Introduction
Acute promyelocytic leukemia (APL) is characterized
by specific chromosomal translocations, typically t(15;17), which
results in the formation of the promyelocytic leukemia-retinoic
acid receptor-α (PML-RARα) fusion gene (1,2).
PML-RARα fusion protein forms homo/heterodimers that sequestrate
RXR and/or PML proteins in a large protein complex and disrupt the
retinoic acid (RA) signal pathway. This specific oncogenic lesion
determines characteristic cell morphology and clinical
presentations, and it also determines the unique response to the
treatment with all-trans retinoic acid (ATRA) or arsenic
agents (3,4). Both drugs have been demonstrated to
target the PML/RARα oncoprotein for proteasome-mediated
degradation. Clinically, ATRA induces complete remissions in ~90%
of newly diagnosed APL, but many patients eventually experience a
relapse and develop ATRA-resistance (5,6).
Arsenic trioxide is also shown to be effective in the treatment of
APL, especially in relapsed APL with ATRA-resistance (7,8).
Arsenic trioxide has dual effects of inducing
differentiation and apoptosis of APL cells. However, there are
issues of availability and cost of arsenic trioxide that limit its
general applications. The development of oral form of arsenic drug
may promote its applications in APL. Arsenic sulfide
(As4S4), also known as realgar, is an oral
arsenic formulation. This oral arsenic drug has been shown to have
similar effect and safety to intravenous arsenic trioxide in the
treatment of newly diagnosed and relapsed/refractory APL or
ATRA-resistance (9). The
therapeutic action of As4S4 is closely
associated with its function of inducing apoptosis. Although it is
known that As4S4 induces cell apoptosis
through degrading PML-RARα fusion protein (10), the definitive molecular mechanisms
of action of As4S4 remain unclear and require
further investigations.
In the present study, we used a comparative
proteomic approach to screen and identify proteins that are
differentially expressed in APL cells induced by
As4S4. By using two-dimensional gel
electrophoresis (2-DE) followed by a matrix-assisted laser
desorption/ionization-time-of-flight mass spectrometry (MALDI-TOF
MS) analysis, we identified prohibitin (PHB) among the
differentially expressed proteins. PHB was significantly
upregulated in ATRA-resistance APL cells (NB4-R1) by
As4S4 treatment. Further studies of
PHB-knockdown and PHB-overexpression indicate a functional role of
PHB in As4S4-induced apoptosis of NB4-R1
cells.
Materials and methods
Cell culture
The ATRA-resistance human APL cell line (NB4-R1),
received from Shanghai Institute of Hematology, (Shanghai, China)
was maintained in cultures with RPMI-1640 medium (Gibco-BRL,
Carlsbad, CA, USA) supplemented with 10% heated-inactivated fetal
bovine serum (FBS) at 37ºC in a humidified incubator containing 5%
CO2.
Cell viability assay
Cytotoxicity of As4S4 (Xi'an
Traditional Chinese Drug Company, Xi'an, China) was assessed by
using MTT assay (Sigma, St. Louis, MO, USA) (11). The absorbance was measured at 570
nm using a universal microplate reader (Model ELx800; BioTek
Instruments, Inc., Winooski, VT, USA). Experiments were performed
in triplicate.
Apoptosis evaluation
Transmission electron microscopy (TEM) and flow
cytometric analysis (FCM) were performed to evaluate cell
apoptosis. After the various treatments, the cell samples were
examined under a JEM-100SX electron microscope (JEOL, Ltd., Tokyo,
Japan) and were analyzed in a FACSCalibur flow cytometer
(Becton-Dickinson, San Jose, CA, USA) and CellQuest software,
respectively. All experiments were performed in triplicate.
2-DE and image analysis
Total cellular proteins were prepared from NB4-R1
cells before and after As4S4 treatment.
