Introduction
In recent decades, an integrated approach to
malignant cell treatment that includes surgery, chemotherapy
(1), radiotherapy, gene therapy
(2,3) and immune therapy (4), has become a reasonable therapeutic
strategy. Oncolytic virotherapy using agents such as Newcastle
disease virus (NDV) is one of the new biological strategies for
gene (2) and immune therapy
(4–6) and has been tested against many
different cancers, including gastric cancer (4), skin tumors (7) and solid cancers (8).
NDV, which is a member of the Paramyxoviridae
family, is a non-segmented negative-strand RNA virus (NNSV). The
NDV genes encode six major structural proteins, including
nucleoprotein (NP), phosphoprotein (P), matrix protein (M), fusion
protein (F), hemagglutinin-neuraminidase (HN) and large (L)
RNA-dependent RNA polymerase, in the following order:
3′-NP-P-M-F-HN-L-5′. Oncolysis (9), apoptosis (10,11),
and enhanced innate immunity (4,12,13)
are the known mechanisms by which NDV kills malignant cells.
The rabies virus (RV), which is a member of
Rhabdoviridae family, is an enveloped, bullet-shaped virus
that is also associated with host cell apoptosis (13,14).
Some clinical trials as early as the 1950s and 1960s demonstrated
the effectiveness of RV in treating melanomatosis (15).
Autophagy is a catabolic and highly conservative
cellular process. Its basic role is to recycle cellular components
during nutritional starvation and other stressful conditions
(16) and is considered a
cytoprotective event (17).
Despite its protective effects, autophagy can also induce a type of
cell death (18) called ‘type II
programmed cell death’ (19).
Whether autophagy contributes to cell death or to protection during
chemotherapy remains controversial (20). However, increasing evidence has
demonstrated that autophagy represents cell death (21). Additionally, many chemicals, such
as avicin D (22) and arsenic
trioxide (23), as well as
cellular stresses (24), and
mitochondrial dysfunctions (25)
can contribute to autophagy. Autophagy is a known potential
anticancer strategy. Previous studies have shown that recombinant
NDV-expressing rabies virus glycoprotein (rL-RVG) has the ability
to spread from cell to cell without the help of trypsin (26), to alter its self-replication, and
to induce cell death via the apoptosis pathway (27). The present study examined the
contribution of autophagy to apoptosis induced by NDV or rL-RVG and
raised a hypothesis regarding cross-talk among autophagy,
apoptosis, mitochondrial dysfunction and endoplasmic reticulum
stress (ERs).
Materials and methods
Materials
The NDV strain LaSota, rL-RVG, and anti-NDV antibody
were provided by Harbin Veterinary Research Institute, Chinese
Academy of Agricultural Sciences. The human cancer cell lines
SGC-7901 and AGS were purchased from the Cancer Cell Repository
(Shanghai Cell Bank, 2010-02-20) and ATCC (MD, USA), respectively.
3-[4,5-dimehyl-2-thiazolyl]-2,5-diphenyl-2H-tetrazolium bromide
(MTT) was purchased from Amresco (PA, USA). 3-Methyladenine (3-MA)
and Hoechst 33342 were obtained from Sigma-Aldrich (St. Louis, MO,
USA). The siRNA specific for human beclin-1 was from GenePharma
(Shanghai, China). All PCR primers were purchased from Shanghai
Sangon Biological Engineering Technology & Services Co., Ltd.
(Shanghai, China). TRIzol and Lipofectamine 2000 were from
Invitrogen (CA, USA). Rabbit polyclonal anti-caspases 3, 8 and 9,
anti-bax, anti-beclin-1, anti-caspase 12, anti-HSP90, and
anti-cytochrome c; and mouse monoclonal anti-Bcl-2 and
anti-grp 78 antibodies were from Boster (Wuhan, China). Rabbit
monoclonal anti-LC3 antibody was from Epitomics (Burlingame, CA,
USA), and mouse monoclonal anti-rabies virus was obtained from
Santa Cruz (CA, USA). HRP-conjugated goat anti-rabbit,
HRP-conjugated goat anti-mouse, and FITC-conjugated goat anti-mouse
antibodies were purchased from CWBio (Shanghai, China).
HRP-conjugated rabbit anti-chicken antibody was from EarthOx Life
Science (Millbrae, CA, USA), and Cy3-conjugated rabbit anti-chicken
antibody was purchased from KPL (CA, USA). The PVDF membrane and
Luminata were provided by Millipore (MA, USA). DMEM, trypsin, and
EDTA-2Na were offered by Gibco (NY, USA). Fetal bovine serum (FBS)
was supplied by Hyclone (UT, USA). A mitochondrial transmembrane
potential analysis kit was from KeyGen Biotech (Nanjing, China).
All other supplies for cell culture were obtained from Costar
Corning (NY, USA).
Cell culture and interference test
SGC-7901 and AGS cells were maintained in DMEM
medium with 10% (v/v) FBS, antibiotics (100 U/ml penicillin and 100
U/ml streptomycin) at 37°C with 5% CO2 and 100%
humidity. When the cells reached ~50–70% confluence, the cells were
infected with NDV or rL-RVG.
3-MA application
When the SGC and AGS cells grown in 6-well plates
reached ~50–70% confluence, the cells were treated with 2.5 mM
3-MA. The next day, the cells were infected with NDV and rL-RVG at
a multiplicity of infection (MOI) of 10. At 24 h after being
infected, the cells were harvested and analyzed by western
blotting.
siRNA application
When the SGC and AGS cells grown in 6-well plates
reached ~50–70% confluence, the cells were transfected with 50 nM
siRNA (beclin-1) according to the manufacturer’s instructions. The
next day, the cells were infected with NDV and rL-RVG at a MOI of
10. At 24 h after being infected, the cells were harvested and
analyzed by western blotting.
MTT analysis
SGC-7901 or AGS cells (1×104) were
cultured in 96-well plates for 24 h with 10% FBS at 37°C with 5%
CO2. On the second day, the cells were incubated in
serum-free DMEM with rL-RVG or NDV at concentrations of
10−2, 10−3, 10−4 and
10−5. After 1 h, 10% FBS was added to each well, and
then the cells were cultured as described previously, with a
phosphate-buffered saline (PBS) group as the negative control. On
the third day, each well was treated with 20 μl MTT (5 mg/ml, PBS)
and incubated for an additional 4 h. Then, 150 μl of DMSO was added
into each well, and the cells were shaken for 10 min. Finally, the
absorbance was measured in triplicate using a standard
spectrophotometer. The dose-response curve for cell survival was
made based on data from the MTT assay. Simultaneously, the
morphological changes in the infected cells were monitored under a
microscope.
Immunofluorescence analysis
Cells were cultured in 24-well plates for 24 h as
previously described, infected with NDV or rL-RVG at a MOI of 10,
or treated with PBS as a negative control. Then, at 24 h
post-infection, the cells were fixed in 4% paraformaldehyde at 4°C
overnight, followed by immunofluorescence staining with antibodies
against NDV and RV and Hoechst 33342 staining. These stained SGC
and AGS cells were monitored using immunofluorescence
microscopy.
