Introduction
Cinnamaldehyde exhibits various cellular effects,
including antitumor, anti-angiogenesis and anti-inflammatory
activities (1–3). Several studies have shown that the
cinnamaldehyde derivatives, 2′-hydroxycinnamaldehyde (HCA) and
2′-benzoyloxycinnamaldehyde (BCA), exhibit antitumor activities by
inducing cell cycle arrest and the production of reactive oxygen
species (ROS) in a variety of human cancer cells, including breast,
leukemia, ovarian, lung and colon cancer (4–8). In
addition, HCA causes ER stress through the inhibition of the
proteasome pathway and mitochondrial perturbation, which are
associated with the induction of apoptosis in SW620 colon cancer
cells (9). Our previous study
demonstrated the potential effects of HCA and BCA on SCC-15 and
Hep-2 human oral cancer cells (10). BCA is a well-known derivative of
HCA that induces the expression and nuclear translocation of EGR1
and the expression of its target genes, including activating
transcription factor 3 (ATF3), NSAID-activated gene 1 protein
(NAG-1), and growth arrest and DNA-damage-inducible protein α
(GADD45A) in prostate cancer cells (11). BCA has shown therapeutic
selectivity in a K-ras-transformed animal model through the
downregulation of antioxidants (12). We have also shown that BCA mainly
exerts its anti-inflammatory effects through the inhibition of the
JNK pathway, thereby leading to AP-1 transcriptional activity
(13). However, little is known
about the mechanistic relationship between autophagy and
cinnamaldehyde derivative-induced apoptosis.
Oral squamous cell carcinoma (OSCC) is an aggressive
disease that is histologically characterized as hyperplasia,
dysplasia, carcinoma in situ and invasive oral cancers
(14,15). Patients with premalignant oral
lesions have an increased risk of developing OSCC and molecular
processes, such as tumor suppressor gene inactivation or oncogene
activation, leading to progression of precancerous lesions to
invasive oral cancers (14,16).
Finding a novel pharmacologic agent that can effectively halt the
process of oral carcinogenesis is one of successful oral cancer
chemoprevention tools and a variety of agents are being tested to
prevent progression to invasive cancer (14,16).
In the present study, we demonstrated that HCA- or
BCA-induced apoptosis was independent of p53 status in
p53-wild-type SGT cells and p53-mutant YD-10B cells. We also
investigated the roles of autophagy in cinnamaldehyde
derivative-induced apoptosis in human head and neck cancer cells
and showed autophagy participates in the regulation of cell death
by enhancing apoptosis. These findings provide new information on
the relationship between autophagy and apoptosis in HCA treated
p53-wild-type and p53-mutant oral cancer cells.
Materials and methods
Cell culture and reagents
The salivary gland adenocarcinoma cell line (SGT,
p53-wild-type) and oral squamous cell carcinoma cell line (YD-10B,
p53-mutant) were maintained in Dulbecco's modified Eagle's medium
(DMEM) or RPMI-1640 medium, respectively. The YD-10B cell line
contains a point mutation in p53 at codon 236 of exon 7, resulting
in the change of codon 236 from TAC to TAA (17). Both cell lines were cultured in
medium supplemented with 10% fetal bovine serum (FBS), 100 units/ml
of penicillin, and 100 μg/ml of streptomycin and were maintained at
37°C in a humidified incubator with a 5% CO2 atmosphere.
2′-Hydroxycinnamaldehyde (HCA) and 2′-methoxycinnamaldehyde (MCA)
were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
2′-Benzoyloxycinnamaldehyde (BCA) and 2′-acetoxy-cinnamaldehyde
(ACA) were a generous gift of Dr S.H. Hong (Department of
Microbiology, School of Dentistry, Kyungpook National University,
Daegu, Korea).
Cell proliferation assay
The cells were seeded in 12-well plates at a density
of 5×105 cells/ml. The cells were then cultured
overnight and were treated with various concentrations of HCA or
BCA for 24 h. The cell viability was measured using the MTT assay
according to the previously described method (18). In brief, 24 h after treatment, the
cells were washed twice with ice-cold PBS, and 0.25 ml of cell
culture medium and 25 μl of
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
solution (5 mg/ml in PBS) was added. After 3 h of incubation, the
medium was removed and 125 μl of acidisopropanol (0.04 mol/l HCl in
isopropanol) was added. The absorbance was measured at a wavelength
of 570 nm, and the results were plotted as the means ± SD of three
separate experiments.
Cell cytotoxicity assay
Cell cytotoxicity was evaluated by measuring the
activity of lactate dehydrogenase (LDH) after treatment with HCA or
BCA using a CytoTox96® Non-Radioactive Cytotoxicity
Assay kit (Promega, Madison, WI, USA) according to the
manufacturer's instruction. The optical density was assessed at a
wavelength of 490 nm.
Western blotting
Cells were treated with various concentrations of
HCA or BCA for the indicated time periods. The cells were then
washed with PBS and harvested in lysis buffer. Samples containing
equal amounts of protein were loaded into each lane of an
SDS-polyacrylamide gel for electrophoresis and were subsequently
transferred onto a PVDF membrane. The membranes were blocked and
then incubated with antibodies. Antibodies against Bak1 (#3814),
Bcl-2 (#2876), Bid (#2002), LC3B (#3868), caspase-7 (#9492) and
p-p53 (#9284) were purchased from Cell Signaling Technology
(Beverly, MA, USA). The p53 (sc-126), caspase-3 (sc-7148),
caspase-9 (sc-7885) and PARP (sc-7150) antibodies were from Santa
Cruz Biotechnology and antibodies against β-actin (A1978) were
purchased from Sigma-Aldrich (St. Louis, MO, USA).
Cell death evaluation
Apoptosis was measured using a cell death detection
ELISA kit (Roche Molecular Biochemicals, Indianapolis, IN, USA).
The relative apoptosis, which correlates with absorption at 405 nm
with a reference wavelength of 490 nm, was measured according to
the manufacturer's instructions.
Annexin V-FITC/PI double staining
For the cell cycle analysis, cells were harvested
and fixed with 70% ethanol for 1 h at 4°C. After washing with cold
PBS, the cells were incubated with DNase-free RNase for 30 min at
37°C. The specific binding of Annexin V-FITC/PI was performed by
incubating the cells for 15 min at room temperature in a binding
buffer (10 mM HEPES, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2)
containing saturated concentrations of Annexin V-FITC and PI.
Apoptotic cells were visualized with a Nikon Eclipse E800 automated
fluorescence microscope (Nikon, Tokyo, Japan). For the flow
cytometric analysis, the cells were measured with a FACSCalibur
flow cytometer (BD Biosciences, San Jose, CA, USA) using CellQuest
software.