Protein extraction was performed by sonication in a sample buffer
(SB) containing 40 mM Tris base, 8 M urea, 2 M thiourea, 4% (w/v)
CHAPS, 1% (w/v) dithiothreitol (DTT), 1 mM EDTA and protease
inhibitor cocktail (Roche Diagnostics Ltd., Mannheim, Germany). For
nuclei enrichment cells were dissolved in 200 μl of lysis buffer
[10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, in
the presence of protease inhibitor cocktail (Sigma), 20 ng/μl DNase
and 20 ng/μl RNase] and incubated on ice for 30 min. After
incubation, NP-40 (Roche) was added at final concentration of 0.5%
(v/v). After centrifugation at 14,000 rpm for 30 min at 4ºC, the
supernatant was used for analysis with the protein concentration
determined by the Bradford method with a commercial Bradford
reagent (Bio-Rad Laboratories, Hercules, CA, USA) (12).
2-DE was performed as described by Görg et al
(13). Briefly, 140 μg of protein
(for silver nitrate staining gels) or 1.4 mg of protein (for
coomassie brilliant blue staining gels) was diluted to 350 μl with
rehydration solution and applied onto 18 cm (pH 3–10) not linear
immobilized pH gradient dry strip (Amersham Pharmacia Biotech,
Uppsala, Sweden). After the strips were rehydrated, isoelectric
focusing was performed in the IPGphor system (Amersham Pharmacia
Biotech) according to the manufacturer's protocol (14). The strips were equilibrated for 15
min in a solution containing 6 M urea, 2% (w/v) SDS, 20 mM DTT, 30%
(w/v) glycerol and 50 mM Tris-HCl (pH 8.8). A second equilibration
was also carried out for 15 min in the same solution except for DTT
replaced by 100 mM iodoacetamide. The second dimension was
performed on 13% SDS-polyacrylamide gradient gels using the PROTEAN
XI Cell (Bio-Rad Laboratories) at 20 mA/gel for 40 min.
Silver nitrate staining according to the protocol of
Lelong et al (15), and
coomassie brilliant blue R-250 (0.05% brilliant blue) was used for
the analytical and preparative gels. The 2-DE images were acquired
using Image scanner (Amersham Pharmacia Biotech). Gel images were
analyzed by the ImageMaster 2D Platinum software (Amersham
Pharmacia Biotech). Spot detection and normalization were performed
by the automated software tools.
MALDI-TOF MS and MALDI-TOF MS/MS
analysis
Differentially expressed spots were manually excised
from 2-DE gels. Gel pieces were destained and digestion. In-gel
digestion was done according to the protocol of Granvogl et
al (16).
MALDI-TOFMS analysis was performed on a Bruker
REFLEX III MALDI-TOF-MS (Bruker-Franzen, Bremen, Germany). Peptides
were desalted by C18 ZipTips (Millipore, Billerica, MA, USA) and
co-crystallized with a solution of 0.5 mg/ml
α-cyano-4-hydroxycinnamic acid dissolved in acetonitrile/0.1% (v/v)
trifluoroacetic acid (TFA) in H2O (1:1) pre-spotted with
a thin layer of 10 mg/ml α-cyano-4-hydroxycinnamic acid dissolved
in ethanol/acetonitrile/0.1% (v/v) TFA in H2O
(49.5:49.5:1). Monoisotopic peptide masses were used to search the
database, allowing a peptide mass accuracy of 0.3 Da and one
partial cleavage. The proteins were identified by peptide mass
fingerprinting (PMF) searching, against the Swiss-Prot databases
and NCBI databases, using the search program Mascot (http://www.matrixscience.com).
The protein spots which were not identified by
MALDI-TOF-MS were analyzed by MA LDI-TOF MS/MS. MALDI-TOF MS/MS
analysis was performed in LIFT mode. Precursor ions were selected
manually. MS/MS spectra were acquired with a minimum of 4000 and a
maximum of 8000 laser shots using the instrument calibration file.
The precursor mass window was set automatically after the precursor
ion selection. Spectra baseline subtraction, smoothing
(Savitsky-Golay) and centroiding was performed by FlexAnalysis
software (version 3.0; Bruker Daltonik GmbH, Bremen, Germany).