RT-PCR analysis
The cells were cultured and infected as described
previously. At 24 h after infection, total RNA was extracted from
the cultured cells using TRIzol reagent. First-strand cDNA
synthesis was performed using oligo(dT) primers and M-MLV reverse
transcriptase. The primer set for the NDV HN gene, the RV G gene
and the human GAPDH gene are shown in Table I. The RT-PCR protocol was as
follows: an initial denaturation at 94°C for 5 min, followed by 30
cycles at 94°C for 30 sec, annealing at 53°C (RVG) or 55°C (NDV and
GAPDH) for 30 sec, and extension at 72°C for 30 sec, with a final
extension at 72°C for 10 min. Then, 5 μl of each PCR product was
loaded onto 1% agarose gels for electrophoresis and visualized with
ethidium bromide. The resulting bands were analyzed using Quantity
One software (Bio-Rad). The primers for PCR are shown in Table I.
 | Table IPCR primers. |
Table I
PCR primers.
| Primer | | Sequence | Product size
(bp) |
|---|
| rL-RVG | Upstream |
5′-AGCCGATGCTCACTACAAG-3′ | 175 |
| Downstream |
5′-CTGGAGGAGGGATGATTGC-3′ | |
| NDV | Upstream |
5′-CTGGACGGTTTGGTGGGAA-3′ | 462 |
| Downstream |
5′-TAATGCGACTGCGGGATGTG-3′ | |
| GAPDH | Upstream |
5′-CAAGGTCATCCATGACAACTTTG-3′ | - |
| Downstream |
5′-GTCCACCACCCTGTTGCTGTAG-3′ | |
Western blot assay
SGC and AGS cells with or without treatment were
lysed in RIPA lysis buffer with a protease inhibitor cocktail
(Santa Cruz). The protein concentration was measured using a BCA
kit (Thermo Fisher Scientific, USA). An equal amount of protein
from each sample was loaded onto a 10% polyacrylamide gel and
separated by electrophoresis. Then, the proteins were transferred
to a polyvinylidene difluoride (PVDF) membrane (Millipore, CA,
USA). The membrane was blocked for 1 h in 5% BSA. Then, the
membrane was incubated with primary antibodies against specific
proteins (i.e., caspases 3, 8 and 9, bcl-2 and bax for apoptosis;
NDV and RVG for infection, Beclin-1 and LC-3 for autophagy;
anti-HSP90, anti-cytochrome c and anti-grp 78 for
endoplasmic reticulum stress; and beta-actin as a control) and then
incubated with HRP-conjugated secondary antibodies. The protein
bands were scanned using a Typhoon 9400 Variable Mode Imager
(Amersham Biosciences, UK) and detected by Pierce ECL Plus
Substrate (Thermo Fisher Scientific).
Transmission electron microscopy
(TEM)
Standard TEM was performed to monitor the
ultrastructure of the cells. Cells were infected with virus as
described previously. At 24 h after infection, the cells were fixed
and embedded in 4% paraformaldehyde and 2.5% glutaraldehyde. Thin
sections were cut and examined at 200 kV using an H-600
transmission electron microscope. Autophagy, as defined by the
presence of double-membraned vacuoles; apoptosis, as characterized
by chromatin margination, pyknosis, and apoptotic bodies; and
endoplasmic reticulum swelling were all observed.
TUNEL assay
Cells were cultured in 24-well plates with slides
and 10% FBS DMEM at 37°C with 5% CO2. Then, the cells
were infected with rL-RVG and NDV within the logarithmic growth
phase and fixed in 4% paraformaldehyde. After fixing, the cells
were stained according to the manufacturer’s instructions. The
slides were observed and imaged under an optical microscope. The
apoptosis index (AI) was calculated as the number of apoptotic
cells/(the number of apoptotic cells + the number of non-apoptotic
cells) × 100%.
Detection of mitochondrial membrane
potential (MMP)
JC-1 fluorescence dye was used to determine the
mitochondrial membrane potential. Cells were cultured in 24-well
plates and treated with NDV or rL-RVG as described above for 24 h,
followed by staining with JC-1 (2.5 μg/ml at 37°C for 30 min). The
cells were monitored using fluorescence microscopy with excitation
wavelengths at 527 nM for green and at 590–600 nM for red. The
changes in the MMP could be accurately assessed by comparing the
ratios of 590–600 nM (red)/527 nM (green).
