Introduction
Colorectal cancer (CRC) is the third most common
cancer in the world with an incidence of 9.4 and 10.1% in men and
women, respectively. Although recent progress in chemotherapy
regimen has increased survival significantly, it remains a major
cause of mortality and morbidity (1).
Different molecular alterations have been described
in CRC such as mutation of the oncogene KRAS or the tumor
suppressor p53 as well as mutation in the TGF-β pathway (2). However, 90% of CRC arise from an
activating mutation of the canonical Wnt pathway. Particularly,
mutation of the adenomatous polyposis coli (APC) gene is believed
to be an initiating event in 80% of sporadic colorectal tumors but
is also responsible for familial adenomatous polyposis (3). This mutation induces the
stabilization of β-catenin resulting in its accumulation in the
nucleus, where it complexes the DNA-binding family TCF/LEF to
activate the transcription of Wnt target genes such as MYC,
CCND1 and AXIN2. Overexpression of Wnt target genes has
been associated with chromosome instability and tumorigenesis
(4). This constant activation of
Wnt signaling then leads to colorectal neoplasia with aberrant
crypt foci especially in the early stages of tumor formation
(5).
Further, high β-catenin/Wnt signaling was previously
linked to cancer stem cell (CSC) properties in colorectal cancer
(6) and pancreatic ductal
adenocarcinoma (7). CSCs are a
rare population of cancer cells exhibiting stem cell traits, such
as self-renewal and tumorigenesis. These stem cells often reside in
a particular niche, have higher resistance to chemotherapy as well
as radiotherapy, and are also capable of invading and migrating to
other tissues (8,9). Therefore, besides the initiation of
the primary tumor, CSCs have been associated with metastasis
formation and cancer relapse (10). Within this context, targeting Wnt
signaling is one of the most promising approaches and several
studies have identified Wnt as a potent anticancer target in CRC
(6–9). However, the main problem resides
today in the difficulty to produce clinical drugs that effectively
target this pathway in cancer cells.
Independently from this, substance P (SP) is an
undecapeptide of the tachykinin family. It is widely distributed
throughout the body and has a wide variety of functions under
physiological conditions (11–13).
It binds to the neurokinin (NK) receptor (NKR) family (NK1R, NK2R
and NK3R), but preferentially to NK1R for which it has a high
affinity, and regulates many biological functions mainly in the
central and peripheral nervous system (14–16).
Recently, it has been reported that the SP/NK1R complex is part of
the tumor microenvironment and is involved in tumor cell
proliferation, angiogenesis and migration (14). The small molecule aprepitant (AP)
is known to cause a variety of therapeutic effects in humans,
partially depending on the dosage in which it is used. In low
doses, AP reduces nausea and vomiting induced by chemotherapy. Of
note, for this indication, AP is approved by the Food and Drug
Administration (FDA). There are currently ongoing clinical trials
investigating the potential to reduce nausea and vomiting in
postoperative patients, including children. In medium doses, AP has
been shown to relieve symptoms of depression (17,18).
In experimental in vitro and in vivo settings, higher
doses of NK1R antagonists induce robust inhibition of tumor growth
and apoptosis in cancer cells of a great variety of adult and
childhood cancers, including colon cancer (19–21).
However, little is known about the intracellular signaling
responsible for the effects. Here, we sought to investigate the
underlying molecular mechanisms in two CRC cell lines. We found a
striking inhibition of canonical Wnt signaling upon treatment with
AP, resulting in potent growth inhibition. Therefore, our
discoveries could have important implementations for the
introduction of new innovative anticancer strategies against colon
cancer.
Materials and methods
Cell culture
We used two colorectal cell lines; DLD1 was obtained
from ATCC (American Type Culture Collection) and LiM6, a derivative
of the LS174T cell line, was a kind gift of Dr Robert S. Bresalier
(MDACC) (22). All cell lines were
grown in RPMI-1640 medium (Invitrogen, Carlsbad, CA, USA)
supplemented with 10% FCS (Invitrogen) and 1%
penicillin/streptomycin (Invitrogen) at 37°C in a humidified
incubator with 5% CO2.
Drugs
Aprepitant (AP), an NK1R antagonist and substance P
(SP), an NK1R agonist were purchased from Selleck Chemicals and
Sigma-Aldrich and were dissolved in DMSO and 0.1 M acetic acid,
respectively.
Proliferation assays
Cell proliferation was assessed using
1-(4,5-dimethylthiazol-2-yl)-3,5-diphenylformazan (MTT) assay. Ten
thousand cells/well were seeded into 96-well plates (NUNC,
Langenselbold, Germany). After 24 h, cells were treated with
increasing doses of AP for 48 h or with DMSO. To assess cell
viability, a final concentration of 0.5 mg/ml (MTT formazan powder,
Sigma-Aldrich diluted in PBS) was first added to each well followed
by 4-h incubation at 37°C. Finally, a lysis solution of 10% SDS and
1 M HCl was added overnight in each well. For the readout, a
multi-scanner microplate reader (Tecan GENios Microplate Reader,
Männedorf, Switzerland) was used to measure the absorbance at 595
nm. Each experiment was realized three times and each condition was
performed in triplicates.
Flow cytometry
DLD1 cells were seeded onto 6-well plates at a
density of 200,000 cells/well. After 24 h, cells were treated for
24 h with 30 μM AP or DMSO. Adherent cells were trypsinized and
pooled together with non-adherent cells, rinsed with PBS and fixed
with ice cold ethanol 70% for 2 h minimum. Cells were washed with
PBS and stained for 30 min with a staining solution: PBS, 0.1%
Triton X-100 (Sigma), 0.2 mg/ml RNAse A (Qiagen, Hilden, Germany)
and 0.02 mg/ml propidium iodide (Sigma-Aldrich). Cells were
analyzed by BD LSRFortessa cell analyzer (BD Biosciences,
Heidelberg, Germany).
Sphere formation culture
Sphere formation medium was prepared as follows:
DMEM-F12 with 1% penicillin/streptomycin, 10 ng/ml human
recombinant βFGF, 20 ng/ml human recombinant EGF, 1% glutamine and
B27 serum-free supplement 1X (all from Invitrogen) subsequently
filtered with a 0.475-μm ultra Cruz syringe filter (Santa Cruz
Biotechnology, Heidelberg, Germany). Parental cells were
trypsinized, rinsed twice with PBS and cell counting was performed
to seed 500 cells onto a 96-well ultra-low attachment plate
(Corning GmbH, Wiesbaden, Germany), each well containing 100 μl of
filtered sphere formation medium. Spheres were cultivated for 8
days and 100 μl of media containing the additives (20 μM AP, 100
ng/ml SP or DMSO) was added every 3 days. Sphere formation ability
was determined under a microscope.
Reverse-phase protein array (RPPA)
Cells were first treated with AP (20 or 40 μM), SP
(100 ng/ml) or DMSO for 24 h, washed with ice-cold PBS, denatured
with a 1% SDS/β-mercaptoethanol lysis buffer solution and stored at
−80°C. The Functional Proteomics RPPA Core Facility at MD Anderson
Cancer Center analyzed the samples as previously described
(23). In brief, probes were
spotted on nitrocellulose slides and 172 different proteins were
analyzed. Densitometry of these spots was performed and values
normalized for protein loading. The values were then transformed to
linear values, which were used for our calculations. The
quantification was realized by calculating the ratios of the linear
protein intensity values of the treated probes with the linear
protein intensity values of their respective control probes.