Quantification of acidic vesicular
organelles
Autophagy was characterized by the formation of
acidic vesicular organelles (AVOs). Cells were seeded in
6-cm2 plates at a density of 5×105 cells/ml.
After treatment with HCA for 24 h, acridine orange (1 μg/ml) was
added to the living cells for 30 min, and the cells were removed
from the plate with trypsin-EDTA and collected in phenol red-free
growth medium. Approximately 1×104 cells were
illuminated with blue (488 nm) excitation light, and the green
(510–530 nm) and red (650 nm) fluorescent emissions were measured
using a flow cytometer (Beckman Coulter, Inc., Brea, CA, USA).
MDC staining
To observe autophagy formation, cells were grown on
glass coverslips for 24 h. After treatment with HCA for 24 h, the
cells were treated with 0.05 mM monodansyl-cadaverine (MDC;
Sigma-Aldrich) at 37°C in 5% CO2 for 10 min. The cells
were then fixed with 4% paraformaldehyde in PBS for 10 min.
Following incubation, the cells were washed three times with PBS
and were immediately analyzed under a fluorescence microscope
(IX-71; Olympus, Tokyo, Japan). The fluorescence was measured at an
excitation wavelength of 380 nm with an emission filter at 530
nm.
siRNA experiment
The siRNA construct for Atg5 was obtained in the
form of select validated siRNA (Bioneer Corp., Daejeon, Korea). The
cells were transfected with 20 nM siRNA using the Lipofectamine
RNAi Max Transfection reagent (Invitrogen, Carlsbad, CA, USA)
according to the manufacturer's instructions. The cells were
harvested 24 h after the transfection. The total cell lysates were
separated by SDS-PAGE and were analyzed by western blot analysis,
as described above.
Statistical analysis
The statistical analysis was performed with data
obtained from three independent experiments. The data are
represented as the means ± SD. The ANOVA and Student's t-test were
applied to determine the statistical significance. P-values
<0.05 were considered to be significant.
Results
Effect of cinnamaldehyde derivatives on
cell proliferation and cytotoxicity in p53-wild-type SGT and
p53-mutant YD-10B cells
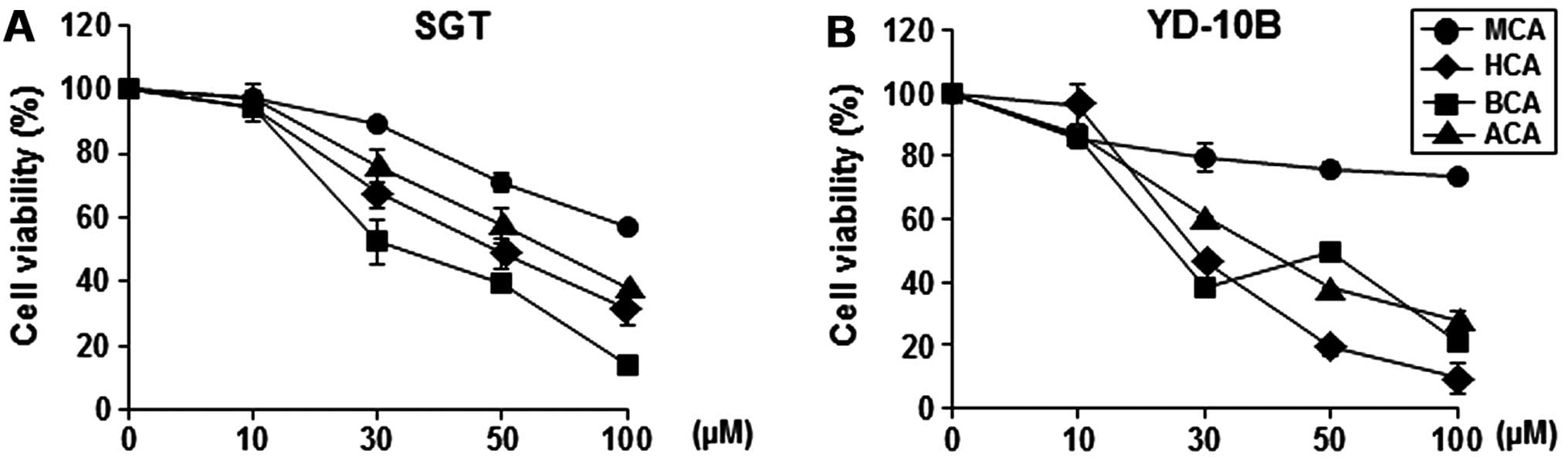
We investigated the cinnamaldehyde derivatives (MCA,
HCA, BCA and ACA) to develop a potential antitumor agent. To assess
the growth-inhibitory effect of these cinnamaldehyde derivatives,
we initially examined their effects on the proliferation of the SGT
and YD-10B cells. As shown in Fig.
1, HCA and BCA showed more potent growth-inhibitory effects
than the other tested cinnamaldehyde derivatives (MCA and ACA)
against SGT and YD-10B cells, with IC50 values ranging
from 30 to 50 μM. In particular, HCA showed the most potent
growth-inhibitory effect in YD-10B cells compared to all other
cinnamaldehyde derivatives. Treatment with 50 μM of HCA reduced the
cell viability by ~81% (Fig.
1B).
