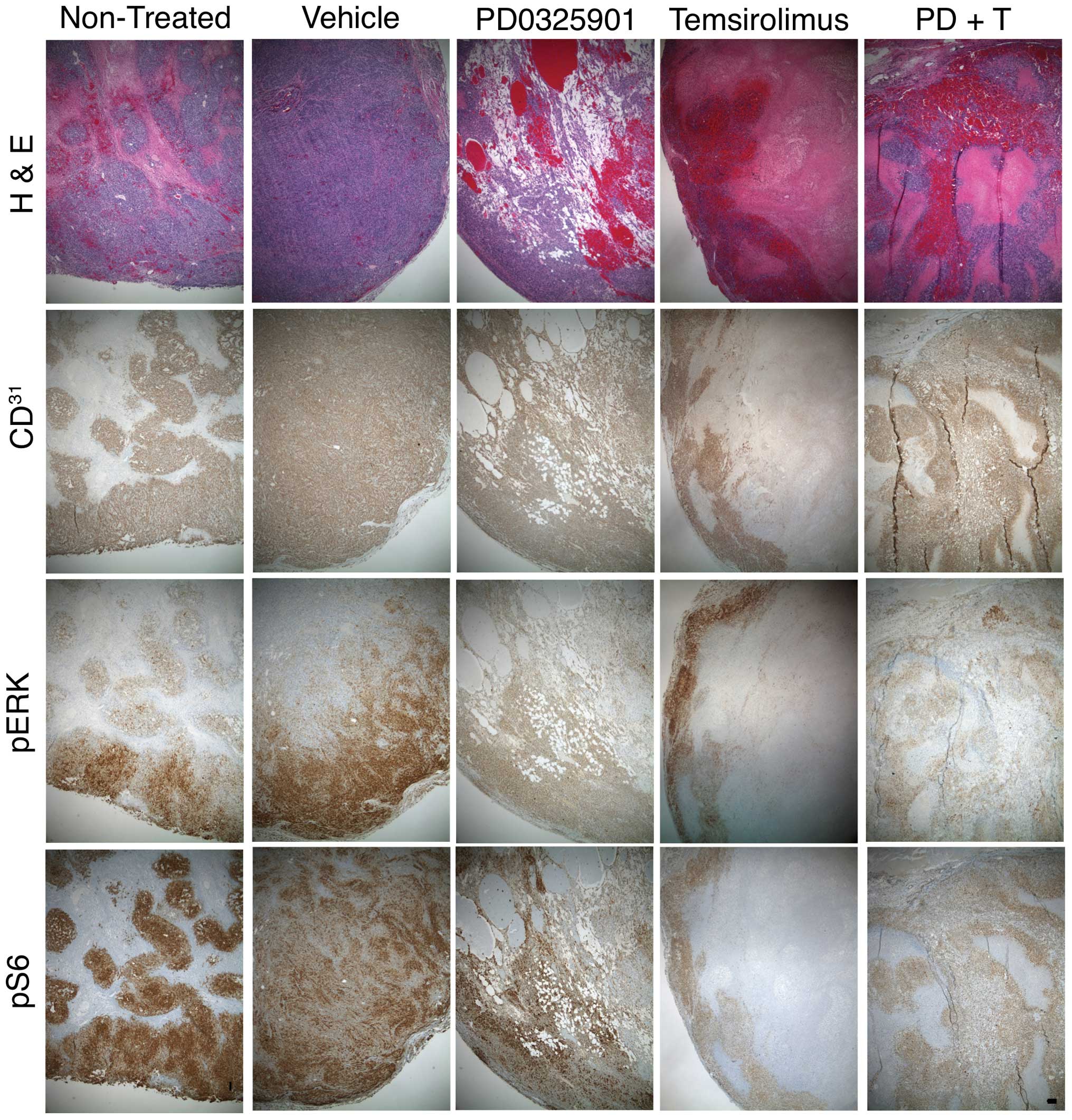
To determine the morphologic consequences of these
treatments, thin sections of formalin-fixed, paraffin-embedded
tumors were evaluated by H&E staining. cAS tumorgrafts showed a
complex architecture as previously described (8). At the tumor periphery,
CD31+ cells were arranged within dense tumor nests and
lined poorly formed vascular channels. In the tumor interior, large
irregular blood vessels were lined with CD31+ cells
that, in places, were multiple cell layers thick. Such tumors
contain large areas of necrosis and fibrin deposition from
intratumor infarcts caused by hemorrhage (Fig. 3).
With PD0325901 treatment, these necrotic regions and
fibrin deposits were replaced by areas containing small, irregular,
perfused vessels lined with CD31+ cells. Temsirolimus
had the opposite effect, producing a tumor interior that was mostly
necrotic and fibrotic, with CD31+ cells lining the tumor
cortex and few areas of CD31+ cells in the tumor
interior. The combination therapy showed a mix of these two
architectures. There was an increase in small, perfused,
CD31+ lined vessels, but the necrotic regions were
increased relative to PD0325901 treatment alone (Fig. 3). These results indicate that the
effects of each drug on tumor architecture were independent of each
other.
To determine the molecular consequences of these
treatments, we next used immunohistochemistry to examine changes in
pERK and pS6 in treated tumorgrafts. PD0325901 reduced pERK
staining intensity at the tumor periphery; there was no noticeable
decrease in the weak pERK1/2 signal in the tumor interior.
