Introduction
Lung cancer is the leading cause of
malignancy-related death worldwide. Non-small cell lung cancer
constitutes ~80% of all cases (1).
The treatment including surgery, radiotherapy and chemotherapy has
changed little for decades. Recently, targeted therapy was
confirmed in a subset of patients with epithelial growth factor
receptor gene mutation or amplification. However, <20% of
patients survive for 5 years (2).
The search for novel treatment modalities is highly required.
Vesicular stomatitis virus (VSV) from the family of
rhab-doviridae is an enveloped, negative-sense RNA virus (3). VSV was suggested to kill selectively
a variety of tumor cell lines and proposed as a promising cancer
treatment (3–5). We and others tested its efficacy
against a panel of both murine and human cancers including ovarian
cancer, colorectal cancer, and lung cancer (6–9).
Notably, although VSV showed promise in ovarian and colorectal
cancer, it achieved limited effects against lung cancer. The lung
cancer intrinsic mechanisms render resistance to VSV.
Cancer cells have defects in the interferon (IFN)
signaling pathway (10). The
defects make the cancer cells vulnerable to viruses including VSV.
VSV induces apoptosis in cancer cells that have defects in the IFN
pathway (11). The apoptosis
pathway plays an important role in mediating the tumoricidal
effects of VSV. Livin was discovered as a member of the inhibitor
of apoptosis family (12–15). It is expressed in a variety of
tumors (16–19), but hardly detectable in the normal
tissue (20). Two splicing
variants have been identified (designated α and β isoforms) for
Livin, which are almost identical except for a 54 bp truncation in
exon 6 (15). The overexpression
of both isoforms block apoptosis induced by TNF-α and anti-CD95
antibody. In this study, we attempted to explore the expression of
Livin in lung adenocarcinoma and its possible relationship to VSV
vulnerability.
Materials and methods
Expression plasmids
We previously constructed the expression plasmid for
the full length Livin-β and the truncated form lacking the first 52
amino acids (tLiv) (21,22). The baculovirus inhibitor of
apoptosis protein repeat (BIR) domain of Livin (BIRLiv, amino acid
53–220) was subcloned into the expression plasmid pVITRO with a
FLAG tag. The carboxyl terminal of Livin (cLiv, amino acid 221–280)
compassing the Really Interesting New Gene domain was subcloned
into the expression plasmid p-EGFP-N1 with green fluorescent
protein fused at its carboxyl terminal. The expression vector for
the second mitochondria-derived activator of caspase (SMAC/Diablo)
was constructed by cloning its sequence into the parental plasmid
pTango-zeo. All the constructs were confirmed by sequencing.
Reagents and cells
Cytotoxic drug cisplatin, caspase-3 inhibitor
z-DEVD-fmk, the colorimetric caspase-3 substrate z-DEVD-pNA, and
antibiotic Geneticin were all purchased from Sigma (Sigma-Aldrich,
St. Louis, MO, USA). Stock preparation of the reagents was stored
at −20°C until use. A549 human lung adenocarcinoma cells, HeLa
cervial cancer cells, MCF-7 breast cancer cells, A2780 ovarian
cancer cells, and HCT116 colon cancer cells were all from the
American Type Culture Collection. Cells were maintained in
Dulbecco's modified Eagle's medium (Sigma-Aldrich) supplemented
with 10% FBS (Invitrogen, Carlsbad, CA, USA), and antibiotics. The
production and titration of VSV was performed as we described
before (6).
Cell transfection
Cells were transfected by using Lipofectamine 2000
transfection reagent (Invitrogen) according to the manufacturer's
instructions. Lipofectamine 2000 was used at a ratio of 2.5 μg/μl
in input plasmid, and Lipofectin-DNA complexes were incubated with
cells for 6 h at 37°C. For stable transfection, cells transfected
with the expression plasmid or control plasmid (empty vector) were
selected in Geneticin (800 μg/ml) for 2 weeks (starting at 24 h
after transfection) and maintained in culture medium supplemented
with Geneticin (400 μg/ml).
Western blot analysis
Proteins were resolved by electrophoresis on 10%
polyacrylamide pre-cast gels (Bio-Rad, Hercules, CA, USA),
electroblotted to Immobilon-P (Millipore, Shanghai, China) and
incubated with block solution (5% non-fat milk, 0.1% Tween-20, in
TBS). Signals were detected with ECL Plus Western Blot Detection
system (GE Healthcare, Beijing, China). The preparation of the
rabbit polyclonal anti-Livin primary antibody was described
previously (21,22), (1:1,000). The rabbit monoclonal
anti-caspase-3 antibody was from Cell Signaling Technology (9665,
Beverly, MA, USA 1:1,000). The rabbit monoclonal anti-SMAC antibody
was from Epitomics (1201-1, Burlingame, CA, USA 1:500). The mouse
monoclonal antibody for β-actin and HRP-conjugated secondary
antibodies were from OriGene (Beijing, China, 1:5,000 and 1:8,000
respectively).
Cell viability
Survival of cells after treatment was quantified by
CCK-8 assay (Dojindo, Shanghai, China) according to the
manufacturer's instructions. Briefly, cells were seeded in 96-well
tissue culture plate, in 100 μl culture medium. CCK-8 labeling
solution was added and the absorbance was measured at a wavelength
of 450 nm with a microplate reader (Bio-Rad, Hercules, CA,
USA).
Apoptosis assay
The apoptosis rate was determined on a flow
cytometry with a propidium iodide (PI)-Annexin V double staining
kit (KeyGen Biotech, Shanghai, China). Briefly, cells were
maintained in complete media. After treatment, cells were washed
with PBS and stained with PI and Annexin V. The fluorescence signal
was detected on a flow cytometer (FACSCalibre,
Becton-Dickinson).
Immunoprecipitation
The immunoprecipitation was performed with an
immunoprecipitation kit (Beyotime, Shanghai, China). Briefly, cell
lysates were incubated with anti-Livin antibody (1:500) at 4°C
overnight. The combined complex was immobilized with protein A+G
agarose, and then the agarose was washed twice with PBS. Finally,
the precipitated protein was eluted with elution buffer. The eluted
Livin protein was then probed with an anti-SMAC antibody.
Terminal deoxynucleotidyl
transferase-mediated dUTP nick end labeling (TUNEL) assay
Cells in a 6-well plate were mounted and sujected to
fluorescent in situ TUNEL assay by using an in situ
apoptotic cell detection kit (Promega, Madison, WI, USA) according
to the manufacturer's protocol. Apoptotic cells were counted under
a light microscope in randomly selected fields.
