Introduction
Most autosomal genes are normally expressed from
both their paternal and maternal alleles. In contrast, imprinted
genes are expressed predominantly and/or exclusively from either of
their paternal or maternal allele (1). For example, certain imprinted genes
such as IGF2, DNMT1 and PEG3 are expressed from their
paternal alleles, while their maternal alleles are silenced.
Conversely, others including RB1, TP73 and CDKN1C are
expressed from their maternal alleles, whereas their paternal
alleles are inactivated. Abnormal regulation of imprinting causes
fetal death or development of diseases such as Beckwith-Wiedemann
syndrome (BWS), Angelman syndrome and Prader-Willi syndrome.
BWS has been considered to be an overgrowth disorder
accompanied by several symptoms including macrosomia, macroglossia
and abdominal wall defects (2,3). In
addition, patients with BWS have been shown to be predisposed to
childhood and embryonal tumors such as Wilms’ tumor and
hepatoblastoma (4–7). Majority of patients with BWS harbor
aberrant alterations at human chromosome 11p15 (3). Both epigenetic and genomic
alterations of the imprinting cluster within this chromosomal
region have been detected in up to 80% of BWS patients as examined
by currently available procedures (8,9). The
imprinting of 11p15.5 region was found to be regulated by two
imprinting control regions, H19DMR located upstream of the
H19 gene and KvDMR within intron 10 of KCNQ1 gene,
which acts as a promoter region of its antisense gene
KCNQ1OT1 (10).
KCNQ1OT1 encodes a paternally expressed long
non-coding RNA, which has been shown to regulate imprinting of
several genes present at 11p15.5 locus in cis (11,12).
Under normal conditions, the maternal and paternal alleles of KvDMR
are methylated and unmethylated, respectively (10). Loss of imprinting of this region,
i.e. loss of methylation of maternal allele, resulted in aberrant
overexpression of KCNQ1OT1, which suppressed the
circumjacent gene expression such as the CDKN1C
(p57KIP2) tumor suppressor gene (13,14).
p57KIP2 is a maternally expressed
imprinted gene encoding a cyclin-dependent protein kinase inhibitor
implicated in the regulation of prenatal and postnatal development
(15). Defects of imprinting at
KvDMR account for the majority (~50%) of molecular aberrations in
BWS patients, which causes the bial-lelic expression of
KCNQ1OT1 and then leads to a significant suppression of
p57KIP2 expression (16,17).
Moreover, loss of methylation at KvDMR in association with reduced
expression level of p57KIP2 was also
observed in a variety of adult tumors including colorectal cancer
and lung cancer (18,19). These findings indicate that
silencing of KCNQ1OT1 might prohibit tumor development
and/or progression.
Pyrrole-imidazole (PI) polyamide has been shown to
be a novel gene-silencing small chemical compound, which binds to a
minor groove of double-strand DNA in a sequence-specific manner
(20). Hairpin polyamide is
derived from distamycin A, which binds to AT-rich DNA sequences
(21). Polyamide containing the
appropriate combination of aromatic amino acids N-methylpyrrole
(Py) and N-Methylimidazole (Im) binds to its target DNA with
affinity and specificity comparable to that of DNA-protein
interaction in cells (22).
According to the previous observations, Im/Py, Py/Im and Py/Py
pairs bind to G-C, C-G, and A-T/T-A, respectively (22).
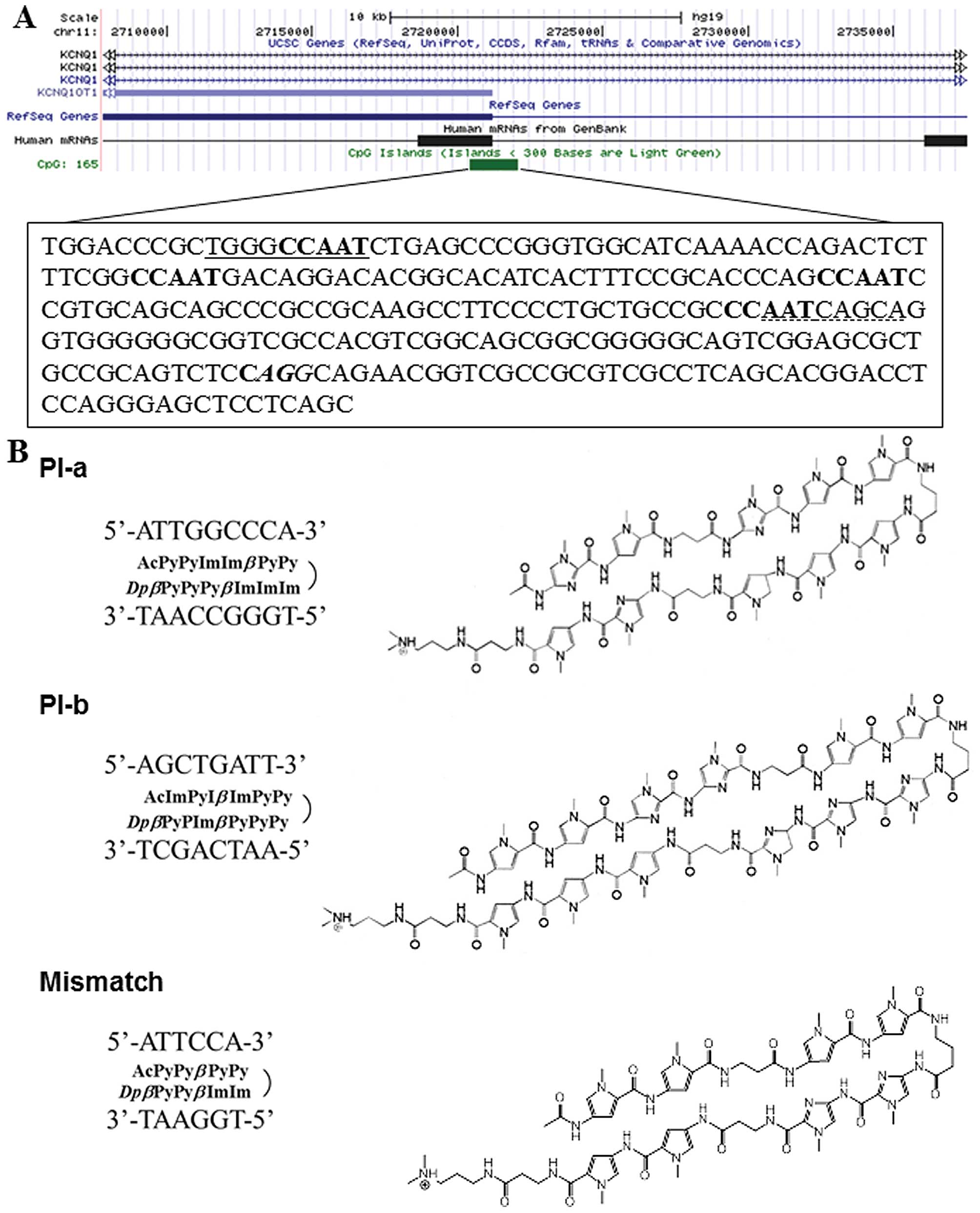
In the present study, we have generated the PI
polyamides targeting the CCAAT box, which is present at KvDMR and
involved in the regulation of KCNQ1OT1 expression (10), and examined whether these compounds
could specifically suppress growth of Wilms’ tumor-derived G401
cells.