Western blot analysis
Cell protein extracts were prepared following
standard procedures. The protein samples (~20 mg) were separated by
SDS-PAGE. After SDS-PAGE, proteins were transferred to
nitrocellulose membranes (Invitrogen, Carlsbad, CA, USA). The
filters were washed, blocked with 5% bovine serum albumin (BSA) in
Tris-buffered saline (25 mM Tris, pH 7.4, 136 mM NaCl, 2.6 mM KCl
and 0.5% Tween-20) for 1 h, and incubated overnight with mouse
anti-PHB antibody diluted to 1:700 (Abcam, Cambridge, MA, USA) at
room temperature. After washing three times with TBST buffer, the
membranes were incubated with the secondary HRP-conjugated goat
anti-mouse IgG Ab (Santa Cruz Biotechnology, Santa Cruz, CA, USA)
at 1:10,000 dilution. Mouse anti-GAPDH antibody (Santa Cruz
Biotechnology) was used to ensure equal loading of samples.
Quantitative real-time PCR (qRT-PCR)
The total RNA from cells was isolated with TRIzol
(Life Technologies, Rockvile, MD, USA) and reverse-transcribed to
cDNA by using the PrimeScript™ RT reagent kit (Takara Bio, Dalian,
China). The cDNA was studied using a CFX96 real-time PCR system
(Bio-Rad Laboratories) with SYBR-Green PCR Master Mix (Takara) to
determine the transcriptional expression of PHB gene. PCR products
were electrophoresed on 1.5% agarose gels. The GAPDH was used for
normalization, relative gene expression was calculated by the
2−ΔΔCt method.
Knockdown and overexpressing of PHB
Lentiviral vector-mediated shRNA targeting human PHB
mRNA (named pGCSIL-GFP-PHB) was previously described (17). The target sequences on the human
PHB gene (GeneBank accession number NM_002634) for RNAi were
designed using an internet application system as follows:
5′-GAGTTCACAGAAGCGGTGGAA3′. A shRNA which had no significant
homology to any known human gene (5′-TTCTCCGAACGTGTCACGT-3′) was
used as a negative control. Oligonucleotides were ligated into the
AgeI and EcoRI sites of pGCSIL-GFP vector (BD
Biosciences, San Jose, CA, USA) to generate a pGCSIL-GFP-PHB, which
was then transformed into E. coli. Positive recombinant
clones were selected by PCR (upstream primer:
5′-CCTATTTCCCATGATTCCTTCATA-3′; downstream primer:
5′-GTAATACGGTTATCCACGCG-3′) and DNA sequencing. The recombinant
lentivirus vector was produced by co-transfecting 293T cells with
the lentivirus expression plasmid and packaging plasmids (pHelper
1.0 and pHelper 2.0) with Lipofectamine 2000 (Invitrogen).
Infectious lentivirus vector was harvested at 48 h
post-transfection and then concentrated. The infectious titer was
determined by the GFP-tagged positive rate in 293T cells. NB4-R1
cells were cultured at a density of 6×105/well in 6-well
plates and infected with lentivirus in RPMI-1640 media containing
10% FBS and 8 μg/ml of polybrene (Sigma), at the multiplicity of
infection (MOI) 20, according to the pre-experimental results.
After 48 h of culture, the transduction efficiency was ascertained
on the basis of GFP expression under a fluorescence microscope. The
knockdown efficiency of PHB was analyzed by real-time quantitative
PCR and western blot analysis. NB4-R1 cells transfected with vector
containing pGCSIL-GFP-PHB were designated as PHB-knockdown
(KD).
The PHB gene overexpression vector (named
pEGFP-N1-3FLAG-PHB) was also established. Briefly, the cDNA
fragment of PHB was amplified using a PCR-based approach (upstream
primer: 5′-CCGCTCGAGATGGCTGCCAAAGT GTTTG; downstream primer:
5′-GGGGTACCGTCTGGGG CAGCTGGAGGAG) from a cDNA library. The PCR
fragment of confirmed sequences was ligated into the XhoI
and KpnI sites of overexpression vector pEGFP-N1-3FLAG (BD
Biosciences). The resultant construct, pEGFP-N1-3FLAG-PHB, was
transformed into E. coli. Positive recombinant clones were
selected by PCR and DNA sequencing (upstream primer:
5′-CGCAAATGGGCGGTAGGCGTG-3′; downstream primer:
5′-CGTCGCCGTCCAGCTCGACCAG-3′). The expression of PHB was analyzed
by real-time quantitative PCR and western blot analysis. The NB4-R1
cell clone transfected with the vector containing
pEGFP-N1-3FLAG-PHB were designated as PHB-overexpression (OE).