Preparation of whole-cell lysates and
cytosolic fraction
For preparation of whole-cell lysates, cells were
lysed in RIPA buffer (150 mM NaCl, 2% Triton X-100, 0.1% SDS, 50 mM
Tris pH 8.0). For western blot analysis, 1% protease inhibitor
cocktail (Sigma) was also added. For preparation of cytosolic
fraction, a nuclear-cytosolic separation kit (Beyotime) was used.
Briefly, cells were harvested and pelleted. The pellets were
resuspended in Buffer A incubated on ice. Buffer B was added to the
solution, and supernatants containing the cytosolic fraction were
obtained by centrifugation. Protein concentrations were determined
by BCA Protein assay (Thermo Fisher, Woburn, MA, USA).
RNAi knock-down
Cells were transfected with siRNA from Invitrogen.
The candidate sequences against Livin were:
AUAGAAGGAGGCCAGACGCAACUCC (BIRC7- HSS149184),
UUUGACCGGAGCAGGAACUGACAGC (BIRC7-HSS149185), and
UGCACACUGUGGACAAAGU CUCUUC (BIRC7-HSS149186). The candidate
sequences against SMAC were: GGCAGAAGCACAGAUAGAATT
(DIABLO-homo-1295), CCGACAAUAUACAAGUUUATT (DIABLO-homo-1013), and
CGGUGUUUCUCAGAAU UGATT-3 (DIABLO-homo-900). The scramble siRNA
sequence (Invitrogen catalog 12935-300) targeting no known coding
sequence was used as a control.
DEVDase activity
The assay was performed on a VersaMAX plate reader
coupled with SOFTMAX software (Molecular Devices) operating in the
end-point or kinetic mode at 37°C. DEVDase acitivity was determined
by using colorimetric pNA substrates (maximal absorbance at 405
nm). Assay buffer was 50 mM Tris, pH 7.4; 0.3% NP-40, and 10 mM
dithiothreitol. Data were recorded every 30 min for various periods
of time as appropriate for each assay.
Real-time PCR
Total RNA was extracted from cells using TRIzol
agent (Invitrogen), followed by reverse transcription with an
iScript cDNA Synthesis kit (Bio-Rad) according to the
manufacturer's instructions. The primers used were: TGACA
GAGGAGGAAGAGGAGGAG (Livin, upstream), AGTCAGC GGCCAGTCATAGAAG
(Livin, downstream), TTCGATGT GTTCTCTGA (SMAC, upstream),
TTGATGTTAAGTCCTG TTG (SMAC, downstream), CCACGAAACTACCTTCAAC TCC
(β-actin, upstream), and GTGATCTCCTTCTGCATCC TGT (β-actin,
downstream). The PCR mix was set up with a SsoFast EvaGreen
Supermix (Bio-Rad). The final concentration of primers was 500 nM
each reaction. Real-time PCR reaction was performed on the CFX96
qPCR systems (Bio-Rad). The PCR reaction constituted 40 thermal
cycles of 95°C for 5 sec, and 60°C for 5 sec. The expression of
Livin or SMAC was calculated with the Gene Expression Macro
(version 6.0) software (Bio-Rad).
Statistical analysis
Statistical analysis was performed with SPSS 19.0
software (IBM inc.). To evaluate statistical significance the
Student's t-test was performed. Results are given as means standard
deviations. All two-sided p-values <0.05 were considered
statistically significant.
Results
The apoptotic effects of VSV on A549
cells
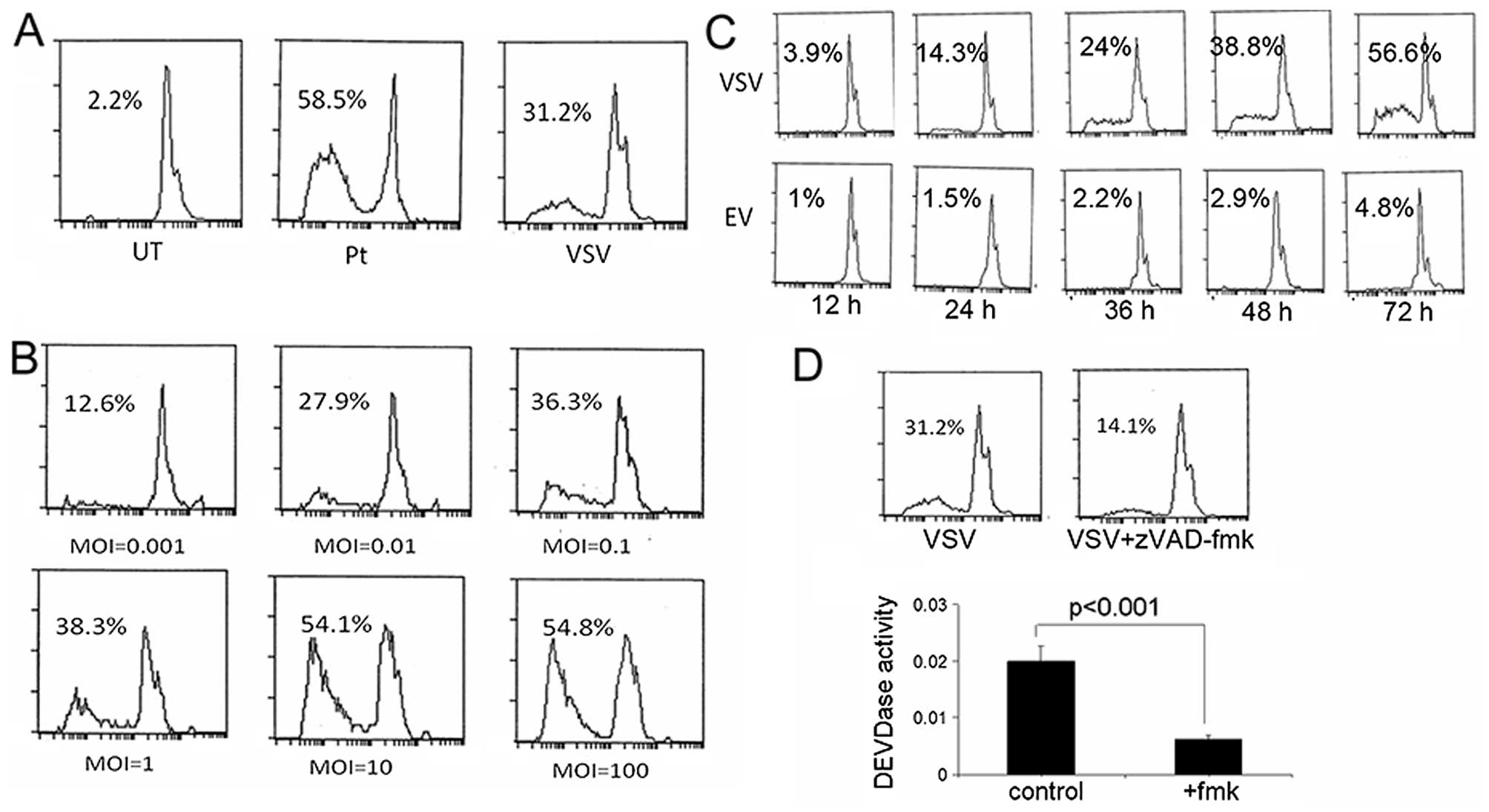
We tested the apoptotic effects of VSV in
vitro and we found VSV induced cell death though to a lesser
extent than the cytotoxic reagent cisplatin which was included as a
positive control (Fig. 1A). In
addition, the effects were both dose- and time-dependent.
