Phosphorylation of proteins by kinases is the most
frequent protein modification and plays a key role in multiple
signal transduction pathways in normal and cancer cells. In recent
years protein kinases have become novel promising candidates for
targeted anticancer therapy. To identify and characterize kinases
as biomarkers for tumor transformation or progression is a major
challenge for clinicians, oncologists, and molecular biologists.
The cytoskeleton-associated serine/threonine kinase
death-associated protein kinase (DAPK) has been described as a
cancer gene chameleon showing functional antagonistic duality in a
cell type and context specific manner (1). Cancer genes are classified according
to whether they function in a dominant or recessive manner.
Dominant cancer genes (oncogenes) are constitutively activated by
gain of function mutations and stimulate cell growth and survival.
For recessive genes (tumor suppressors) the loss of function leads
to the inactivation and loss of cell cycle control and repair
capacity. Mutations in the DAPK gene are very rare. There are many
other mechanisms such as promoter hypermethylation,
autophosphorylation of calmodulin-domain, protein degradation or
inhibitory phosphorylations of the DAPK molecule itself that might
inactivate DAPK. Noteworthy, DAPK can act not only through its
catalytic activity but also triggers multiprotein complexes through
its scaffold function (2). The
broad regulation levels of this kinase let it be involved in many
different cellular functions such as cell death (apoptosis,
anoikis, autophagy), repair and mechanosensing (3–5).
Colorectal cancer (CRC) develops in a multistep
process and specific molecular hits have been defined that are
closely correlated with single morphological alterations along the
carcinogenesis process summarized in the Vogelstein model (6). For sporadic cancer two major
different pathogenetic pathways exist: the chromosomal instability
phenotype (CIN, counts for 85% of tumors) and the microsatellite
instability phenotype (MSI, counts for 15% of tumors), both are
caused by loss of a general genetic stability. In 2012 a
three-group classification system has been reported according to
alterations in known signal transduction pathways: i) WNT and TGFβ
signaling ii) PIK3CA and RAS signaling, and iii) p53 signaling
(7,8). Also epigenetic alterations contribute
to altered gene expression in colorectal cancer (9). In this regard a CpG island methylator
phenotype has been described (CIMP). Moreover, CIMP is included in
different molecular classification systems. Nevertheless its
prognostic predictive role is not clarified due to lack of unified
test systems. There are also other risk factors for the development
of a colorectal carcinoma. Patients with inflammatory bowel disease
(IBD) show an increased risk for tumor development. The
pathogenesis of IBD-associated carcinogenesis is poorly understood.
What we know is that similar to sporadic cancer also IBD-associated
cancer is a consequence of a sequence of single molecular
alterations (10).
Although it is not surprising that many molecular
hits are overlapping in both cancers the major difference is the
frequency and the timing of these molecular alterations (10,11).
For DAPK in sporadic colorectal cancer there is a loss of protein
by promoter hypermethylation already in very small tumors and thus
DAPK loss plays a role at very early steps of the tumor formation
process (12). Moreover, loss of
DAPK in colorectal carcinomas has been associated with higher lymph
node metastasis and poor prognosis (13). In contrast, besides an early
inactivation by promoter methylation in a subset of tumors, DAPK is
remarkably activated in colon cancer in the setting of inflammation
(14). So far only one of the two
major IBD forms has been studied for DAPK expression: ulcerative
colitis (UC) (15). There are no
data on the role of DAPK in Crohn’s disease (CD). Recently, it has
been shown that DAPK may play a role in UC-associated tumor
transformation (16). Pro- as well
as anti-inflammatory functions have been suggested for DAPK,
dependent on the cell type and stimulus.
As the development of colorectal cancer is a
long-term complication of chronic inflammation it would be helpful
for patient management to identify molecular biomarkers that
predict the risk of tumor development as early as possible. DAPK
might be a possible candidate for therapeutic intervention but its
gene chameleon nature needs an ultimate understanding of its
functions and regulation in different cell types under different
inflammatory stimuli.
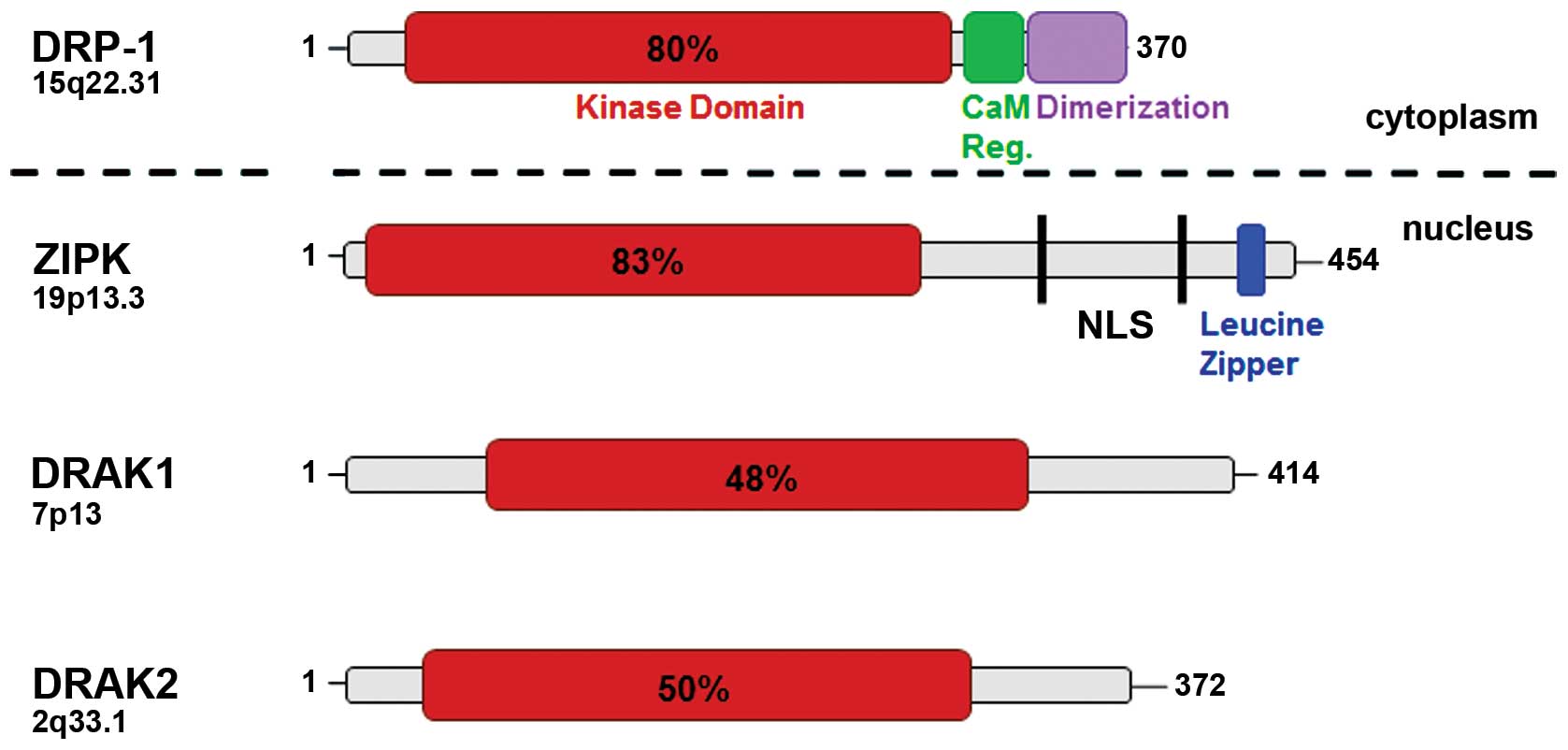
DAPK1 (here referred as DAPK) represents one of the
five members of the DAPK family (4). These molecules differ in size and
subcellular localization (Fig. 1,
Tables I–III). DAPK-related protein 1 (DRP-1,
DAPK2) and zipper-interacting protein kinase (ZIPK, DAPK3) share
the highest homology with approximately 80% at the N-terminus
whereas DAPK related apoptosis inducing kinase 1 and 2 (DRAK1,
DRAK2) have only 50% homology compared with DAPK (17–20).