Western blot analyses
LiM6 and DLD1 cells were first treated with AP (30
μM) or DMSO for 24 h, washed with ice-cold PBS and lysed with a
lysis buffer (0.5% Triton X-100, 1 mM sodium orthovanadate, 1
protease inhibitor cocktail tablet in purified water). The protein
concentration was determined by using the Bio-Rad protein assay
(Bio-Rad Laboratories, Munich, Germany). For each condition, 20 μg
of proteins were loaded on 8–12% Tris-glycine gels (Invitrogen),
separated by electrophoresis and electroblotted onto nitrocellulose
membranes (Amersham Life Sciences, Buckinghamshire, UK). The
membranes were then incubated for 2 h in a blocking solution (5%
BSA in TBS, 0.1% Tween-20), followed by an overnight incubation
with primary antibodies at a 1:1,000 dilution against β-catenin,
LRP5 (low-density lipoprotein receptor-related protein 5), MYC,
PARP [poly(ADP-ribose) polymerase] and β-actin (all Cell Signaling
Technologies, Danvers, MA, USA). Finally, the blots were washed
with TBS-0.1% Tween-20 and incubated for 1 h at room temperature
with a peroxidase-conjugated goat anti-rabbit IgG antibody (Dako,
Hamburg, Germany) at a dilution of 1:2,000. The detection was
realized with an enhanced chemiluminescence reaction (ECL Prime
Western blotting detection; Amersham Life Sciences) and β-actin
served as a loading control.
Immunofluorescence
DLD1 cells were seeded onto 18-mm diameter cover
slips (Thermo Scientific, Braunschweig, Germany) in a 12-well
plates format (NUNC) at a density of 75,000 cells/well. Cells were
treated with 30 μM AP, 70 nM SP or DMSO for 24 h. Cells were then
fixed with 4% paraformaldehyde for 15 min, followed by a
permeabilization step for 15 min with 0.15% Triton X-100 in PBS and
a blocking step for 30 min with 1% BSA in PBS. The cells were then
incubated overnight at 4°C with a rabbit primary antibody against
β-catenin (Cell Signaling Technologies) diluted in blocking
solution at 1:80. After multiple washing steps, cells were
incubated in the dark with a goat anti-rabbit secondary antibody
conjugated with Alexa Fluor 488 (Invitrogen) at a 1:200 dilution
for 1 h. Finally, cover slips were rinsed three times with PBS and
mounted on microscope slides with Vectashield (Vector Laboratories,
Burlingame, CA, USA) containing 4,6-diamidino-2-pheylindole (DAPI).
The images were taken with a Olympus FluoView™ FV1000 confocal
microscope.
RNA isolation and RT-qPCR
Total RNA isolation was realized with TRI-reagent
(Sigma-Aldrich) and 2 μg were used for cDNA synthesis using random
primers (Roche Diagnostics, Penzberg, Germany) and SuperScript II
Reverse Transcriptase (Invitrogen). Each PCR reaction was made with
40 ng of cDNA, 500 nM of the primer pair and SoAdvanced™ Universal
SYBR Green Supermix (Bio-Rad Laboratories) for a final volume of 20
μl. Thermal cycling was realized by the Mastercycler EP gradient S
(Eppendorf, Hamburg, Germany) and consisted of 40 cycles with
denaturation at 95°C for 15 sec, annealing at 55°C for 15 sec and
elongation at 72°C for 30 sec. All experimental conditions were
assessed in duplicate. The primers used were as follows:
CTNNB1 (NM_001904.2) forward, 5′-ACGTCCATGGGTGGGACA-3′;
reverse, 5′-CTAGGATGTGAAGGGCTCCG-3′. AXIN2 (NM_004655.3)
forward, 5′-TATCCAGTGATGCGCTGACG-3′; reverse,
5′-TGTTTCTTACTGCCCACACGAT-3′. MYC (NM_002467.3) forward,
5′-CACCACCAGCAGCGACTCT-3′; reverse, 5′-CAGACTCTGACCTTTTGCCAGG-3′.
FOXM1 (NM_202002.2) forward, 5′-CTCCCGCAGCATCAAGCAA-3′;
reverse, 5′-GCCAGGACGCTGATGGTCTC-3′.
TOP/FOP luciferase reporter assay
To assess luciferase activity we used the
Dual-Luciferase Reporter Assay system (Promega, Madison, WI, USA).
DLD1 and LiM6 cells were seeded in 24-well plates at a density of
35,000 cells/well for 24 h. For the transfection, we used FuGENE HD
reagent (Promega) and αMEM medium. We co-transfected inducible
Firefly luciferase expressing SuperTOP or SuperFOP vectors
(AddGene plasmids nos. 12456 and 12457) and constitutively
Renilla luciferase expressing normalization vector pRL-TK
(Promega) at a ratio of 50:1. The latter vector was used as a
control of transfection efficiency. Simultaneously, the cells were
treated with different concentrations of AP as indicated or with
DMSO. After 24 h of incubation total cell lysate was extracted
using reporter lysis buffer (Promega). Total extract (5 μl), 25 μl
of Luciferase substrate LarII solution (Promega) and 25 μl of Stop
and Glow 1X solution (Promega) were used to assess Firefly
and Renilla luciferase activities, respectively. Finally,
Wnt activity was measured by the ratio of SuperTOP/SuperFOP
luciferase activities each normalized to the respective
Renilla luciferase activities. The results are displayed as
relative light unit (RLU) and each experimental condition was
assessed in triplicate.
Statistical analyses
Results are expressed as the mean ± standard error
of the mean (SEM). All statistical comparisons were made with a
standard t-test and Mann-Whitney U test using biostatistics
software from GraphPad Prism® (La Jolla, CA, USA). The
criterion for significance was p<0.05 and p<0.01 for all
comparisons.