Statistical analysis
The data comparisons were performed using one-way
analysis of variance (ANOVA) in SPSS V17.0 software. Differences
were considered statistically significant at p<0.05. All
experiments were repeated at least three times.
Results
Expression of viral genes and proteins in
infected stomach adenocarcinoma cells
The expression of viral proteins was monitored by
fluorescence microscopy. Almost all of the SGC and AGS cells that
were infected with NDV expressed the NDV HN protein, and almost all
of the SGC and AGS cells infected with rL-RVG expressed both the
NDV HN protein and RVG protein, while neither of these proteins was
present in the control group. RVG and NDV protein expression was
upregulated in the rL-RVG-infected group compared with the
NDV-infected group, as shown in Fig.
1A and B. Western blot analysis confirmed these results, as
shown as Fig. 1C. The PCR products
of the NDV HN and RVG genes were detected to assess NDV HN and RVG
mRNA expression in infected SGC and AGS cells. The results showed
that the RVG gene (175 bp) was expressed in SGC and AGS cells
infected with rL-RVG and that the NDV HN gene (462 bp) was
expressed in the SGC and AGS cell infected groups. In contrast,
neither the RVG gene nor the NDV HN gene was expressed in the
control group (Fig. 1D).
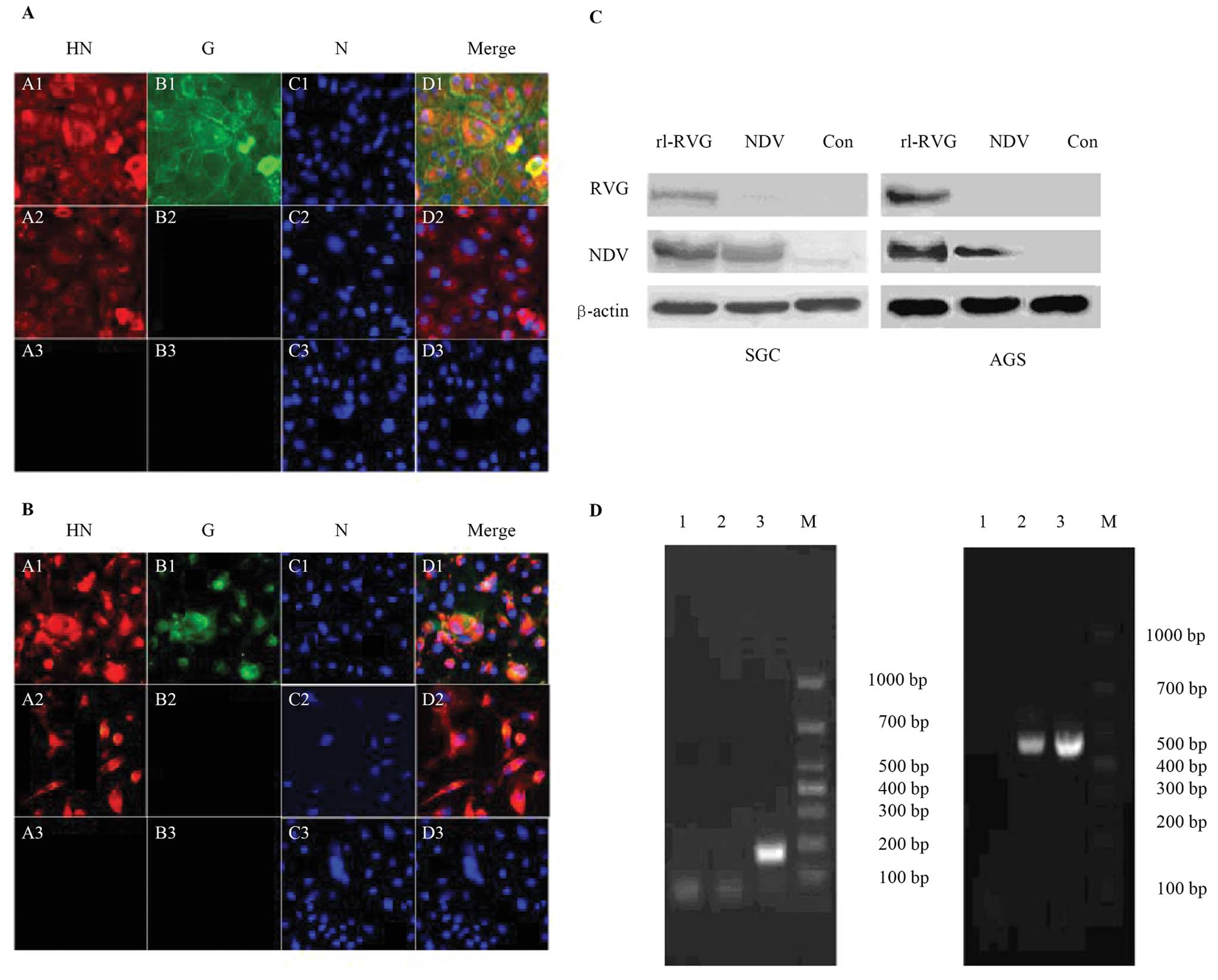 | Figure 1Expression of viral proteins and
genes in infected SGC and AGS cells. RVG and NDV protein expression
was monitored by immunofluorescence (x200 magnification) (A) for
SGC and (B) for AGS. The RVG protein (green) was only expressed in
the rL-RVG-infected group, while the NDV protein (red) was
expressed in both the rL-RVG- and NDV-infected groups. Neither
protein was expressed in the control group. (C) RVG and NDV protein
expression in SGC cells and in AGS cells was detected by western
blotting. The RVG protein was only present in the rL-RVG-infected
group, and the NDV protein was expressed in both the rL-RVG- and
NDV-infected groups. Neither protein was expressed in the PBS
group. (D) NDV and RVG mRNA expression. Lane 1, PBS group; lane 2,
NDV-infected group; lane 3, rL-RVG-infected group; M, marker. (B)
NDV HN mRNA expression. Lane 1, PBS group; lane 2, NDV-infected
group; lane 3, rL-RVG-infected group; M, marker. |
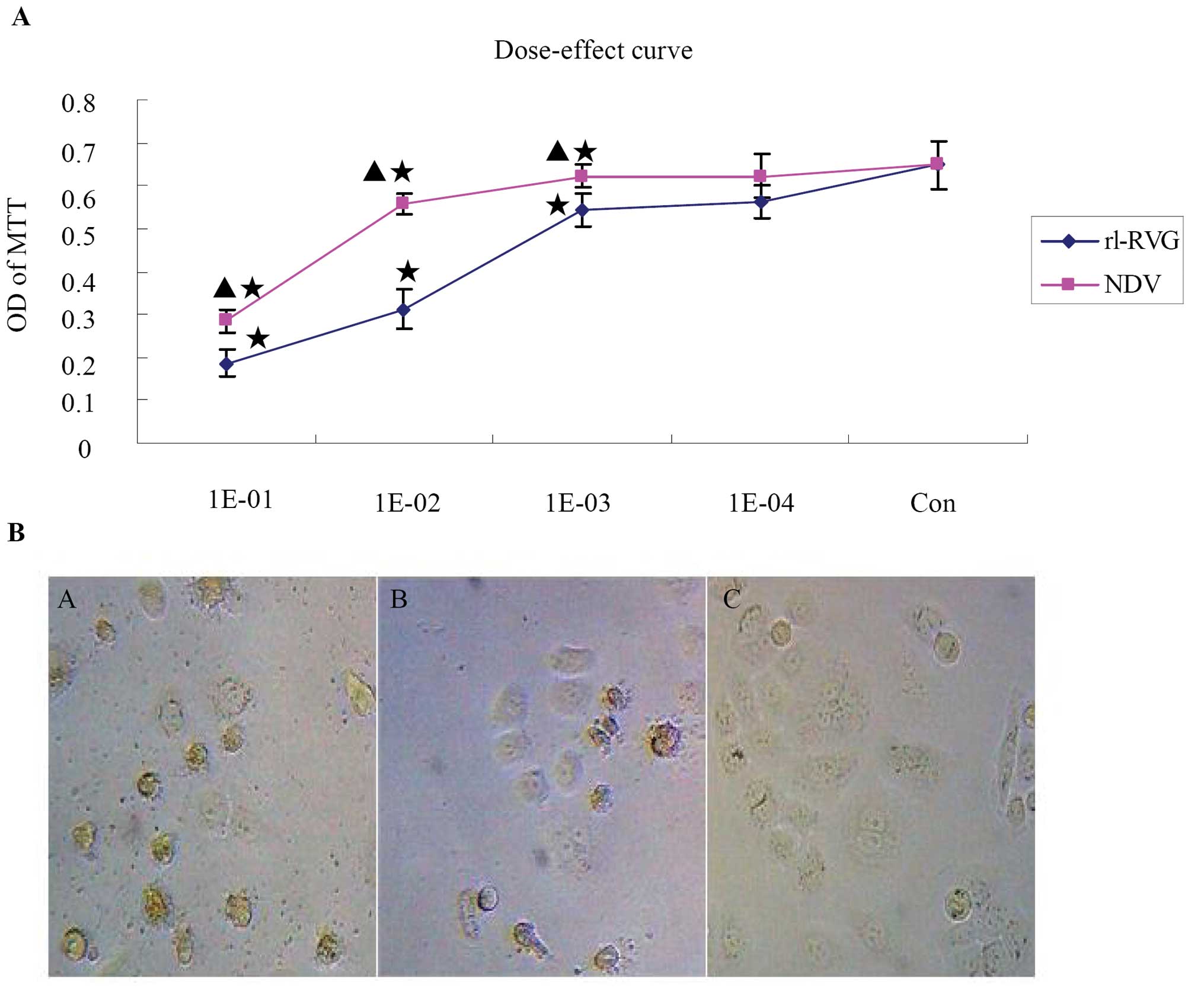
The proliferation changes of SGC and AGS
cells infected with rL-RVG and NDV
The dose-response curves for rL-RVG and NDV in SGC
cells were monitored by MTT. After 24 or 48 h of rL-RVG or NDV
infection, the infected SGC cells were assessed by MTT assay. SGC
cell viability decreased with an increase in virus concentration or
in incubation time. When the cells were infected simultaneously
with NDV and rL-RVG, the OD of MTT increased in both the NDV- and
rL-RVG-infected groups as the virus dilution decreased, and at the
103 dilution, the dose-response curve reached a plateau
compared with the control group. Simultaneously, the OD of MTT in
the rL-RVG-infected group was weaker compared with that in the
NDV-infected group, suggesting that the rL-RVG-infected group had a
greater inhibition ratio. The inhibition ratio increased over time
after infection. Thus, the inhibition ratio caused by NDV and
rL-RVG infection was dependent on both time and dose. The OD values
of MTT in SGC cells after 24 h are shown in Fig. 2A and Table II. Additionally, the morphological
changes in the infected or uninfected cells, as observed by
microscopy, are shown in Fig. 2B;
the cells had a greater decrease in size in the rL-RVG-infected
group compared with the NDV-infected group, and the number of
smaller cells was higher in the rL-RVG-infected group compared with
the NDV-infected group. No changes were observed in the PBS
group.
| Table IIMTT results of the cellular
dose-response curve for SGC cells after 24 h of infection. |
Table II
MTT results of the cellular
dose-response curve for SGC cells after 24 h of infection.