In contrast to the cytoskeleton-associated family members, ZIPK has
also nuclear functions and interacts with transcription factors
such as ATF4 and STAT3 (21). DAPK
is a multi-domain structure protein that exerts its action through
the catalytic activity and phosphorylation of specific substrates
with DAPK-containing motifs or as a scaffold protein by stabilizing
or triggering multiprotein complexes. One of the most important
functional domains is the calmodulin auto-regulatory domain that is
localized inside of the catalytic cleft. Calmodulin binding leads
to changes in conformation which allow DAPK activation via binding
to its substrates (22). Also
auto-phosphorylation at residue Ser308 inhibits the DAPK function
by reducing its affinity to calmodulin (23).
Besides the prominent function of the catalytic
subunit all additional domains such as the ankyrin repeats, the
ROC-COR domain in the cytoskeleton-binding region, and the death
domain have particular function in the concerted action of this
multifunctional protein (Tables I
and II). DAPK contains 8 ankyrin
repeats that determine primarily the localization of DAPK.
Moreover, this region is important for protein-protein
interactions. Thereby a negative DAPK regulator, the
DAPK-interacting protein (DIP1), is binding at the ankyrin repeat
domain. Phosphorylation of Tyr491/Tyr492 by Src tyrosine kinase
within the ankyrin repeat domain leads to inactivation of DAPK
(24) whereas the interaction with
the phosphatase LAR reconstitutes the activity of DAPK (24).
The death domain at the DAPK C-terminus mediates
DAPK’s function in Fas- and TNF-induced cell death (3). It interacts with microtubule affinity
regulating kinases (MAPK1/2) that phosphorylate tau and thereby
destabilize microtubules (27).
ERK is known to phosphorylate DAPK at Ser735 in the death domain
increasing its catalytic activity. The death domain-mediated
phosphorylation of the TSC2 protein leads to autophagy induction
(28). Also the interaction of the
transmembrane receptor UNC5H is mediated through the death domain.
UNC5H recruits DAPK and PP2A to the lipid rafts where Ser308 is
then dephosphorylated leading finally to an increase in activity.
There are death domain-mediated interactions that result in a
destabilization of DAPK such as the binding with KLHL20, an adaptor
for the Cullin3 ligase, which promotes the proteasomal degradation
of DAPK.
The cellular level of DAPK can be regulated
manifold. On the transcriptional level promoter hypermethylation
has been described that strongly correlates with DAPK protein loss
(21,29,30).
The promoter of DAPK has a high density of CpG islands and motifs
for a number of transcription factors are located within these
regions such as for NFκB, E2F1 or AP1 (21). For colon tumors, the literature
reports a wide range of 5–80% methylation frequency possibly caused
by investigating different CpG islands in different studies. So far
there is no systematic study comparing the significance of
different CpG islands for protein expression. Despite the high
frequency of hypermethylated tumors DAPK is not included in the
CIMP phenotype gene panel.
DAPK can be transcriptionally inhibited by the
pro-inflammatory transcription factors STAT3 and NFκB (16,31,32).
In addition, DAPK mRNA expression can be triggered by p53 (33), C/EBP-β (34), HSF1 (35), and SMAD (36). Whereas C/EBP-β binding depends on
IFNγ exposure, the binding of SMAD to the corresponding motifs is
triggered by TGF-β. In general, DAPK might be upregulated
transcriptionally in response to DNA damage (21,37).
Recently, miRNAs (miR-103, miR-107) have been
identified to target DAPK 3′UTR. High miR-103 and miR-107
expression was correlated with high level of metastases and poor
survival in colorectal cancer patients which is in agreement with
DAPK’s role as a metastasis suppressor (13). A data base search in silico
predicted also some additional miRNAs that might play a role in
DAPK regulation (21). However,
experimental evidence for these miRNAs is lacking.
The stability of DAPK is regulated
post-translationally by two different intracellular proteolysis
systems (Table III). One is the
ubiquitin proteasome system with HSC70-interacting protein (CHIP)
that forms the complex between DAPK and HSP90 (39), DIP1 that interacts with the ankyrin
repeat domain of DAPK (40) or the
KLHL20 protein that acts as an adaptor for Cullin3-based E3-ligases
and interacts with the death domain of DAPK (41). Several reports show that selective
mechanisms exist for reducing cellular DAPK levels by directed
targeting degradation of active DAPK (39). The other degradation system is the
autophagocytic/lysosomal system. Here, the tuberous sclerosis
complex (TSC) formed by its two proteins TSC1 (hamartin) and TSC2
(tuberin) inhibits the activation of mammalian target of rapamycin
complex 1 (mTOR). Binding of the death domain to TSC2 leads either
to phosphorylation of TSC2 by DAPK, a dissociation of the complex
and mTOR activation or a reduction in DAPK levels directly by TSC2
via a post-translational mechanism (42). Finally, there is a non-ubiquitin,
non-autophagic pathway for DAPK regulation which is dependent on
cathepsin B. Cathepsin B binds to C-terminus region between the
cytoskeleton-binding domain and the death domain and leads to a
decrease in DAPK expression (43).
In addition to the multitude of DAPK upstream
regulators controlling its catalytic activity via phosphorylation
events and also its structural stability as mentioned above,
further DAPK-binding proteins grouped as the DAPK interactome have
been discovered (44) (Tables I and III). Only the minority of these
interaction partners has been identified to be DAPK substrates.
Thus, it is assumed that the specific binding of these proteins
itself to certain consensus DAPK phosphorylation motifs (45) might be sufficient to trigger
functional DAPK signaling. Effects of binding of CaM, cathepsin B,
DIP1, Hsp90, LAR, Src, TSC2 and UNC5H2 to DAPK were discussed
above.
Localized to the actin cytoskeleton, DAPK
prominently interacts with cytoskeleton-associated proteins. In
2010, Ivanovska et al found first that DAPK has a scaffold
function to the LIMK/cofilin complex under TNF treatment which
indicates a novel cytoskeleton-associated mechanism of TNF-induced
DAPK-dependent actin remodeling and apoptosis in colorectal cancer
cells (2). Henshall et al
demonstrated that the interaction of actin and TNFR-1 with DAPK in
rat brain is involved in the recruitment of DAPK to cell death
signaling complexes including TNFR-1 and another DAPK interaction
partner FADD (46). Actin binding
was shown to cause structural rearrangement of microfilaments.
Further on, they suggest that 14-3-3 binding modifies DAPK effects
in epileptic brain injury. MAP1B was identified as a positive
cofactor in DAPK-mediated autophagy including vesicle formation and
membrane blebbing. In addition, beclin-1 activation by DAPK and
further protein-protein interaction also was found to trigger
autophagy (47). ERK enhances
death-promoting effects by DAPK Ser735 phosphorylation (48) whereas Ser289 phosphorylation by RSK
has a reducing effect on apoptotic activity of DAPK (49). Targeted by PKD, DAPK was found to
mediate JNK signaling and caspase-independent cell death upon
oxidative stress (50). ZIPK and
DAPK were shown to functionally cooperate in causing cell death
(51).