Results
NK1R blocking leads to the regulation of
specific proteins involved in different signaling pathways
We have recently described that upon NK1R blockage,
growth of hepatoblastoma cells could be inhibited in vitro
and in vivo (19). Here, we
treated colon cancer cell lines LiM6 (β-catenin mutation)
(24) and DLD1 (APC
mutation) (25) with increasing
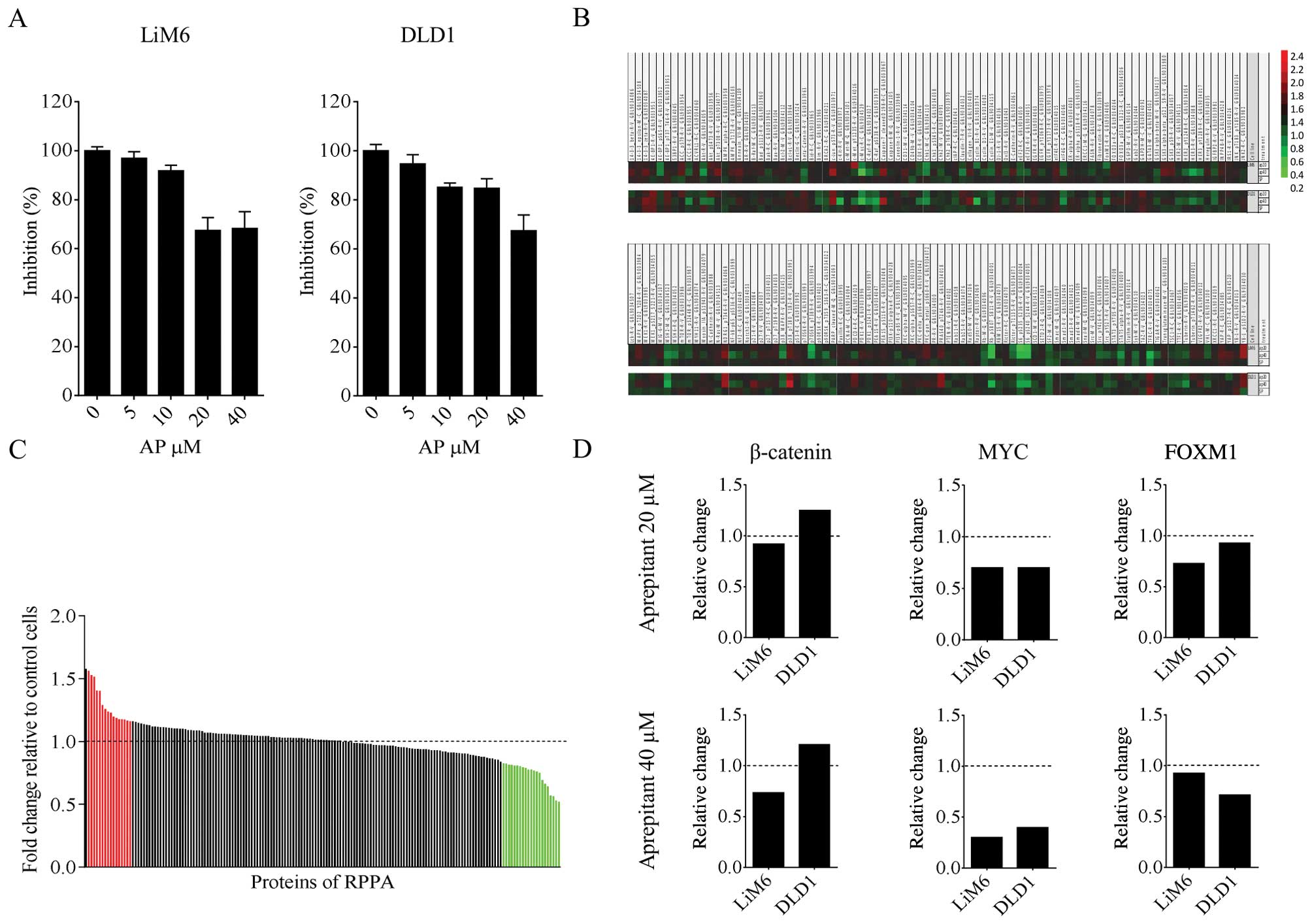
doses of AP for 48 h and then performed standard MTT assays. We
observed a dose-dependent inhibition of cell growth in both cell
lines (Fig. 1A).
In order to have a better understanding of the
downstream molecular mechanisms responsible for these observed
effects, we used reverse phase protein array (RPPA) analysis. We
treated human CRC cells LiM6 and DLD1 with 20 or 40 μM AP for 24 h
and screened for changes in protein expression level of 172
proteins involved in multiple signaling pathways that are typically
altered in cancer (23). Values
from 20 and 40 μM were put into relation to the control value of
each cell line and the pathway molecule (Fig. 1B).
To scrutinize the regulation pattern in the two cell
lines, we calculated the mean of the values relative to the control
of 20 plus 40 μM conditions for each molecule and displayed the
results in Fig. 1C. The cut off
values are specified in the figure legend. Only a small number of
proteins exhibited an upregulation (red) or downregulation (green)
common in the two cell lines. Interestingly, we found that
important proteins associated with Wnt signaling were
downregulated, including the Wnt target MYC and the β-catenin
interacting factor FOXM1 (Fig.
1D). Taken together, these findings suggest inhibition of Wnt
signaling through inhibition of NK1R. Of note, in these early
experiments β-catenin, one key component of the Wnt signaling
pathway, was not downregulated in DLD1 (Fig. 1C and D).
NK1R antagonism inhibits the AKT/mTOR
pathway
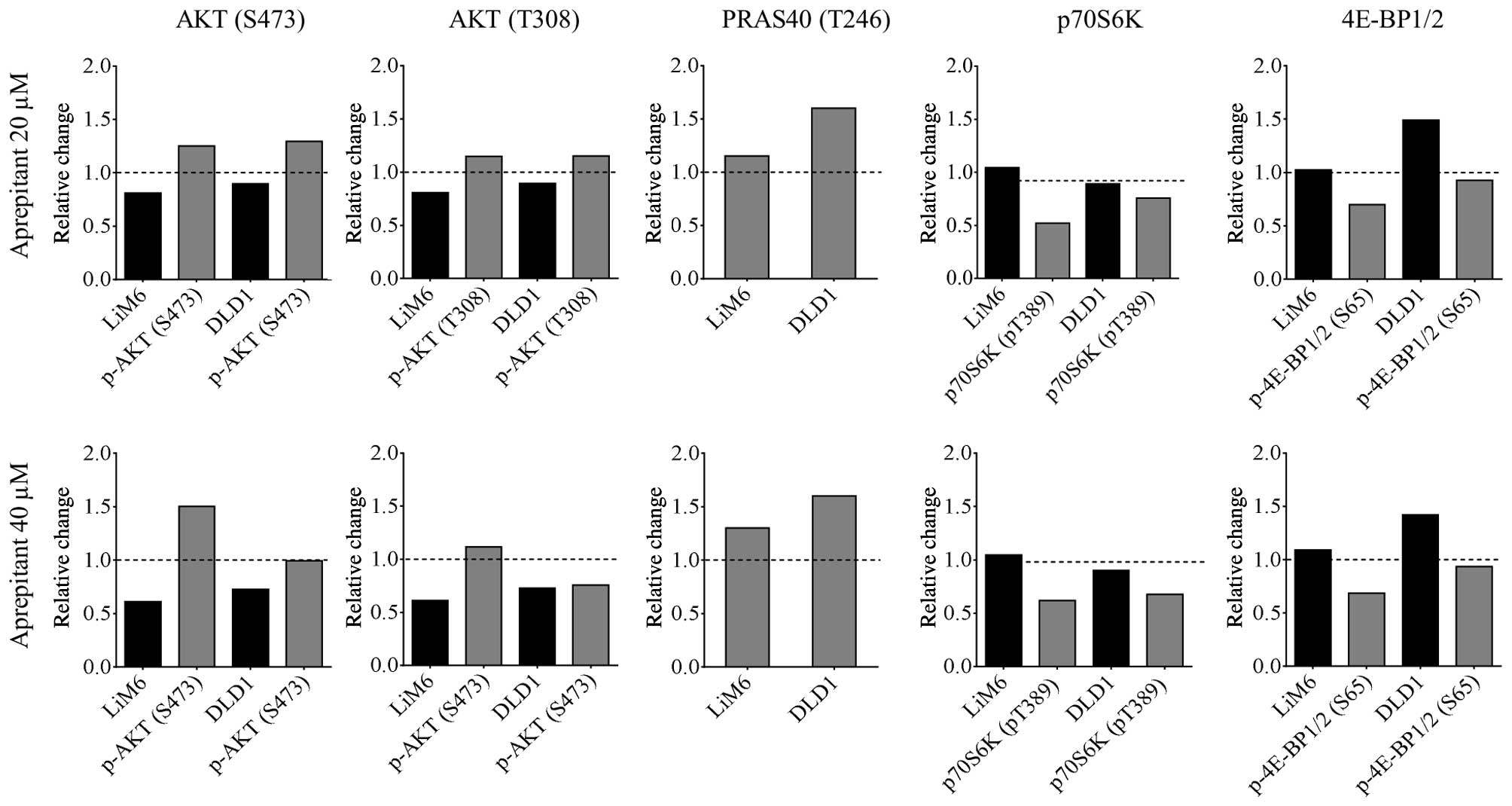
Apart from Wnt signaling, we also found the AKT/mTOR
pathway to be inhibited (Fig. 2,
right panels) at the level of its downstream proteins
phospho-4EBP1/2 and phospho-p70S6K (grey bars) compared to their
total forms (black bars). Total AKT was also dose-dependently
downregulated upon AP treatment in the two cell lines, whereas no
clear trend could be extracted from its phospho-form at Thr308. In
contrast, phosphorylation of AKT at Ser473 was upregulated
(Fig. 2, left panels) along with
its substrate PRAS40 at Thr246, a member and repressor of the TORC1
complex (26). These results
indicate that apart from decreased Wnt signaling, inhibition of
NK1R might lead to additional downregulation of the AKT/mTOR
signaling pathway.