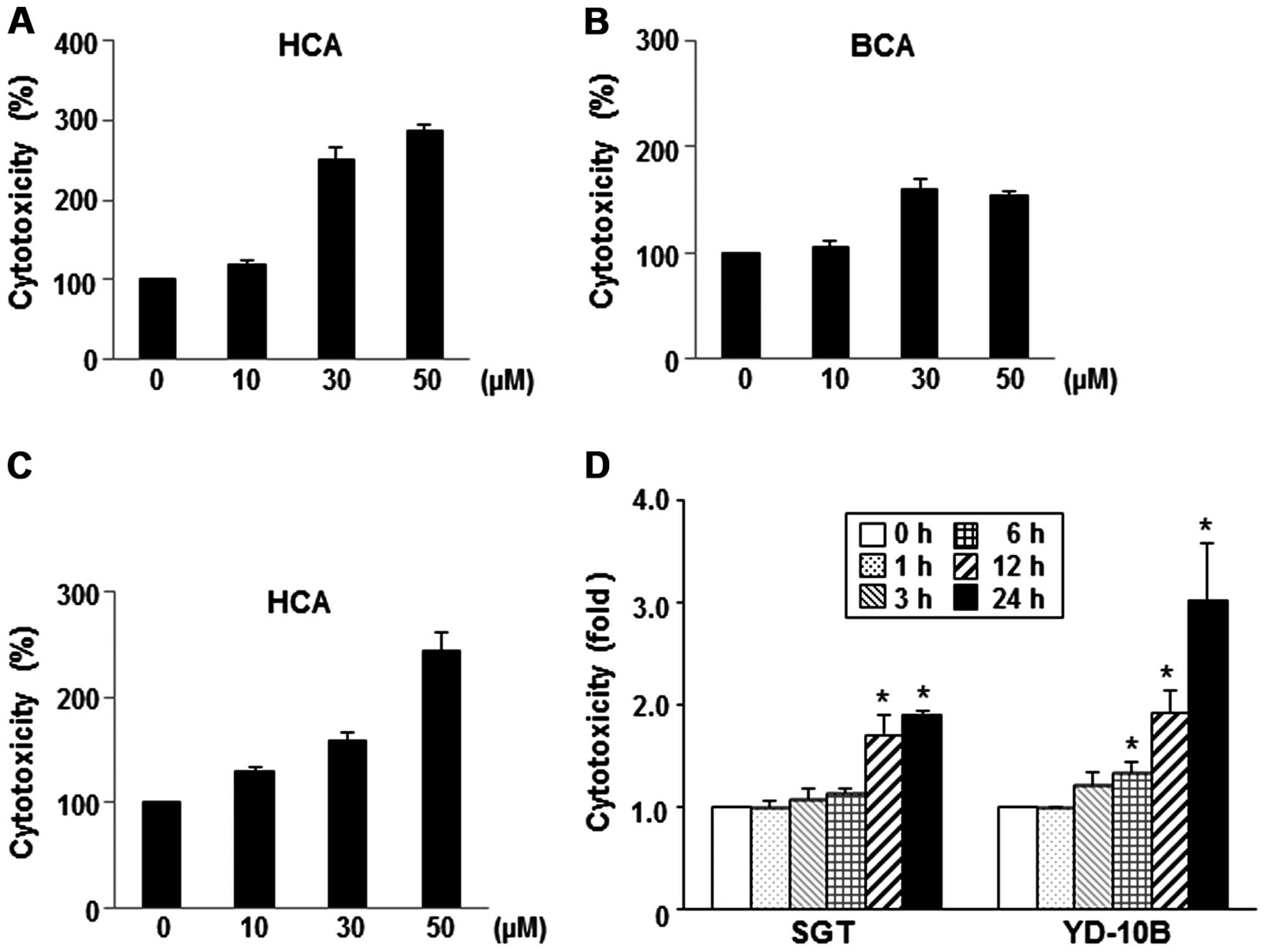
To evaluate the dose-dependent cytotoxic effects of
HCA and BCA, the cells were incubated with varying concentrations
of HCA or BCA for 24 h. As shown in Fig. 2A and B, HCA and BCA caused
dose-dependent cell cytotoxicity compared to the untreated SGT
cells. When the YD-10B cells were treated with HCA for 24 h, the
cell cytotoxicity also increased in a dose-dependent manner
(Fig. 2C). A constant
concentration of HCA (50 μM) was applied to SGT and YD-10B cells
for different time periods, and the cytotoxicity was measured
relative to untreated control cells. Fig. 2D showed that HCA induced high
cytotoxicity between 12 and 24 h in both SGT and YD-10B cells.
Similar effects were noted in the BCA-treated cells (data not
shown).
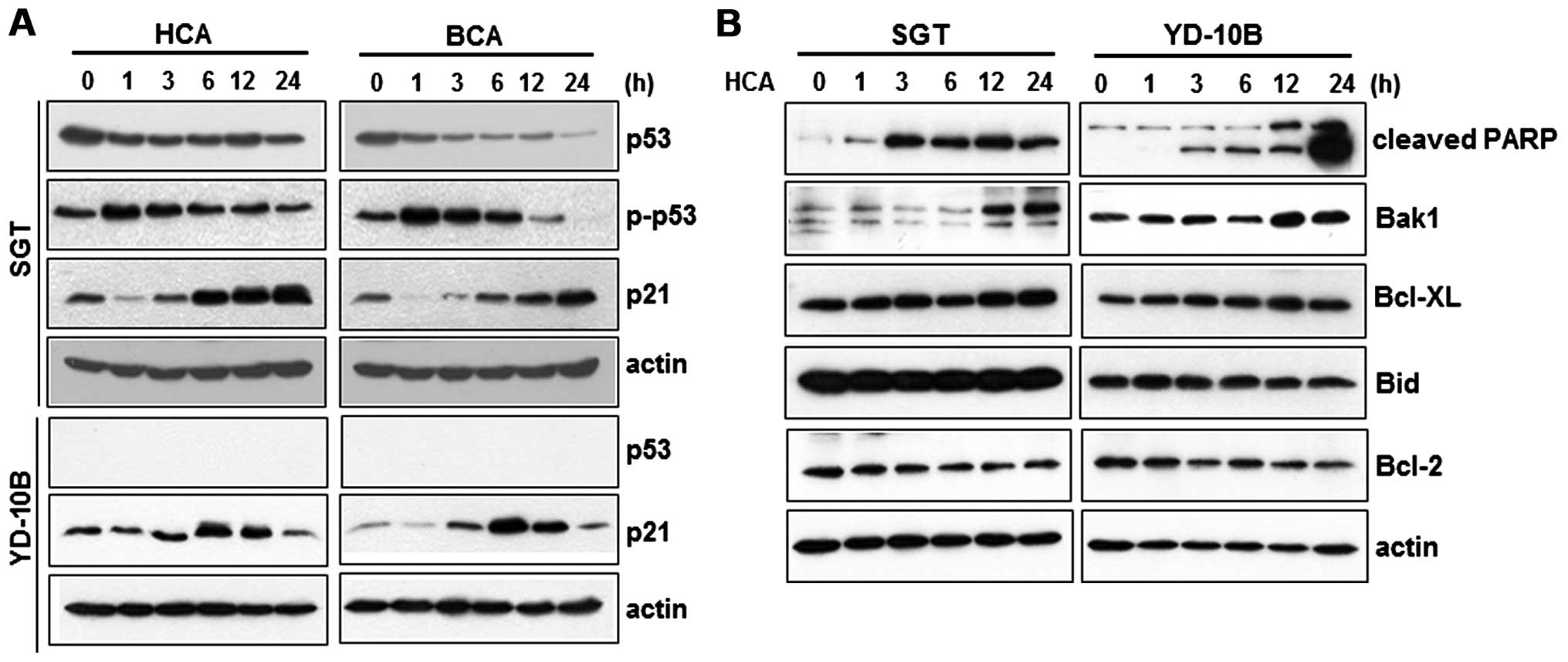
HCA treatment leads to the upregulation
of p53-independent p21 and Bak1
It has been reported that p53 modulates the
activation and oligomerization of Bax/Bak, which leads to
mitochondria-mediated cell death (19,20).