A list of DAPK interacting partners and DAPK
substrates with consensus DAPK motif (RxxS/T and KR/RxxS/T) is
given in Tables I and III. It has to be mentioned that the
prediction of DAPK substrates based only on phosphorylation sites
is not a sufficient tool since some phosphorylation sites do not
represent the specific DAPK motif. Nevertheless, the following
proteins have been verified to be DAPK substrates: MLC and
tropomyosin-1, MCM3, CaM, P21, P53, S6, Syntaxin-1A (52–58),
TAU (MAPT) (27). MLC, ZIPK,
Beclin1, HSF1, and tropomyosin-1 link DAPK to cell death associated
membrane blebbing, cell motility and stress fiber formation
(52,53).
Inflammatory bowel diseases (IBD) comprise a
heterogeneous set of gastrointestinal (GI) tract disorders which
are grouped into two major entities, namely CD and UC (59,60).
IBD typically affect children and young adults and the chronically
relapsing inflammation of the GI-tract can cause a high individual
and socioeconomic disease burden for people suffering from IBD.
Despite considerable progress in IBD therapy during the past years,
treatment options are still limited and all potent therapeutics
bear the risk of relevant side effects, e.g. by suppressing immune
effector functions resulting in increased susceptibility to
infections.
The precise etiology of IBD has not been clarified
yet, but it is well accepted that multiple factors are involved in
the pathogenesis. Both CD and UC are characterized by dysregulated
immune-responses in genetically predisposed individuals influenced
by the microbiome and additional environmental cues (59,60).
In addition, defects of the intestinal epithelial barrier might
precede and influence the onset of IBD (61).
Recent genetic and immunological investigations have
revealed important insights into molecular players involved in the
pathogenesis of IBD (62).
Genome-wide association studies (GWAS) have linked susceptibility
to IBD with single nucleotide polymorphisms (SNP) at more than 150
gene loci which are markedly enriched in genes involved in primary
immunodeficiency as well as immune-mediated diseases (63). The contribution of a single risk
locus to the individual’s IBD risk appears low, and the majority of
these loci is shared by both UC and CD.
Various innate and adaptive immune cells including
macrophages, T effector cells, regulatory T cells or innate
lymphoid cells have been implicated in intestinal inflammation and
IBD pathogenesis (62–64). Moreover, current IBD therapies are
primarily directed against imbalanced immune responses in IBD
patients with strategies targeting the expansion and/or homing of
pro-inflammatory T cell lymphocytes and the T cell-macrophage axis
(59,60,65–67).
Notably, DAPK is well-known for being involved in modulating pro-
and anti-inflammatory immune responses in macrophage and T cell
studies suggesting a potential role in IBD (68).
There is also growing evidence that defects of the
intestinal epithelial barrier may trigger and influence intestinal
inflammation in IBD patients (61). Noteworthy, we were able to
demonstrate that DAPK can act as negative regulator of STAT3 in
IECs suggesting an important role for DAPK in barrier function and
potentially during IBD pathogenesis (16).
Cytokines are central players of the immunological
crosstalk between different lamina propria cells and they can also
shape barrier function by signaling from immune cell subsets to the
intestinal epithelium (69,70).
It is well-known that the expression of multiple cytokines is
elevated in the intestine during ongoing gut inflammation (59,60,64).
In addition, functional studies in experimental models have
revealed that cytokines can potently influence the course of
intestinal inflammation (59,60,64).
Such studies are further supported by genetic evidence from GWAS in
IBD patients correlating single nucleotide polymorphisms with DNA
loci containing genes associated with cytokine signaling (63).
Tumor necrosis factor alpha (TNF-α) is a
pro-inflammatory key molecule promoting the perpetuation of chronic
intestinal inflammation in IBD, and anti TNF-α therapies are potent
treatment options within current IBD-therapies for a substantial
portion of IBD patients (59,60,71).
Of note, several studies reported that DAPK is crosslinked with
TNF-receptor signaling and NF-κB activation providing further
evidence for a potential therapeutic importance of DAPK in IBD.
However, it was demonstrated that DAPK can process apparently
opposing roles when either inhibiting or promoting inflammation
(16,35,68,72–74).
Thus, further analyses with careful characterization of cell type
specific actions and context-dependent influences are needed before
DAPK might be considered a therapeutic target candidate in IBD.
Besides TNF-α, several other pro-inflammatory cytokines have been
associated with intestinal inflammation (64). Some of them such as IL-1β, IL-6,
IL-17 and IL-18 are also known to be modulated by DAPK (16,75–77).
In addition, DAPK is required for the formation of the NLRP3
inflammasome which is a key regulator for the expression of
pro-inflammatory cytokines including IL-1β and IL-18 by macrophages
and has been linked to IBD (75,78,79).
TGF-β receptor signaling is another pathway that
seems to play a critical role in IBD. Notably, there is evidence
that chronic intestinal inflammation in IBD patients is perpetuated
by T effector cells expressing high levels of SMAD7 rendering them
less susceptible towards suppression by regulatory T cells and
TGF-β signaling (80). Moreover,
SMAD7 inhibition by antisense oligonucleotides has evolved as
promising therapeutic strategy in patients with CD (81). DAPK is also connected to TGF-β
signal transduction via other SMAD-protein family members (63,82,83).
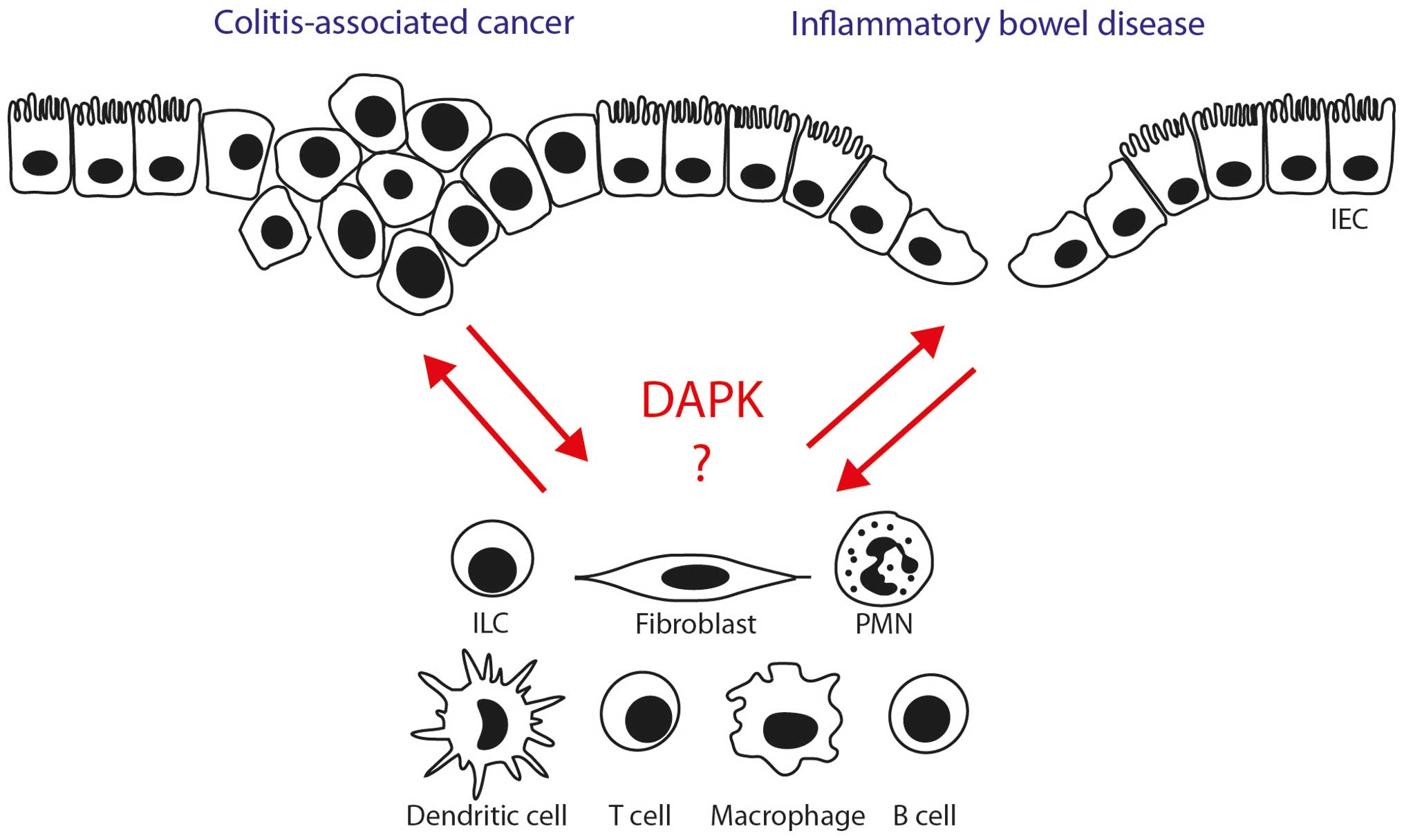
Thus, several lines of evidence suggest that
targeting DAPK might influence the course of intestinal
inflammation via modulation of immune cell activity and intestinal
epithelial barrier function (Fig.