PD0325901 did not appear to change pS6 staining intensity.
Temsirolimus reduced pS6 staining at the cortex but the residual
interior signal was still present. Temsirolimus produced no
decrease of pERK staining intensity in viable cells. Tumors from
mice treated with PD0325901 plus temsirolimus showed reduced pERK
and pS6 staining (Fig. 3). These
results indicate that each drug effectively inhibited its intended
target, but their combination did not enhance their effects on
these targets.
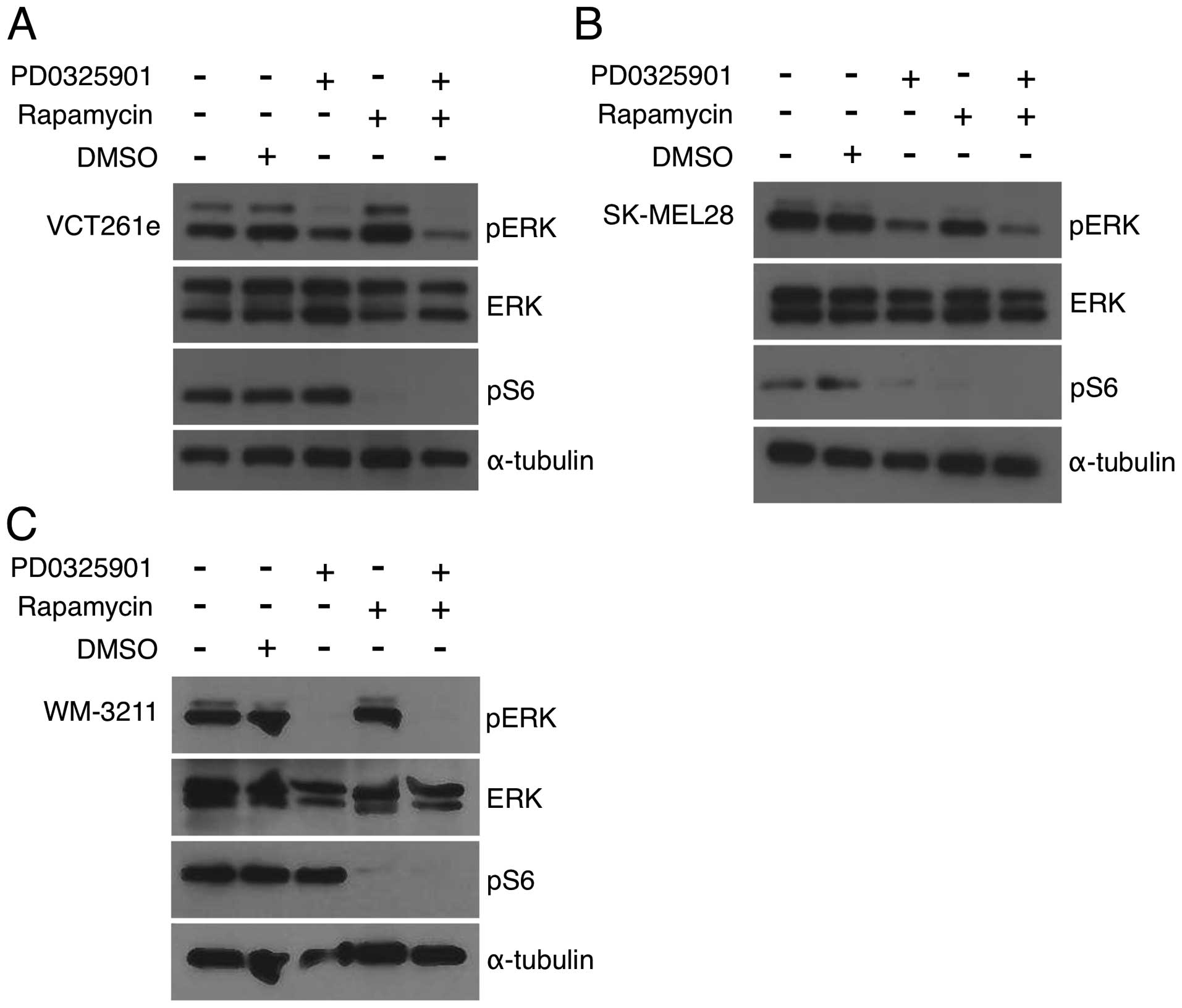
Recent studies of rhabdomyosarcoma have concluded
that combined inhibition of MEK and mTOR is synergic because of
anti-counteractive interaction: each drug blocks reciprocal
activation of the other pathway (31,32).
To determine whether the same is true for AS, we performed
immunoblots with antibodies against phosphorylated ERK and S6.
Canine angiosarcoma primary cell isolates were treated with each
drug alone or with a 4:1 (PD0325901:rapamycin) ratio (Fig. 1A). Treatment with 40 nM PD0325901
alone resulted in no effect or minimal reduction of pERK, which is
consistent with PD0325901 at this dose having a minimal effect on
cell viability. Rapamycin treatment at 100 nM was sufficient to
decrease pS6 to nearly undetectable levels in cAS primary isolates
(Fig. 4A) and to reduce the pS6
signal to a level consistent with pathway inhibition (Fig. 4A and B). The addition of rapamycin
with PD0325901 resulted in only a minor reduction of pERK (Fig. 4A). Similar results were observed in
other cAS primary cells (data not shown). SK-Mel28 cells were
treated alone and PD0325901 and rapamycin at a 4:5 molar ratio.