Transfection at the multiplicity of infection (MOI) of 100 caused
death in half of all cells (Fig.
1B). At the MOI of 1, the cell death increased from 3.9% at 12
h post-transfection to 56.6% at 78 h (Fig. 1C). We explored the relationship
between caspase activity and death-inducing effects of VSV.
Notably, inhibition of caspase-3 with an exogenous inhibitor
(zVAD-fmk) significantly abrogated the death-inducing effects of
VSV (Fig. 1D). These results
indicated the death-inducing effects of VSV were
caspase-dependent.
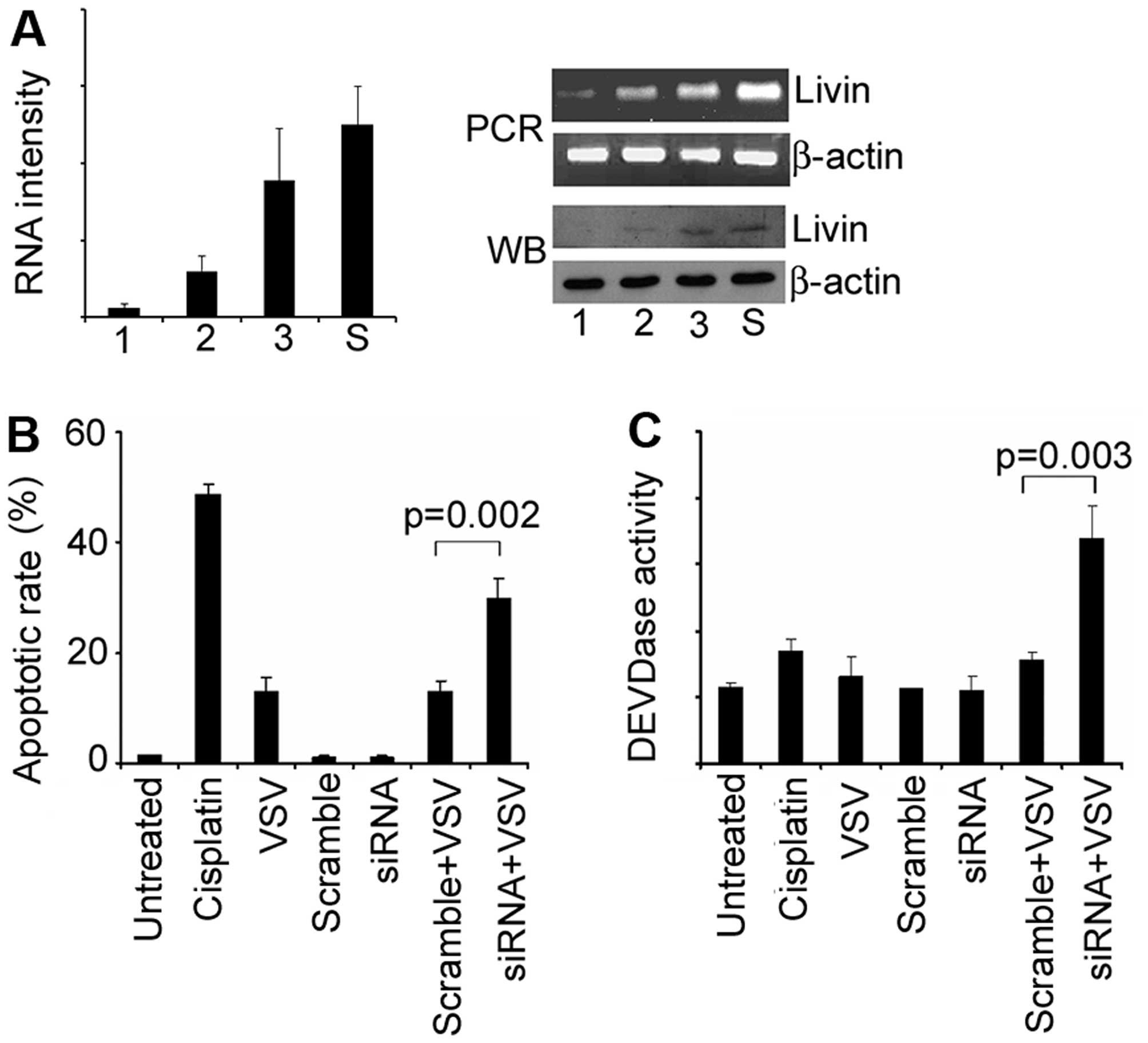
The endogenous expression of Livin in
A549 cells and its relationship with VSV treatment
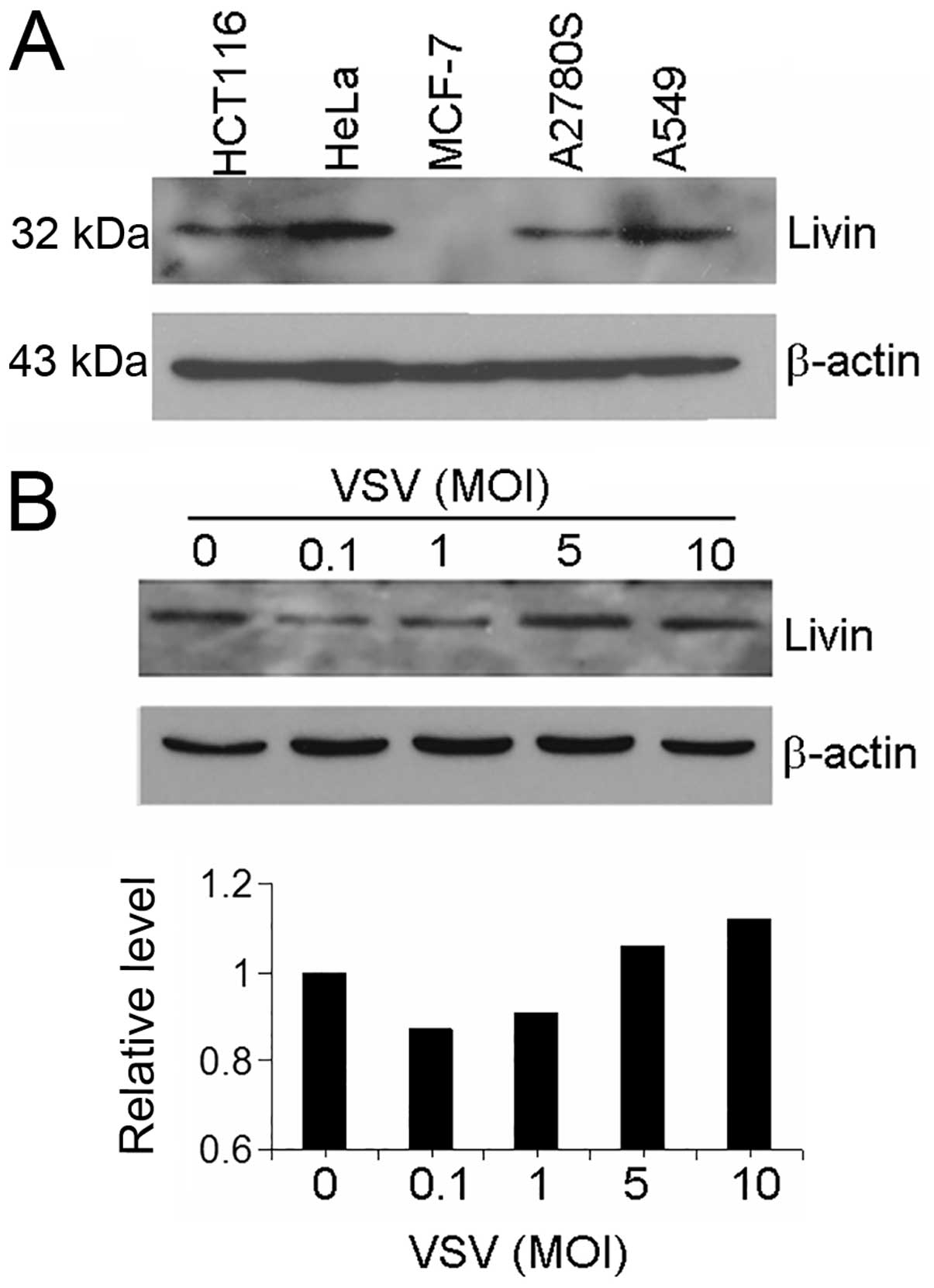
The expression of Livin in various cancer cells were
detected with western blot analysis (Fig. 2A). Among the cancer cells, the HeLa