Materials and methods
Cell lines and culture conditions
Human fibroblasts-derived BWS6 and BWS9 cells, which
have been established from Beckwith-Wiedemann syndrome patients,
were kindly provided by Dr M.J. Higgins (Roswell Park Cancer
Institute), and Human Wilms’ tumor G401 cells were purchased from
Human Science (Kyoto, Japan). Both BWS6 and BWS9 cells were
maintained in DMEM (Nakarai Tesque, Kyoto, Japan) supplemented with
10% heat-inactivated fetal bovine serum (FBS) (Nichirei Bioscience,
Tokyo, Japan) and 100 IU/ml of penicillin/streptomycin/glutamine
(PSG) (Life Technologies, Carlsbad, CA, USA). G401 cells were
cultured in McCoy’s 5A (Life Technologies) supplemented with 10%
heat-inactivated FBS and 100 IU/ml of PSG. Cells were grown at 37°C
in a CO2 incubator with a humidified atmosphere
containing 5% CO2 and 95% air.
Synthesis of PI polyamides targeting
human KCNQ1OT1
PI polyamides used in this study were synthesized by
a machine-assisted automatic synthesis system, PSSM-8 peptide
synthesizer (Shimadzu, Kyoto, Japan) as previously described
(23,24). Two PI polyamides termed PI-a and
PI-b were designed to span the boundary of 2 of 4 CCAAT boxes
within human KCNQ1OT1 promoter region (Fig. 1). Mismatch PI polyamide, which does
not recognize CCAAT sequence, was also designed and produced. After
synthesis and purification, these PI polyamides were dissolved in
ultra pure water at a final concentration of 1 mM as stock
solutions.
Gel retardation assay
FITC-labeled oligonucleotides containing target
sequences of PI polyamides were synthesized by Eurofin Genomics
(Tokyo, Japan). Sequences of the oligonucleotides are as follows:
the oligonucleotide containing PI-a target sequence,
5′-GAGTCTAACCGGGTCGCCCTTTTGGGCGAC CCGGTTAGACTC-3′ and the
oligonucleotide containing PI-b target sequence, 5′-GGTGGTCGAACTAA
CCCGCTTTTGCGGGTTAGTCGACCACC-3′. To generate hairpin structure of
double-strand DNA, 1 μM of oligonucleotides dissolved in an
annealing buffer (5 M NaCl, 500 mM EDTA, 1 M Tris-HCl pH 8.0) were
boiled at 100°C for 10 min, and then left at room temperature to
gradually cool them down. The resultant FITC-labeled hairpin
oligonucleotides were incubated in the presence or absence of 1 or
3 μM of PI polyamides for 1 h at room temperature. The reaction
mixtures were separated by electrophoresis through 20%
polyacrylamide gel in Tris-borate-EDTA buffer and then visualized
with the ImageQant LAS4000 (Fujifilm, Tokyo, Japan).
Analysis of the intracellular
distribution of PI polyamides
BWS6 and G401 cells were maintained in the presence
or absence of FITC-labeled PI polyamides for 24 h. After washing in
ice-cold phosphate-buffered saline (PBS, Nakarai Tesque), cells
were fixed in 4% formaldehyde solution (Wako Pure Chemical
Industries, Osaka, Japan) for 30 min, and then stained with
4′,6-diamidino-2-phenylindole dihydrochloride (DAPI, Sigma-Aldrich,
St. Louis, MO, USA). Finally, cells were observed under
fluorescence microscopy Axiovert 2000 (Carl Zeiss Microscopy,
Oberkochen, Germany).
Quantitative real-time RT-PCR
Seventy-two hours after incubation in the presence
or absence of PI polyamides, cells were washed in ice-cold PBS and
lysed in TRIzol reagent (Life Technologies). Then, total RNA was
prepared according to the manufacturer’s instructions, and 500 ng
of total RNA was treated with DNaseI (Qiagen, Hilden, Germany),
followed by reverse transcription using Prime Script (Takara). The
resultant cDNA was subjected to quantitative real-time RT-PCR
analysis using Dimer Eraser (Takara). The primers used in this
analysis are as follows: KCNQ1OT1 (forward: 5′-AATGGG
GATGTGAGGATCAGG-3′, reverse: 5′-TGACCCCAGTG GAATATGTGC-3′); GAPDH
(forward: 5′-GCACCGTCAA GGCTGAGAAC-3′, reverse: 5′-TGGTGAAGACGCCAGT
GGA-3′). GAPDH was used as an internal control.
Cell proliferation assay
G401 cells were seeded at 2×103
cells/96-well microplates. Then cells were exposed to the indicated
concentrations of PI polyamides or left untreated. At the indicated
time points after treatment, cell viability was measured by
standard WST-8 assay (Nacalai tesque).