NK1R antagonism induces apoptosis and a
G2 arrest
It is known that NK1R antagonists induce apoptosis
in human hepatoblastoma cancer cells and other cancers (19,20).
Therefore, as a next step, we investigated whether this held true
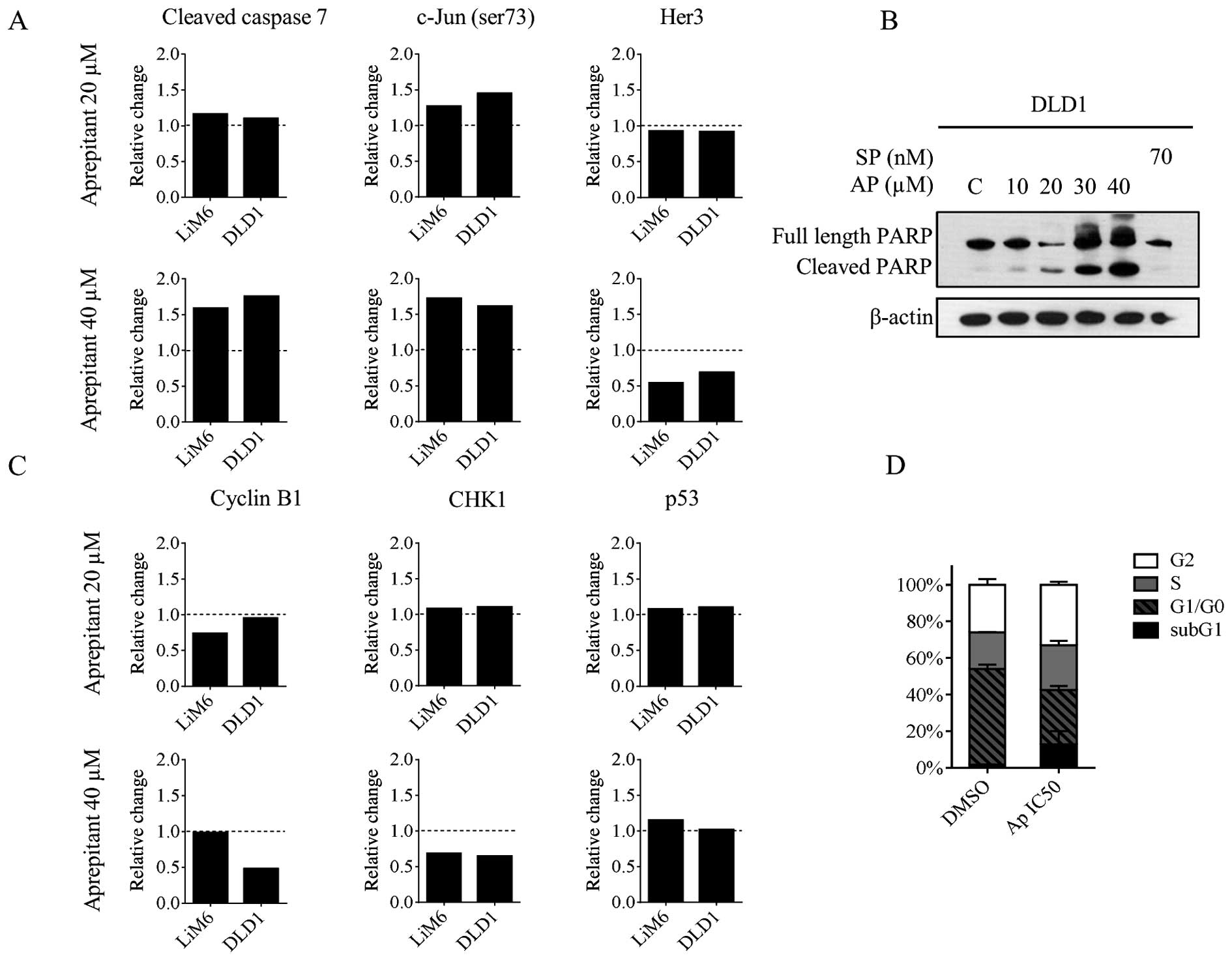
in our setup. First, we screened our data obtained by RPPA for
alterations in apoptosis markers. We found that cleaved caspase 7
and phospho-c-Jun (Ser73) were upregulated upon AP treatment
(Fig. 3A), two well known
mediators of apoptosis (27,28).
In order to characterize apoptosis in more detail,
we treated DLD1 cells for 24 h with increasing concentration of AP
and performed western blot analysis for PARP which executes the
apoptotic process (29). We found
a dose-dependent increase of cleaved PARP indicating an activation
of apoptosis, whereas SP had no effect (Fig. 3B). Further, Her3 was found
down-regulated (Fig. 3A, right
panels) and targeting this receptor in colorectal cancer cells has
been described to induce a G2 arrest as well as apoptosis (30). We then analyzed the effect of AP on
the expression of cell cycle regulators. In our RPPA data, we found
cyclin B1 and CHK1 to be downregulated, whereas p53 was slightly
upregulated (Fig. 3C). These
proteins are known to regulate the G2-M transition (31). To investigate more in depth the
growth inhibition observed in DLD1 and to confirm our RPPA data, we
treated the cells for 24 h with 30 μM AP and analyzed the cell
cycle profile by propidium iodide staining and FACS analysis. As
shown in Fig. 3D, AP induced a G2
arrest (33% compared with 26% for DMSO-treated cells, white bar)
and an increase of cells in the subG1 (12.7% compared with 1.63%
for DMSO-treated cells, black bar) phase indicating either late
apoptosis or necrosis.
NK1R antagonism inhibits the canonical
Wnt signaling
After analyzing the RPPA results above, we found
strong evidence that AP might induce a reduction of Wnt activity.
To validate this hypothesis, we then carried out Super TOP/FOP
(STF) assays in non-treated cells to assess baseline Wnt activity
in DLD1 and LiM6 cells and compared their activity to the
pancreatic cancer cell line L3.6pl, a cell line known to express
little Wnt. As expected, we found that both colon cancer cell lines
displayed high levels of Wnt activity with a ~500-fold increased
activity compared to L3.6pl (Fig.
4A).