and A549 cells contained high expression level of Livin, while the
MCF-7 breast cancer cells had barely any expression. Then we asked
whether the transfection of VSV had effects on Livin expression. At
low MOI (MOI=0.1), the transfection downregulated the expression of
Livin below the baseline level. The expression became stronger with
the increasing dose of VSV (MOI from 0.1 to 10, Fig. 2B).
Overexpression of Livin and its fragments
render A549 cells resistant to VSV-induced cell death
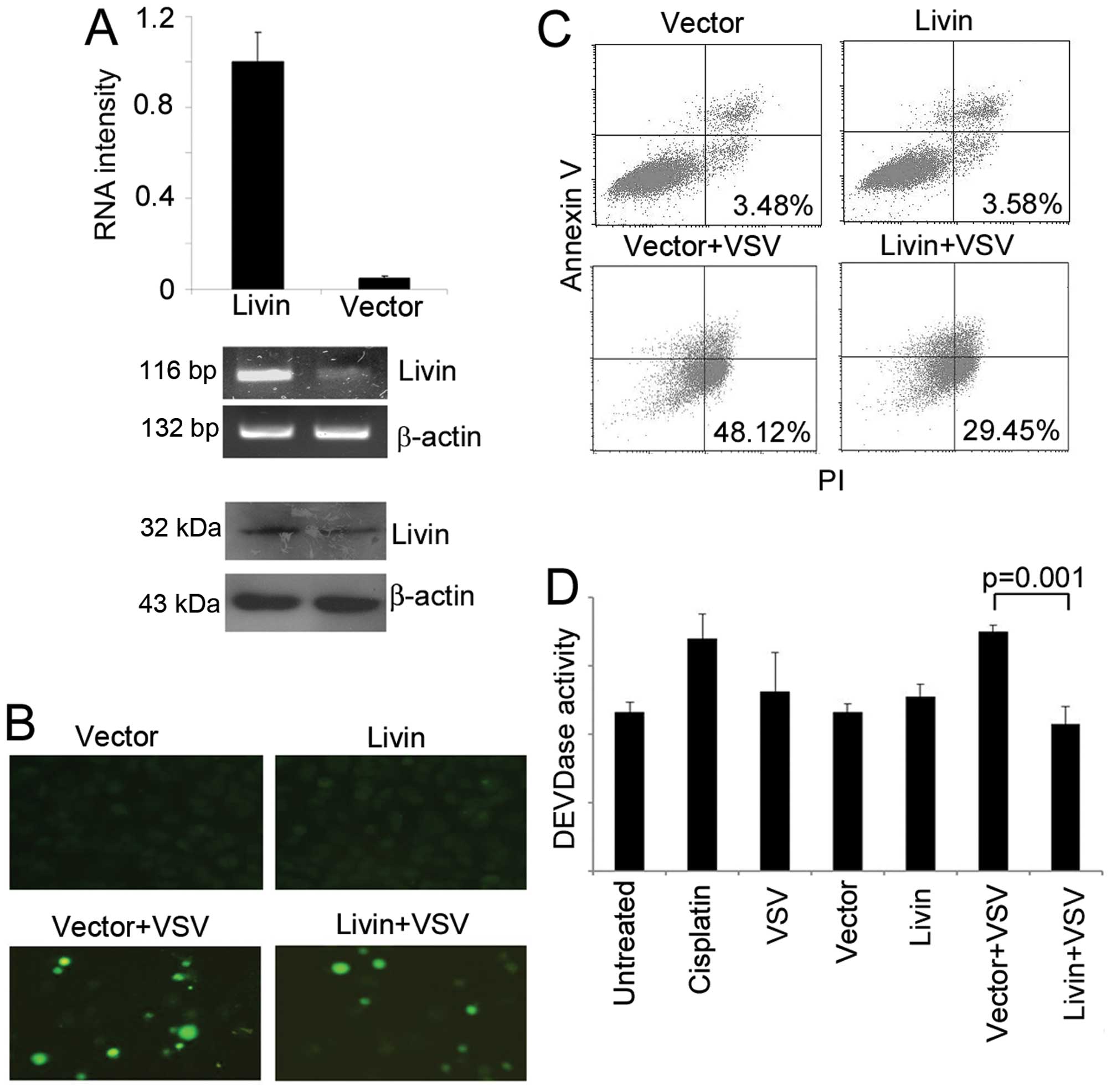
We exogenously elevated the expression of Livin by
transfection of an expression plasmid of full-length Livin. The
overexpression was confirmed by real-time PCR, half-quantitative
PCR, and western blot analysis (Fig.