PD0325901-treated SK-MEL-28 reduced pERK and pS6 levels (Fig. 4B). WM-3211 cells were treated with
PD0325901 and rapamycin alone and at a 4:1 molar combination. While
PD0325901 and rapamycin were sufficient to reduce pERK and pS6
levels, respectively, combination treated did not further reduce
phosphorylation levels (Fig. 4C).
While we see no evidence of direct reciprocal activation, these
data suggest that mTOR inhibition sensitizes cells to even small
reductions in MEK signaling.
PD0325901 and rapamycin largely results
in individual pathway inhibition
Since combinatorial MEK and mTOR inhibition showed
no reciprocal activation or synergistic decrease in ERK and S6
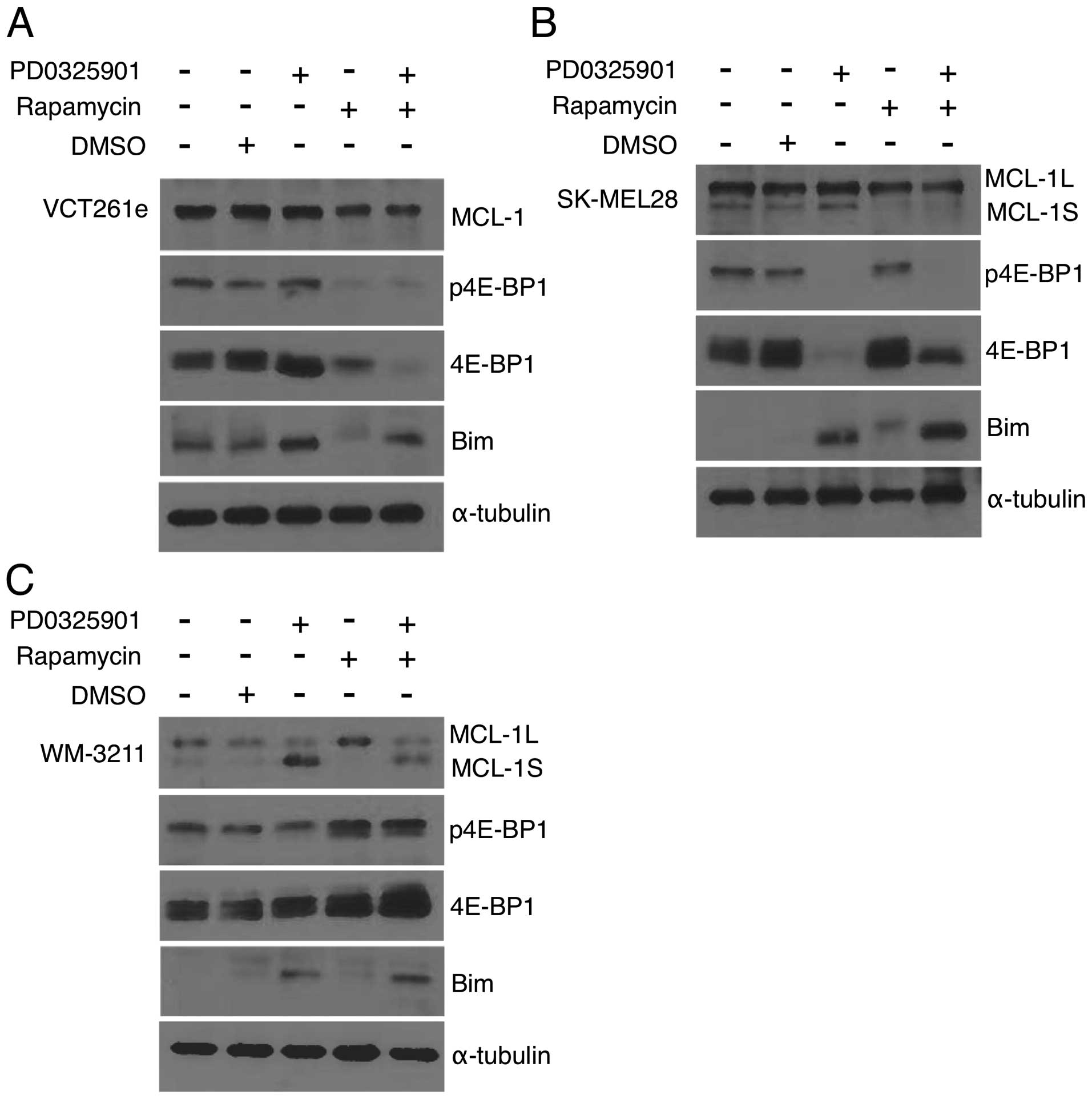
phosphorylation, we next looked at levels of 4E-BP1.
Phosphorylation of 4E-BP1 is reported to be regulated through both
the MEK and mTOR pathways in MEK driven tumors (33,34).
4E-BP1 inhibits the 5′ mRNA cap recognition of eIF4F complex
repressing translation (35).
Phosphorylation of 4E-BP1 impedes the binding of 4E-BP1 to eIF4E
allowing for eIF4F complex formation and translation initiation
(36). For these experiments cells
were treated with drugs at the previously determined optimal ratio.