| Dilution | rL-RVG | NDV | t-value | p-value |
|---|
| 101 |
0.18656±0.029626 |
0.283727±0.026238 | 3921 | 0.017 |
| 102 |
0.312080±0.04746 |
0.55666±0.024781 | 11.521 | <0.001 |
| 103 |
0.54400±0.037857 |
0.62300±0.028032 | 3.461 | 0.02 |
| 104 |
0.56130±0.038402 |
0.61522±0.61522 | 4.971 | 0.08 |
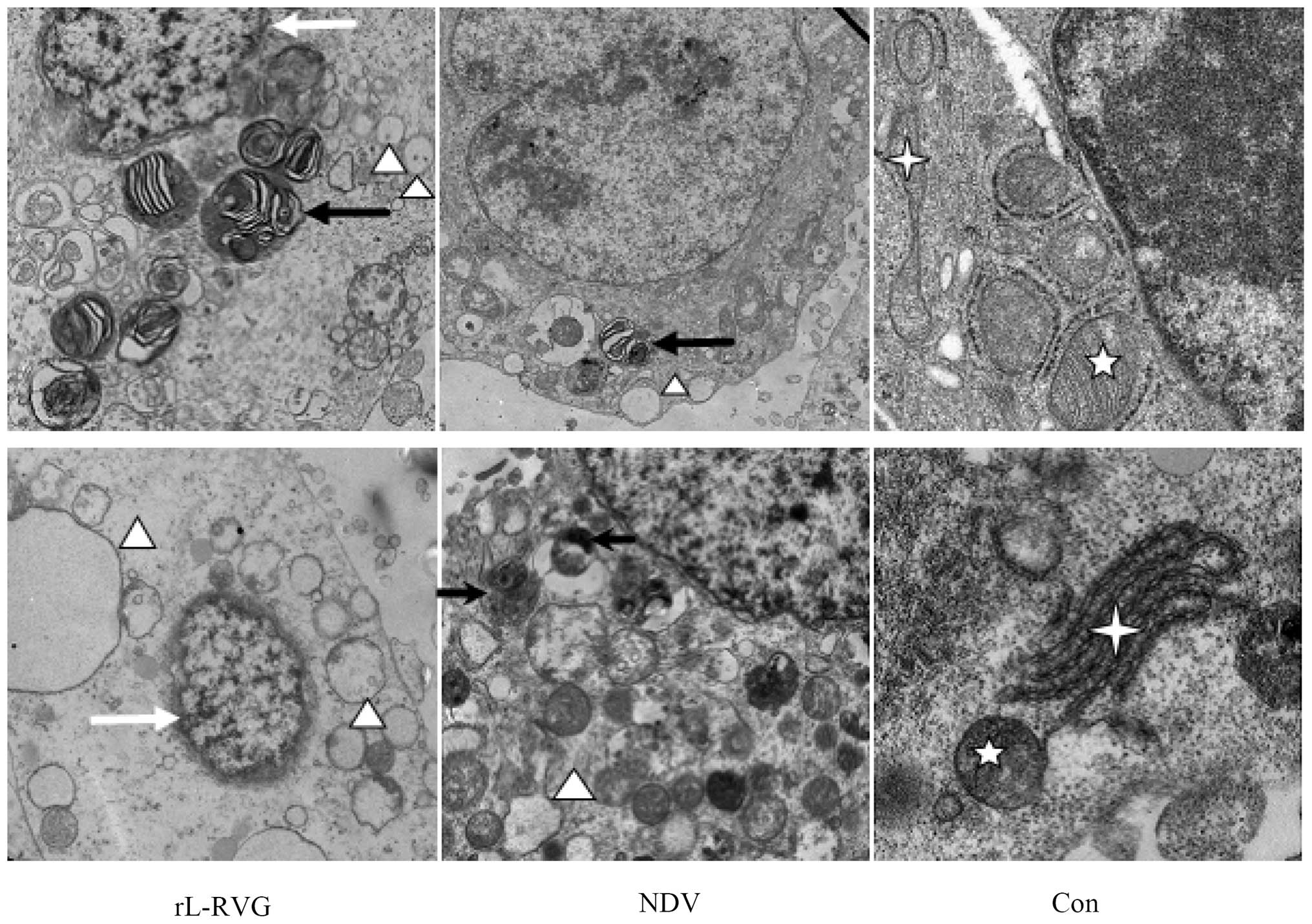
rL-RVG and NDV infection affected the
ultrastructure of SGC and AGS cells
We monitored the cells by TEM to assess the
ultrastructure of the cells after infection directly. The cells
that were infected with NDV or rL-RVG showed increased autophagy
(black arrow), apoptosis (white arrow) and ERs (white triangle),
with a larger effect observed in the L-RVG group compared with the
NDV-infected group, as shown in Fig.
3.
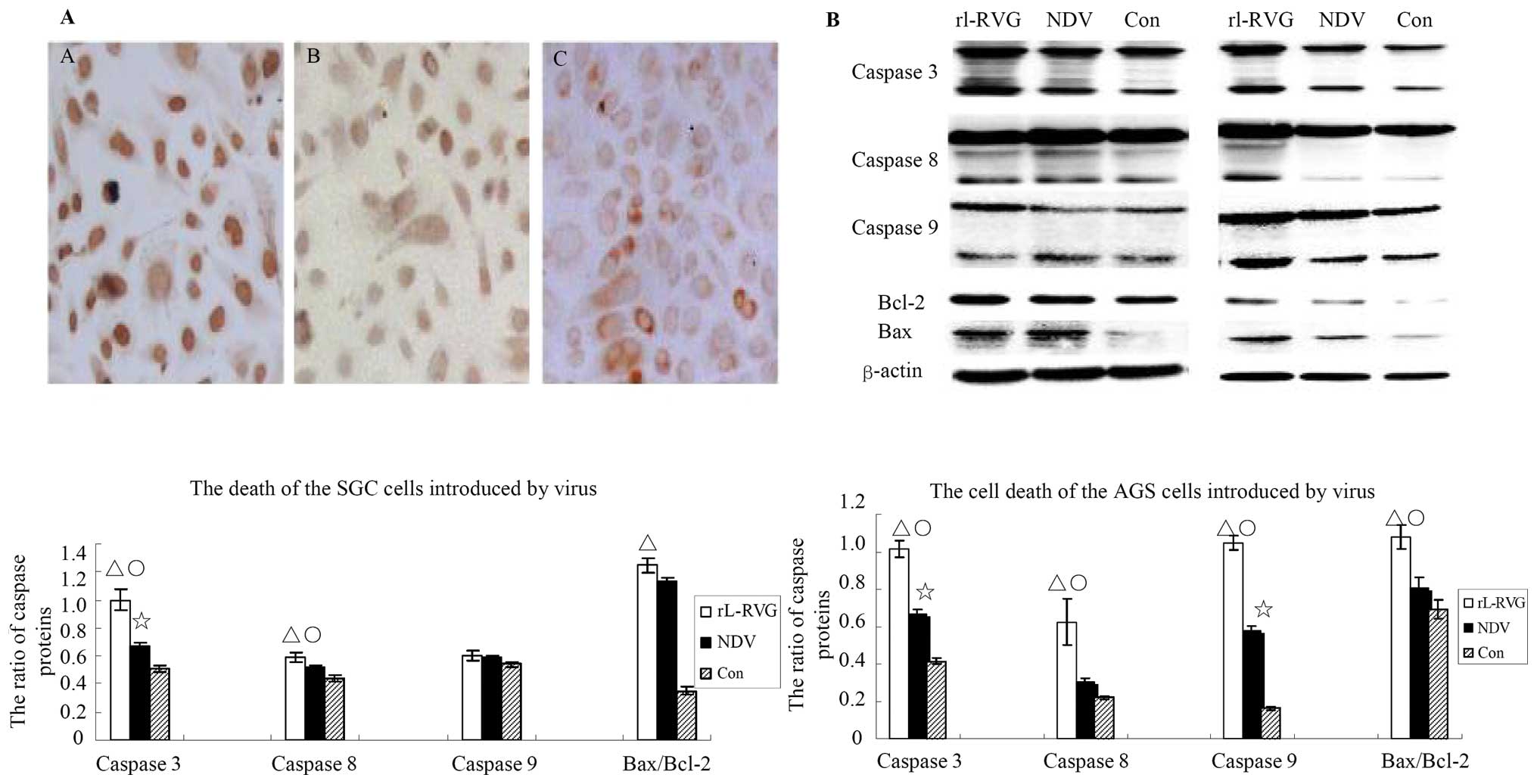
NDV and rL-RVG induce apoptosis in SGC
and AGS cells
Apoptosis was monitored by TUNEL assay; apoptosis in
the virus-infected SGC cells was obvious compared with the control
cells, and the number of apoptotic cells in the rL-RVG-infected
groups was greater than that in the NDV-infected group, as shown in
Fig. 4A. Furthermore, to assess
the apoptotic cell death pathway caused by NDV and rL-RVG
infection, the expression of the apoptosis-associated proteins
caspase 3, 8 and 9, bcl-2, and bax in the SGC and AGS cells was
examined routinely at 24 h after being infected with NDV or rL-RVG.