2). However, direct evidence for a critical role of DAPK is
limited so far indicating the need for further studies
investigating the cell type specific function of DAPK during
intestinal inflammation.
DAPK is involved in several forms of cell death
including apoptosis, autophagy and anoikis suggesting a potential
role in colitis-associated cancer (CAC) (44,84).
Although DAPK is often considered a tumor suppressor, pro-survival
roles have also been reported (44,84).
In particular, there is evidence that DAPK might exert divergent
functions under inflammatory conditions (15,16).
IBD patients with longstanding inflammation of the
colon are at increased risk for CRC (85). This risk is associated with the
duration and anatomic extent of colitis and presence of other
inflammatory diseases such primary sclerosing cholangitis (86–88).
In fact, recommendations for colon cancer screening (time of
initial screening colonoscopy and surveillance intervals) in IBD
patients are stricter than for the general population (89).
CRC can be grouped into different entities with
sporadic CRC being the most frequent subtype. IBD patients
including UC patients as well as CD patients with colonic
involvement are particular prone to CAC which can differ from
classical sporadic CRC in various features. Sporadic CRC
classically develops from normal mucosa via adenomatous polyps to
CRC spanning over many years undergoing the adenoma - carcinoma
sequence by accumulating sequential gene alterations including APC,
KRAS and p53 (90–92). In CAC, similar genetic alterations
are found, but a different order of hits including early p53
mutations could pave the way for direct progression to CAC skipping
the stage of adenomatous precursor lesions (93,94).
CAC can show typical morphological features including flat tumor
growth from multiple foci (90).
Previous work reported positive feedback mechanisms between DAPK
and p53 indicating potential functional relevance of DAPK for CAC
growth control (33,95). In addition, our studies have
provided direct evidence for the interaction of DAPK with p38 MAPK
and STAT3 signaling in inflammation-associated colorectal cancer
cells (14,16).
The composition of the local microenvironment can
further influence the tumor development. Of note, elevated levels
of inflammatory cytokines and growth factors are typically found in
CAC allowing for tumor growth promotion in experimental CAC models
(96–99). As DAPK modulates the expression
and/or signaling of some of these molecules such as IL-1β and
IL-18, it is tempting to speculate that DAPK might also influence
CAC development by interfering with the proinflammatory cytokine
signaling. Moreover, elevated levels of reactive oxygen and
nitrogen species typically found with chronic inflammation could
further promote DNA damage and DAPK expression during CAC
development (21,100).
As in sporadic CRC, epigenetic alterations are often
present in CAC and may influence tumor development. DAPK belongs to
the genes that are frequent targets of hypermethylation in CRC, and
aberrant methylation of DAPK in long-standing UC and CAC was
demonstrated (15,101).
In summary, several findings suggest that DAPK could
modify molecular mechanisms of colitis-associated tumorigenesis. To
date however, it has not been clarified which particular context is
critical for rendering DAPK1 either a tumor suppressor or oncogenic
molecule in tumor epithelial and/or tumor stromal cells. Upcoming
analyses could provide important functional insights and might put
DAPK1 on stage as a target for CAC therapy.
The role of DAPK in the intestine has not been
sufficiently elucidated so far. DAPK is expressed and can be
activated by numerous cell types including intestinal epithelial
cells (IECs) as well as innate and adaptive immune cells typically
populating the gut such as macrophages and T cells, respectively.
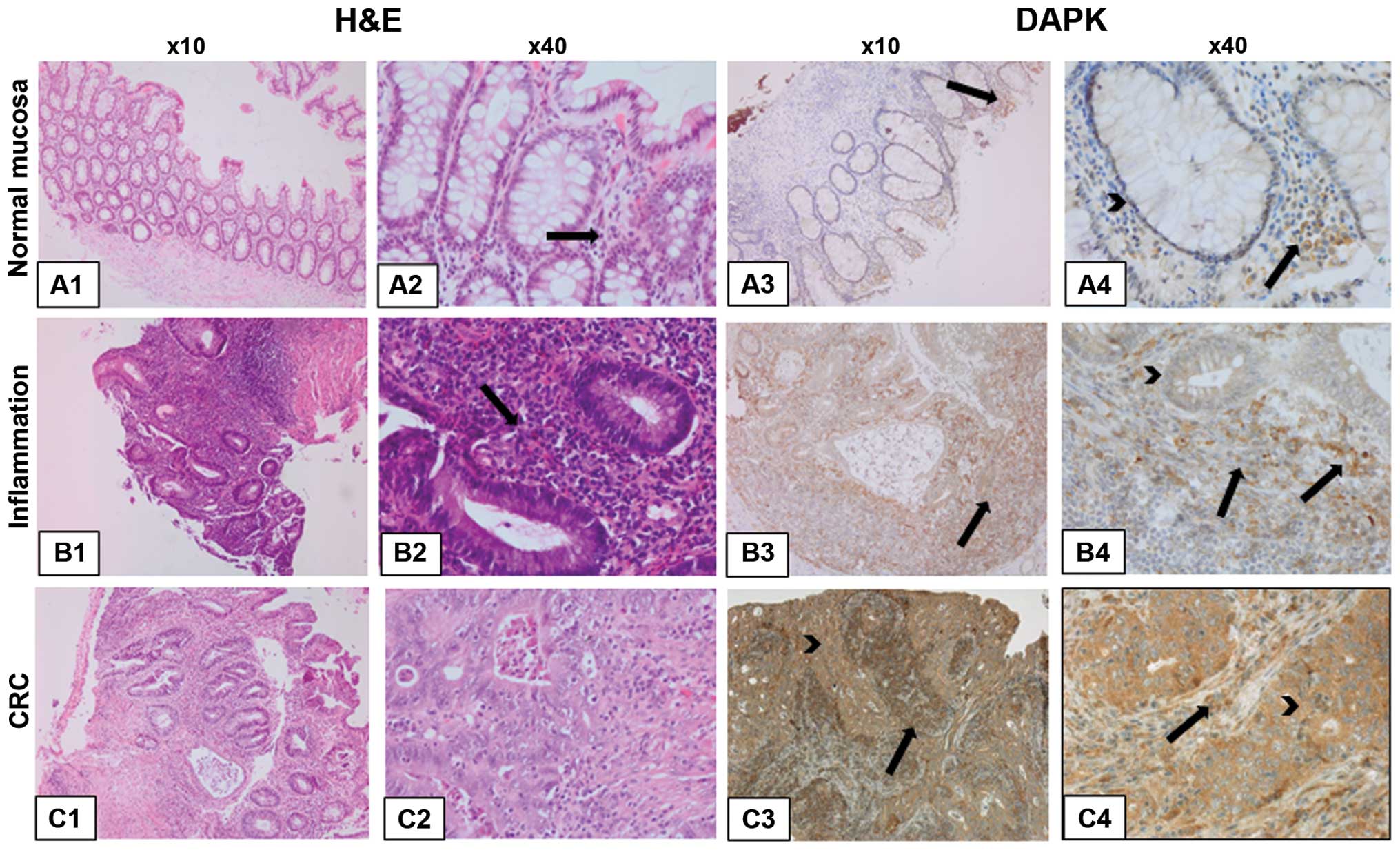
The immunohistochemical DAPK expression in single stages of
UC-associated carcinogenesis in regard to different cell types is
demonstrated exemplarily in Fig.