Fluorescene-activated cell sorting
(FACS)
G401 cells were seeded at 5×104
cells/2-cm diameter dishes. Then, cells were treated with or
without the indicated PI polyamides. Seventy-two hours after
treatment, cells were washed in ice-cold PBS, treated with
trypsin/EDTA, and 2×105 cells were subsequently stained
with Annexin V-FITC plus propidium iodide using Annexin V-FITC
Apoptosis Detection kit (Abcam, Bristol, UK) according to the
manufacturer’s protocols. Finally, cells were sorted by FACSCalibur
flow cytometer with FL-1 (Annexin V channels) and FL-2 (PI
channels), and then analyzed by CellQuest (BD Biosciences,
Rockville, MD, USA). As a positive control, cells were treated with
10 μM of H2O2 for 9 h to induce cell
death.
Statistical analysis
Statistical significance was calculated using the
Student’s t-test. Results were considered as significant with
P-value of <0.05.
Results
Specific binding of the PI polyamides to
their target double-strand DNA
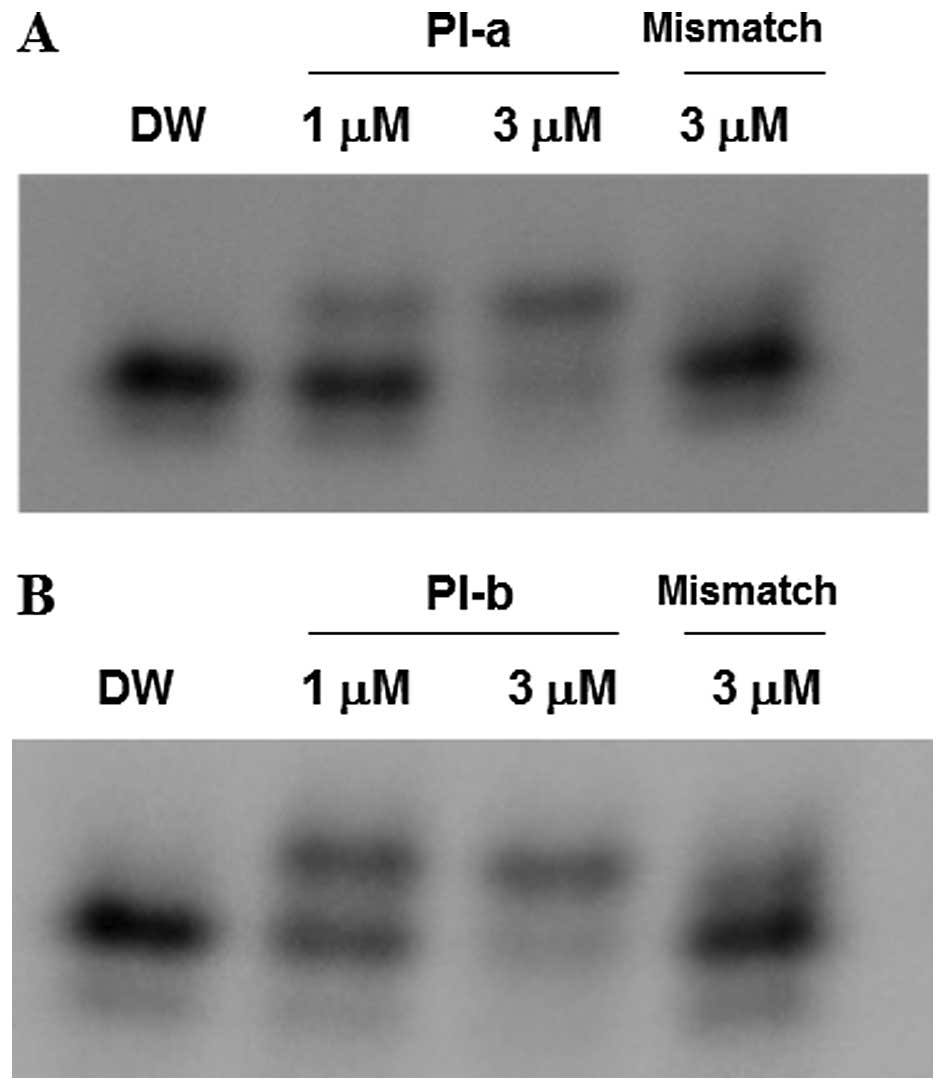
To verify whether the synthesized PI polyamides
(termed PI-a and PI-b) could bind to their target DNA sequences,
gel mobility shift assays were performed. For this purpose, the
oligonucleotide containing target sequence of PI-a was incubated
with DW, PI-a or non-specific PI polyamide (termed mismatch), and
then reaction mixtures were analyzed by poly-acrylamide gel
electrophoresis. As shown in Fig.
2A, a clearly retarded band was observed in the presence of
PI-a but not of mismatch PI polyamide. Similarly, an
oligonucleotide/PI-b complex was detectable in the reaction mixture
containing PI-b and its target DNA sequence, whereas mismatch PI
polyamide failed to bind to oligonucleotide containing the target
DNA of PI-b (Fig. 2B). These
results indicate that both PI-a and PI-b can specifically recognize
and bind to their target DNA sequences in vitro.