For VCT261e cAS primary cell isolates, 4E-BP1 was unaffected by MEK
inhibition. In contrast, rapamycin caused a marked reduction in
total 4E-BP1 levels (Fig. 5A).
This effect was more pronounced in cells treated with both agents.
The opposite was observed in SK-MEL-28 cells (Fig. 5B). MEK inhibition reduced 4E-BP1
levels, while rapamycin had no effect. Combined treatment with both
drugs caused a partial reduction in 4E-BP1 levels and a loss of
phosphorylation. In WM-3211 cells, neither drug markedly altered
4E-BP1 expression or phosphorylation (Fig. 5C).
Pro-survival MCL-1 protein levels were analyzed.
MCL-1 is cooperatively regulated by MEK and mTOR in the MEK driven
OCM1A melanoma cell line (33),
and mTOR is known to induce cell survival through upregulation of
MCL-1 protein (37). MCL-1 can
exist in two splice variants. MCL-1L is known as a pro-survival
protein. MCL-1S is hypothesized to bind and inhibit MCL-1L to block
cell survival. Only one splice form was detected in canine cells.
In VCT261e cells, expression of MCL-1 was unaffected by PD0325901
and modestly reduced by rapamycin (Fig. 5A). A similar result was achieved in
SK-MEL-28 cells where rapamycin reduced levels of both the MCL-1L
and MCL-1S (Fig. 5B). However, in
WM-3211 cells while PD0325901 alone induced expression of MCL-1S
only and rapamycin alone induced the expression of MCL-1L (Fig. 5C), the combination treatment did
not have a marked effect.
Finally, we examined the expression of pro-apoptotic
Bim. For all three cell types we observed PD0325901, alone or in
combination with rapamycin, induced Bim expression (Fig. 5A–C) while rapamycin had no effect.
With the exception of 4E-BP1, there appears to be no cooperative
modulation of these pathways. In fact, two independent prodeath
pathways were present. Rapamycin decreased pro-survival MCL-1, and
PD0325901 increased pro-apoptotic Bim expression (Fig. 5A). Collectively, our data indicate
in these cell lines that although PD0325901 induces expression of
the pro-apoptotic protein Bim and rapamycin reduces levels of the
pro-survival protein MCL-1L, there is no evidence that these two
inhibitors have a common target.
Discussion
Since angiosarcomas are rare compared to other
cancers, such as breast and lung cancer, they are relatively
understudied. Perhaps this is why so few advances have been made in
the treatment of angiosarcomas in the past 20 years. Our most
effective weapon against angiosarcoma is still a surgeon’s scalpel.
Radiation therapy or chemotherapies such as doxorubicin can delay
progression, but they cannot prevent it. One reason for our slow
progress is the lack of a clear molecular target. Angiosarcoma has
no single identifiable genomic cause; mutations in several
different genes have been reported. Most recently, Bejhati et
al reported mutations in the angiogenesis-related genes
PTPRB and PLCG1 in 10/39 and 3/34 tumors,
respectively (3). In addition,
constitutive activation of KRAS-2 (4–6) and
of VEGF receptor-2 (7) has been
documented. Of note, several of these signal through the MEK/ERK
signaling pathway (38–41).
Recently, we published a study showing that human
and canine angiosarcomas express focal to widespread active ERK and
are sensitive to MEK inhibition (8). Thus, targeting MEK signaling may be
an effective therapy. The goal of this follow-up study was to
identify drugs or compounds that synergize with the MEK inhibitor
PD0325901 in order to develop a more effective treatment. A recent
study highlighting the potential benefits of combination therapies
for oncology stated that intratumor heterogeneity, the rapid
evolution of bypass mechanisms, and genomic instability lessen the
likelihood that monotherapies will provide sustained patient
benefit (42). The authors
conclude, and we agree, that combination therapy is the future for
treating oncology patients.
A recent survey of clinical articles (43) involving drug combinations found
that the term synergy is frequently used without an appropriate
understanding of either the underlying concept or of the
computational approaches to evaluate it, only 20% of preclinical
research articles used appropriate methods. This is a concern since
the misinterpretation of this concept can adversely impact the
formulation of drug combinations in clinical studies. Each of the
computational approaches we can use to evaluate drug interactions
has its strengths and weaknesses (23,44,45).
We chose the methods of Chou and Talalay (23) because they are commonly used to
objectively evaluate synergy and because the software needed to
make the calculations is freely available. We used cell viability
data to calculate the combination index (CI), which is used to
objectively evaluate whether two drugs interact in an additive,
synergic, or antagonistic fashion. A CI value <1 is considered
synergic and a value >1 is considered antagonistic. A CI of 1 is
additive.
Using this approach, we have discovered that
melanoma and angiosarcoma were insensitive to mTOR inhibition.
Treatment with nanomolar levels of mTOR inhibitor, however,
rendered these cells as sensitive to MEK inhibition as melanoma
with mutant BRAF. This effect was also seen in vivo,
treatment of tumorgrafts with MEK plus mTOR inhibitors was more
effective than monotherapy. Furthermore, MEK-insensitive WM-3211
cells responded to MEK inhibitors when treated simultaneously with
nanomolar amounts of rapamycin. This shows that a low dose of an
mTOR inhibitor can dramatically enhance the response to MEK
inhibition and potentially widen the applications of MEK-targeted
therapy.