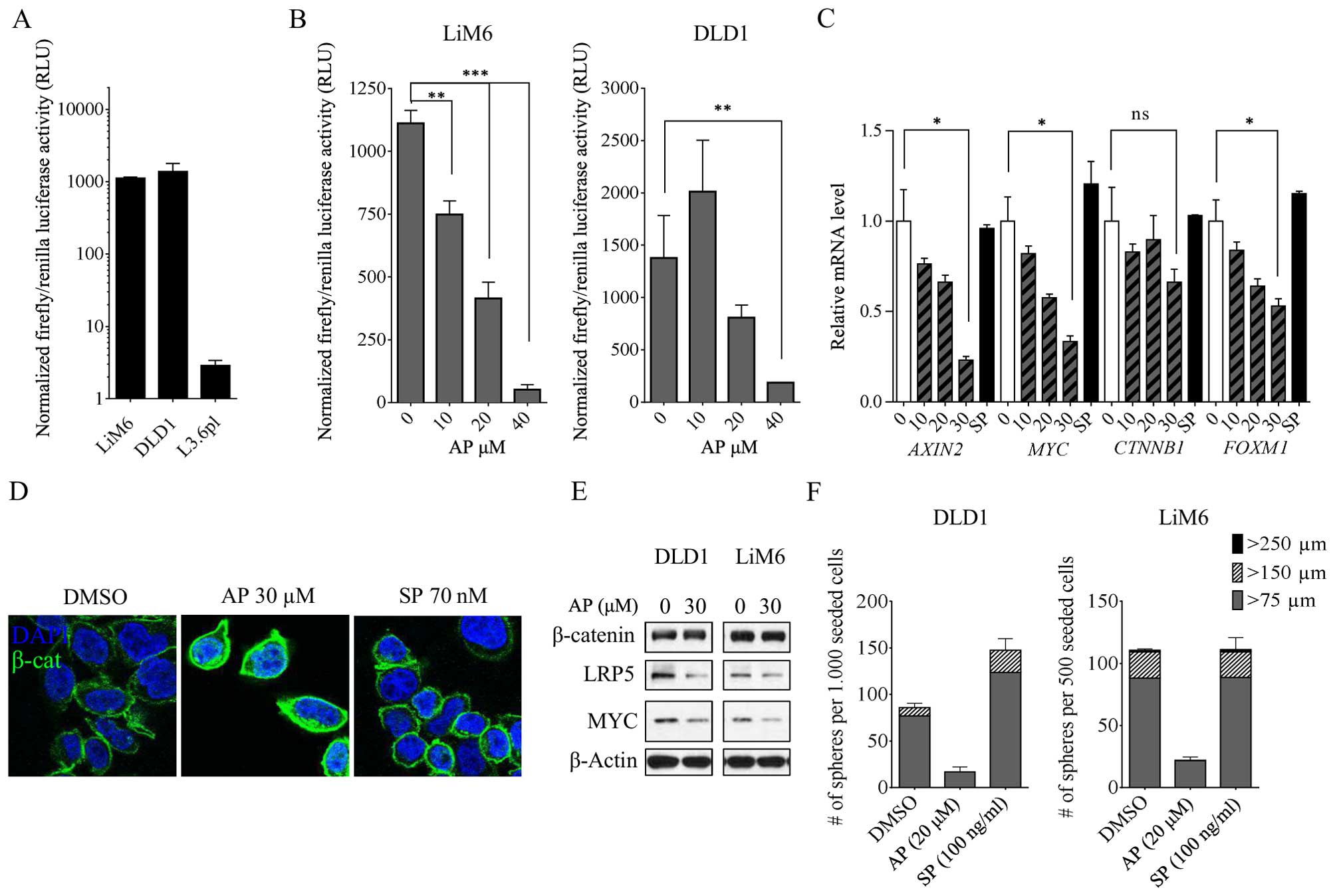 | Figure 4AP induces a robust inhibition of Wnt
activity and promotes membrane-bound β-catenin in colorectal cancer
cells. (A) Baseline Wnt activity was assessed using the
SuperTOP/FOP reporter system. Cells were transfected with SuperTOP
(non-mutated TCF/LEF binding sites) or SuperFOP (mutated TCF/LEF
binding sites) for 24 h. Results are presented by the ratio of
SuperTOP/SuperFOP Firefly luciferase and normalized to
Renilla luciferase. (B) Ratio of SuperTOP/FOP (A) after 24 h
of treatment with different concentrations of AP (10, 20 or 40 μM)
or DMSO (0) in LiM6 and DLD1. (C) qRT-PCR of Wnt target genes
(AXIN2, MYC, CTNNB1) and Wnt-associated gene (FOXM1).
Cells were treated with increasing doses of AP (10, 20 or 30 μM,
gray and black columns), SP (black columns) or DMSO (white columns)
for 24 h. (D) Immunofluorescent staining of β-catenin (green) in
DLD1 and nuclear staining with DAPI (blue). Cells were treated with
30 μM AP, 70 nM SP or DMSO for 24 h. The staining was examined by
confocal microscopy. (E) Selected proteins of the Wnt pathway were
validated by western blot analysis (β-catenin, LRP5, MYC). Cells
were treated with 30 μM AP or DMSO (0) for 24 h followed by
extraction of total cell lysates. (F) Sphere-forming assays (SFA)
in DLD1 and LiM6 with simultaneous treatment with AP or SP. Shown
are the number of spheres per 500 seeded cells in an ultra-low
attachment 96-well plate after 10 days. Sphere sizes were evaluated
and categorized as follows: >250 μm (black), >150 μm (black
and grey) and >75 μm (grey) n=3. |
Next, we treated the colon cancer cells with
increasing doses of AP. After a one-time treatment, we discovered a
robust inhibition of Wnt activity in both colorectal cell lines
(Fig. 4B). These results were
corroborated by qRT-PCR data in DLD1. The Wnt target gene
MYC was dose-dependently downregulated upon AP treatment
along with AXIN2 and FOXM1 (Fig. 4C). Intriguingly, in contrast to the
RPPA data, CTNNB1 was also downregulated, although the trend
was not as convincing as observed in the other genes. Thus, instead
of targeting β-catenin for degradation, AP might induce a decrease
of Wnt activity by sequestrating it away from the nucleus resulting
in decreased TCF/β-catenin activity, ultimately leading to a lower
Wnt target gene expression.
To further investigate this hypothesis and the
down-regulation of the β-catenin/Wnt signaling pathway following AP
treatment, we cultured cells with 30 μM AP or 70 nM SP for 24 h,
stained them for β-catenin and analyzed them with confocal
microscopy. At the membrane, β-catenin is required for cell
adhesion where it complexes with E-cadherin. Consequently, despite
being protected from degradation, β-catenin is not available for
transducing a signal to the nucleus in this state (32). We observed a strong accumulation of
membrane-bound β-catenin upon AP treatment (Fig. 4D). However, SP stimulation did not
seem to affect β-catenin/Wnt signaling when compared to the
control.
Finally, we treated the cells with 30 μM AP and
performed western blot analysis. We found that the Wnt co-receptor
LRP5 as well as the Wnt target MYC had markedly lower expression
upon AP treatment compared to the respective control. In the same
experiment, we analyzed the expression of β-catenin following AP
treatment. Similar to the RPPA data, we did not observe any
striking changes of total β-catenin in whole cell lysates of LiM6
or DLD1 (Fig. 4E). Taken together,
these data demonstrate that targeting the NK1R in colon cancer cell
lines markedly reduces Wnt-signaling.
In a next step, we investigated whether this held
true not only in differentiated colon cancer cell lines, but also
in colon cancer stem cells (CSCs). We grew colorectal CSC-like
cells as described (33) and
assessed sphere formation ability (SFA) in both DLD1 and LiM6
(Fig. 4F). Upon AP treatment, we
discovered a striking decrease, both in sphere number and size.
Interestingly, activation of the SP/NK1R system with recombinant SP
significantly increased the SFA of DLD1 CSCs in number and size,
whereas in LiM6, we were not able to find differences between
SP-stimulated cells and control (Fig.
4F).
Taken together, these experiments show that
inhibition of the SP/NK1R system with AP decreased the canonical
Wnt signaling in colorectal cancer cells, likely by arresting
β-catenin in its membrane-bound localization. Also, inhibition of
the SP/NK1R system inhibited the growth of anoikis-resistant
CSC-like colorectal spheres.