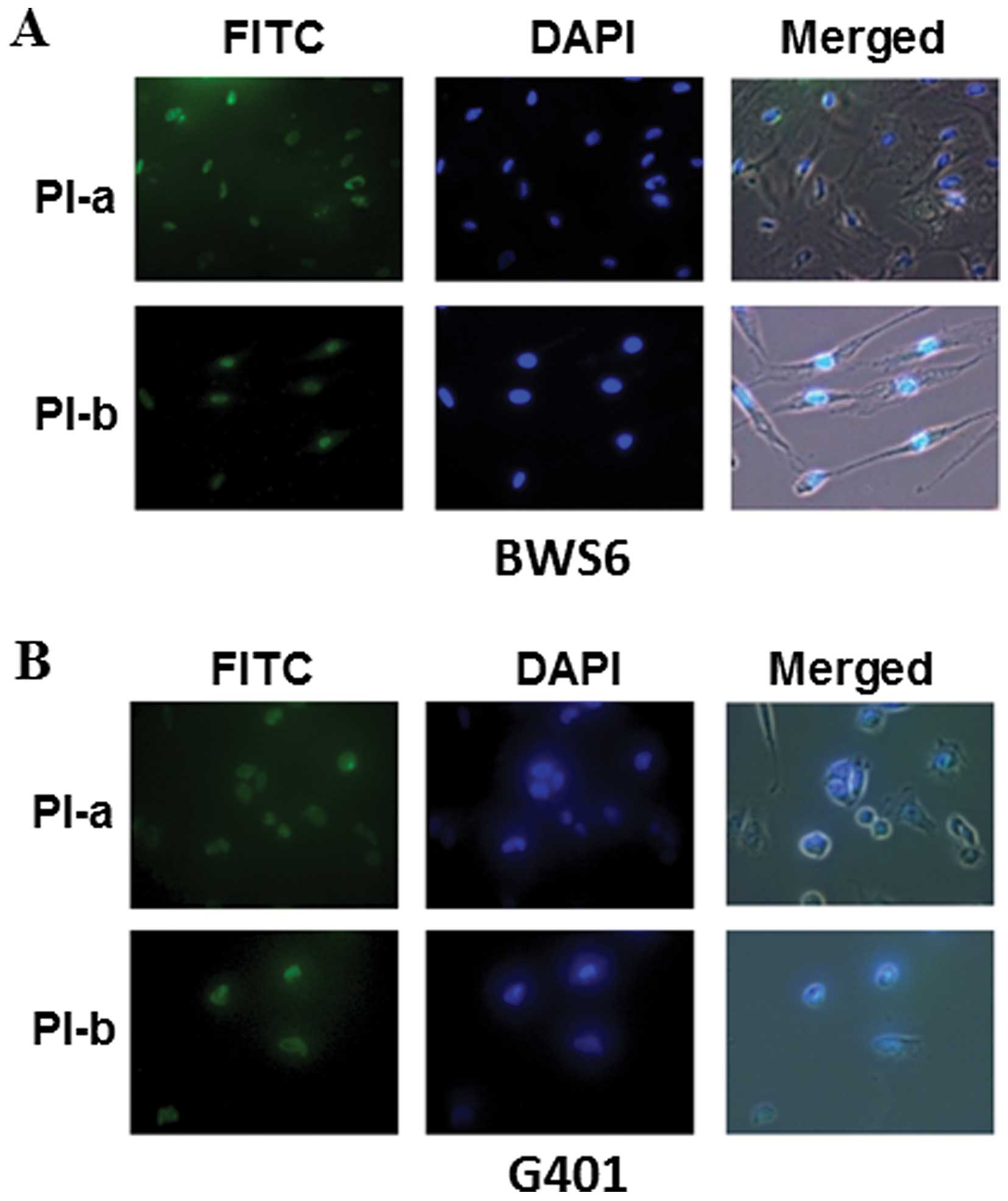
Nuclear distribution of PI
polyamides
Since the PI polyamides could be expected to exert
gene silencing function through the direct interaction with their
target DNA sequence in cells, it should be required to confirm
their nuclear distribution. To adequately address this issue, we
generated FITC-conjugated PI-a and PI-b. Human fibroblasts-derived
from a BWS patient BWS6 cells and Wilms’ tumor-derived G401 cells
were cultured in fresh medium containing PI-a or PI-b. Twenty-four
hours after incubation, cells were fixed, stained with DAPI and
observed under a confocal microscope. As seen in Fig. 3, a significant nuclear access of
FITC-labeled PI-a and PI-b was detected under our experimental
conditions. These observations strongly suggest that PI-a and PI-b
display nuclear localization without any drug delivery systems such
as adenovirus.
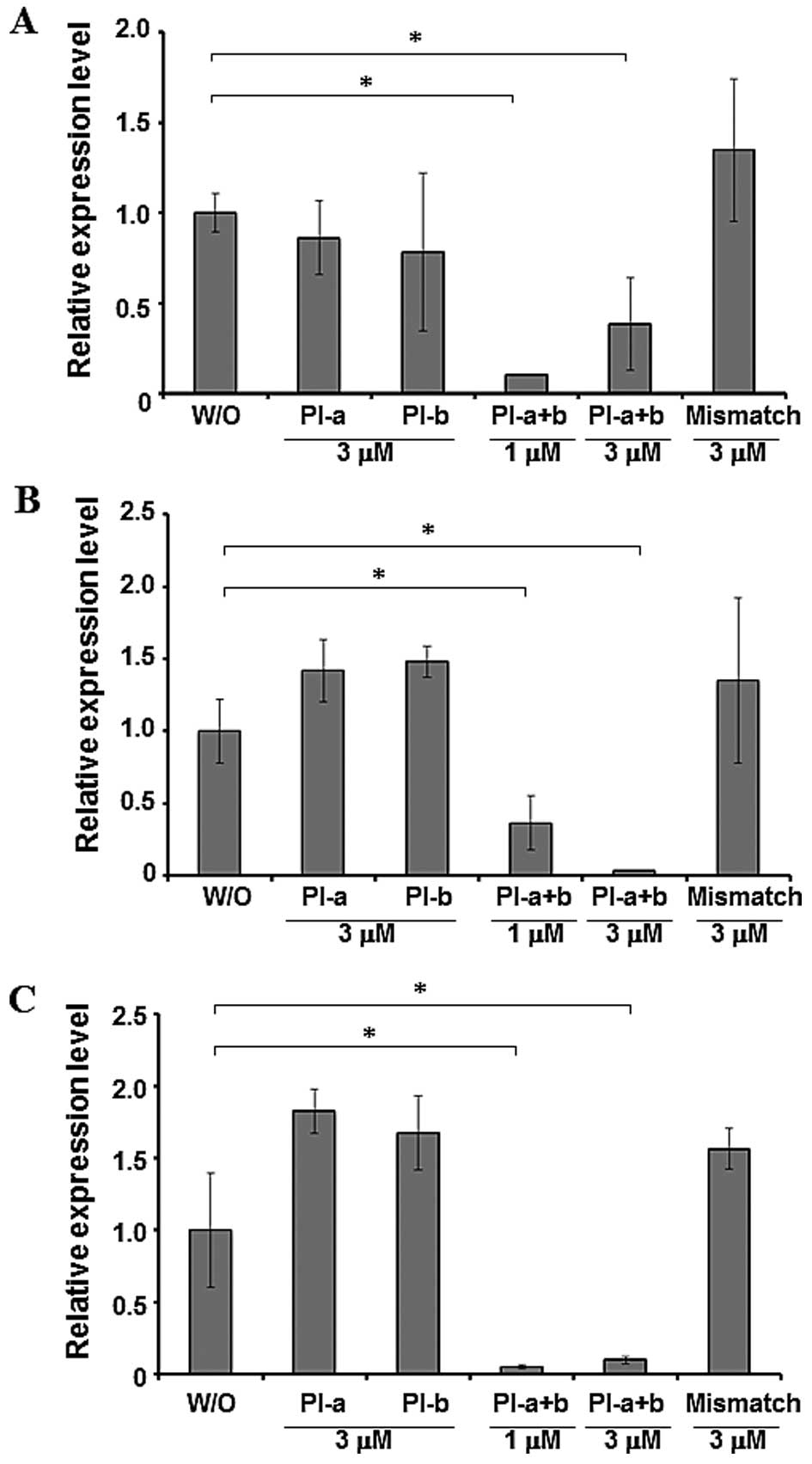
Combinatory treatment with PI-a and PI-b
efficiently suppresses KCNQ1OT1 transcription
Next, we examined whether PI-a and/or PI-b could
downregulate the transcription of their target gene
KCNQ1OT1. To this end, BWS6, BWS9 and G401 cells were
incubated in the presence of the indicated concentrations of PI-a,
PI-b, PI-a plus PI-b or mismatch PI polyamide. Seventy-two hours
after treatment, total RNA was prepared and the expression level of
KCNQ1OT1 was analyzed by quantitative real-time PCR.