3.
DAPK-positive tumor-associated macrophages have been
localized in close proximity with apoptotic colorectal cancer cells
suggesting direct crosstalk between macrophages and tumor
epithelial cells in the intestine (102). Based on studies with purified
primary leukocytes and immune cell lines, DAPK might also be
involved in the functional regulation of immune cell populations
during chronic intestinal inflammation (68). For macrophages, inhibition of
inflammation was shown via IFN-γ activated inhibitor of translation
(GAIT) complex (103). In
addition, macrophages can produce a variety of pro-inflammatory
cytokines that are partly controlled by DAPK, e.g. via functional
assembly of the NLRP3 inflammasome and activation of caspase-1
(75).
In the adaptive arm of the immune system, DAPK was
shown to block the nuclear translocation of ERK1/2 in T lymphocytes
(104). Further work demonstrated
decreased T cell proliferation and IL-2 production upon stimulation
by the T cell receptor (74)
indicating that DAPK can interfere with T cell activation which
might have important implications for chronic inflammatory diseases
such as IBD.
Thus, several pieces of evidence suggest potential
contributions of DAPK in the regulation of gut inflammation and
intestinal homeostasis. However, further studies are needed to
clarify the dominant effects of DAPK in different innate and
adaptive immune cell subsets as well as non-immune cells populating
the bowel wall during chronic gut inflammation.
Regarding direct evidence for a role of DAPK in
intestinal inflammation there are many open questions: Which immune
cells in the intestinal lamina propria are controlled by DAPK? Are
there differential effects on subsets of T helper cell populations
including Th1, Th17, and regulatory T cells? What is the role of
DAPK in B cells? Are there different effects on macrophage subsets
including M1 and M2 macrophages? Which role does DAPK play in
non-immune stromal cells such as fibroblasts? How does DAPK
interact with signals from the microbiome?
Providing answers to the above questions may help in
better understanding of how DAPK controls the function of gut cell
populations associated with the pathogenesis of IBD and CRC
(Fig. 2). Perspectively, novel
insights into molecular disease mechanisms and potential key
checkpoints might facilitate the design of new therapeutic
approaches for IBD and CRC. Therefore, further studies are awaited
that directly reveal the role of DAPK in intestinal homeostasis,
intestinal inflammation and CRC.
The authors would like to thank the
Interdisciplinary Center of Clinical Research (IZKF) at the
University Clinics Erlangen (project D21 to R.S.-S. and C.N.), the
Emerging Fields Initiative at the FAU Erlangen-Nürnberg (to C.N.
and R.S.-S.), and the Deutsche Forschungsgemeinschaft (DFG grant
Ne1927 to C.N.) for financial support.
|
1
|
Schneider-Stock R: Death-associated kinase
(DAPK): a cancer ‘gene chameleon’. Apoptosis. 19:2852014.
View Article : Google Scholar
|
|
2
|
Ivanovska J, Tregubova A, Mahadevan V,
Chakilam S, Gandesiri M, Benderska N, Ettle B, Hartmann A, Söder S,
Ziesché E, et al: Identification of DAPK as a scaffold protein for
the LIMK/cofilin complex in TNF-induced apoptosis. Int J Biochem
Cell Biol. 45:1720–1729. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cohen O, Inbal B, Kissil JL, Raveh T,
Berissi H, Spivak-Kroizaman T, Feinstein E and Kimchi A: DAP-kinase
participates in TNF-alpha- and Fas-induced apoptosis and its
function requires the death domain. J Cell Biol. 146:141–148. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bialik S and Kimchi A: The
death-associated protein kinases: Structure, function, and beyond.
Annu Rev Biochem. 75:189–210. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gautel M: Cytoskeletal protein kinases:
Titin and its relations in mechanosensing. Pflugers Arch.
462:119–134. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Brenner H, Kloor M and Pox CP: Colorectal
cancer. Lancet. 383:1490–1502. 2014. View Article : Google Scholar
|
|
7
|
Wu WK, Wang XJ, Cheng AS, Luo MX, Ng SS,
To KF, Chan FK, Cho CH, Sung JJ and Yu J: Dysregulation and
crosstalk of cellular signaling pathways in colon carcinogenesis.
Crit Rev Oncol Hematol. 86:251–277. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jass JR: Classification of colorectal
cancer based on correlation of clinical, morphological and
molecular features. Histopathology. 50:113–130. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Draht MX, Riedl RR, Niessen H, Carvalho B,
Meijer GA, Herman JG, van Engeland M, Melotte V and Smits KM:
Promoter CpG island methylation markers in colorectal cancer: The
road ahead. Epigenomics. 4:179–194. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Feagins LA, Souza RF and Spechler SJ:
Carcinogenesis in IBD: Potential targets for the prevention of
colorectal cancer. Nat Rev Gastroenterol Hepatol. 6:297–305. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Itzkowitz SH and Yio X: Inflammation and
cancer IV. Colorectal cancer in inflammatory bowel disease: The
role of inflammation. Am J Physiol Gastrointest Liver Physiol.
287:G7–G17. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mittag F, Kuester D, Vieth M, Peters B,
Stolte B, Roessner A and Schneider-Stock R: DAPK promotor
methylation is an early event in colorectal carcinogenesis. Cancer
Lett. 240:69–75. 2006. View Article : Google Scholar
|
|
13
|
Chen HY, Lee YR and Chen RH: The functions
and regulations of DAPK in cancer metastasis. Apoptosis.
19:364–370. 2014. View Article : Google Scholar
|
|
14
|
Bajbouj K, Poehlmann A, Kuester D, Drewes
T, Haase K, Hartig R, Teller A, Kliche S, Walluscheck D, Ivanovska
J, et al: Identification of phosphorylated p38 as a novel
DAPK-interacting partner during TNFalpha-induced apoptosis in
colorectal tumor cells. Am J Pathol. 175:557–570. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kuester D, Guenther T, Biesold S, Hartmann
A, Bataille F, Ruemmele P, Peters B, Meyer F, Schubert D, Bohr UR,
et al: Aberrant methylation of DAPK in long-standing ulcerative
colitis and ulcerative colitis-associated carcinoma. Pathol Res
Pract. 206:616–624. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chakilam S, Gandesiri M, Rau TT, Agaimy A,
Vijayalakshmi M, Ivanovska J, Wirtz RM, Schulze-Luehrmann J,
Benderska N, Wittkopf N, et al: Death-associated protein kinase
controls STAT3 activity in intestinal epithelial cells. Am J
Pathol. 182:1005–1020. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kawai T, Matsumoto M, Takeda K, Sanjo H
and Akira S: ZIP kinase, a novel serine/threonine kinase which
mediates apoptosis. Mol Cell Biol. 18:1642–1651. 1998.PubMed/NCBI
|
|
18
|
Kögel D, Plöttner O, Landsberg G,
Christian S and Scheidtmann KH: Cloning and characterization of
Dlk, a novel serine/threonine kinase that is tightly associated
with chromatin and phosphorylates core histones. Oncogene.