The inactivation of the β-catenin/Wnt
pathway is independent of the initial Wnt baseline activity
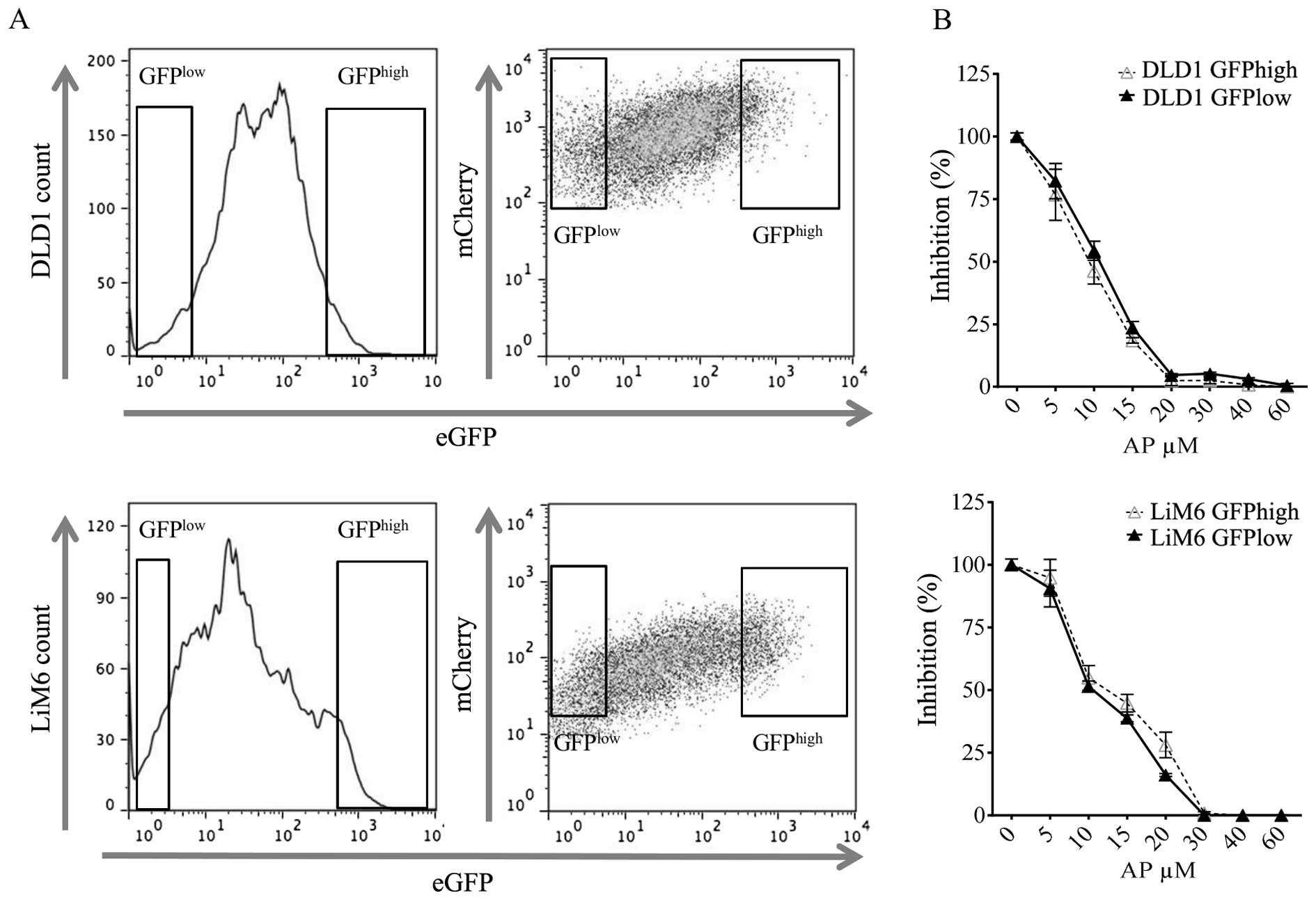
In order to better understand the inhibitory effect
on Wnt signaling, we sought to investigate whether differences
exist regarding the response rate to AP treatment within cell
populations that are constitutively active regarding the
β-catenin/Wnt pathway. Such cells with high constitutive Wnt
activation have been described to have higher stemness properties
(6). To make our findings
comparable to those from Vermeulen et al, we transduced the
colorectal cell lines LiM6 and DLD1 with a 7xTCF-eGFP/mCherry
(7TGC) lentiviral construct (34)
allowing us to separate cells with different Wnt activities by
single cell sorting via FACS (Fig.
5A). Wnt activity in cells with the same level of mCherry
intensity then correlates with their GFP intensity. After
separation, we independently cultured Wnthigh and
Wntlow expressing cells and subsequently treated these
cells with increasing doses of AP for 48 h (Fig. 5B). Interestingly, we did not
observe a difference in cell survival for any of the doses tested
between cells with high or low Wnt activity. These findings suggest
that the apoptosis-inducing property of NK1R targeting is
maintained even in cells with supposedly increased cancer stemness
potential (Wnthigh). Furthermore, our data indicate that
in a particular cell line or population, the observed inhibitory
effects caused by NK1R antagonism are independent of the initial
Wnt baseline activation.
Discussion
It is well established that aberrant activation of
Wnt signaling is a key event in colorectal tumorigenesis.
Therefore, recent efforts have focused on developing new molecules
that block this pathway. For this purpose, the use of
antibody-based therapy, small molecules (35) or other direct and indirect
inhibitors (36) have all been
studied in depth, but to date no component exists that has
succeeded at a clinical level. In this study, we found that
aprepitant (AP), an NK1R antagonist currently approved by the FDA
for the treatment of chemotherapy-induced nausea and vomiting,
triggered a robust inhibition of Wnt/β-catenin signaling activity
and a growth inhibition in colorectal cancer cells. Although
β-catenin was not clearly downregulated at the protein level or
mRNA level, we found that AP induced an accumulation in its
membrane-bound state, where it cannot execute its function as a
transcriptional co-activator. Consequently, we found a significant
inhibition of Wnt/β-catenin activity as measured by the luciferase
reporter assay.
The cell lines used in our study were DLD1 and LiM6,
which harbor APC and β-catenin mutations,
respectively. Consequently, the molecules upstream of the
destruction complex, the latter being composed of AXIN, adenomatous
polyposis coli (APC), glycogen synthase kinase 3β (GSK3β), casein
kinase 1 (CK1) and protein phosphatase 2A (PP2A) (37), should not be needed for pathway
activation/modulation. Taking this into account, it appears that AP
acts either at the level of β-catenin or downstream of it.
Therefore, we hypothesized that the inhibition of Wnt activity was
due either to the loss of β-catenin translocation into the nucleus
or by inhibition of β-catenin binding to the transcription factors
TCF/LEF. In this sense, it has been demonstrated that β-catenin
activation directly correlates with FOXM1 and nestin expression, a
stem cell marker (38). Moreover,
it has been suggested that FOXM1 might be crucial for
Wnt/β-catenin activity. This could be demonstrated in FOXM1
knockout mice where Wnt/β-catenin signaling activity was
significantly reduced in FOXM1−/− colon tumors
(39). In accordance with these
data, in our study we found FOXM1 to be significantly downregulated
following treatment with AP.
We performed immunof luorescence stainings for
β-catenin to detect its cellular localization following NK1R
inhibition. Intriguingly, we discovered strong accumulation of
membrane-bound β-catenin reflecting an inhibition of Wnt activity.
As mentioned above, one explanation for this could be
downregulation of FOXM1 expression.
Another, perhaps more grounded hypothesis is that AP
induces an arrest in its membrane-bound localization, which then
subsequently inhibits its function as a transcriptional
co-activator by binding to TCF/LEF. Indeed, while free cytosolic
β-catenin is normally required for signal transduction,
membrane-bound β-catenin is sequestrated for cell adhesion and
consequently protected from degradation. Unfortunately, our data do
not permit us to extrapolate by which mechanisms β-catenin
accumulates at the membrane. A possible explanation suggested by
previous reports (40) is that
E-cadherin phosphorylation promotes cell adhesion by increasing the
E-cadherin/β-catenin affinity. Particularly the cytoplasmic domain
of E-cadherin contains several phosphorylation sites for casein
kinase II (CKII) and GSK3β.