To examine the importance of p53 in HCA- or BCA-induced cell growth
inhibition, we performed western blot analysis using anti-p53 and
anti-p21 antibodies. Cells were treated with HCA or BCA for various
time periods, and the cell lysates were prepared. As shown in
Fig. 3A, HCA or BCA treatment
caused significant p21 induction as well as p53 phosphorylation in
SGT cells. p21 induction was also observed in YD-10B cells, whereas
p53 phosphorylation was not detected. These results clearly showed
that HCA and BCA can inhibit cell growth in YD-10B cells through
the p53-independent induction of p21, suggesting that p53 does not
play an essential role in cell growth inhibition in YD-10B
cells.
Bcl-2 family proteins also play a critical role in
the regulation of the mitochondria-dependent cell death pathway
(21). To investigate whether HCA
affects the levels of pro-apoptotic proteins (Bax and Bak1),
BH3-only protein (Bid), and anti-apoptotic proteins (Bcl-2 and
Bcl-XL), SGT and YD-10B cells were treated with HCA and western
blot analysis was performed. We observed the time-dependent
upregulation of Bak1 in SGT and YD-10B cells, while the level of
anti-apoptotic protein Bcl-2 was decreased in the cells treated
with HCA (Fig. 3B).
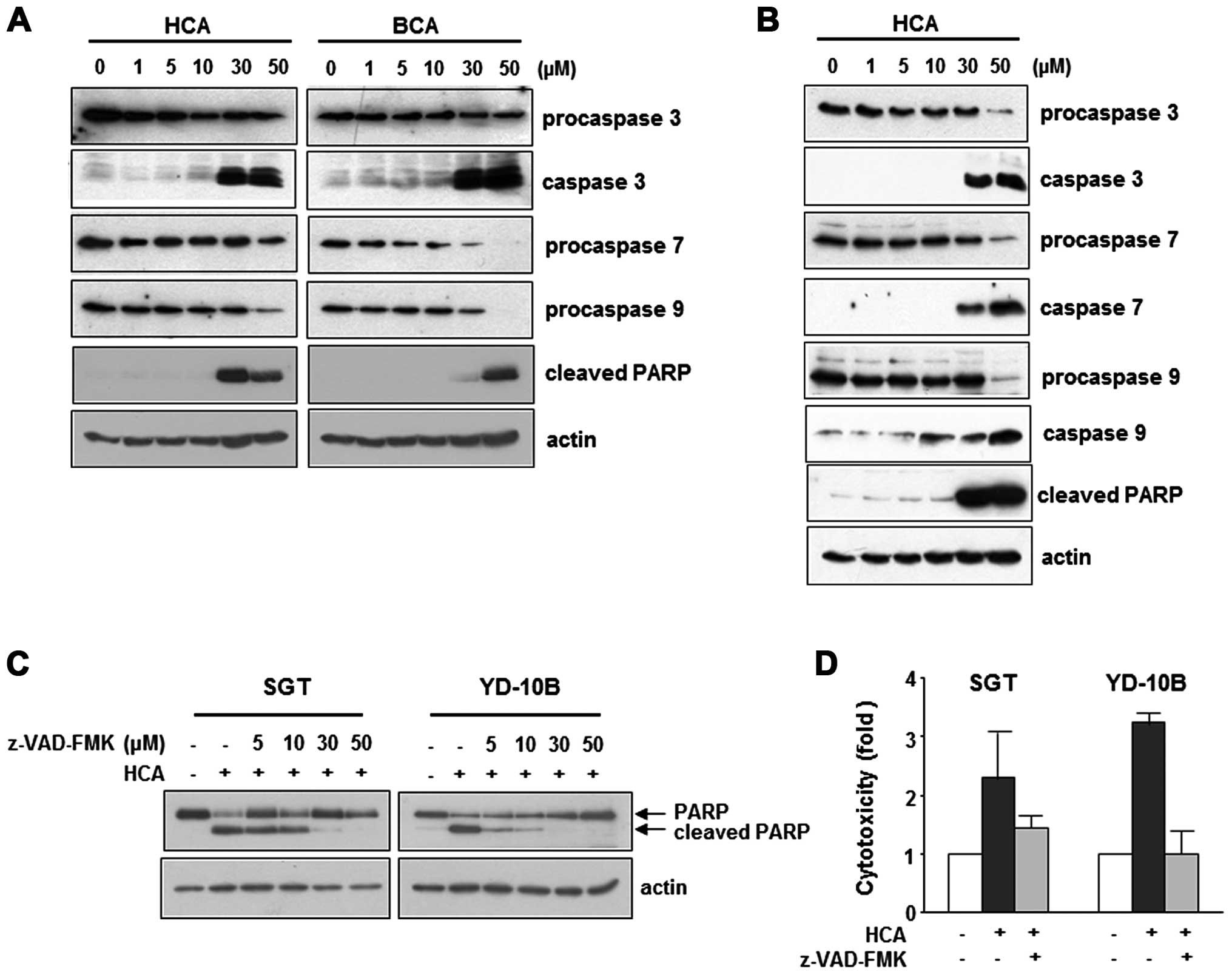
HCA induces caspase-dependent apoptotic
cell death
To address whether HCA induces apoptosis in SGT and
YD-10B cells, both cell types were treated with HCA for 24 h and
then assessed for caspase activity. As shown in Fig. 4A, HCA reduced the level of
procaspase-3, -7 and -9 in SGT cells in a dose-dependent manner. In
YD-10B cells, the protein levels of procaspase-3, -7 and -9 were
also decreased by HCA treatment (Fig.
4B). HCA largely increased the levels of cleaved PARP in both
SGT and YD-10B cells (Fig. 4A and
B).
To confirm the role of HCA in caspase-mediated
apoptosis, SGT and YD-10B cells were treated with z-VAD-FMK (a
pan-caspase inhibitor) before exposure to HCA. Importantly, our
data showed that z-VAD-FMK inhibited the HCA-induced PARP
activation in both cell lines (Fig.
4C). Moreover, z-VAD-FMK substantially protected the cells
against HCA-induced cytotoxicity (Fig.
4D). Collectively, these findings suggest that HCA induces the
mitochondrial caspase-dependent apoptotic pathway in SGT and YD-10B
cells.