17:2645–2654. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Inbal B, Shani G, Cohen O, Kissil JL and
Kimchi A: Death-associated protein kinase-related protein 1, a
novel serine/threonine kinase involved in apoptosis. Mol Cell Biol.
20:1044–1054. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kawai T, Nomura F, Hoshino K, Copeland NG,
Gilbert DJ, Jenkins NA and Akira S: Death-associated protein kinase
2 is a new calcium/calmodulin-dependent protein kinase that signals
apoptosis through its catalytic activity. Oncogene. 18:3471–3480.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Benderska N and Schneider-Stock R:
Transcription control of DAPK. Apoptosis. 19:298–305. 2014.
View Article : Google Scholar
|
|
22
|
Dagher R, Peng S, Gioria S, Fève M, Zeniou
M, Zimmermann M, Pigault C, Haiech J and Kilhoffer MC: A general
strategy to characterize calmodulin-calcium complexes involved in
CaM-target recognition: DAPK and EGFR calmodulin binding domains
interact with different calmodulin-calcium complexes. Biochim
Biophys Acta. 1813.1059–1067. 2011.
|
|
23
|
de Diego I, Kuper J, Bakalova N, Kursula P
and Wilmanns M: Molecular basis of the death-associated protein
kinase-calcium/calmodulin regulator complex. Sci Signal. 3:ra62010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang WJ, Kuo JC, Ku W, Lee YR, Lin FC,
Chang YL, Lin YM, Chen CH, Huang YP, Chiang MJ, et al: The tumor
suppressor DAPK is reciprocally regulated by tyrosine kinase Src
and phosphatase LAR. Mol Cell. 27:701–716. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bialik S and Kimchi A: Biochemical and
functional characterization of the ROC domain of DAPK establishes a
new paradigm of GTP regulation in ROCO proteins. Biochem Soc Trans.
40:1052–1057. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Carlessi R, Levin-Salomon V, Ciprut S,
Bialik S, Berissi H, Albeck S, Peleg Y and Kimchi A: GTP binding to
the ROC domain of DAP-kinase regulates its function through
intra-molecular signalling. EMBO Rep. 12:917–923. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kim BM, You MH, Chen CH, Lee S, Hong Y,
Hong Y, Kimchi A, Zhou XZ and Lee TH: Death-associated protein
kinase 1 has a critical role in aberrant tau protein regulation and
function. Cell Death Dis. 5:e12372014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Stevens C, Lin Y, Harrison B, Burch L,
Ridgway RA, Sansom O and Hupp T: Peptide combinatorial libraries
identify TSC2 as a death-associated protein kinase (DAPK) death
domain-binding protein and reveal a stimulatory role for DAPK in
mTORC1 signaling. J Biol Chem. 284:334–344. 2009. View Article : Google Scholar
|
|
29
|
Kissil JL, Feinstein E, Cohen O, Jones PA,
Tsai YC, Knowles MA, Eydmann ME and Kimchi A: DAP-kinase loss of
expression in various carcinoma and B-cell lymphoma cell lines:
Possible implications for role as tumor suppressor gene. Oncogene.
15:403–407. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Leung RC, Liu SS, Chan KY, Tam KF, Chan
KL, Wong LC and Ngan HY: Promoter methylation of death-associated
protein kinase and its role in irradiation response in cervical
cancer. Oncol Rep. 19:1339–1345. 2008.PubMed/NCBI
|
|
31
|
Shanmugam R, Gade P, Wilson-Weekes A,
Sayar H, Suvannasankha A, Goswami C, Li L, Gupta S, Cardoso AA, Al
Baghdadi T, et al: A noncanonical Flt3ITD/NF-κB signaling pathway
represses DAPK1 in acute myeloid leukemia. Clin Cancer Res.
18:360–369. 2012. View Article : Google Scholar
|
|
32
|
Hayakawa J, Mittal S, Wang Y, Korkmaz KS,
Adamson E, English C, Ohmichi M, McClelland M and Mercola D:
Identification of promoters bound by c-Jun/ATF2 during rapid
large-scale gene activation following genotoxic stress. Mol Cell.
16:521–535. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Martoriati A, Doumont G, Alcalay M,
Bellefroid E, Pelicci PG and Marine JC: dapk1, encoding an
activator of a p19ARF-p53-mediated apoptotic checkpoint, is a
transcription target of p53. Oncogene. 24:1461–1466. 2005.
View Article : Google Scholar
|
|
34
|
Gade P, Roy SK, Li H, Nallar SC and
Kalvakolanu DV: Critical role for transcription factor C/EBP-beta
in regulating the expression of death-associated protein kinase 1.
Mol Cell Biol. 28:2528–2548. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Benderska N, Ivanovska J, Rau TT,
Schulze-Luehrmann J, Mohan S, Chakilam S, Gandesiri M, Ziesché E,
Fischer T, Söder S, et al: DAPK-HSF1 interaction as a new positive
feedback loop for TNF-induced apoptosis in colorectal cancer cells.
J Cell Sci. 127:5273–5287. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Massagué J, Seoane J and Wotton D: Smad
transcription factors. Genes Dev. 19:2783–2810. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gandesiri M, Chakilam S, Ivanovska J,
Benderska N, Ocker M, Di Fazio P, Feoktistova M, Gali-Muhtasib H,
Rave-Fränk M, Prante O, et al: DAPK plays an important role in
panobinostat-induced autophagy and commits cells to apoptosis under
autophagy deficient conditions. Apoptosis. 17:1300–1315. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jin Y and Gallagher PJ: Antisense
depletion of death-associated protein kinase promotes apoptosis. J
Biol Chem. 278:51587–51593. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang L, Nephew KP and Gallagher PJ:
Regulation of death-associated protein kinase. Stabilization by
HSP90 hetero-complexes. J Biol Chem. 282:11795–11804. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jin Y, Blue EK, Dixon S, Shao Z and
Gallagher PJ: A death-associated protein kinase (DAPK)-interacting
protein, DIP-1, is an E3 ubiquitin ligase that promotes tumor
necrosis factor-induced apoptosis and regulates the cellular levels
of DAPK. J Biol Chem. 277:46980–46986. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lee YR, Yuan WC, Ho HC, Chen CH, Shih HM
and Chen RH: The Cullin 3 substrate adaptor KLHL20 mediates DAPK
ubiquitination to control interferon responses. EMBO J.
29:1748–1761. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gallagher PJ and Blue EK:
Post-translational regulation of the cellular levels of DAPK.
Apoptosis. 19:306–315. 2014. View Article : Google Scholar
|
|
43
|
Lin Y, Stevens C and Hupp T:
Identification of a dominant negative functional domain on DAPK-1
that degrades DAPK-1 protein and stimulates TNFR-1-mediated
apoptosis. J Biol Chem. 282:16792–16802. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lin Y, Hupp TR and Stevens C:
Death-associated protein kinase (DAPK) and signal transduction:
Additional roles beyond cell death. FEBS J. 277:48–57. 2010.
View Article : Google Scholar
|
|
45
|
Benderska N, Chakilam S, Hugle M,
Ivanovska J, Gandesiri M, Schulze-Luhrmann J, Bajbouj K, Croner R
and Schneider-Stock R: Apoptosis signalling activated by TNF in the
lower gastrointestinal tract--review. Curr Pharm Biotechnol.