A third explanation for our findings involves a
mechanism consisting in a direct or indirect inhibition of the
TCF/β-catenin complex by AP. Previous reports have shown that
several small molecules specifically disrupt the TCF7L2/β-catenin
complex and thus trigger growth inhibition in colorectal cells
in vitro and in vivo (41,42).
The described mechanism of inhibition downstream of
β-catenin is particularly interesting, because this form of
targeting Wnt signaling could potentially avoid some side effects
that have been claimed to be unique to inhibition upstream of
β-catenin. For example, with the inhibition downstream of
β-catenin, physiologic complex formation such as
E-cadherin/β-catenin, which ensures proper cell adhesion in normal
cells, is not disrupted. Nevertheless, inhibition downstream of
β-catenin will nonetheless target Wnt target genes, which are the
main actors of tumorigenesis. As suspected, we discovered a
downregulation at the mRNA level and/or protein level of several
important target genes such as MYC, AXIN2 and
CCND1.
Another interesting finding of our study was the
fact that treatment with AP resulted in growth inhibition of cells
independent of their constitutional Wnt activity. For example, we
observed the same therapeutic effect after separating colon cancer
cells in Wnthigh and Wntlow expressing cells.
Furthermore, when we treated the pancreatic cancer cell line
L3.6pl, a cell line known to express minimal Wnt, we also observed
a strong growth inhibition (Ilmer et al, unpublished data).
This can potentially have several explanations. Given the critical
role of Wnt signaling for any cancer cell, it is likely that
abrogation of Wnt signaling will have detrimental effects on the
cell, regardless of its constitutional Wnt activity. On the other
hand, these results could be explained with the fact that, most
likely, Wnt inhibition is not the only mechanism by which AP
inhibits cancer cells. The latter would be the more encouraging
explanation, since it is known that cancer cells have enormous
potential to escape anticancer agents, especially if they rely on
one particular pathway. Indeed, in our study, along with inhibition
of Wnt signaling, we found the AKT/mTOR signaling pathway to be
downregulated significantly.
AKT/mTOR signaling pathway is known to be activated
in cancer cells and is involved in tumorigenesis. Thus, the
inhibition of AKT/mTOR by itself is undergoing thorough
investigation as a promising candidate for future anticancer
strategies. Interestingly, in accordance to our data, mTOR
inhibition following NK1R blockage has been described for other
cancers (43). Given that in our
study we did not only see robust inhibition of AKT/mTOR, but also
significant inhibition on Wnt signaling, we wonder whether the two
pathways counter-affect each other and if a defined, inverse
cross-link is activated between the two following AP treatment.
Crosslinks between the AKT/mTOR and Wnt pathways have been
described. For example, in the absence of β-catenin, GSK3β can be
inhibited to activate mTOR. Inversely, Wnt signaling can activate
TSC2, which negatively regulates mTOR (44). In our study, we found mainly a
downregulation of total AKT and its downstream target p70S6K,
4E-BP1/2 indicating an overall pathway inhibition. Unlike our
expectation, p-AKT (Ser473) and PRAS40 (Thr246) were found to be
upregulated. This phenomenon has already been described following
the inhibition of the mTORC1 and is believed to be mediated through
mTORC2 complex. It has been suggested that upregulation of p-AKT at
Ser473 could be responsible for the development of drug resistance
(45). Unfortunately, at this
point we cannot extract from our data insight into whether
treatment with AP activates a defined cross-link between the two
pathways or if the downstream effects are completely independent
from one another.
Taken together, our results indicate that AP
inhibits cell growth of colorectal as well as CSC-like cancer
cells. Upon NK1R inhibition, we found two pivotal cancer pathways
to be repressed: Wnt and AKT/mTOR. Therefore, we here highlight for
the first time insight into the intracellular mechanisms triggered
by AP treatment in colorectal cancer cells. These findings could be
of great importance for the generation of an effective anticancer
therapy against colorectal and other cancers.
Acknowledgements
Michael Berger and Matthias Ilmer were supported by
postdoctoral stipends of the German Academic Exchange Program
(DAAD). Michael Berger was additionally funded by the
Friedrich-Baur foundation Munich, Münchener Medizinische
Wochenschrift, as well as the Förderung für Forschung und Lehre of
LMU University. This study was supported in part by grants of the
Alliance of Cardiovascular Researchers to Eckhard Alt. Roland
Kappler obtained funding from the Bettina Bräu foundation, Munich,
and the Gänseblümchen-Voerde foundation, Voerde.
References
|
1
|
Boyle P and Langman JS: ABC of colorectal
cancer: Epidemiology. BMJ. 321:805–808. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fearon ER: Molecular genetics of
colorectal cancer. Annu Rev Pathol. 6:479–507. 2011. View Article : Google Scholar
|
|
3
|
Fearnhead NS, Britton MP and Bodmer WF:
The ABC of APC. Hum Mol Genet. 10:721–733. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Giles RH, van Es JH and Clevers H: Caught
up in a Wnt storm: Wnt signaling in cancer. Biochim Biophys Acta.
1653:1–24. 2003.PubMed/NCBI
|
|
5
|
Fodde R, Smits R and Clevers H: APC,
signal transduction and genetic instability in colorectal cancer.
Nat Rev Cancer. 1:55–67. 2001. View
Article : Google Scholar
|
|
6
|
Vermeulen L, De Sousa E Melo F, van der
Heijden M, Cameron K, de Jong JH, Borovski T, Tuynman JB, Todaro M,
Merz C, Rodermond H, et al: Wnt activity defines colon cancer stem
cells and is regulated by the microenvironment. Nat Cell Biol.
12:468–476. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ilmer M, Boiles AR, Regel I, Yokoi K,
Michalski CW, Wistuba II, Rodriguez J, Alt E and Vykoukal J: RSPO2
enhances canonical Wnt signaling to confer stemness-associated
traits to susceptible pancreatic cancer cells. Cancer Res.
75:1883–1896. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Prud’homme GJ: Cancer stem cells and novel
targets for antitumor strategies. Curr Pharm Des. 18:2838–2849.
2012. View Article : Google Scholar
|
|
9
|
Reya T, Morrison SJ, Clarke MF and
Weissman IL: Stem cells, cancer, and cancer stem cells. Nature.
414:105–111. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nagler C, Zänker KS and Dittmar T: Cell
fusion, drug resistance and recurrence CSCs. Adv Exp Med Biol.
714:173–182. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Quartara L and Maggi CA: The tachykinin
NK1 receptor. Part II: Distribution and pathophysiological roles.
Neuropeptides. 32:1–49. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hökfelt T, Pernow B and Wahren J:
Substance P: A pioneer amongst neuropeptides. J Intern Med.
249:27–40. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Varty GB, Cohen-Williams ME and Hunter JC:
The antidepressant-like effects of neurokinin NK1 receptor
antagonists in a gerbil tail suspension test. Behav Pharmacol.
14:87–95. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rosso M, Muñoz M and Berger M: The role of
neurokinin-1 receptor in the microenvironment of inflammation and
cancer. Sci World J. 2012:3814342012.
|
|
15
|
Muñoz M and Coveñas R: Neurokinin-1
receptor: A new promising target in the treatment of cancer. Discov
Med. 10:305–313. 2010.PubMed/NCBI
|
|
16
|
Muñoz M, Rosso M and Coveñas R: A new
frontier in the treatment of cancer: NK-1 receptor antagonists.