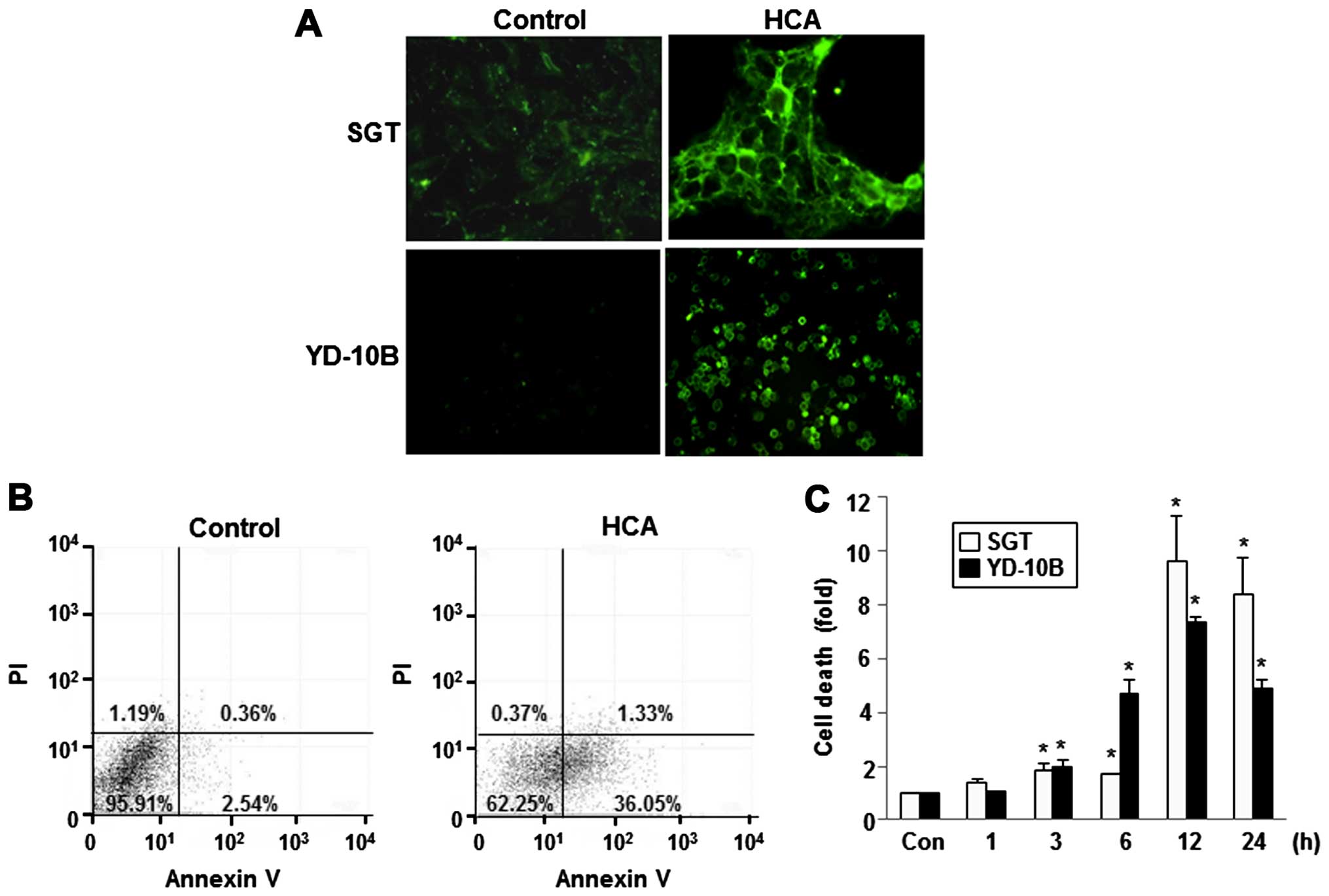
To confirm these results, apoptotic cells were
quantified using the Annexin V-FITC/PI double staining assay. After
treatment with 50 μM HCA for 24 h, apoptosis was observed by
visualizing the green fluorescence in SGT and YD-10B cells using
fluorescence microscopy. As shown in Fig. 5A, intense fluorescence was observed
in the HCA-treated cells. After HCA treatment, a number of
apoptotic cells were significantly increased in both cell lines. An
increased number of Annexin V-positive cells (early apoptosis) was
detected in the flow cytometric analysis after HCA treatment,
indicating the onset of apoptosis in HCA-treated cells (Fig. 5B). In addition, a cell death
detection ELISA clearly showed that treatment with 50 μM HCA
induced cell death in a time-dependent manner in both SGT and
YD-10B cells (Fig. 5C).
HCA-induced autophagy regulates
apoptosis
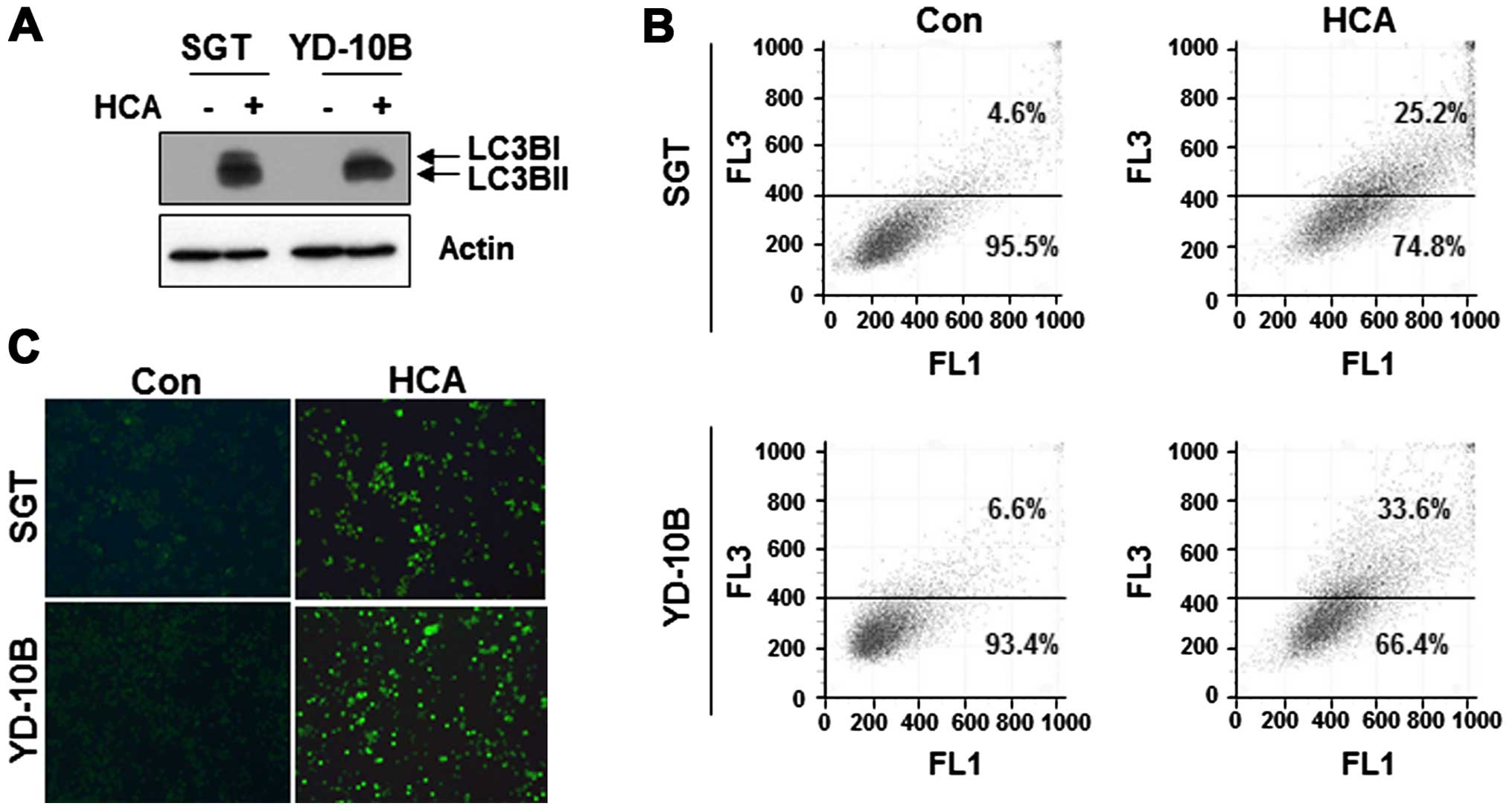
We next questioned whether HCA induces autophagy in
SGT and YD-10B cells. To address this issue, the cells were treated
with 50 μM HCA for 24 h, and the level of LC3B, a marker for
autophagy, was detected by western blot analysis. As shown in
Fig. 6A, the levels of LC3B were
increased in both HCA-treated cell lines.