Combinations of MEK and mTOR inhibitors have been
tested in several carcinomas, including lung cancer (46–48),
melanoma (49), colorectal cancer
(50), and pancreatic cancer
(51), but their combined effect
on sarcomas have only been reported for rhabdomysarcoma (31,32).
MEK 1 and 2 have essential roles in fundamental cellular activities
including cell survival, proliferation, motility, and
differentiation, as well as in vital activities such as
angiogenesis and immune response (52,53).
Similarly, the PI3K/AKT/mTOR survival pathway regulates diverse
processes such as cell proliferation, differentiation, metabolism,
cytoskeletal organization, apoptosis, and cancer-cell survival
(54). Mechanistically, two drugs
can show synergy because they have anti-counteractive actions,
complementary actions, or facilitating actions (55). Because of the diverse functions of
the MEK1/2 and the PI3K/AKT/mTOR pathways, it is not clear how
inhibitors of each pathway will interact or synergize.
Understanding the biologic mechanisms underlying synergy is
important to help identify biomarkers of response as well as novel,
efficacious combinations.
Recent studies in rhabdomyosarcoma have concluded
that combined inhibition of MEK and mTOR is synergic because of
anti-counteractive interaction: each drug blocks reciprocal
activation of the other pathway (31,32).
However, these results are based on in vitro effects and do
not take into account in vivo tumor:stromal interactions.
Moreover, the data (immunoblots) are qualitative and cannot be used
objectively to determine whether the effects are synergic,
additive, or antagonistic. In angiosarcomas we see no convincing
evidence by immuno-blotting or immunohistochemistry that either
drug promotes activation of the other pathway. Instead, our data
indicate that independent pathways are affected. In vitro,
PD0325901 alone increases the pro-apoptotic protein Bim while
treatment with rapamycin decreases pro-survival MCL-1. In
vivo, PD0325901 results in vascular changes, and temsirolimus
affects survival. Based on this, we hypothesize that these pathways
signal independently to promote angiosarcoma growth and
vascularization.
An alternative possibility is that these signaling
pathways converge on a common, not-yet-identified target or
activity that is required for angiosarcoma progression. For
example, several studies indicate that each of these pathways
regulates angiogenesis. Endothelial AKT overexpression increases
in vivo angiogenesis (56),
and rapamycin has been reported to inhibit tumor angiogenesis in
xenografts (57). Similarly,
constitutive expression of MEK1 in fibroblasts elevates expression
of VEGF mRNA through the binding of the transcription factors Sp1
and AP-2 to its promoter region (58). In addition, the treatment of
endothelial cells with VEGF causes activation of both ERK 1 and 2
(59). Anthrax lethal factor, a
protease that inactivates MEK1 and 2 (60) as well as mitogen-activated protein
kinases 3, 4, 6 and 7 (61),
substantially inhibits vascularization in mouse xenograft studies
(62) and in models of retinal
angiogenesis (63,64). Thus, the PI3K/AKT/mTOR and MEK/ERK
signaling pathways may converge on an angiogenesis-related target
required for angiosarcoma progression.
Acknowledgements
We thank Lisa Turner for technical assistance, David
Nadziejka for writing assistance, and Dr J. Bromberg-White and Agni
Naidu for helpful discussion and comments on the manuscript. This
project was supported by the AKC Canine Health Foundation. We also
acknowledge financial support from the National Institutes of
Health/National Cancer Institute (RC2CA148149) and individual
supporters of Van Andel Research Institute through the Purple
Community and Annual Giving programs.
References
|
1
|
Andersen N, Froman R, Kitchell B and
Duesbery N: Angiosarcoma, clinical and molecular aspects. Soft
Tissue Sarcoma. Derbel F: I-Tech Education and Publishing; Rijeka,
Croatia: pp. 149–174. 2011
|
|
2
|
Young RJ, Brown NJ, Reed MW, Hughes D and
Woll PJ: Angiosarcoma. Lancet Oncol. 11:983–991. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Behjati S, Tarpey PS, Sheldon H,
Martincorena I, Van Loo P, Gundem G, Wedge DC, Ramakrishna M, Cooke
SL, Pillay N, et al: Recurrent PTPRB and PLCG1 mutations in
angiosarcoma. Nat Genet. 46:376–379. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Marion MJ, Froment O and Trépo C:
Activation of Ki-ras gene by point mutation in human liver
angiosarcoma associated with vinyl-chloride exposure. Mol Carcinog.
4:450–454. 1991. View Article : Google Scholar
|
|
5
|
Przygodzki RM, Finkelstein SD, Keohavong
P, Zhu D, Bakker A, Swalsky PA, Soini Y, Ishak KG and Bennett WP:
Sporadic and Thorotrast-induced angiosarcomas of the liver manifest
frequent and multiple point mutations in K-ras-2. Lab Invest.
76:153–159. 1997.PubMed/NCBI
|
|
6
|
Weihrauch M, Bader M, Lehnert G, Koch B,
Wittekind C, Wrbitzky R and Tannapfel A: Mutation analysis of
K-ras-2 in liver angiosarcoma and adjacent nonneoplastic liver
tissue from patients occupationally exposed to vinyl chloride.