Curr Med Chem. 17:504–516. 2010. View Article : Google Scholar
|
|
17
|
Kramer MS1, Winokur A, Kelsey J, Preskorn
SH, Rothschild AJ, Snavely D, Ghosh K, Ball WA, Reines SA, Munjack
D, et al: Demonstration of the efficacy and safety of a novel
substance P (NK1) receptor antagonist in major depression.
Neuropsychopharmacology. 29:385–392. 2004. View Article : Google Scholar
|
|
18
|
Kramer MS, Cutler N, Feighner J,
Shrivastava R, Carman J, Sramek JJ, Reines SA, Liu G, Snavely D,
Wyatt-Knowles E, et al: Distinct mechanism for antidepressant
activity by blockade of central substance P receptors. Science.
281:1640–1645. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Berger M, Neth O, Ilmer M, Garnier A,
Salinas-Martín MV, de Agustín Asencio JC, von Schweinitz D, Kappler
R and Muñoz M: Hepatoblastoma cells express truncated neurokinin-1
receptor and can be growth inhibited by aprepitant in vitro and in
vivo. J Hepatol. 60:985–994. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Muñoz M, González-Ortega A, Salinas-Martín
MV, Carranza A, Garcia-Recio S, Almendro V and Coveñas R: The
neurokinin-1 receptor antagonist aprepitant is a promising
candidate for the treatment of breast cancer. Int J Oncol.
45:1658–1672. 2014.PubMed/NCBI
|
|
21
|
Muñoz M and Rosso M: The NK-1 receptor
antagonist aprepitant as a broad spectrum antitumor drug. Invest
New Drugs. 28:187–193. 2010. View Article : Google Scholar
|
|
22
|
Bresalier RS, Niv Y, Byrd JC, Duh QY,
Toribara NW, Rockwell RW, Dahiya R and Kim YS: Mucin production by
human colonic carcinoma cells correlates with their metastatic
potential in animal models of colon cancer metastasis. J Clin
Invest. 87:1037–1045. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hennessy BT, Lu Y, Gonzalez-Angulo AM,
Carey MS, Myhre S, Ju Z, Davies MA, Liu W, Coombes K,
Meric-Bernstam F, et al: A technical assessment of the utility of
reverse phase protein arrays for the study of the functional
proteome in non-microdissected human breast cancers. Clin
Proteomics. 6:129–151. 2010. View Article : Google Scholar
|
|
24
|
Brabletz T, Jung A, Reu S, Porzner M,
Hlubek F, Kunz-Schughart LA, Knuechel R and Kirchner T: Variable
beta-catenin expression in colorectal cancers indicates tumor
progression driven by the tumor environment. Proc Natl Acad Sci
USA. 98:10356–10361. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang J, Zhang W, Evans PM, Chen X, He X
and Liu C: Adenomatous polyposis coli (APC) differentially
regulates beta-catenin phosphorylation and ubiquitination in colon
cancer cells. J Biol Chem. 281:17751–17757. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wiza C, Nascimento EB and Ouwens DM: Role
of PRAS40 in Akt and mTOR signaling in health and disease. Am J
Physiol Endocrinol Metab. 302:E1453–E1460. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bossy-Wetzel E, Bakiri L and Yaniv M:
Induction of apoptosis by the transcription factor c-Jun. EMBO J.
16:1695–1709. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Leppä S and Bohmann D: Diverse functions
of JNK signaling and c-Jun in stress response and apoptosis.
Oncogene. 18:6158–6162. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lazebnik YA, Kaufmann SH, Desnoyers S,
Poirier GG and Earnshaw WC: Cleavage of poly(ADP-ribose) polymerase
by a proteinase with properties like ICE. Nature. 371:346–347.
1994. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gespach C: Increasing potential of HER3
signaling in colon cancer progression and therapy. Clin Cancer Res.
18:917–919. 2012. View Article : Google Scholar
|
|
31
|
Taylor WR and Stark GR: Regulation of the
G2/M transition by p53. Oncogene. 20:1803–1815. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Orsulic S, Huber O, Aberle H, Arnold S and
Kemler R: E-cadherin binding prevents beta-catenin nuclear
localization and beta-catenin/LEF-1-mediated transactivation. J
Cell Sci. 112:1237–1245. 1999.PubMed/NCBI
|
|
33
|
Lonardo E, Hermann PC, Mueller MT, Huber
S, Balic A, Miranda-Lorenzo I, Zagorac S, Alcala S,
Rodriguez-Arabaolaza I, Ramirez JC, et al: Nodal/Activin signaling
drives self-renewal and tumorigenicity of pancreatic cancer stem
cells and provides a target for combined drug therapy. Cell Stem
Cell. 9:433–446. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fuerer C and Nusse R: Lentiviral vectors
to probe and manipulate the Wnt signaling pathway. PLoS One.
5:e93702010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lepourcelet M, Chen YN, France DS, Wang H,
Crews P, Petersen F, Bruseo C, Wood AW and Shivdasani RA:
Small-molecule antagonists of the oncogenic Tcf/beta-catenin
protein complex. Cancer Cell. 5:91–102. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Moon RT, Kohn AD, De Ferrari GV and Kaykas
A: WNT and beta-catenin signalling: Diseases and therapies. Nat Rev
Genet. 5:691–701. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Klaus A and Birchmeier W: Wnt signalling
and its impact on development and cancer. Nat Rev Cancer.
8:387–398. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gong A and Huang S: FoxM1 and
Wnt/β-catenin signaling in glioma stem cells. Cancer Res.
72:5658–5662. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yoshida Y, Wang IC, Yoder HM, Davidson NO
and Costa RH: The forkhead box M1 transcription factor contributes
to the development and growth of mouse colorectal cancer.
Gastroenterology. 132:1420–1431. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lickert H, Bauer A, Kemler R and Stappert
J: Casein kinase II phosphorylation of E-cadherin increases
E-cadherin/beta-catenin interaction and strengthens cell-cell
adhesion. J Biol Chem. 275:5090–5095. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gonsalves FC, Klein K, Carson BB, Katz S,
Ekas LA, Evans S, Nagourney R, Cardozo T, Brown AM and DasGupta R:
An RNAi-based chemical genetic screen identifies three
small-molecule inhibitors of the Wnt/wingless signaling pathway.
Proc Natl Acad Sci USA. 108:5954–5963. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tian W, Han X, Yan M, Xu Y, Duggineni S,
Lin N, Luo G, Li YM, Han X, Huang Z, et al: Structure-based
discovery of a novel inhibitor targeting the β-catenin/Tcf4
interaction. Biochemistry. 51:724–731. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mayordomo C, García-Recio S, Ametller E,
Fernández-Nogueira P, Pastor-Arroyo EM, Vinyals L, Casas I, Gascón
P and Almendro V: Targeting of substance P induces cancer cell
death and decreases the steady state of EGFR and Her2. J Cell
Physiol. 227:1358–1366. 2012. View Article : Google Scholar
|
|
44
|
Inoki K, Ouyang H, Zhu T, Lindvall C, Wang
Y, Zhang X, Yang Q, Bennett C, Harada Y, Stankunas K, et al: TSC2
integrates Wnt and energy signals via a coordinated phosphorylation
by AMPK and GSK3 to regulate cell growth. Cell. 126:955–968. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Breuleux M, Klopfenstein M, Stephan C,
Doughty CA, Barys L, Maira SM, Kwiatkowski D and Lane HA: Increased
AKT S473 phosphorylation after mTORC1 inhibition is rictor
dependent and does not predict tumor cell response to PI3K/mTOR
inhibition. Mol Cancer Ther. 8:742–753. 2009. View Article : Google Scholar : PubMed/NCBI
|