13:2248–2258. 2012. View Article : Google Scholar
|
|
46
|
Henshall DC, Araki T, Schindler CK,
Shinoda S, Lan JQ and Simon RP: Expression of death-associated
protein kinase and recruitment to the tumor necrosis factor
signaling pathway following brief seizures. J Neurochem.
86:1260–1270. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zalckvar E, Berissi H, Mizrachy L,
Idelchuk Y, Koren I, Eisenstein M, Sabanay H, Pinkas-Kramarski R
and Kimchi A: DAP-kinase-mediated phosphorylation on the BH3 domain
of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and
induction of autophagy. EMBO Rep. 10:285–292. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chen CH, Wang WJ, Kuo JC, Tsai HC, Lin JR,
Chang ZF and Chen RH: Bidirectional signals transduced by DAPK-ERK
interaction promote the apoptotic effect of DAPK. EMBO J.
24:294–304. 2005. View Article : Google Scholar :
|
|
49
|
Anjum R, Roux PP, Ballif BA, Gygi SP and
Blenis J: The tumor suppressor DAP kinase is a target of
RSK-mediated survival signaling. Curr Biol. 15:1762–1767. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Eisenberg-Lerner A and Kimchi A: DAP
kinase regulates JNK signaling by binding and activating protein
kinase D under oxidative stress. Cell Death Differ. 14:1908–1915.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Shani G, Marash L, Gozuacik D, Bialik S,
Teitelbaum L, Shohat G and Kimchi A: Death-associated protein
kinase phosphorylates ZIP kinase, forming a unique kinase hierarchy
to activate its cell death functions. Mol Cell Biol. 24:8611–8626.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kuo JC, Lin JR, Staddon JM, Hosoya H and
Chen RH: Uncoordinated regulation of stress fibers and focal
adhesions by DAP kinase. J Cell Sci. 116:4777–4790. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Houle F, Poirier A, Dumaresq J and Huot J:
DAP kinase mediates the phosphorylation of tropomyosin-1 downstream
of the ERK pathway, which regulates the formation of stress fibers
in response to oxidative stress. J Cell Sci. 120:3666–3677. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Bialik S, Berissi H and Kimchi A: A high
throughput proteomics screen identifies novel substrates of
death-associated protein kinase. Mol Cell Proteomics. 7:1089–1098.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Schumacher AM, Schavocky JP, Velentza AV,
Mirzoeva S and Watterson DM: A calmodulin-regulated protein kinase
linked to neuron survival is a substrate for the
calmodulin-regulated death-associated protein kinase. Biochemistry.
43:8116–8124. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Fraser JA and Hupp TR: Chemical genetics
approach to identify peptide ligands that selectively stimulate
DAPK-1 kinase activity. Biochemistry. 46:2655–2673. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Schumacher AM, Velentza AV, Watterson DM
and Dresios J: Death-associated protein kinase phosphorylates
mammalian ribosomal protein S6 and reduces protein synthesis.
Biochemistry. 45:13614–13621. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Tian JH, Das S and Sheng ZH:
Ca2+-dependent phosphorylation of syntaxin-1A by the
death-associated protein (DAP) kinase regulates its interaction
with Munc18. J Biol Chem. 278:26265–26274. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Danese S and Fiocchi C: Ulcerative
colitis. N Engl J Med. 365:1713–1725. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Baumgart DC and Sandborn WJ: Crohn’s
disease. Lancet. 380:1590–1605. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Atreya R and Neurath MF: IBD pathogenesis
in 2014: Molecular pathways controlling barrier function in IBD.
Nat Rev Gastroenterol Hepatol. 12:67–68. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Strober W, Fuss I and Mannon P: The
fundamental basis of inflammatory bowel disease. J Clin Invest.
117:514–521. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Jostins L, Ripke S, Weersma RK, Duerr RH,
McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et
al: International IBD Genetics Consortium (IIBDGC): Host-microbe
interactions have shaped the genetic architecture of inflammatory
bowel disease. Nature. 491:119–124. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Neurath MF: Cytokines in inflammatory
bowel disease. Nat Rev Immunol. 14:329–342. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Feagan BG, Rutgeerts P, Sands BE, Hanauer
S, Colombel JF, Sandborn WJ, Van Assche G, Axler J, Kim HJ, Danese
S, et al: GEMINI 1 Study Group: Vedolizumab as induction and
maintenance therapy for ulcerative colitis. N Engl J Med.
369:699–710. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Sandborn WJ, Feagan BG, Rutgeerts P,
Hanauer S, Colombel JF, Sands BE, Lukas M, Fedorak RN, Lee S,
Bressler B, et al: GEMINI 2 Study Group: Vedolizumab as induction
and maintenance therapy for Crohn’s disease. N Engl J Med.
369:711–721. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Atreya R, Zimmer M, Bartsch B, Waldner MJ,
Atreya I, Neumann H, Hildner K, Hoffman A, Kiesslich R, Rink AD, et
al: Antibodies against tumor necrosis factor (TNF) induce T-cell
apoptosis in patients with inflammatory bowel diseases via TNF
receptor 2 and intestinal CD14+ macrophages.
Gastroenterology. 141:2026–2038. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Lai MZ and Chen RH: Regulation of
inflammation by DAPK. Apoptosis. 19:357–363. 2014. View Article : Google Scholar
|
|
69
|
Backert I, Koralov SB, Wirtz S, Kitowski
V, Billmeier U, Martini E, Hofmann K, Hildner K, Wittkopf N, Brecht
K, et al: STAT3 activation in Th17 and Th22 cells controls
IL-22-mediated epithelial host defense during infectious colitis. J
Immunol. 193:3779–3791. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Pickert G, Neufert C, Leppkes M, Zheng Y,
Wittkopf N, Warntjen M, Lehr HA, Hirth S, Weigmann B, Wirtz S, et
al: STAT3 links IL-22 signaling in intestinal epithelial cells to
mucosal wound healing. J Exp Med. 206:1465–1472. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Atreya R, Neumann H, Neufert C, Waldner
MJ, Billmeier U, Zopf Y, Willma M, App C, Münster T, Kessler H, et
al: In vivo imaging using fluorescent antibodies to tumor necrosis
factor predicts therapeutic response in Crohn’s disease. Nat Med.
20:313–318. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Jin Y, Blue EK and Gallagher PJ: Control
of death-associated protein kinase (DAPK) activity by
phosphorylation and proteasomal degradation. J Biol Chem.
281:39033–39040. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Yoo HJ, Byun HJ, Kim BR, Lee KH, Park SY
and Rho SB: DAPk1 inhibits NF-κB activation through TNF-α and
INF-γ-induced apoptosis. Cell Signal. 24:1471–1477. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Chuang YT, Fang LW, Lin-Feng MH, Chen RH
and Lai MZ: The tumor suppressor death-associated protein kinase
targets to TCR-stimulated NF-kappa B activation. J Immunol.
180:3238–3249. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Chuang YT, Lin YC, Lin KH, Chou TF, Kuo
WC, Yang KT, Wu PR, Chen RH, Kimchi A and Lai MZ: Tumor suppressor
death-associated protein kinase is required for full IL-1β
production. Blood. 117:960–970. 2011. View Article : Google Scholar
|
|
76
|
Turner-Brannen E, Choi KY, Arsenault R,
El-Gabalawy H, Napper S and Mookherjee N: Inflammatory cytokines
IL-32 and IL-17 have common signaling intermediates despite
differential dependence on TNF-receptor 1. J Immunol.