3A). The overexpression made the cells more resistant to VSV
treatment, as showed by qualitative TUNEL staining, and
quantitative flow cytometry (Fig. 3B
and C). The overexpression also decreased the caspase-3
activity when challenged with VSV (Fig. 3D). To further confirm the
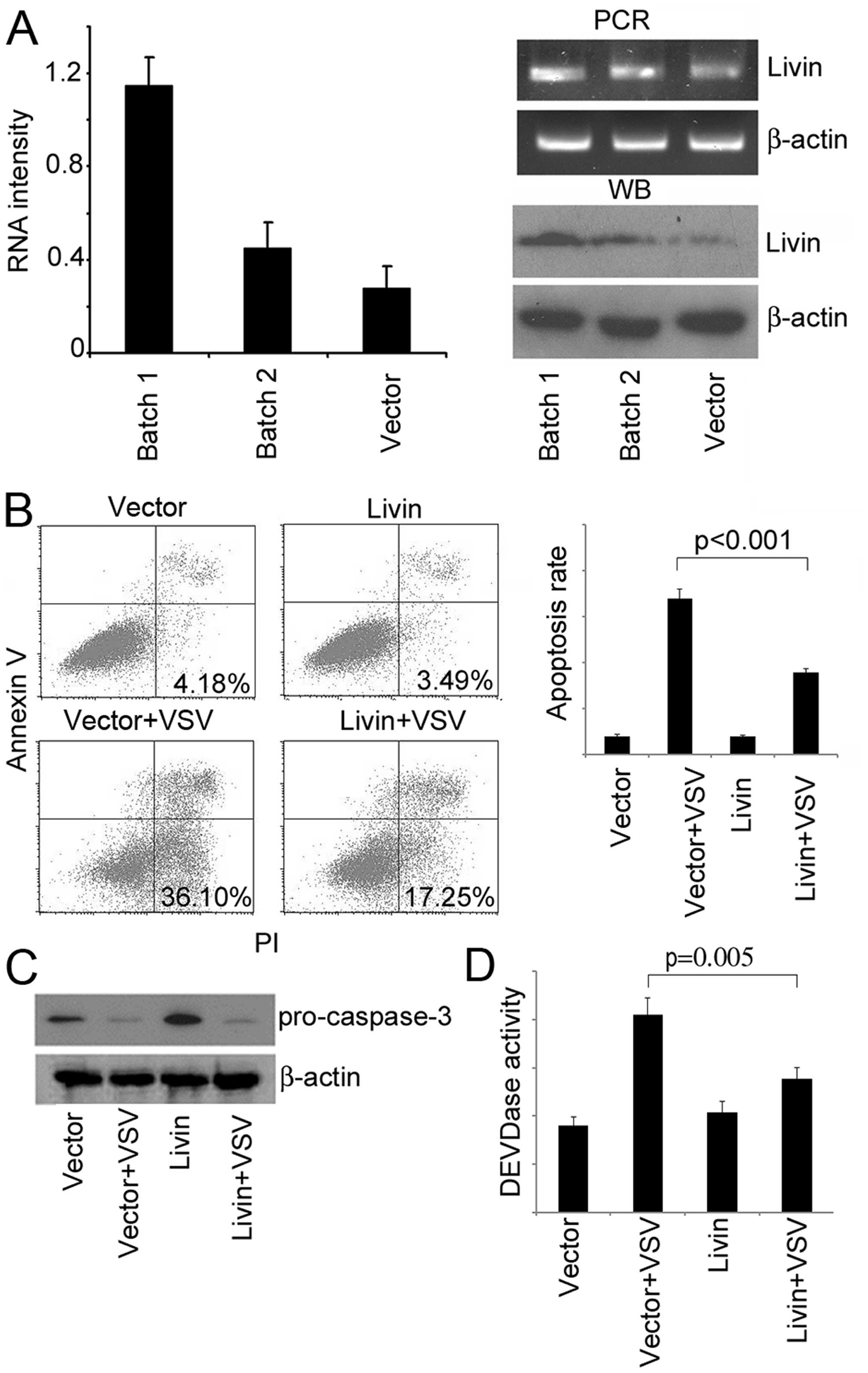
observation, we made stable transfectants of Livin (Fig. 4A). Similar observations were
obtained as the transient transfection (Fig. 4B–D).
We next asked which domain was responsible for the
anti-apoptotic effects. We prepared several vectors for the
different segments of Livin (Fig.
5A). When introduced into cells, the vector for the BIR domain
exerted an anti-apoptotic effects together with the full-length
Livin (Fig. 5B). The other
segments had no obvious effects on the VSV sensitivity. These
observations suggested a possible role of the BIR domain for the
anti-apoptotic effects of Livin against VSV challenge.