Environ Mol Mutagen. 40:36–40. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Antonescu CR, Yoshida A, Guo T, Chang NE,
Zhang L, Agaram NP, Qin LX, Brennan MF, Singer S and Maki RG: KDR
activating mutations in human angiosarcomas are sensitive to
specific kinase inhibitors. Cancer Res. 69:7175–7179. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Andersen NJ, Nickoloff BJ, Dykema KJ,
Boguslawski EA, Krivochenitser RI, Froman RE, Dawes MJ, Baker LH,
Thomas DG, Kamstock DA, et al: Pharmacologic inhibition of MEK
signaling prevents growth of canine hemangiosarcoma. Mol Cancer
Ther. 12:1701–1714. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bromberg-White JL, Andersen NJ and
Duesbery NS: MEK genomics in development and disease. Brief Funct
Genomics. 11:300–310. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hoshino R, Chatani Y, Yamori T, Tsuruo T,
Oka H, Yoshida O, Shimada Y, Ari-i S, Wada H, Fujimoto J, et al:
Constitutive activation of the 41-/43-kDa mitogen-activated protein
kinase signaling pathway in human tumors. Oncogene. 18:813–822.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Platz A, Egyhazi S, Ringborg U and Hansson
J: Human cutaneous melanoma; a review of NRAS and BRAF mutation
frequencies in relation to histogenetic subclass and body site. Mol
Oncol. 1:395–405. 2008. View Article : Google Scholar
|
|
12
|
Shimo T, Matsumura S, Ibaragi S, Isowa S,
Kishimoto K, Mese H, Nishiyama A and Sasaki A: Specific inhibitor
of MEK-mediated cross-talk between ERK and p38 MAPK during
differentiation of human osteosarcoma cells. J Cell Commun Signal.
1:103–111. 2007. View Article : Google Scholar
|
|
13
|
Benini S, Manara MC, Cerisano V,
Perdichizzi S, Strammiello R, Serra M, Picci P and Scotlandi K:
Contribution of MEK/MAPK and PI3-K signaling pathway to the
malignant behavior of Ewing’s sarcoma cells: Therapeutic prospects.
Int J Cancer. 108:358–366. 2004. View Article : Google Scholar
|
|
14
|
Ding Y, Boguslawski EA, Berghuis BD, Young
JJ, Zhang Z, Hardy K, Furge K, Kort E, Frankel AE, Hay RV, et al:
Mitogen-activated protein kinase kinase signaling promotes growth
and vascularization of fibrosarcoma. Mol Cancer Ther. 7:648–658.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sodhi A, Montaner S, Patel V, Zohar M,
Bais C, Mesri EA and Gutkind JS: The Kaposi’s sarcoma-associated
herpes virus G protein-coupled receptor up-regulates vascular
endothelial growth factor expression and secretion through
mitogen-activated protein kinase and p38 pathways acting on
hypoxia-inducible factor 1alpha. Cancer Res. 60:4873–4880.
2000.PubMed/NCBI
|
|
16
|
Rinehart J, Adjei AA, Lorusso PM,
Waterhouse D, Hecht JR, Natale RB, Hamid O, Varterasian M, Asbury
P, Kaldjian EP, et al: Multicenter phase II study of the oral MEK
inhibitor, CI-1040, in patients with advanced non-small-cell lung,
breast, colon, and pancreatic cancer. J Clin Oncol. 22:4456–4462.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Haura EB, Ricart AD, Larson TG, Stella PJ,
Bazhenova L, Miller VA, Cohen RB, Eisenberg PD, Selaru P, Wilner
KD, et al: A phase II study of PD-0325901, an oral MEK inhibitor,
in previously treated patients with advanced non-small cell lung
cancer. Clin Cancer Res. 16:2450–2457. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
LoRusso PM, Krishnamurthi SS, Rinehart JJ,
Nabell LM, Malburg L, Chapman PB, DePrimo SE, Bentivegna S, Wilner
KD, Tan W, et al: Phase I pharmacokinetic and pharmacodynamic study
of the oral MAPK/ERK kinase inhibitor PD-0325901 in patients with
advanced cancers. Clin Cancer Res. 16:1924–1937. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim KB, Kefford R, Pavlick AC, Infante JR,
Ribas A, Sosman JA, Fecher LA, Millward M, McArthur GA, Hwu P, et
al: Phase II study of the MEK1/MEK2 inhibitor Trametinib in
patients with metastatic BRAF-mutant cutaneous melanoma previously
treated with or without a BRAF inhibitor. J Clin Oncol. 31:482–489.
2013. View Article : Google Scholar
|
|
20
|
Moore RD and Chaisson RE: Natural history
of HIV infection in the era of combination antiretroviral therapy.
AIDS. 13:1933–1942. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Al-Lazikani B, Banerji U and Workman P:
Combinatorial drug therapy for cancer in the post-genomic era. Nat
Biotechnol. 30:679–692. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zumla A, Nahid P and Cole ST: Advances in
the development of new tuberculosis drugs and treatment regimens.