To confirm the presence of autophagy, the
HCA-treated cells were stained with acridine orange and the
formation of characteristic acidic vesicular organelles (AVOs) was
quantified by flow cytometry. As shown in Fig. 6B, the number of AVOs was increased
in HCA-treated cells compare with untreated control cells. In the
MDC staining assay, MDC-labeled vacuoles were weakly detected in
the control cells. However, in the HCA-treated cells, the number of
MDC-labeled cells was largely increased (Fig. 6C). Taken together, these results
suggest that HCA induced autophagy in SGT and YD-10B cells.
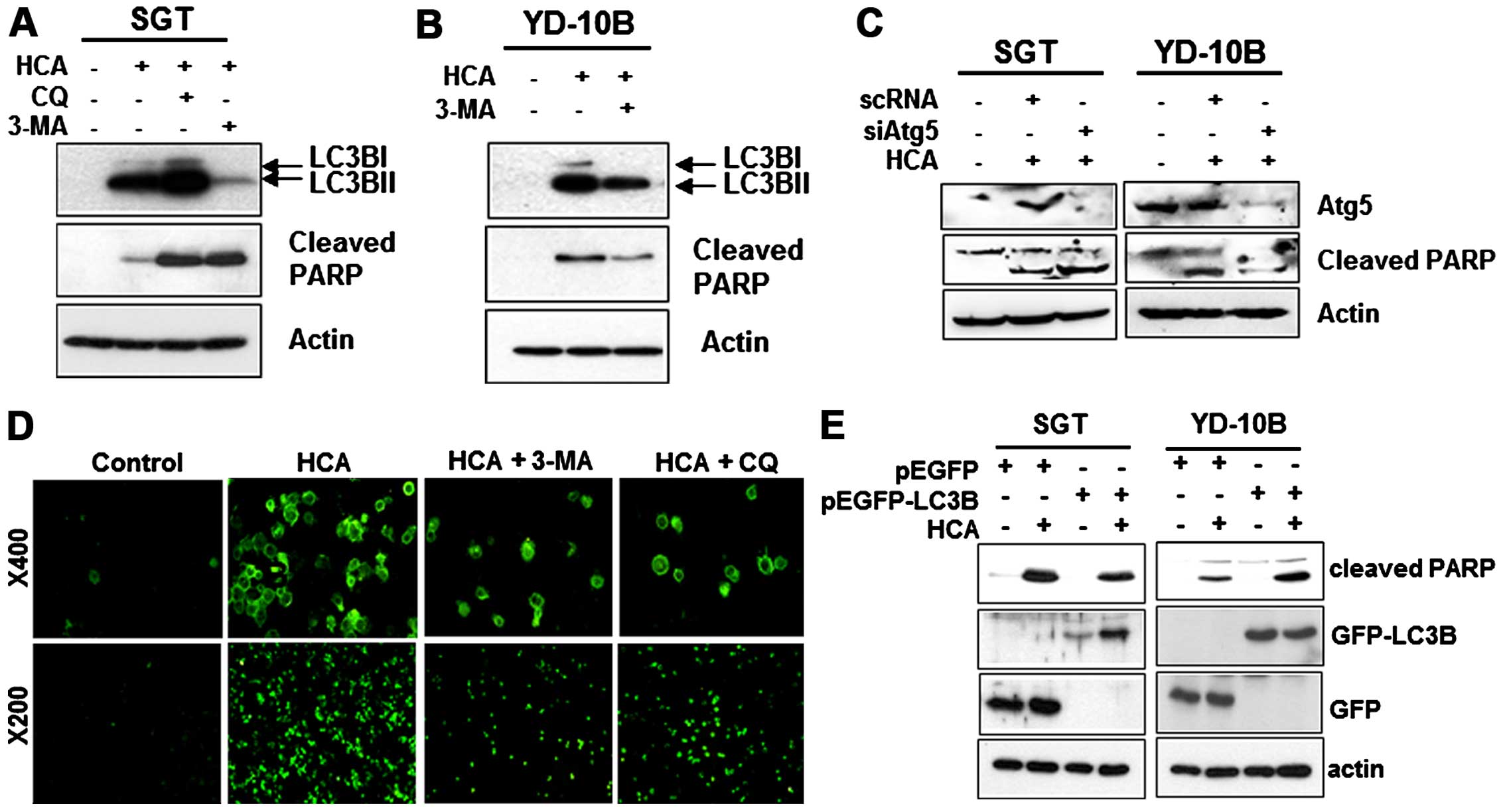
We subsequently examined the relationship between
apoptosis and autophagy in HCA-treated cells. To investigate
potential cross-talk between HCA-induced autophagy and apoptosis,
we examined whether HCA-induced apoptosis can be inhibited by an
autophagy inhibitor. SGT and YD-10B cells were incubated with 50 μM
HCA for 24 h in the presence or absence of the autophagy
inhibitors, chloroquine (CQ) or 3-methyladenine (3-MA), and
apoptosis was evaluated by detecting the level of cleaved PARP. As
shown in Fig. 7A, HCA induced PARP
cleavage in SGT cells. Notably, the HCA-induced PARP activation
strongly increased with the addition of CQ or 3-MA, even though the
level of LC3B decreased in the 3-MA treated SGT cells. In YD-10B
cells, the HCA-induced LC3B level was also suppressed by the
addition of 3-MA (Fig. 7B).