Unexpectedly, a significant downregulation of KCNQ1OT1 was not
detected in all cells examined after PI-a or PI-b single treatment
(Fig. 4). Notably, the combinatory
treatment of cells with PI-a and PI-b resulted in a marked decrease
in the expression level of KCNQ1OT1. As expected, mismatch
PI polyamide alone had no detectable effect on the expression level
of KCNQ1OT1.
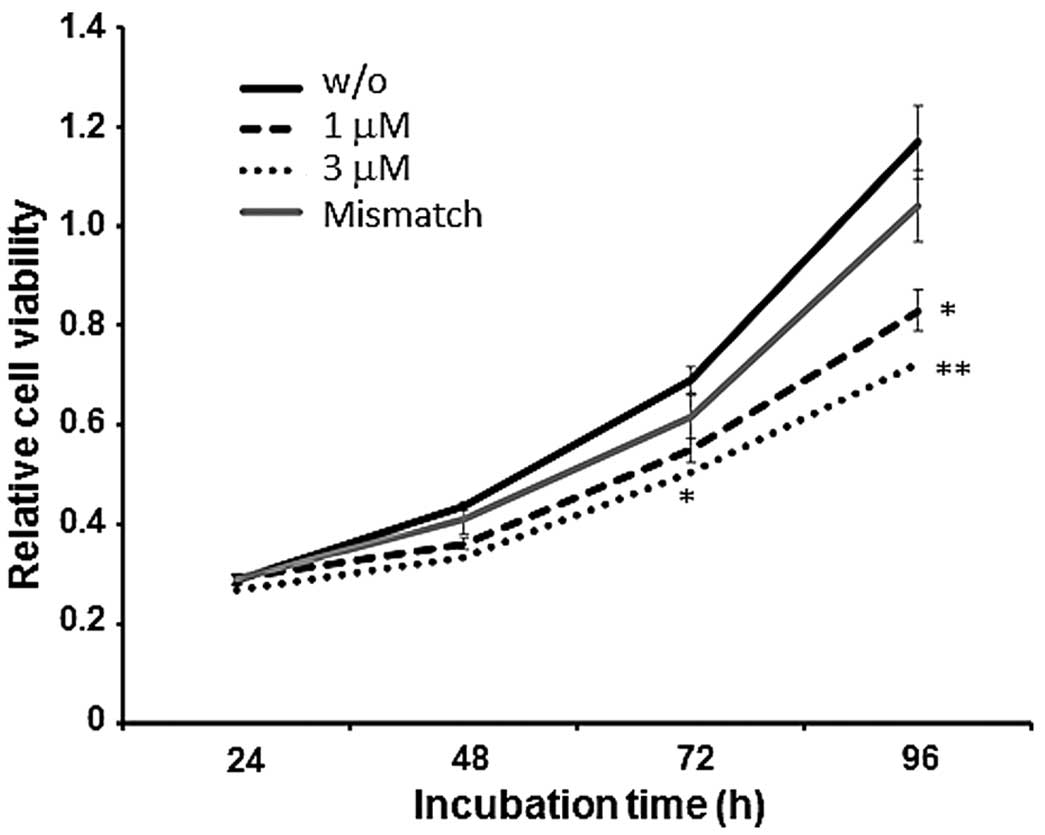
Reduction in cell viability of G401 cells
exposed to a combined treatment with PI-a and PI-b
Since it has been described that a higher
KCNQ1OT1 level is tightly associated with tumor cell growth
(25), these observations prompted
us to examine whether the simultaneous treatment of PI-a and PI-b
could affect cell proliferation of G401 cells. For this purpose,
G401 cells were exposed to the indicated concentrations of PI-a
plus PI-b or mismatch PI polyamide. At the indicated time points
after treatment, cells were subjected to the standard WST8 assay to
assess their cell viability. As shown in Fig. 5, the combinatory treatment of PI-a
and PI-b caused a remarkable suppression of G401 cell
proliferation, whereas mismatch PI polyamide had a negligible
effect on their viability. As expected, PI-a or PI-b single
treatment did not affect G401 cell proliferation (data not shown).
These results imply that the PI polyamide-mediated silencing of
KCNQ1OT1 expression might contribute to the suppression of
G401 cell proliferation.