Nat Rev Drug Discov. 12:388–404. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: The combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gerstung M, Eriksson N, Lin J, Vogelstein
B and Beerenwinkel N: The temporal order of genetic and pathway
alterations in tumorigenesis. PLoS One. 6:e271362011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lee CS, Dykema KJ, Hawkins DM, Cherba DM,
Webb CP, Furge KA and Duesbery NS: MEK2 is sufficient but not
necessary for proliferation and anchorage-independent growth of
SK-MEL-28 melanoma cells. PLoS One. 6:e171652011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Woodman SE, Trent JC, Stemke-Hale K, Lazar
AJ, Pricl S, Pavan GM, Fermeglia M, Gopal YN, Yang D, Podoloff DA,
et al: Activity of dasatinib against L576P KIT mutant melanoma:
Molecular, cellular, and clinical correlates. Mol Cancer Ther.
8:2079–2085. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Solit DB, Garraway LA, Pratilas CA, Sawai
A, Getz G, Basso A, Ye Q, Lobo JM, She Y, Osman I, et al: BRAF
mutation predicts sensitivity to MEK inhibition. Nature.
439:358–362. 2006. View Article : Google Scholar
|
|
28
|
Frost P, Moatamed F, Hoang B, Shi Y, Gera
J, Yan H, Frost P, Gibbons J and Lichtenstein A: In vivo antitumor
effects of the mTOR inhibitor CCI-779 against human multiple
myeloma cells in a xenograft model. Blood. 104:4181–4187. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hidalgo M, Buckner JC, Erlichman C,
Pollack MS, Boni JP, Dukart G, Marshall B, Speicher L, Moore L and
Rowinsky EK: A phase I and pharmacokinetic study of temsirolimus
(CCI-779) administered intravenously daily for 5 days every 2 weeks
to patients with advanced cancer. Clin Cancer Res. 12:5755–5763.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yuan R, Kay A, Berg WJ and Lebwohl D:
Targeting tumorigenesis: Development and use of mTOR inhibitors in
cancer therapy. J Hematol Oncol. 2:452009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Guenther MK, Graab U and Fulda S:
Synthetic lethal interaction between PI3K/Akt/mTOR and Ras/MEK/ERK
pathway inhibition in rhabdomyosarcoma. Cancer Lett. 337:200–209.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Renshaw J, Taylor KR, Bishop R, Valenti M,
De Haven Brandon A, Gowan S, Eccles SA, Ruddle RR, Johnson LD,
Raynaud FI, et al: Dual blockade of the PI3K/AKT/mTOR (AZD8055) and
RAS/MEK/ERK (AZD6244) pathways synergistically inhibits
rhabdomyosarcoma cell growth in vitro and in vivo. Clin Cancer Res.
19:5940–5951. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ho AL, Musi E, Ambrosini G, Nair JS,
Deraje Vasudeva S, de Stanchina E and Schwartz GK: Impact of
combined mTOR and MEK inhibition in uveal melanoma is driven by
tumor genotype. PLoS One. 7:e404392012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
She QB, Halilovic E, Ye Q, Zhen W,
Shirasawa S, Sasazuki T, Solit DB and Rosen N: 4E-BP1 is a key
effector of the oncogenic activation of the AKT and ERK signaling
pathways that integrates their function in tumors. Cancer Cell.
18:39–51. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gingras AC, Kennedy SG, O’Leary MA,
Sonenberg N and Hay N: 4E-BP1, a repressor of mRNA translation, is
phosphorylated and inactivated by the Akt(PKB) signaling pathway.
Genes Dev. 12:502–513. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ruggero D and Sonenberg N: The Akt of
translational control. Oncogene. 24:7426–7434. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mills JR, Hippo Y, Robert F, Chen SM,
Malina A, Lin CJ, Trojahn U, Wendel HG, Charest A, Bronson RT, et
al: mTORC1 promotes survival through translational control of
Mcl-1. Proc Natl Acad Sci USA. 105:10853–10858. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Leevers SJ and Marshall CJ: Activation of
extracellular signal-regulated kinase, ERK2, by p21ras oncoprotein.
EMBO J. 11:569–574. 1992.PubMed/NCBI
|
|
39
|
Graells J, Vinyals A, Figueras A, Llorens
A, Moreno A, Marcoval J, Gonzalez FJ and Fabra A: Overproduction of
VEGF concomitantly expressed with its receptors promotes growth and
survival of melanoma cells through MAPK and PI3K signaling. J
Invest Dermatol. 123:1151–1161. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xu S, Huo J, Lee KG, Kurosaki T and Lam
KP: Phospholipase Cgamma2 is critical for Dectin-1-mediated
Ca2+ flux and cytokine production in dendritic cells. J
Biol Chem. 284:7038–7046. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang Q, Yu C, Peng S, Xu H, Wright E,
Zhang X, Huo X, Cheng E, Pham TH, Asanuma K, et al: Autocrine VEGF
signaling promotes proliferation of neoplastic Barrett’s epithelial
cells through a PLC-dependent pathway. Gastroenterology.
146:461–472 e6. 2014. View Article : Google Scholar
|
|
42
|
Renovanz M and Kim EL: Intratumoral
heterogeneity, its contribution to therapy resistance and
methodological caveats to assessment. Front Oncol. 4:1422014.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ocana A, Amir E, Yeung C, Seruga B and
Tannock IF: How valid are claims for synergy in published clinical
studies? Ann Oncol. 23:2161–2166. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Greco WR, Faessel H and Levasseur L: The
search for cytotoxic synergy between anticancer agents: A case of
Dorothy and the ruby slippers? J Natl Cancer Inst. 88:699–700.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhao L, Au JL and Wientjes MG: Comparison
of methods for evaluating drug-drug interaction. Front Biosci
(Elite Ed). 2:241–249. 2010. View
Article : Google Scholar
|
|
46
|
Engelman JA, Chen L, Tan X, Crosby K,
Guimaraes AR, Upadhyay R, Maira M, McNamara K, Perera SA, Song Y,
et al: Effective use of PI3K and MEK inhibitors to treat mutant
Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med.