Livin inhibits the apoptotic effects of
VSV by binding SMAC in the cytosol
We observed Livin modulated the apoptotic effects of
VSV through the inhibition of caspase activity, and the BIR domain
was critical for the anti-apoptotic effects. We reasoned that Livin
inhibited caspase activity due to SMAC binding with its BIR domain,
which is a possible mechanistic explanation for its anti-apoptotic
effects. Firstly, we tested the possible involvement of SMAC in the
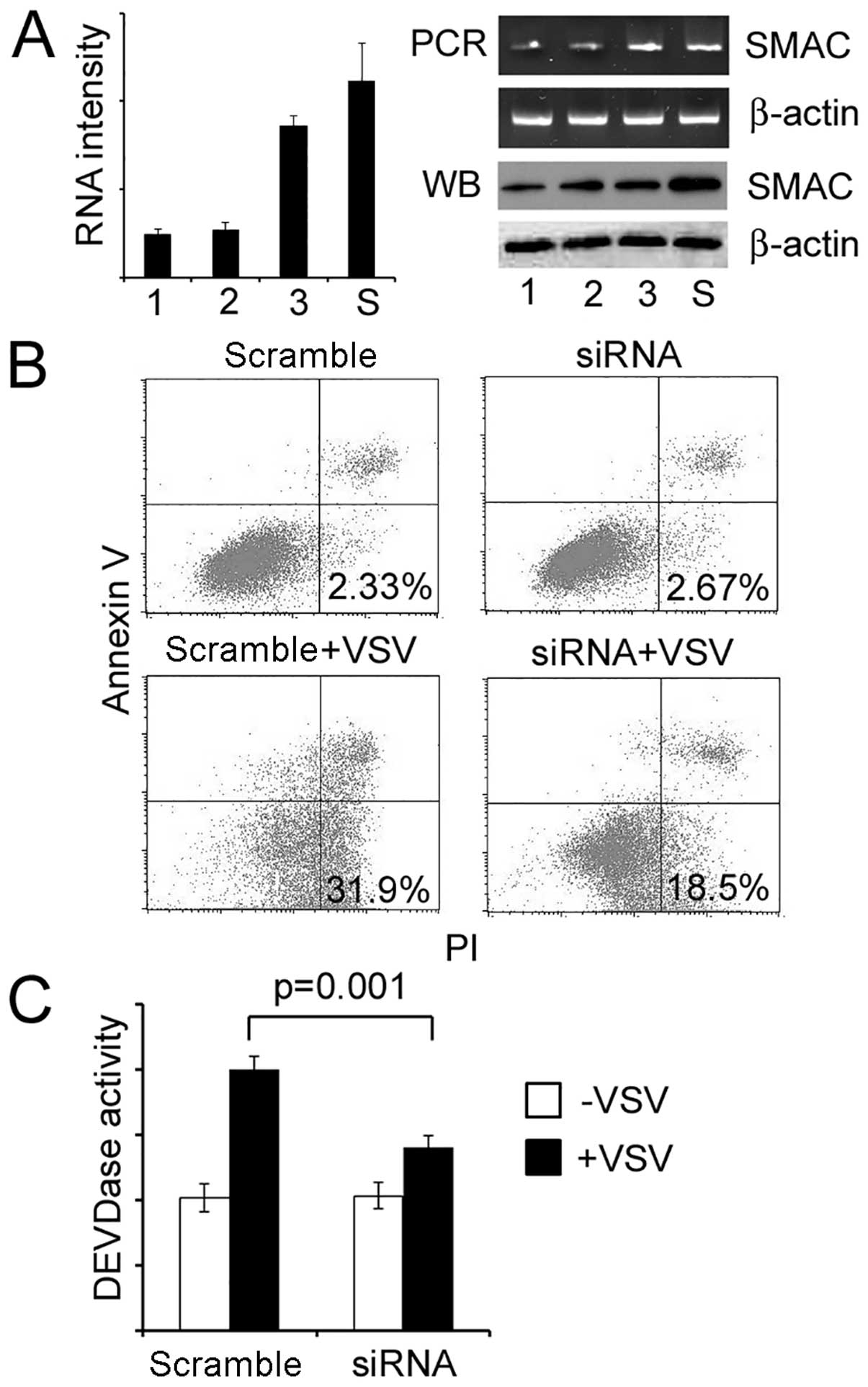
VSV treatment. SMAC was efficiently knocked down with a selected
siRNA sequence (Fig. 6A), and the
cells became resistant to VSV challenge (Fig. 6B and C). This observation was alike
to that of the overexpression of Livin and suggested a possible
relationship between SMAC and Livin.
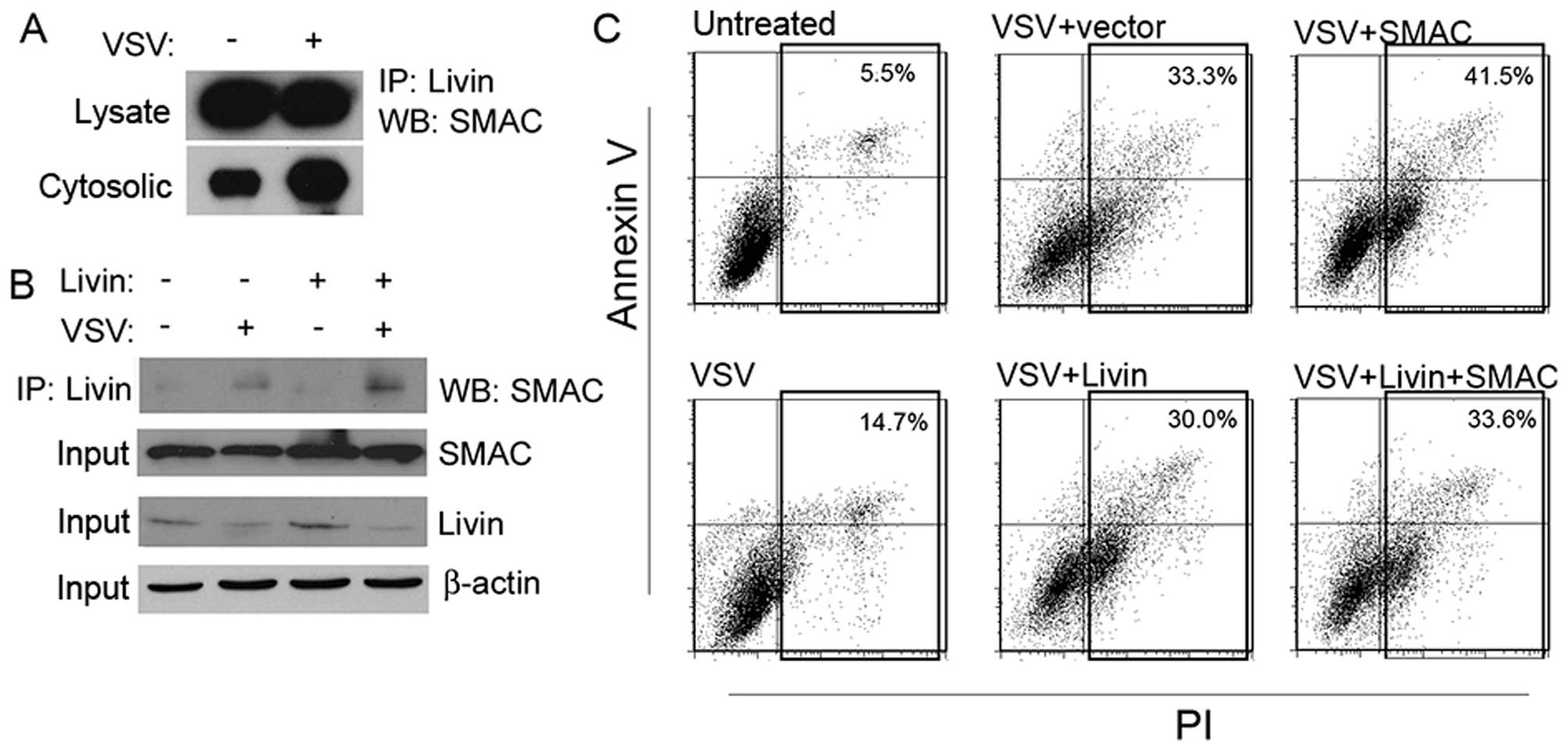
To further confirm that Livin was associated with
SMAC, we performed a co-IP experiment. Livin bound to SMAC in both
the whole-cell lysates and the cytosolic fraction (Fig. 7A). Overexpression of Livin led to a
lager amount of SMAC bound in the cytosolic fraction, but not in
the whole-cell lysates (Fig. 7B).