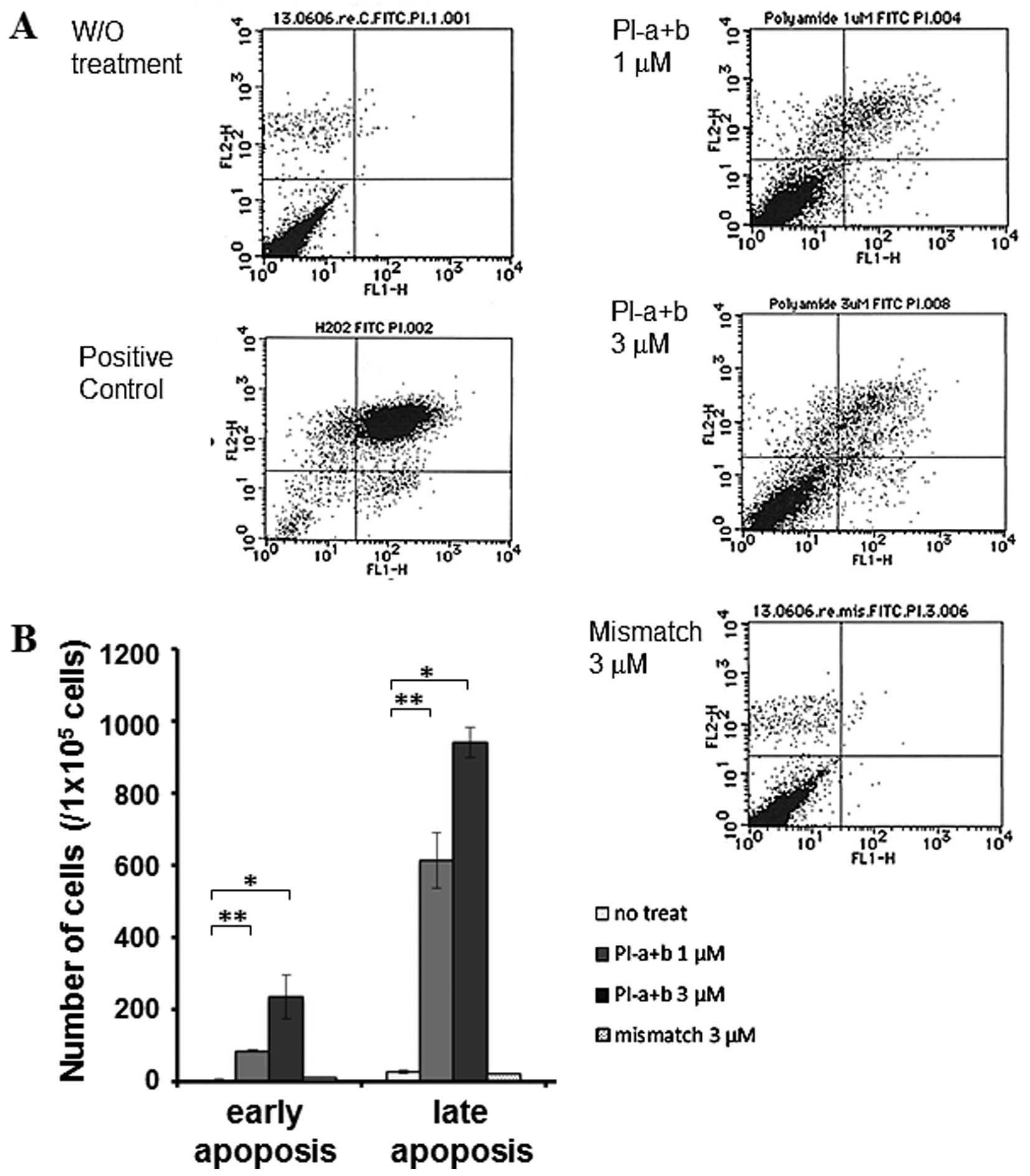
G401 cells undergo cell death in response
to PI-a plus PI-b
Based on our above-mentioned results, we asked
whether a combined treatment with PI-a plus PI-b in G401 cells
could induce cell death. To this end, we used PI and Annexin V
double staining experiments followed by FACS analysis. It has been
established that Annexin V-positive cells undergo early apoptotic
cell death (26). G401 cells were
treated with the indicated concentrations of PI-a plus PI-b or with
mismatch PI polyamide. As a positive control, G401 cells were
exposed to H2O2 for 9 h. It is well-known
that Annexin V-positive/ PI-negative cells and Annexin
V-positive/PI-positive cells undergo early apoptosis and late
apoptosis/necrosis, respectively (27). As seen in Fig. 6A, a large number of cells exposed
to H2O2 displayed Annexin
V-positive/PI-positive, indicating that cells undergo late
apoptosis in response to H2O2. Apoptotic
cells were barely detectable in untreated or mismatch-treated
cells. In contrast, substantial amounts of cells were enriched in
early and late apoptotic fractions. Average number of cells showing
early apoptosis, late apoptosis or necrosis is depicted as a graph
in Fig. 6B. According to our
present results, the combinatory treatment of cells with PI-a and
PI-b markedly induced apoptosis in a dose-dependent manner. In
addition, the relatively small number of Annexin
V-negative/PI-positive cells was observed in each experiment.
Discussion
In the present study, we successfully produced
pyrrole-imidazole (PI) polyamides (PI-a and PI-b) against CCAAT
boxes within the promoter region of oncogenic KCNQ1OT1 gene.
Based on our present results, the combined treatment with PI-a and
PI-b in Wilms’ tumor-derived G401 cells significantly reduced
KCNQ1OT1 gene expression and also their cell viability,
which might be caused by the induction of apoptosis.
The molecular targeting anticancer drugs, which
could block oncogenic signaling pathways, would be powerful and
promising cancer therapeutics. Recent high-throughput technologies
make it much easier to identify critical genes implicated in the
initiation and/or progression of a variety of cancers, which are
aberrantly expressed and/or activated in cancer cells (28). Although several antisense oligo DNA
and siRNA against these cancer-related specific genes have been
developed, their poor cellular uptake and also delivery to the
appropriate organs or tissues remain major obstacles for their
clinical application (29). Since
it has been shown that PI polyamides are efficiently localized into
cell nucleus without any specific drug delivery systems such as
virus vehicles both in vitro and in vivo (30,31),
these observations prompted us to develop gene-specific silencing
PI polyamides. For example, we have previously described that PI
polyamide recognizing AP-1-binding site located within the promoter
region of matrix metalloproteinase (MMP)-9 gene massively
reduces its expression level and also inhibits malignant phenotypes
of cancer cells both in vitro and in vivo (30).