186:7127–7135. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Nakav S, Cohen S, Feigelson SW, Bialik S,
Shoseyov D, Kimchi A and Alon R: Tumor suppressor death-associated
protein kinase attenuates inflammatory responses in the lung. Am J
Respir Cell Mol Biol. 46:313–322. 2012. View Article : Google Scholar
|
|
78
|
Bauer C, Duewell P, Mayer C, Lehr HA,
Fitzgerald KA, Dauer M, Tschopp J, Endres S, Latz E and Schnurr M:
Colitis induced in mice with dextran sulfate sodium (DSS) is
mediated by the NLRP3 inflammasome. Gut. 59:1192–1199. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Schoultz I, Verma D, Halfvarsson J,
Törkvist L, Fredrikson M, Sjöqvist U, Lördal M, Tysk C, Lerm M,
Söderkvist P, et al: Combined polymorphisms in genes encoding the
inflammasome components NALP3 and CARD8 confer susceptibility to
Crohn’s disease in Swedish men. Am J Gastroenterol. 104:1180–1188.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Fantini MC, Rizzo A, Fina D, Caruso R,
Sarra M, Stolfi C, Becker C, Macdonald TT, Pallone F, Neurath MF
and Monteleone G: Smad7 controls resistance of colitogenic T cells
to regulatory T cell-mediated suppression. Gastroenterology.
136:1308–1316. e1–3. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Monteleone G, Fantini MC, Onali S, Zorzi
F, Sancesario G, Bernardini S, Calabrese E, Viti F, Monteleone I,
Biancone L and Pallone F: Phase I clinical trial of Smad7 knockdown
using antisense oligonucleotide in patients with active Crohn’s
disease. Mol Ther. 20:870–876. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Jang CW, Chen CH, Chen CC, Chen JY, Su YH
and Chen RH: TGF-beta induces apoptosis through Smad-mediated
expression of DAP-kinase. Nat Cell Biol. 4:51–58. 2002. View Article : Google Scholar
|
|
83
|
MacDonald TT, Monteleone I, Fantini MC and
Monteleone G: Regulation of homeostasis and inflammation in the
intestine. Gastroenterology. 140:1768–1775. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Shiloh R, Bialik S and Kimchi A: The DAPK
family: a structure-function analysis. Apoptosis. 19:286–297. 2014.
View Article : Google Scholar
|
|
85
|
Ekbom A, Helmick C, Zack M and Adami HO:
Ulcerative colitis and colorectal cancer. A population-based study.
N Engl J Med. 323:1228–1233. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Mathy C, Schneider K, Chen YY, Varma M,
Terdiman JP and Mahadevan U: Gross versus microscopic pancolitis
and the occurrence of neoplasia in ulcerative colitis. Inflamm
Bowel Dis. 9:351–355. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Rutter M, Saunders B, Wilkinson K, Rumbles
S, Schofield G, Kamm M, Williams C, Price A, Talbot I and Forbes A:
Severity of inflammation is a risk factor for colorectal neoplasia
in ulcerative colitis. Gastroenterology. 126:451–459. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Broomé U, Lindberg G and Löfberg R:
Primary sclerosing cholangitis in ulcerative colitis - a risk
factor for the development of dysplasia and DNA aneuploidy?
Gastroenterology. 102:1877–1880. 1992.
|
|
89
|
Van Assche G, Dignass A, Bokemeyer B,
Danese S, Gionchetti P, Moser G, Beaugerie L, Gomollón F, Häuser W,
Herrlinger K, et al: European Crohn’s and Colitis Organisation:
Second European evidence-based consensus on the diagnosis and
management of ulcerative colitis part 3: Special situations. J
Crohn’s Colitis. 7:1–33. 2013. View Article : Google Scholar
|
|
90
|
Ullman TA and Itzkowitz SH: Intestinal
inflammation and cancer. Gastroenterology. 140:1807–1816. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Vogelstein B, Fearon ER, Hamilton SR, Kern
SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM and Bos
JL: Genetic alterations during colorectal-tumor development. N Engl
J Med. 319:525–532. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Fearon ER: Molecular genetics of
colorectal cancer. Annu Rev Pathol. 6:479–507. 2011. View Article : Google Scholar
|
|
93
|
Hussain SP, Amstad P, Raja K, Ambs S,
Nagashima M, Bennett WP, Shields PG, Ham AJ, Swenberg JA, Marrogi
AJ, et al: Increased p53 mutation load in noncancerous colon tissue
from ulcerative colitis: A cancer-prone chronic inflammatory
disease. Cancer Res. 60:3333–3337. 2000.PubMed/NCBI
|
|
94
|
Redston MS, Papadopoulos N, Caldas C,
Kinzler KW and Kern SE: Common occurrence of APC and K-ras gene
mutations in the spectrum of colitis-associated neoplasias.
Gastroenterology. 108:383–392. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Michie AM, McCaig AM, Nakagawa R and
Vukovic M: Death-associated protein kinase (DAPK) and signal
transduction: Regulation in cancer. FEBS J. 277:74–80. 2010.
View Article : Google Scholar
|
|
96
|
Grivennikov S, Karin E, Terzic J, Mucida
D, Yu GY, Vallabhapurapu S, Scheller J, Rose-John S, Cheroutre H,
Eckmann L, et al: IL-6 and Stat3 are required for survival of
intestinal epithelial cells and development of colitis-associated
cancer. Cancer Cell. 15:103–113. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Neufert C, Becker C, Türeci Ö, Waldner MJ,
Backert I, Floh K, Atreya I, Leppkes M, Jefremow A, Vieth M, et al:
Tumor fibroblast-derived epiregulin promotes growth of
colitis-associated neoplasms through ERK. J Clin Invest.
123:1428–1443. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Salcedo R, Worschech A, Cardone M, Jones
Y, Gyulai Z, Dai RM, Wang E, Ma W, Haines D, O’hUigin C, et al:
MyD88-mediated signaling prevents development of adenocarcinomas of
the colon: Role of interleukin 18. J Exp Med. 207:1625–1636. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Popivanova BK, Kitamura K, Wu Y, Kondo T,
Kagaya T, Kaneko S, Oshima M, Fujii C and Mukaida N: Blocking
TNF-alpha in mice reduces colorectal carcinogenesis associated with
chronic colitis. J Clin Invest. 118:560–570. 2008.PubMed/NCBI
|
|
100
|
D’Incà R, Cardin R, Benazzato L, Angriman
I, Martines D and Sturniolo GC: Oxidative DNA damage in the mucosa
of ulcerative colitis increases with disease duration and
dysplasia. Inflamm Bowel Dis. 10:23–27. 2004. View Article : Google Scholar
|
|
101
|
Goel A and Boland CR: Epigenetics of
colorectal cancer. Gastroenterology. 143:1442–1460. e12012.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Schneider-Stock R, Kuester D, Ullrich O,
Mittag F, Habold C, Boltze C, Peters B, Krueger S, Hintze C, Meyer
F, et al: Close localization of DAP-kinase positive
tumour-associated macrophages and apoptotic colorectal cancer
cells. J Pathol. 209:95–105. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Mukhopadhyay R, Ray PS, Arif A, Brady AK,
Kinter M and Fox PL: DAPK-ZIPK-L13a axis constitutes a
negative-feedback module regulating inflammatory gene expression.
Mol Cell. 32:371–382. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Kamal M, Pawlak A, BenMohamed F,
Valanciuté A, Dahan K, Candelier M, Lang P, Guellaën G and Sahali
D: C-mip interacts with the p85 subunit of PI3 kinase and exerts a
dual effect on ERK signaling via the recruitment of Dip1 and DAP
kinase. FEBS Lett. 584:500–506. 2010. View Article : Google Scholar
|