However, unlike SGT cells, the HCA-induced PARP cleavage in YD-10B
cells was inhibited by 3-MA treatment, indicating that autophagy
induces apoptosis in these cells. A siRNA experiment against Atg5
was performed to inhibit the formation of the autophagosome.
Fig. 7A and B show that Atg5
knock-down increased the levels of cleaved PARP in SGT cells and
decreased the level of cleaved PARP in YD-10B cells (Fig. 7C). Taken together, these results
suggest that autophagy might contribute to apoptosis in head and
neck cancer cells treated with HCA. The inhibition of apoptosis by
autophagy inhibitors was confirmed by visualizing the green
fluorescence in YD-10B cells (Fig.
7D). We then examined whether the overexpression of LC3B
affects HCA-mediated apoptosis. For this experiment, the cells were
transfected with LC3B expressing vectors and the correlation
between autophagy and apoptosis was assessed by detecting the
levels of cleaved PARP. Notably, the overexpression of LC3B
decreased the levels of cleaved PARP in SGT cells compared with the
HCA-treated control. However, the overexpression of LC3B increased
the levels of cleaved PARP in YD-10B cells (Fig. 7E). These results strongly suggest
that autophagy may actively contribute to the HCA-induced apoptosis
in YD-10B cells and negatively regulates HCA-induced apoptosis in
SGT cells.
Discussion
In the present study, we showed that the
cinnamaldehyde derivatives, HCA and BCA, have potent
anti-proliferative and cytotoxic activity against p53-wild-type
(SGT) and p53-mutant (YD-10B) head and neck cancer cells. We also
observed that the HCA- and BCA-treated cells have apoptotic
morphologies, reductions in the pro-forms of caspase-3, -7 and -9,
and increased levels of PARP cleavage, implying the induction of
apoptosis via the mitochondrial caspase-dependent signaling
pathway. Our findings are consistent with other reports showing
that HCA and BCA trigger apoptosis in MDA-MB-231 breast cancer
cells and SW620 colon cancer cells by activating caspase-3 and PARP
while suppressing the expression of anti-apoptotic proteins such as
Bcl-XL and Bcl-2 (4).
Genetic defects and mutations in the tumor
suppressor gene p53 have been observed in various types of human
malignancies (22). Many studies
have demonstrated that p53 induces the expression of genes involved
in cell cycle control, DNA repair, and apoptosis in response to DNA
damage (22,23). We observed that HCA treated SGT
cells (p53-wild-type) exhibited p53 activation. Consistent with the
role of p53 as a cell cycle regulator, we also observed the
increased expression of p21 in SGT cells. Notably, p53 expression
was not observed following HCA treatment in p53-mutant YD-10B
cells; instead, the accumulation of p21 was observed. Although the
cellular stress-induced upregulation of p53 has been used as an
indicator of p53-dependent apoptosis in cancer cells (23,24),
our data demonstrated that HCA and BCA exhibit similar cytotoxic
effects in p53-wild-type SGT cells and p53-mutant YD-10B cells
which suggests a p53-independent cytotoxic mechanism.
Bcl-2 family members and mitochondria are important
targets of p53 (25). Generally,
the apoptotic pathway is activated by changes in the balance
between anti- and pro-apoptotic members of the Bcl-2 family. Upon
activation, Bak and Bax, the pro-apoptotic members of the Bcl-2
family, oligomerize and permeabilize the outer mitochondrial
membrane, resulting in the release of cytochrome c, the
activation of caspase-9 and subsequent PARP cleavage. Our data
showed that HCA mediates apoptosis via a pathway involving the
modulation of Bak1 and Bcl-2 as well as the initiator caspase
proteins of the intrinsic pathway, caspase-3, -7 and -9. HCA
induced the expression of Bak1 and decreased the expression of
Bcl-2 in SGT and YD-10B cells. The levels of Bcl-XL and Bid
remained unchanged. Previous studies have shown that phospho-p53
functionally re-organizes the Bak/Bcl-XL complex and activates Bak
(20,24,25).
We could not find any different activation patterns of Bcl-2 family
members in either the p53-wild-type (SGT) or p53-mutant type
(YD-10B) cells. Additionally, HCA showed growth-inhibitory activity
as well as apoptosis induction in both cell lines, suggesting HCA
is a potent anticancer agent that induces mitochondria-mediated
apoptosis regardless of the p53 status.
Autophagy has been shown to engage in a complex
interplay with apoptosis. It has been suggested that the autophagic
response observed in cells treated with diverse cytotoxic agents is
involved in protecting cells from apoptosis. Alternatively,
autophagy may be associated with a mechanism contributing to
apoptosis (26–28). Despite these studies, the
relationship between autophagy and apoptosis in head and neck
cancer cells remains poorly understood.
We found that HCA-induced autophagy leads to the
regulation of the apoptotic pathway. 3-MA, a specific inhibitor of
the early autophagic process, strongly induced the activation of
PARP in HCA-treated SGT cells. However, the suppression of LC3B by
3-MA blocked the HCA-induced PARP activation in YD-10B cells. In
addition, HCA-induced apoptosis was inhibited by the overexpression
of LC3B in SGT cells, while promoted in YD-10B cells. These
findings suggest that the HCA-induced autophagy is associated with
the mechanism contributing to apoptosis in SGT and YD-10B
cells.
In conclusion, we demonstrated for the first time
that HCA induces autophagy and apoptosis in head and neck cancer
cells. Both HCA and BCA can trigger the intracellular cell death
pathway in a p53-independent manner. However, HCA-induced apoptosis
was alternatively regulated by LC3B-mediated autophagy, i)
apoptosis in HCA-treated YD-10B cells was blocked by the addition
of 3-MA, ii) HCA-induced apoptosis in SGT cells was increased by
3-MA. Therefore, our results suggest HCA as a potential therapeutic
candidate for human head and neck cancer.
Acknowledgements
The present study was supported by the National
Research Foundation of Korea (NRF) grant funded by the Korea
government MSIP (no. 2008-0062283).
References
|
1
|
Jeong HW, Han DC, Son KH, Han MY, Lim JS,
Ha JH, Lee CW, Kim HM, Kim HC and Kwon BM: Antitumor effect of the
cinnamaldehyde derivative CB403 through the arrest of cell cycle
progression in the G2/M phase. Biochem Pharmacol. 65:1343–1350.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chao LK, Hua KF, Hsu HY, Cheng SS, Lin IF,
Chen CJ, Chen ST and Chang ST: Cinnamaldehyde inhibits
pro-inflammatory cytokines secretion from monocytes/macrophages
through suppression of intracellular signaling. Food Chem Toxicol.