In our results, single application of either PI-a or
PI-b did not affect the expression level of KCNQ1OT1,
however, the treatment with the combination of these two PI
polyamides downregulated KCNQ1OT1. At the present moment, we
could not find good explanation for this discrepancy. Previously,
Du et al reported that an introduced point mutation in each
of four CCAAT box sequences reduced promoter activity modestly but
not completely (10,32). The authors also showed data
suggesting that simultaneous targeting of two CCAAT box regions of
KCNQ1OT1 promoter could be also involved in the transcriptional
regulation of this gene. Those findings indicate that blocking just
one CCAAT box is not enough to suppress the gene expression
level.
Of note, we found for the first time that the
combinatory treatment of our PI polyamides significantly
downregulates oncogenic KCNQ1OT1 gene transcription, and its
downregulation leads to the suppression of cancerous cell
proliferation as well as the induction of apoptosis. It has been
shown that KCNQ1OT1 has the ability to regulate the
imprinting status of the circumjacent genes, such as ASCL2,
TSPAN32, CD81, TSSC4, p57KIP2,
SLC22A18 and PHLDA2 of the same allele (13,33).
Among them, p57KIP2 has been considered to
be one of tumor suppressor gene products implicated in BWS
(34). Accumulating evidence
suggests that p57KIP2 is one of Cip/Kip family of
cyclin-dependent protein kinase inhibitor and blocks cell
proliferation by inhibiting cell cycle progression (35). Additionally, p57KIP2 has
the ability to promote apoptosis and prohibit tissue invasion as
well as metastasis (36,37). Therefore, we sought to examine
whether the combinatory treatment of the PI polyamides could affect
the expression level of the above-mentioned circumjacent genes
including p57KIP2. Unexpectedly, the
expression level of these genes remained unchanged even in the
presence of the PI polyamides (data not shown). At present, we do
not know how the combined treatment with the two PI polyamides
could suppress cancerous cell proliferation and also induce
apoptosis. Further study is required to adequately address this
issue.
Collectively, our present results indicate that the
above reported PI polyamides targeting KCNQ1OT1 promotes
apoptosis in Wilms’ tumor G401 cells through a significant
downregulation of KCNQ1OT1 transcription, and support our
hypothesis that PI polyamide-mediated gene silencing might be a
novel attractive strategy for anticancer therapy.
Acknowledgements
This work was supported by the Academic Frontier
Project for 2006 Project for Private Universities, a matching fund
subsidy from MEXT (to H.N.), and MEXT-Supported Program for the
Strategic Research Foundation at Private Universities (2011–2015)
to K.F., K.S., T.U., N.F., M.S., T.K., and H.N.
References
|
1
|
Reik W and Walter J: Genomic imprinting:
Parental influence on the genome. Nat Rev Genet. 2:21–32. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Choufani S, Shuman C and Weksberg R:
Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet.
154C:343–354. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jacob KJ, Robinson WP and Lefebvre L:
Beckwith-Wiedemann and Silver-Russell syndromes: Opposite
developmental imbalances in imprinted regulators of placental
function and embryonic growth. Clin Genet. 84:326–334. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Weksberg R, Nishikawa J, Caluseriu O, Fei
YL, Shuman C, Wei C, Steele L, Cameron J, Smith A, Ambus I, et al:
Tumor development in the Beckwith-Wiedemann syndrome is associated
with a variety of constitutional molecular 11p15 alterations
including imprinting defects of KCNQ1OT1. Hum Mol Genet.
10:2989–3000. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
DeBaun MR and Tucker MA: Risk of cancer
during the first four years of life in children from The
Beckwith-Wiedemann Syndrome Registry. J Pediatr. 132:398–400. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rump P, Zeegers MP and van Essen AJ: Tumor
risk in Beckwith-Wiedemann syndrome: A review and meta-analysis. Am
J Med Genet A. 136:95–104. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Higashimoto K, Soejima H, Saito T, Okumura
K and Mukai T: Imprinting disruption of the CDKN1C/KCNQ1OT1 domain:
The molecular mechanisms causing Beckwith-Wiedemann syndrome and
cancer. Cytogenet Genome Res. 113:306–312. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Scott RH, Douglas J, Baskcomb L, Nygren
AO, Birch JM, Cole TR, Cormier-Daire V, Eastwood DM, Garcia-Minaur
S, Lupunzina P, et al: Methylation-specific multiplex
ligation-dependent probe amplification (MS-MLPA) robustly detects
and distinguishes 11p15 abnormalities associated with overgrowth
and growth retardation. J Med Genet. 45:106–113. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Weksberg R, Shuman C and Beckwith JB:
Beckwith-Wiedemann syndrome. Eur J Hum Genet. 18:8–14. 2010.
View Article : Google Scholar :
|
|
10
|
Du M, Zhou W, Beatty LG, Weksberg R and
Sadowski PD: The KCNQ1OT1 promoter, a key regulator of genomic
imprinting in human chromosome 11p15.5. Genomics. 84:288–300. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kanduri C: Kcnq1ot1: A chromatin
regulatory RNA. Semin Cell Dev Biol. 22:343–350. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mohammad F, Pandey GK, Mondal T, Enroth S,
Redrup L, Gyllensten U and Kanduri C: Long noncoding RNA-mediated
maintenance of DNA methylation and transcriptional gene silencing.
Development. 139:2792–2803. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mancini-Dinardo D, Steele SJ, Levorse JM,
Ingram RS and Tilghman SM: Elongation of the Kcnq1ot1 transcript is
required for genomic imprinting of neighboring genes. Genes Dev.
20:1268–1282. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chiesa N, De Crescenzo A, Mishra K, Perone
L, Carella M, Palumbo O, Mussa A, Sparago A, Cerrato F, Russo S, et
al: The KCNQ1OT1 imprinting control region and non-coding RNA: New
properties derived from the study of Beckwith-Wiedemann syndrome
and Silver-Russell syndrome cases. Hum Mol Genet. 21:10–25. 2012.