We next performed a co-transfection. Overexpression of Livin made
cells resistant to VSV, as showed before. However, the
simultaneously overexpressed SMAC compromised its anti-apoptotic
effects (Fig. 7C).
Knock-down of Livin make cells vulnerable
to VSV
Three pairs of candidate siRNA sequences were
screened for the knockdown of endogenous Livin. The most efficient
sequence 1 was selected based on results from both real-time PCR
and western blot analysis (Fig.
8A) for further experiments. Knock-down of Livin made cells
more sensitive to VSV treatment (Fig.
8B) with elevated caspase-3 activity (Fig. 8C). These results indicated Livin as
critical for the resistance of VSV.
Discussion
VSV was proposed as a promising treatment modality
for cancer (3–9). To fully realize its potential in
cancer treatment, a deeper insight of its mechanisms is highly
required. Previous studies focused on the IFN pathway as an
explanation for its selective tumoricidal effects (11). Evasion of apoptosis is considered a
hallmark of cancer cells (23),
and overcoming the evasion is critical for cancer treatment
(24). We believe the apoptosis
pathway must play a role in mediating the therapeutic effects of
VSV.
Our proposal is supported by previous reports
(25–27). In one study, a small molecular
BCL-2 inhibitor significantly sensitized tumor cells to VSV
(28). This study highlighted that
endogenous apoptosis inhibitors were associated with VSV
sensitivity. The apoptosis pathway is negatively regulated by
endogenous factors such as the Bcl-2 or IAP family (29). However, few studies have been
conducted to test the contribution of IAP to the apoptosis by
VSV.
Previously, we reported VSV alone achieved mild
effects on A549 xenogeneic transplant tumor model (6). Other groups later reported similar
results. One study reported that compared with hepatocellular
carcinoma Hepa-G2 cells and hepatoma Huh-7 cells, the VSV induced
less apoptosis rates in A549 cells at the same MOI (30). These studies indicated lung
adenocarcinoma was resistant to VSV treatment. The present study
determined the expression of Livin in A549 cells to assess whether
the expression of the anti-apoptotic factor Livin would contribute
to the resistance to VSV in this cell line.
In our experiment, we found the Livin expression was
associated with VSV infection. This suggested Livin might be
involved in the VSV-induced apoptosis. As anticipated, the
overexpression of Livin made cells resistant to VSV treatment.
Finally, we further confirmed its role in the VSV treatment by
knock-down of Livin. This emphasized Livin was important for A549
cells to resist VSV treatment. Our study shows that the members of
the IAP family exert impact on the efficacy of VSV treatment.
There are controversies regarding the mechanism by
which Livin inhibits apoptosis. Initially it was thought Livin
could bind and suppress the downstream effector caspase-3, or −9,
which was a canonical mechanism used by other IAPs such as XIAP
(13). However, later experiments
failed to prove this notion. Vucic et al reported Livin only
weakly inhibited caspase-9 activity with inhibition constant Ki
~3–5 μM. Also, it was argued that Livin might regulate apoptosis by
sequestering SMAC from XIAP (31).
In our study, we found Livin overexpression led to resistance to
VSV together with reduced caspase-3 activity. We wanted to dissect
the relationship between Livin and SMAC in the condition of VSV
treatment. We found the knock-down of SMAC achieved similar effects
with those of Livin overexpression. The co-IP also confirmed the
binding of Livin to SMAC in the cytosol. The simultaneous
transfection of SMAC abrogated the effects of Livin expression.
These results strongly argued Livin inhibited VSV-induced apoptosis
by binding and inhibiting SMAC.
We detected the binding of Livin to SMAC by the
co-IP experiment. SMAC was sequestered from XIAP by the membrane of
mitochondria in resting state and released into the cytosol upon
apoptosis simuli (32). In our
study the Livin-bound SMAC increased in the cytosol but not the
whole-cell lysates. We also found the expression and release of
SMAC remained consistent before and after VSV treatment (data not
shown). Thus, we reasoned the elevated cytosolic SMAC by the VSV
was binded with Livin and lost its ability to neutralize XIAP. This
might be the mechanistic explanation for the reduced apoptosis by
Livin. In the whole-cell lysates, the amount of SMAC remained
unchanged before and after Livin overexpression.
In conclusion, our study explored the relationship
between SMAC and Livin and the impact of this relationship on the
VSV sensitivity in lung adenocarcinoma cells. Our results suggested
the important role of Livin and its partner molecule in the process
of VSV treatment. Our study helps to deepen our knowledge of the
molecular events after VSV transfection and improve the therapeutic
effects of VSV.
Acknowledgements
This study was supported by National Natural Science
Foundation of China (nos. 82172684 and 81200640).