Accumulations of cleaved caspase 3, 8 and 9 and of bax/bcl-2 were
found in SGC and AGS cells infected with virus. The expression
levels of cleaved caspase 3 and 8 and bax/bcl2 proteins were
upregulated in the rL-RVG-infected group compared with the other
two groups. The expression levels of these proteins were higher in
the NDV-infected group compared with those in the control group. In
the SGC cells, the expression levels of cleaved caspase 8 protein
did not differ between the NDV-infected group and the control
group. Additionally, the expression levels of cleaved caspase 9
protein did not differ among all the three groups, and the protein
ratio of bax to bcl2 did not differ between the NDV-infected group
and the control group. In the AGS cells, the expression levels of
cleaved caspase 8 protein did not differ between the NDV-infected
group and the control group, and the protein ratio of bax to bcl-2
did not differ between the NDV-infected group and the control
group, as shown in Fig. 4B.
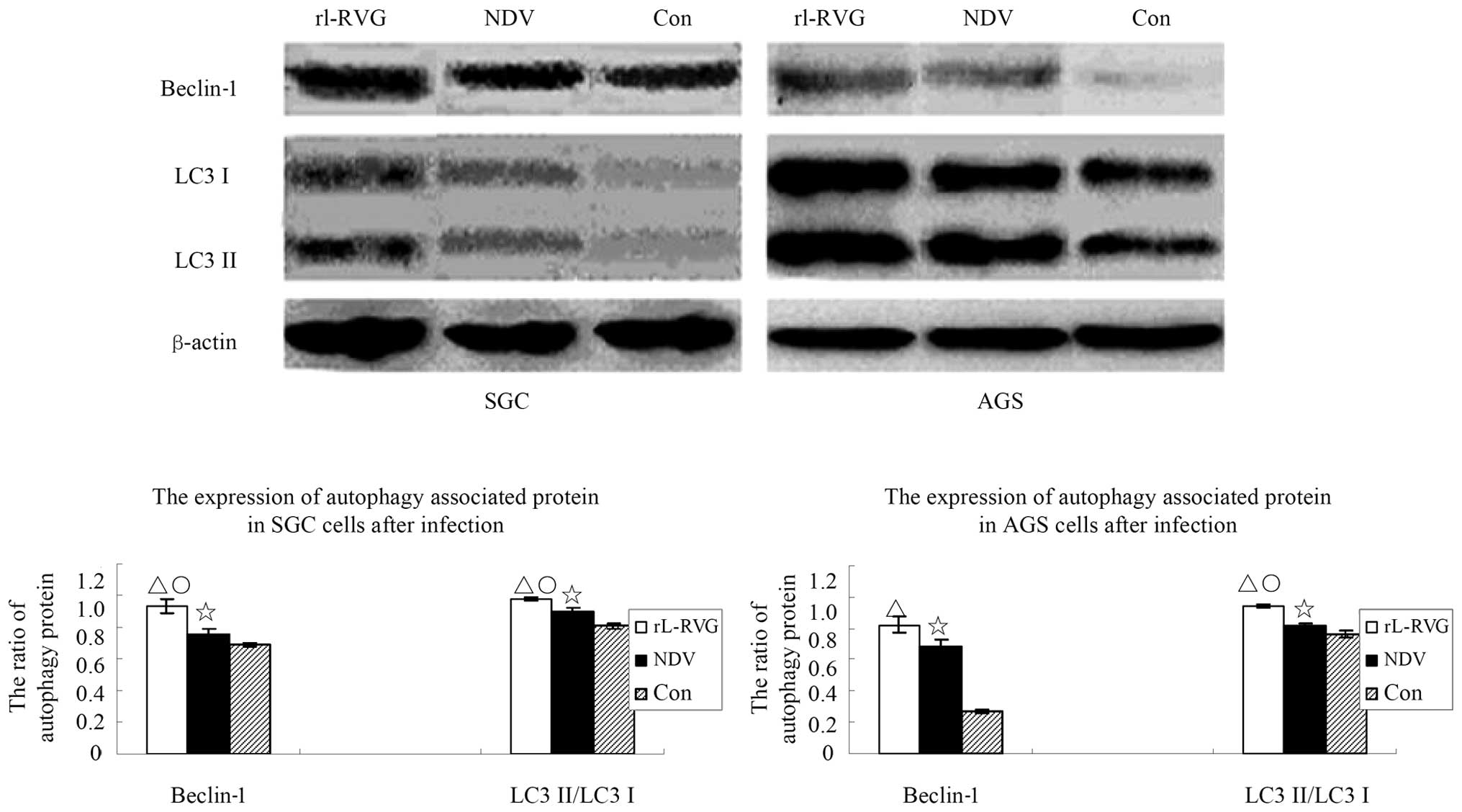
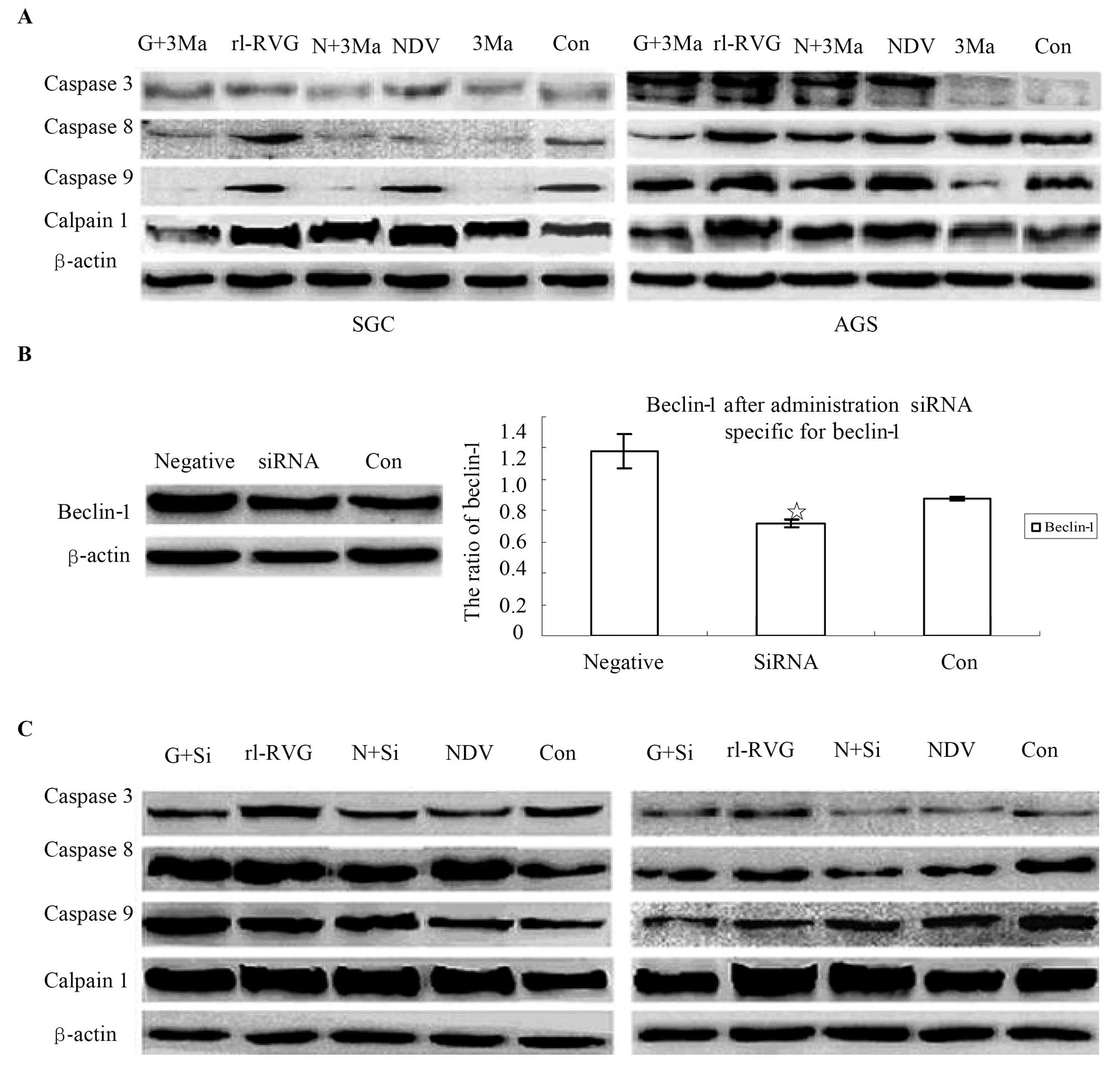
NDV and rL-RVG induce significant
autophagy in SGC and AGS cells
Because TEM analysis indicated that autophagy
significantly increased after SGC cells were infected with virus,
we detected the autophagy activity of cells with or without NDV or
rL-RVG infection. The ratio of lipid-bound LC3-II, which is
converted to LC3-I, the soluble LC3-I is associated with the
formation of autophagosomes and can indicate autophagic activity.