View Article : Google Scholar
|
|
15
|
Zhang P, Wong C, DePinho RA, Harper JW and
Elledge SJ: Cooperation between the Cdk inhibitors p27(KIP1) and
p57(KIP2) in the control of tissue growth and development. Genes
Dev. 12:3162–3167. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee MP, DeBaun MR, Mitsuya K, Galonek HL,
Brandenburg S, Oshimura M and Feinberg AP: Loss of imprinting of a
paternally expressed transcript, with antisense orientation to
KVLQT1, occurs frequently in Beckwith-Wiedemann syndrome and is
independent of insulin-like growth factor II imprinting. Proc Natl
Acad Sci USA. 96:5203–5208. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Diaz-Meyer N, Day CD, Khatod K, Maher ER,
Cooper W, Reik W, Junien C, Graham G, Algar E, Der Kaloustian VM,
et al: Silencing of CDKN1C (p57KIP2) is associated with
hypomethylation at KvDMR1 in Beckwith-Wiedemann syndrome. J Med
Genet. 40:797–801. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nakano S, Murakami K, Meguro M, Soejima H,
Higashimoto K, Urano T, Kugoh H, Mukai T, Ikeguchi M and Oshimura
M: Expression profile of LIT1/KCNQ1OT1 and epigenetic status at the
KvDMR1 in colorectal cancers. Cancer Sci. 97:1147–1154. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Soejima H, Nakagawachi T, Zhao W,
Higashimoto K, Urano T, Matsukura S, Kitajima Y, Takeuchi M,
Nakayama M, Oshimura M, et al: Silencing of imprinted CDKN1C gene
expression is associated with loss of CpG and histone H3 lysine 9
methylation at DMR-LIT1 in esophageal cancer. Oncogene.
23:4380–4388. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dervan PB: Molecular recognition of DNA by
small molecules. Bioorg Med Chem. 9:2215–2235. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Arcamone F, Penco S, Orezzi P, Nicolella V
and Pirelli A: Structure and synthesis of distamycin A. Nature.
203:1064–1065. 1964. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dervan PB and Edelson BS: Recognition of
the DNA minor groove by pyrrole-imidazole polyamides. Curr Opin
Struct Biol. 13:284–299. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bando T, Narita A, Saito I and Sugiyama H:
Molecular design of a pyrrole-imidazole hairpin polyamides for
effective DNA alkylation. Chemistry. 8:4781–4790. 2002. View Article : Google Scholar
|
|
24
|
Murty MS and Sugiyama H: Biology of
N-methylpyrrole-N-methylimidazole hairpin polyamide. Biol Pharm
Bull. 27:468–474. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wan J, Huang M, Zhao H, Wang C, Zhao X,
Jiang X, Bian S, He Y and Gao Y: A novel tetranucleotide repeat
polymorphism within KCNQ1OT1 confers risk for hepatocellular
carcinoma. DNA Cell Biol. 32:628–634. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vermes I, Haanen C, Steffens-Nakken H and
Reutelingsperger C: A novel assay for apoptosis. Flow cytometric
detection of phosphatidylserine expression on early apoptotic cells
using fluorescein labelled Annexin V. J Immunol Methods. 184:39–51.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Aubry JP, Blaecke A, Lecoanet-Henchoz S,
Jeannin P, Herbault N, Caron G, Moine V and Bonnefoy JY: Annexin V
used for measuring apoptosis in the early events of cellular
cytotoxicity. Cytometry. 37:197–204. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dobbelstein M and Moll U: Targeting
tumour-supportive cellular machineries in anticancer drug
development. Nat Rev Drug Discov. 13:179–196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Farooqi AA, Rehman ZU and Muntane J:
Antisense therapeutics in oncology: Current status. Onco Targets
Ther. 7:2035–2042. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang X, Nagase H, Watanabe T, Nobusue H,
Suzuki T, Asami Y, Shinojima Y, Kawashima H, Takagi K, Mishra R, et
al: Inhibition of MMP-9 transcription and suppression of tumor
metastasis by pyrrole-imidazole polyamide. Cancer Sci. 101:759–766.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Matsuda H, Fukuda N, Ueno T, Katakawa M,
Wang X, Watanabe T, Matsui S, Aoyama T, Saito K, Bando T, et al:
Transcriptional inhibition of progressive renal disease by gene
silencing pyrrole-imidazole polyamide targeting of the transforming
growth factor-β1 promoter. Kidney Int. 79:46–56. 2011. View Article : Google Scholar
|
|
32
|
Du M, Beatty LG, Zhou W, Lew J, Schoenherr
C, Weksberg R and Sadowski PD: Insulator and silencer sequences in
the imprinted region of human chromosome 11p15.5. Hum Mol Genet.
12:1927–1939. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fitzpatrick GV, Soloway PD and Higgins MJ:
Regional loss of imprinting and growth deficiency in mice with a
targeted deletion of KvDMR1. Nat Genet. 32:426–431. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Arima T, Kamikihara T, Hayashida T, Kato
K, Inoue T, Shirayoshi Y, Oshimura M, Soejima H, Mukai T and Wake
N: ZAC, LIT1 (KCNQ1OT1) and p57KIP2 (CDKN1C) are in an imprinted
gene network that may play a role in Beckwith-Wiedemann syndrome.
Nucleic Acids Res. 33:2650–2660. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
LaBaer J, Garrett MD, Stevenson LF,
Slingerland JM, Sandhu C, Chou HS, Fattaey A and Harlow E: New
functional activities for the p21 family of CDK inhibitors. Genes
Dev. 11:847–862. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jin RJ, Lho Y, Wang Y, Ao M, Revelo MP,
Hayward SW, Wills ML, Logan SK, Zhang P and Matusik RJ:
Down-regulation of p57Kip2 induces prostate cancer in the mouse.
Cancer Res. 68:3601–3608. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Guo H, Lv Y, Tian T, Hu TH, Wang WJ, Sui
X, Jiang L, Ruan ZP and Nan KJ: Downregulation of p57 accelerates
the growth and invasion of hepatocellular carcinoma.
Carcinogenesis. 32:1897–1904. 2011. View Article : Google Scholar : PubMed/NCBI
|