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Reck M, Heigener DF, Mok T, Soria JC and
Rabe KF: Management of non-small-cell lung cancer: Recent
developments. Lancet. 382:709–719. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stojdl DF, Lichty B, Knowles S, Marius R,
Atkins H, Sonenberg N and Bell JC: Exploiting tumor-specific
defects in the interferon pathway with a previously unknown
oncolytic virus. Nat Med. 6:821–825. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Balachandran S, Porosnicu M and Barber GN:
Oncolytic activity of vesicular stomatitis virus is effective
against tumors exhibiting aberrant p53, Ras, or myc function and
involves the induction of apoptosis. J Virol. 75:3474–3479. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ebert O, Shinozaki K, Huang TG, Savontaus
MJ, García-Sastre A and Woo SL: Oncolytic vesicular stomatitis
virus for treatment of orthotopic hepatocellular carcinoma in
immune-competent rats. Cancer Res. 63:3605–3611. 2003.PubMed/NCBI
|
|
6
|
Li Q, Wei YQ, Wen YJ, Zhao X, Tian L, Yang
L, Mao YQ, Kan B, Wu Y, Ding ZY, et al: Induction of apoptosis and
tumor regression by vesicular stomatitis virus in the presence of
gemcitabine in lung cancer. Int J Cancer. 112:143–149. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lin X, Chen X, Wei Y, Zhao J, Fan L, Wen
Y, Wu H and Zhao X: Efficient inhibition of intraperitoneal human
ovarian cancer growth and prolonged survival by gene transfer of
vesicular stomatitis virus matrix protein in nude mice. Gynecol
Oncol. 104:540–546. 2007. View Article : Google Scholar
|
|
8
|
Du XB, Lang JY, Xu JR, Lu Y, Wen YJ, Zhao
JM, Diao P, Yuan ZP, Yao B, Fan LY, et al: Vesicular stomatitis
virus matrix protein gene enhances the antitumor effects of
radiation via induction of apoptosis. Apoptosis. 13:1205–1214.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Stewart JH IV, Ahmed M, Northrup SA,
Willingham M and Lyles DS: Vesicular stomatitis virus as a
treatment for colorectal cancer. Cancer Gene Ther. 18:837–849.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wong LH, Krauer KG, Hatzinisiriou I,
Estcourt MJ, Hersey P, Tam ND, Edmondson S, Devenish RJ and Ralph
SJ: Interferon-resistant human melanoma cells are deficient in
ISGF3 components, STAT1, STAT2, and p48-ISGF3gamma. J Biol Chem.
272:28779–28785. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Moussavi M, Fazli L, Tearle H, Guo Y, Cox
M, Bell J, Ong C, Jia W and Rennie PS: Oncolysis of prostate
cancers induced by vesicular stomatitis virus in PTEN knockout
mice. Cancer Res. 70:1367–1376. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gyrd-Hansen M and Meier P: IAPs: From
caspase inhibitors to modulators of NF-kappaB, inflammation and
cancer. Nat Rev Cancer. 10:561–574. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vucic D, Stennicke HR, Pisabarro MT,
Salvesen GS and Dixit VM: ML-IAP, a novel inhibitor of apoptosis
that is preferentially expressed in human melanomas. Curr Biol.
10:1359–1366. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lin JH, Deng G, Huang Q and Morser J:
KIAP, a novel member of the inhibitor of apoptosis protein family.
Biochem Biophys Res Commun. 279:820–831. 2000. View Article : Google Scholar
|
|
15
|
Ashhab Y, Alian A, Polliack A, Panet A and
Ben Yehuda D: Two splicing variants of a new inhibitor of apoptosis
gene with different biological properties and tissue distribution
pattern. FEBS Lett. 495:56–60. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tanabe H, Yagihashi A, Tsuji N, Shijubo Y,
Abe S and Watanabe N: Expression of survivin mRNA and livin mRNA in
non-small-cell lung cancer. Lung Cancer. 46:299–304. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gazzaniga P, Gradilone A, Giuliani L,
Gandini O, Silvestri I, Nofroni I, Saccani G, Frati L and Aglianò
AM: Expression and prognostic significance of LIVIN, SURVIVIN and
other apoptosis-related genes in the progression of superficial
bladder cancer. Ann Oncol. 14:85–90. 2003. View Article : Google Scholar
|
|
18
|
Choi J, Hwang YK, Sung KW, Lee SH, Yoo KH,
Jung HL, Koo HH, Kim HJ, Kang HJ, Shin HY, et al: Expression of
Livin, an antiapoptotic protein, is an independent favorable
prognostic factor in childhood acute lymphoblastic leukemia. Blood.
109:471–477. 2007. View Article : Google Scholar
|
|
19
|
Xiang Y, Yao H, Wang S, Hong M, He J, Cao
S, Min H, Song E and Guo X: Prognostic value of Survivin and Livin
in nasopharyngeal carcinoma. Laryngoscope. 116:126–130. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gong J, Chen N, Zhou Q, Yang B, Wang Y and
Wang X: Melanoma inhibitor of apoptosis protein is expressed
differentially in melanoma and melanocytic naevus, but similarly in
primary and metastatic melanomas. J Clin Pathol. 58:1081–1085.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ding ZY, Liu GH, Olsson B and Sun XF:
Upregulation of the antiapoptotic factor Livin contributes to
cisplatin resistance in colon cancer cells. Tumour Biol.
34:683–693. 2013. View Article : Google Scholar
|
|
22
|
Liu GH, Wang C and Ding ZY: Overexpression
of the truncated form of Livin reveals a complex interaction with
caspase-3. Int J Oncol. 42:2037–2045. 2013.PubMed/NCBI
|
|
23
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Luo J, Solimini NL and Elledge SJ:
Principles of cancer therapy: Oncogene and non-oncogene addiction.
Cell. 136:823–837. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kopecky SA and Lyles DS: Contrasting
effects of matrix protein on apoptosis in HeLa and BHK cells
infected with vesicular stomatitis virus are due to inhibition of
host gene expression. J Virol. 77:4658–4669. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gadaleta P, Perfetti X, Mersich S and
Coulombié F: Early activation of the mitochondrial apoptotic
pathway in Vesicular Stomatitis virus-infected cells. Virus Res.
109:65–69. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gaddy DF and Lyles DS: Oncolytic vesicular
stomatitis virus induces apoptosis via signaling through PKR, Fas,
and Daxx. J Virol. 81:2792–2804. 2007. View Article : Google Scholar :
|
|
28
|
Tumilasci VF, Olière S, Nguyên TL, Shamy
A, Bell J and Hiscott J: Targeting the apoptotic pathway with BCL-2
inhibitors sensitizes primary chronic lymphocytic leukemia cells to
vesicular stomatitis virus-induced oncolysis. J Virol.
82:8487–8499. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Igney FH and Krammer PH: Death and
anti-death: Tumour resistance to apoptosis. Nat Rev Cancer.
2:277–288. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Schache P, Gürlevik E, Strüver N, Woller
N, Malek N, Zender L, Manns M, Wirth T, Kühnel F and Kubicka S: VSV
virotherapy improves chemotherapy by triggering apoptosis due to
proteasomal degradation of Mcl-1. Gene Ther. 16:849–861. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Vucic D, Franklin MC, Wallweber HJ, Das K,
Eckelman BP, Shin H, Elliott LO, Kadkhodayan S, Deshayes K,
Salvesen GS, et al: Engineering ML-IAP to produce an
extraordinarily potent caspase 9 inhibitor: Implications for
Smac-dependent anti-apoptotic activity of ML-IAP. Biochem J.
385:11–20. 2005. View Article : Google Scholar :
|
|
32
|
Wu H, Tschopp J and Lin SC: Smac mimetics
and TNFalpha: A dangerous liaison? Cell. 131:655–658. 2007.
View Article : Google Scholar : PubMed/NCBI
|