46:220–231. 2008. View Article : Google Scholar
|
|
3
|
Kwon BM, Lee SH, Cho YK, Bok SH, So SH,
Youn MR and Chang SI: Synthesis and biological activity of
cinnamaldehydes as angiogenesis inhibitors. Bioorg Med Chem Lett.
7:2473–2476. 1997. View Article : Google Scholar
|
|
4
|
Han DC, Lee MY, Shin KD, Jeon SB, Kim JM,
Son KH, Kim HC, Kim HM and Kwon BM: 2′-benzoyloxycinnamaldehyde
induces apoptosis in human carcinoma via reactive oxygen species. J
Biol Chem. 279:6911–6920. 2004. View Article : Google Scholar
|
|
5
|
Lee CW, Hong DH, Han SB, Park SH, Kim HK,
Kwon BM and Kim HM: Inhibition of human tumor growth by 2′-hydroxy-
and 2′-benzoyloxycinnamaldehydes. Planta Med. 65:263–266. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Koh WS, Yoon SY, Kwon BM, Jeong TC, Nam KS
and Han MY: Cinnamaldehyde inhibits lymphocyte proliferation and
modulates T-cell differentiation. Int J Immunopharmacol.
20:643–660. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ka H, Park HJ, Jung HJ, Choi JW, Cho KS,
Ha J and Lee KT: Cinnamaldehyde induces apoptosis by ROS-mediated
mitochondrial permeability transition in human promyelocytic
leukemia HL-60 cells. Cancer Lett. 196:143–152. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lee CW, Lee SH, Lee JW, Ban JO, Lee SY,
Yoo HS, Jung JK, Moon DC, Oh KW and Hong JT:
2-hydroxycinnamaldehyde inhibits SW620 colon cancer cell growth
through AP-1 inactivation. J Pharmacol Sci. 104:19–28. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hong SH, Kim J, Kim JM, Lee SY, Shin DS,
Son KH, Han DC, Sung YK and Kwon BM: Apoptosis induction of
2′-hydroxycin-namaldehyde as a proteasome inhibitor is associated
with ER stress and mitochondrial perturbation in cancer cells.
Biochem Pharmacol. 74:557–565. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kim SA, Sung YK, Kwon BM, Yoon JH, Lee H,
Ahn SG and Hong SH: 2′-Hydroxycinnamaldehyde shows antitumor
activity against oral cancer in vitro and in vivo in a rat tumor
model. Anticancer Res. 30:489–494. 2010.PubMed/NCBI
|
|
11
|
Kang HS, Ock J, Lee HJ, Lee YJ, Kwon BM
and Hong SH: Early growth response protein 1 upregulation and
nuclear translocation by 2′-benzoyloxycinnamaldehyde induces
prostate cancer cell death. Cancer Lett. 329:217–227. 2013.
View Article : Google Scholar
|
|
12
|
Ock J, Lee HA, Ismail IA, Lee HJ, Kwon BM,
Suk K, Lee WH and Hong SH: Differential antiproliferation effect of
2′-benzoyloxy-cinnamaldehyde in K-ras-transformed cells via
downregulation of thiol antioxidants. Cancer Sci. 102:212–218.
2011. View Article : Google Scholar
|
|
13
|
Kwon JY, Hong SH, Park SD, Ahn SG, Yoon
JH, Kwon BM and Kim SA: 2′-Benzoyloxycinnamaldehyde inhibits nitric
oxide production in lipopolysaccharide-stimulated RAW 264.7 cells
via regulation of AP-1 pathway. Eur J Pharmacol. 696:179–186. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Young MR, Levingston C and Johnson SD:
Cytokine and adipokine levels in patients with premalignant oral
lesions or in patients with oral cancer who did or did not receive
1α,25-dihydroxyvitamin D3 treatment upon cancer diagnosis. Cancers
(Basel). 7:1109–1124. 2015. View Article : Google Scholar
|
|
15
|
Moon SM, Ahn MY, Kwon SM, Kim SA, Ahn SG
and Yoon JH: Homeobox C5 expression is associated with the
progression of 4-nitroquinoline 1-oxide-induced rat tongue
carcinogenesis. J Oral Pathol Med. 41:470–476. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
William WN Jr and El-Naggar AK: A novel
target for oral cancer chemoprevention? Notch quite, yet…. Cancer
Prev Res (Phila). 8:262–265. 2015. View Article : Google Scholar
|
|
17
|
Lee EJ, Kim J, Lee SA, Kim EJ, Chun YC,
Ryu MH and Yook JI: Characterization of newly established oral
cancer cell lines derived from six squamous cell carcinoma and two
mucoepidermoid carcinoma cells. Exp Mol Med. 37:379–390. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim SA, Kim YC, Kim SW, Lee SH, Min JJ,
Ahn SG and Yoon JH: Antitumor activity of novel indirubin
derivatives in rat tumor model. Clin Cancer Res. 13:253–259. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang L, Yu J, Park BH, Kinzler KW and
Vogelstein B: Role of BAX in the apoptotic response to anticancer
agents. Science. 290:989–992. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pietsch EC, Perchiniak E, Canutescu AA,
Wang G, Dunbrack RL and Murphy ME: Oligomerization of BAK by p53
utilizes conserved residues of the p53 DNA binding domain. J Biol
Chem. 283:21294–21304. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Oda E, Ohki R, Murasawa H, Nemoto J,
Shibue T, Yamashita T, Tokino T, Taniguchi T and Tanaka N: Noxa, a
BH3-only member of the Bcl-2 family and candidate mediator of
p53-induced apoptosis. Science. 288:1053–1058. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Soussi T and Lozano G: p53 mutation
heterogeneity in cancer. Biochem Biophys Res Commun. 331:834–842.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Vogelstein B, Lane D and Levine AJ:
Surfing the p53 network. Nature. 408:307–310. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Oren M: Decision making by p53: Life,
death and cancer. Cell Death Differ. 10:431–442. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Vaseva AV and Moll UM: The mitochondrial
p53 pathway. Biochim Biophys Acta. 1787:414–420. 2009. View Article : Google Scholar
|
|
26
|
Levy JM and Thorburn A: Targeting
autophagy during cancer therapy to improve clinical outcomes.
Pharmacol Ther. 131:130–141. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen S, Rehman SK, Zhang W, Wen A, Yao L
and Zhang J: Autophagy is a therapeutic target in anticancer drug
resistance. Biochim Biophys Acta. 1806:220–229. 2010.PubMed/NCBI
|
|
28
|
Choi KS: Autophagy and cancer. Exp Mol
Med. 44:109–120. 2012. View Article : Google Scholar : PubMed/NCBI
|