In a preliminary experiment, we found that the autophagic activity
peaked at least two times after infection from 30 min to 48 h. The
first peak occurred at the 3rd hour after infection, while the
second peak occurred at 24 h after infection (data not shown).
Considering that autophagy protects against infection, we suggested
that the second peak time contributes to cell death. In addition to
monitoring the ratio of LC3-II to LC3-I in cells at 24 h
post-infection, we also monitored another autophagy marker,
beclin-1, in these cells. As shown in Fig. 5. the expression of the
autophagy-associated protein beclin-1 and LC3 in the SGC cells of
the rL-RVG-infected group showed a significant difference compared
with the other two groups (△,○p<0.05); the
expression of the autophagy-associated protein in the NDV-infected
group showed a significant difference compared with the control
group (⋆p<0.05). The trend is the same in the
expression of beclin-1 and LC3 in the AGS cells and the same trend
exists among the three group. However, beclin-1 in the AGS cells
showed no significant differences between the rL-RVG and the
NDV-infected group (p>0.05).
NDV and rL-RVG cause endoplasmic
reticulum stress in the SGC cells
Recent evidence has demonstrated that ERs is a type
of trigger for autophagy. In the present study, TEM images showed
infected cells with endoplasmic reticulum swelling, indicating that
the SGC cells had ERs. Therefore, we monitored the expression of
the following proteins associated with ERs: grp-78, HSP-90 and
cytochrome c. The expression of the grp-78, and HSP-90
proteins in SGC cells showed significant differences among the
three groups (△,⋆,○p<0.05), and the expression of the
cytochrome c protein in the control groups was higher
compared with that in the two infected groups
(△,⋆p<0.05). However, no significant
difference was observed between the rL-RVG and NDV-infected groups
(p>0.05). These results suggested that the infected cells
remained under ERs.
Autophagy contributes to SGC and AGS
apoptosis induced by NDV and rL-RVG
Recent research has indicated that autophagy can
have both cytoprotective and cytotoxic effects (28,29).
3-MA, which is an inhibitor of autophagy, was administered to SGC
and AGS cells with or without NDV or rL-RVG infection to determine
whether autophagy contributes to the apoptosis of NDV- and
rL-RVG-infected cells. The expression of the apoptosis-associated
proteins caspase 3, 8 and 9 and calpain 1, which is related to
autophagy and apoptosis (30),
decreased in SGC and AGS cells with 3-MA treatment relative to
control cells without 3-MA treatment. The effect was greater in the
rL-RVG-infected groups than in the NDV-infected groups, as shown in
Fig. 7A. Because 3-MA is a
non-specific autophagy suppressor, it inhibits both PI3K-III (an
autophagy inducer) and PI3K-I (an autophagy suppressor). We
silenced beclin-1, which is an autophagy regulator, to confirm the
3-MA results. The potency of the siRNA treatment was determined by
western blotting, as shown in Fig.
7B. We found that the expression of the beclin-1 protein
decreased significantly in SGC cells upon beclin-1 siRNA treatment.
Therefore, we monitored the apoptosis-associated proteins caspase
3, 8 and 9 and calpain 1, and the results were similar to the 3-MA
treatment experiment, as shown in Fig.
7C.
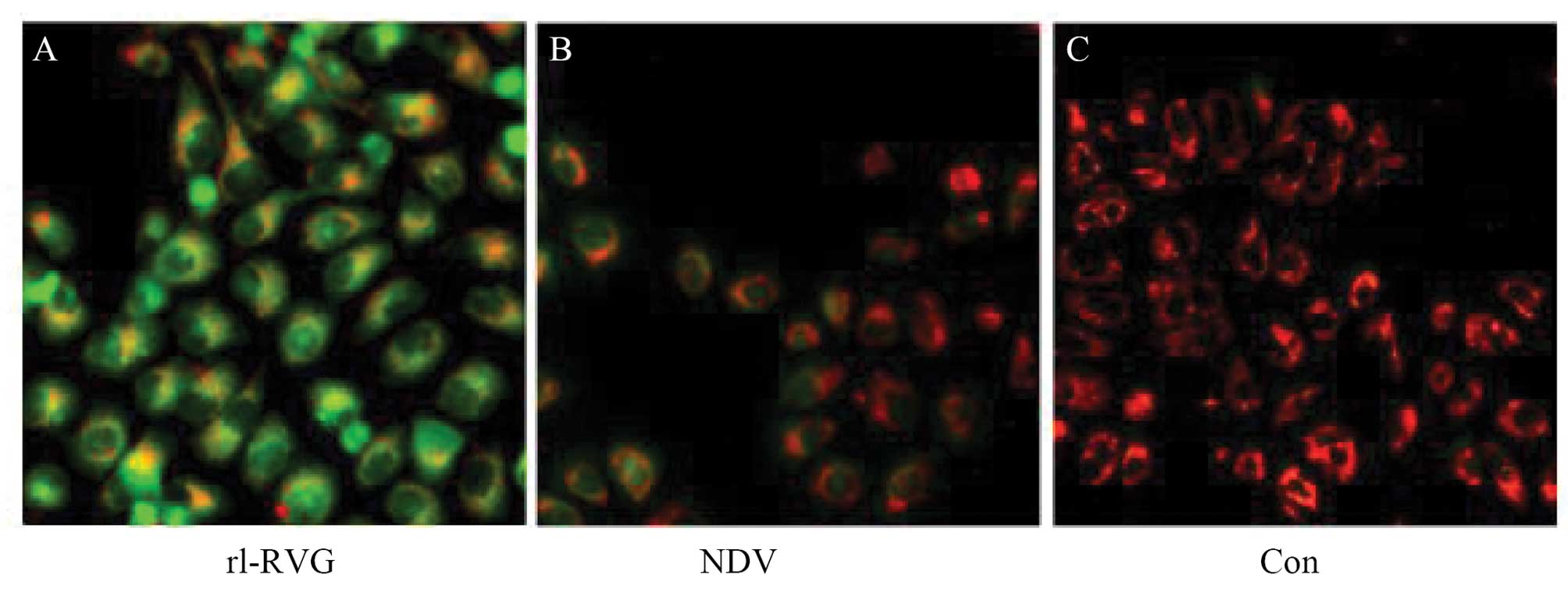
NDV and rL-RVG induce mitochondrial
dysfunction
Mitochondria have essential roles in autophagy
(31). We monitored the MMP with
JC-1 staining. The greater the intensity of green in a cell, the
more dysfunctional the mitochondria are. We found that the red
fluorescence intensity decreased and the green fluorescence
intensity increased in these cells. The ratio of red to green
significantly decreased in the NDV- and rL-RVG-infected groups
compared with the control group, and the ratio of red to green the
in the rL-RVG-infected group decreased more than that in
NDV-infected group, as shown in Fig.
8.
Discussion
NDV, which is a member of the oncolytic virus
family, has the ability to inhibit malignant cells via multiple
mechanisms (15). In recent
decades, NDV has been shown to induce tumor cell death via its
oncolytic effects (9). NDV can
also induce the immune system to eliminate tumor cells (6), and NDV can infect tumor cells more
effectively compared with normal cells (12). The rL-RVG virus spreads more easily
within cells (26), and RVG has a
potential oncolytic effect (13,14);
therefore, we propose that the recombinant virus rL-RVG has much
more antitumor potency.
In the present study, we found that rL-RVG caused
stomach adenocarcinoma SGC-7901 and AGS cell death, which is
consistent with previous studies (27). Interestingly, in addition to
apoptosis, both NDV and rL-RVG can induce autophagy, ERs and
mitochondrial dysfunction. The results of the present study
indicated that the net effects of autophagy, mitochondrial
dysfunction and ERs contribute to tumor cell death. We considered
that the observed mitochondrial dysfunction, ERs, autophagy, and
apoptosis, indicate possible cross-talk with other pathways.