14:1351–1356. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Faber AC, Li D, Song Y, Liang MC, Yeap BY,
Bronson RT, Lifshits E, Chen Z, Maira SM, García-Echeverría C, et
al: Differential induction of apoptosis in HER2 and EGFR addicted
cancers following PI3K inhibition. Proc Natl Acad Sci USA.
106:19503–19508. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Jokinen E, Laurila N and Koivunen JP:
Alternative dosing of dual PI3K and MEK inhibition in cancer
therapy. BMC Cancer. 12:6122012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Posch C, Moslehi H, Feeney L, Green GA,
Ebaee A, Feichtenschlager V, Chong K, Peng L, Dimon MT, Phillips T,
et al: Combined targeting of MEK and PI3K/mTOR effector pathways is
necessary to effectively inhibit NRAS mutant melanoma in vitro and
in vivo. Proc Natl Acad Sci USA. 110:4015–4020. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhang YJ, Tian XQ, Sun DF, Zhao SL, Xiong
H and Fang JY: Combined inhibition of MEK and mTOR signaling
inhibits initiation and progression of colorectal cancer. Cancer
Invest. 27:273–285. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Chang Q, Chen E and Hedley DW: Effects of
combined inhibition of MEK and mTOR on downstream signaling and
tumor growth in pancreatic cancer xenograft models. Cancer Biol
Ther. 8:1893–1901. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Depeille PE, Ding Y, Bromberg-White JL and
Duesbery NS: MKK signaling and vascularization. Oncogene.
26:1290–1296. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Mansfield PJ, Shayman JA and Boxer LA:
Regulation of polymorphonuclear leukocyte phagocytosis by myosin
light chain kinase after activation of mitogen-activated protein
kinase. Blood. 95:2407–2412. 2000.PubMed/NCBI
|
|
54
|
Yuan TL and Cantley LC: PI3K pathway
alterations in cancer: Variations on a theme. Oncogene.
27:5497–5510. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Jia J, Zhu F, Ma X, Cao Z, Li Y and Chen
YZ: Mechanisms of drug combinations: Interaction and network
perspectives. Nat Rev Drug Discov. 8:111–128. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Phung TL, Ziv K, Dabydeen D, Eyiah-Mensah
G, Riveros M, Perruzzi C, Sun J, Monahan-Earley RA, Shiojima I,
Nagy JA, et al: Pathological angiogenesis is induced by sustained
Akt signaling and inhibited by rapamycin. Cancer Cell. 10:159–170.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Guba M, von Breitenbuch P, Steinbauer M,
Koehl G, Flegel S, Hornung M, Bruns CJ, Zuelke C, Farkas S,
Anthuber M, et al: Rapamycin inhibits primary and metastatic tumor
growth by antiangiogenesis: Involvement of vascular endothelial
growth factor. Nat Med. 8:128–135. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Milanini J, Viñals F, Pouysségur J and
Pagès G: p42/p44 MAP kinase module plays a key role in the
transcriptional regulation of the vascular endothelial growth
factor gene in fibroblasts. J Biol Chem. 273:18165–18172. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
D’Angelo G, Struman I, Martial J and
Weiner RI: Activation of mitogen-activated protein kinases by
vascular endothelial growth factor and basic fibroblast growth
factor in capillary endothelial cells is inhibited by the
antiangiogenic factor 16-kDa N-terminal fragment of prolactin. Proc
Natl Acad Sci USA. 92:6374–6378. 1995. View Article : Google Scholar
|
|
60
|
Duesbery NS, Webb CP, Leppla SH, Gordon
VM, Klimpel KR, Copeland TD, Ahn NG, Oskarsson MK, Fukasawa K,
Paull KD, et al: Proteolytic inactivation of MAP-kinase-kinase by
anthrax lethal factor. Science. 280:734–737. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Vitale G, Bernardi L, Napolitani G, Mock M
and Montecucco C: Susceptibility of mitogen-activated protein
kinase kinase family members to proteolysis by anthrax lethal
factor. Biochem J. 352:739–745. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Duesbery NS, Resau J, Webb CP, Koochekpour
S, Koo HM, Leppla SH and Vande Woude GF: Suppression of
ras-mediated transformation and inhibition of tumor growth and
angiogenesis by anthrax lethal factor, a proteolytic inhibitor of
multiple MEK pathways. Proc Natl Acad Sci USA. 98:4089–4094. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Bromberg-White JL, Boguslawski E and
Duesbery NS: Perturbation of mouse retinal vascular morphogenesis
by anthrax lethal toxin. PLoS One. 4:e69562009. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Bromberg-White JL, Boguslawski E, Hekman
D, Kort E and Duesbery NS: Persistent inhibition of oxygen-induced
retinal neovascularization by anthrax lethal toxin. Invest
Ophthalmol Vis Sci. 52:8979–8992. 2011. View Article : Google Scholar : PubMed/NCBI
|