The autophagy pathway cross-talks with the apoptosis
pathway; virus-induced autophagy can also contribute to apoptosis
(32). Autophagy might also be a
mechanism that contributes to virus-induced cell death (33). Although autophagy can promote tumor
survival in some cases (29,34),
autophagy can also be fatal to malignant cells (35). Autophagy and apoptosis work
together in multiple ways to determine the fate of tumor cells
(28,36). In the present study, both autophagy
and apoptosis increased in the cells after viral infection. In the
last decade, many researchers have confirmed that Bcl-2 plays an
important role in both apoptosis and autophagy (37–39).
Beclin-1 has an N-terminal BH3 domain, which this protein a
subgroup of the Bcl-2 family (39,40);
therefore, beclin-1 can participate in the activity of the
Bcl-2/Bcl-xL complex and inhibit the formation of autophagosomes
(41). We found that the
virus-infected cells showed increase beclin-1 expression, thus
decreasing the interaction between Beclin-1 and Bcl-2/Bcl-xL and
promoting autophagy and apoptosis. In addition, caspase 8 and
caspase 12 play important roles in autophagy (41,42).
We also found that the expression of apoptosis-associated proteins
increased in cells after viral infection. We suggest that rL-RVG
and NDV cause tumor cell death via both autophagy and
apoptosis.
Autophagy and ERs are related (43); both excess autophagy and excess ERs
can cause cell death (44), and
both moderate autophagy and moderate ERs are adaptive mechanisms
when cells are under severe stress (45). Autophagy can recycle the organelles
and provide the basic energy and material for cell survival
(16,46). In addition, Meng et al
(47) and Meng et al (48),
reported that autophagy can enhance NDV replication in tumor cells.
Considering that NDV and rL-RVG introduced massive autophagy in the
present study, we propose that the autophagy induced by NDV and
rL-RVG allowed the viruses to increase replication and that these
increased amounts of the viruses induced even more autophagy, thus
becoming a positive feedback loop, with more and more virus
initiating the unfolded protein response and causing ERs. Finally,
autophagy and ERs reach a certain threshold, inducing cell
death.
Additionally, autophagy and mitochondrial
dysfunction work together to cause cell death. Mitochondria
contribute to autophagy during cell death and disease by the
mitochondrial permeability transition pathway (46,49)
and by reactive oxygen species (ROS) generation (50). The protein calpain, which is a
member of the calmodulin family, can degrade ATG-5, and cleaved
ATG-5 can anchor to mitochondria, causing cytochrome c
release, followed by mitochondrial dysfunction (30). In the present study, we found that
the viruses caused excess autophagy and mitochondrial dysfunction,
indicating that these viruses participate in cell death. The above
results suggest that autophagy and mitochondrial dysfunction
contribute to cell death. Although autophagy dysfunction may result
in abnormal mitochondrial function (51), further experiments will be required
to establish cross-talk among autophagy, mitochondria and oxidative
stress pathways.
In conclusion, this study demonstrated that NDV and
rL-RVG induce stomach adenocarcinoma cell death via autophagy and
apoptosis, in association with ERs and mitochondrial dysfunction.
Cell death may involve multiple pathways. Although the causal
relations among autophagy, apoptosis and ERs remain unclear, rL-RVG
may become a powerful candidate for antitumor treatments.
Acknowledgements
The authors would like to thank Professor Zhigao Bu
and Dr Jinying Ge from the State Key Laboratory of Veterinary
Biotechnology, Harbin Veterinary Research Institute, Chinese
Academy of Agricultural Sciences, Harbin, China for supplying us
with the recombinant Newcastle disease. The authors would also like
to thank Dr Zhijian Zhang and Aihua Gong from Jiangsu University
(Jiangsu, China) for kindly providing suggestions for the
experiments that were performed. This study was supported by the
Social Development Technological Support Projects of Zhenjiang,
Jiangsu, China (grant no. SH2014046).
References
|
1
|
Chen J, Shen W, Xia J, Xu R, Zhu M and Xu
M: Effect of S-1 maintenance chemotherapy following DCF regimen in
patients with advanced gastric cancer. Nan Fang Yi Ke Da Xue Xue
Bao. 34:1057–1060. 2014.(In Chinese). PubMed/NCBI
|
|
2
|
Khalighinejad N, Hariri H, Behnamfar O,
Yousefi A and Momeni A: Adenoviral gene therapy in gastric cancer:
A review. World J Gastroenterol. 14:180–184. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Janke M, Peeters B, de Leeuw O, Moorman R,
Arnold A, Fournier P and Schirrmacher V: Recombinant Newcastle
disease virus (NDV) with inserted gene coding for GM-CSF as a new
vector for cancer immunogene therapy. Gene Ther. 14:1639–1649.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Beutner U, Lorenz U, Illert B, Rott L,
Timmermann W, Vollmers HP, Müller-Hermelink HK, Thiede A and
Ulrichs K: Neoadjuvant therapy of gastric cancer with the human
monoclonal IgM antibody SC-1: Impact on the immune system. Oncol
Rep. 19:761–769. 2008.PubMed/NCBI
|
|
5
|
Garg AD and Agostinis P: ER stress,
autophagy and immunogenic cell death in photodynamic
therapy-induced anti-cancer immune responses. Photochem Photobiol
Sci. 13:474–487. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hossain A, Radwan FF, Doonan BP, God JM,
Zhang L, Bell PD and Haque A: A possible cross-talk between
autophagy and apoptosis in generating an immune response in
melanoma. Apoptosis. 17:1066–1078. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Puhlmann J, Puehler F, Mumberg D, Boukamp
P and Beier R: Rac1 is required for oncolytic NDV replication in
human cancer cells and establishes a link between tumorigenesis and
sensitivity to oncolytic virus. Oncogene. 29:2205–2216. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pecora AL, Rizvi N, Cohen GI, Meropol NJ,
Sterman D, Marshall JL, Goldberg S, Gross P, O’Neil JD, Groene WS,
et al: Phase I trial of intravenous administration of PV701, an
oncolytic virus, in patients with advanced solid cancers. J Clin
Oncol. 20:2251–2266. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sinkovics JG and Horvath JC: Newcastle
disease virus (NDV): Brief history of its oncolytic strains. J Clin
Virol. 16:1–15. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang S, Liu W, Cui H, Sun S and Wang J: In
vitro induction of apoptosis in tumor cells by inactivated NDV and
IAV. Cancer Biother Radiopharm. 22:200–205. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yaacov B, Eliahoo E, Lazar I, Ben-Shlomo
M, Greenbaum I, Panet A and Zakay-Rones Z: Selective oncolytic
effect of an attenuated Newcastle disease virus (NDV-HUJ) in lung
tumors. Cancer Gene Ther. 15:795–807. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lam HY, Yeap SK, Rasoli M, Omar AR, Yusoff
K, Suraini AA and Alitheen NB: Safety and clinical usage of
newcastle disease virus in cancer therapy. J Biomed Biotechnol.
2011:7187102011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lay S, Préhaud C, Dietzschold B and Lafon
M: Glycoprotein of nonpathogenic rabies viruses is a major inducer
of apoptosis in human jurkat T cells. Ann NY Acad Sci.
1010:577–581. 2003. View Article : Google Scholar
|
|
14
|
Préhaud C, Lay S, Dietzschold B and Lafon
M: Glycoprotein of nonpathogenic rabies viruses is a key
determinant of human cell apoptosis. J Virol. 77:10537–10547. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vähä-Koskela MJ, Heikkilä JE and Hinkkanen
AE: Oncolytic viruses in cancer therapy. Cancer Lett. 254:178–216.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Klionsky DJ and Emr SD: Autophagy as a
regulated pathway of cellular degradation. Science. 290:1717–1721.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rubinsztein DC, Codogno P and Levine B:
Autophagy modulation as a potential therapeutic target for diverse
diseases. Nat Rev Drug Discov. 11:709–730. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Carroll RG and Martin SJ: Autophagy in
multiple myeloma: What makes you stronger can also kill you. Cancer
Cell. 23:425–426. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kroemer G and Levine B: Autophagic cell
death: The story of a misnomer. Nat Rev Mol Cell Biol. 9:1004–1010.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ahn JH and Lee M: Autophagy-dependent
survival of mutant B-Raf melanoma cells selected for resistance to
apoptosis induced by inhibitors against oncogenic B-Raf. Biomol
Ther (Seoul). 21:114–120. 2013. View Article : Google Scholar
|
|
21
|
Martín V, Sanchez-Sanchez AM,
Puente-Moncada N, Gomez-Lobo M, Alvarez-Vega MA, Antolín I and
Rodriguez C: Involvement of autophagy in melatonin-induced
cytotoxicity in glioma-initiating cells. J Pineal Res. 57:308–316.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xu ZX, Liang J, Haridas V, Gaikwad A,
Connolly FP, Mills GB and Gutterman JU: A plant triterpenoid,
avicin D, induces autophagy by activation of AMP-activated protein
kinase. Cell Death Differ. 14:1948–1957. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cheng J, Wei HL, Chen J and Xie B:
Antitumor effect of arsenic trioxide in human K562 and K562/ADM
cells by autophagy. Toxicol Mech Methods. 22:512–519.
2012.PubMed/NCBI
|
|
24
|
Kulich I and Žárský V: Autophagy-related
direct membrane import from ER/cytoplasm into the vacuole or
apoplast: A hidden gateway also for secondary metabolites and
phytohormones? Int J Mol Sci. 15:7462–7474. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shailasree S, Venkataramana M, Niranjana
SR and Prakash HS: Cytotoxic effect of p-coumaric acid on
neuroblastoma, N2a cell via generation of reactive oxygen species
leading to dysfunction of mitochondria inducing apoptosis and
autophagy. Mol Neurobiol. 51:119–130. 2015. View Article : Google Scholar
|
|
26
|
Ge J, Wang X, Tao L, Wen Z, Feng N, Yang
S, Xia X, Yang C, Chen H and Bu Z: Newcastle disease virus-vectored
rabies vaccine is safe, highly immunogenic, and provides
long-lasting protection in dogs and cats. J Virol. 85:8241–8252.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yan Y, Liang B, Zhang J, Liu Y and Bu X:
Apoptotic induction of lung adenocarcinoma A549 cells infected by
recombinant RVG Newcastle disease virus (rLRVG) in vitro. Mol Med
Rep. 11:317–326. 2015.
|
|
28
|
Chen W, Sun Y, Liu K and Sun X: Autophagy:
A double-edged sword for neuronal survival after cerebral ischemia.
Neural Regen Res. 9:1210–1216. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gong A, Ye S, Xiong E, Guo W, Zhang Y,
Peng W, Shao G, Jin J, Zhang Z, Yang J, et al: Autophagy
contributes to ING4-induced glioma cell death. Exp Cell Res.
319:1714–1723. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yousefi S, Perozzo R, Schmid I, Ziemiecki
A, Schaffner T, Scapozza L, Brunner T and Simon HU:
Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis.
Nat Cell Biol. 8:1124–1132. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yuzefovych LV, LeDoux SP, Wilson GL and
Rachek LI: Mitochondrial DNA damage via augmented oxidative stress
regulates endoplasmic reticulum stress and autophagy: Crosstalk,
links and signaling. PLoS One. 8:e833492013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zorn U, Dallmann I, Grosse J, Kirchner H,
Poliwoda H and Atzpodien J: Induction of cytokines and cytotoxicity
against tumor cells by Newcastle disease virus. Cancer Biother.
9:225–235. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tsuchihara K, Fujii S and Esumi H:
Autophagy and cancer: Dynamism of the metabolism of tumor cells and
tissues. Cancer Lett. 278:130–138. 2009. View Article : Google Scholar
|
|
34
|
Kimura T, Takabatake Y, Takahashi A and
Isaka Y: Chloroquine in cancer therapy: A double-edged sword of
autophagy. Cancer Res. 73:3–7. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mariño G, Martins I and Kroemer G:
Autophagy in Ras-induced malignant transformation: Fatal or vital?
Mol Cell. 42:1–3. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gong JS and Kim GJ: The role of autophagy
in the placenta as a regulator of cell death. Clin Exp Reprod Med.
41:97–107. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Luo S and Rubinsztein DC: BCL2L11/BIM: A
novel molecular link between autophagy and apoptosis. Autophagy.
9:104–105. 2013. View Article : Google Scholar :
|
|
38
|
Saeki K, Yuo A, Okuma E, Yazaki Y, Susin
SA, Kroemer G and Takaku F: Bcl-2 down-regulation causes autophagy
in a caspase-independent manner in human leukemic HL60 cells. Cell
Death Differ. 7:1263–1269. 2000. View Article : Google Scholar
|
|
39
|
Lomonosova E and Chinnadurai G: BH3-only
proteins in apoptosis and beyond: An overview. Oncogene. 27(Suppl
1): S2–S19. 2008. View Article : Google Scholar
|
|
40
|
Pattingre S, Tassa A, Qu X, Garuti R,
Liang XH, Mizushima N, Packer M, Schneider MD and Levine B: Bcl-2
antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell.
122:927–939. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cho DH, Jo YK, Hwang JJ, Lee YM, Roh SA
and Kim JC: Caspase-mediated cleavage of ATG6/Beclin-1 links
apoptosis to autophagy in HeLa cells. Cancer Lett. 274:95–100.
2009. View Article : Google Scholar
|
|
42
|
Lin CJ, Lee CC, Shih YL, Lin CH, Wang SH,
Chen TH and Shih CM: Inhibition of mitochondria- and endoplasmic
reticulum stress-mediated autophagy augments temozolomide-induced
apoptosis in glioma cells. PLoS One. 7:e387062012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jheng JR, Ho JY and Horng JT: ER stress,
autophagy, and RNA viruses. Front Microbiol. 5:3882014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Digaleh H, Kiaei M and Khodagholi F: Nrf2
and Nrf1 signaling and ER stress crosstalk: Implication for
proteasomal degradation and autophagy. Cell Mol Life Sci.
70:4681–4694. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Tian J, Hu X and Qu Q: Effect and
mechanism of endoplasmic reticulum stress on cisplatin resistance
in ovarian carcinoma]. Zhonghua Zhong Liu Za Zhi. 36:324–328.
2014.(In Chinese). PubMed/NCBI
|
|
46
|
Suzuki SW, Onodera J and Ohsumi Y:
Starvation induced cell death in autophagy-defective yeast mutants
is caused by mitochondria dysfunction. PLoS One. 6:e174122011.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Meng C, Zhou Z, Jiang K, Yu S, Jia L, Wu
Y, Liu Y, Meng S and Ding C: Newcastle disease virus triggers
autophagy in U251 glioma cells to enhance virus replication. Arch
Virol. 157:1011–1018. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Meng G, Xia M, Wang D, Chen A, Wang Y,
Wang H, Yu D and Wei J: Mitophagy promotes replication of oncolytic
Newcastle disease virus by blocking intrinsic apoptosis in lung
cancer cells. Oncotarget. 5:6365–6374. 2014.PubMed/NCBI
|
|
49
|
Marzetti E, Csiszar A, Dutta D, Balagopal
G, Calvani R and Leeuwenburgh C: Role of mitochondrial dysfunction
and altered autophagy in cardiovascular aging and disease: From
mechanisms to therapeutics. Am J Physiol Heart Circ Physiol.
305:H459–H476. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wu JJ, Quijano C, Chen E, Liu H, Cao L,
Fergusson MM, Rovira II, Gutkind S, Daniels MP, Komatsu M, et al:
Mitochondrial dysfunction and oxidative stress mediate the
physiological impairment induced by the disruption of autophagy.
Aging (Albany, NY). 1:425–437. 2009.
|
|
51
|
Lee J, Giordano S and Zhang J: Autophagy,
mitochondria and oxidative stress: Cross-talk and redox signalling.
Biochem J. 441:523–540. 2012. View Article : Google Scholar :
|