Statistical analysis
The results are expressed as mean ± standard
deviation values of three experiments performed in duplicate.
Statistical analysis was carried out by one-way analysis of
variance. Newman-Keuls test was used for the identification of
statistically significant differences in spot volume percentage
among different samples. Differences were considered statistically
significant when P<0.05.
Results
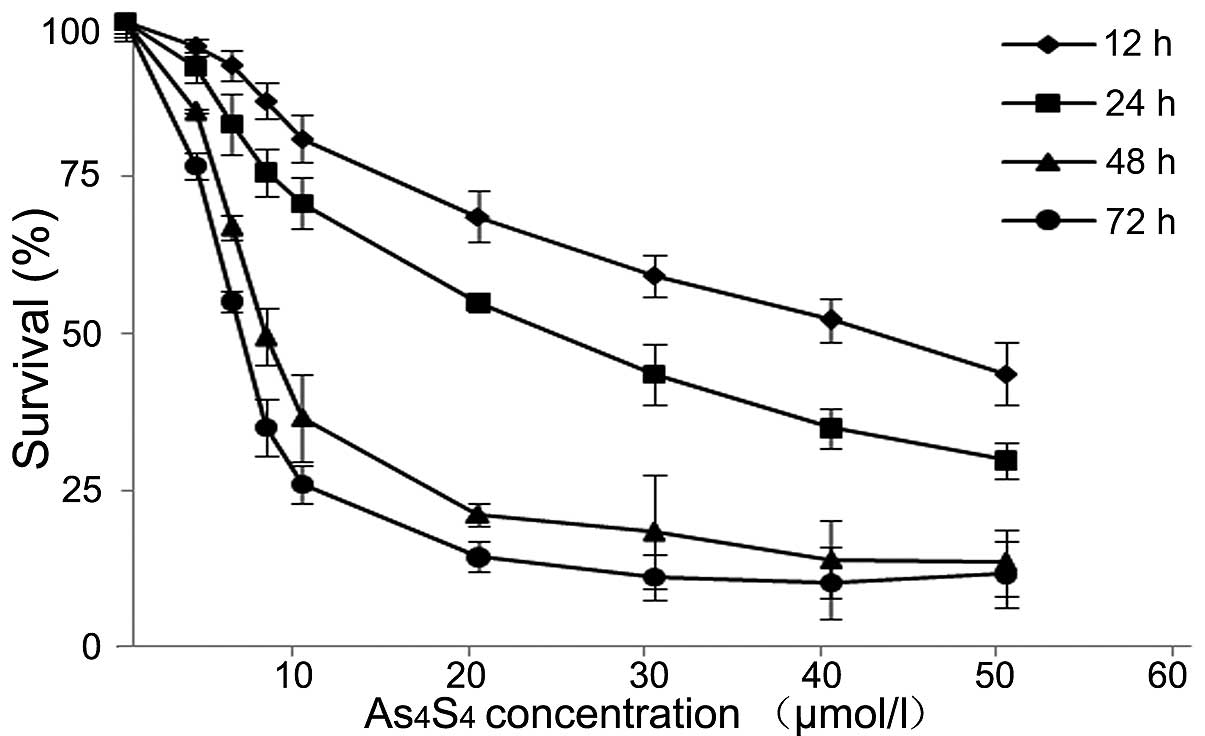
As4S4 inhibits the
growth of ATRA-resistant NB4-R1 cells
We started with MTT assay to evaluate the
cytotoxicity of As4S4 on ATRA-resistant
NB4-R1 cells. The results demonstrated that
As4S4 inhibited the growth of NB4-R1 cells in
a dose-and time-dependent manner (Fig.
1). The IC50 values of As4S4
were determined at 43.04±0.11 μM for 12 h, 25.07±0.27 μM for 24 h,
9.70±0.13 μM for 48 h and 6.38±0.09 μM for 72 h in culture. The
concentration of 25 μM, the IC50 of
As4S4 at 24 h, was chosen for subsequent
experiments.
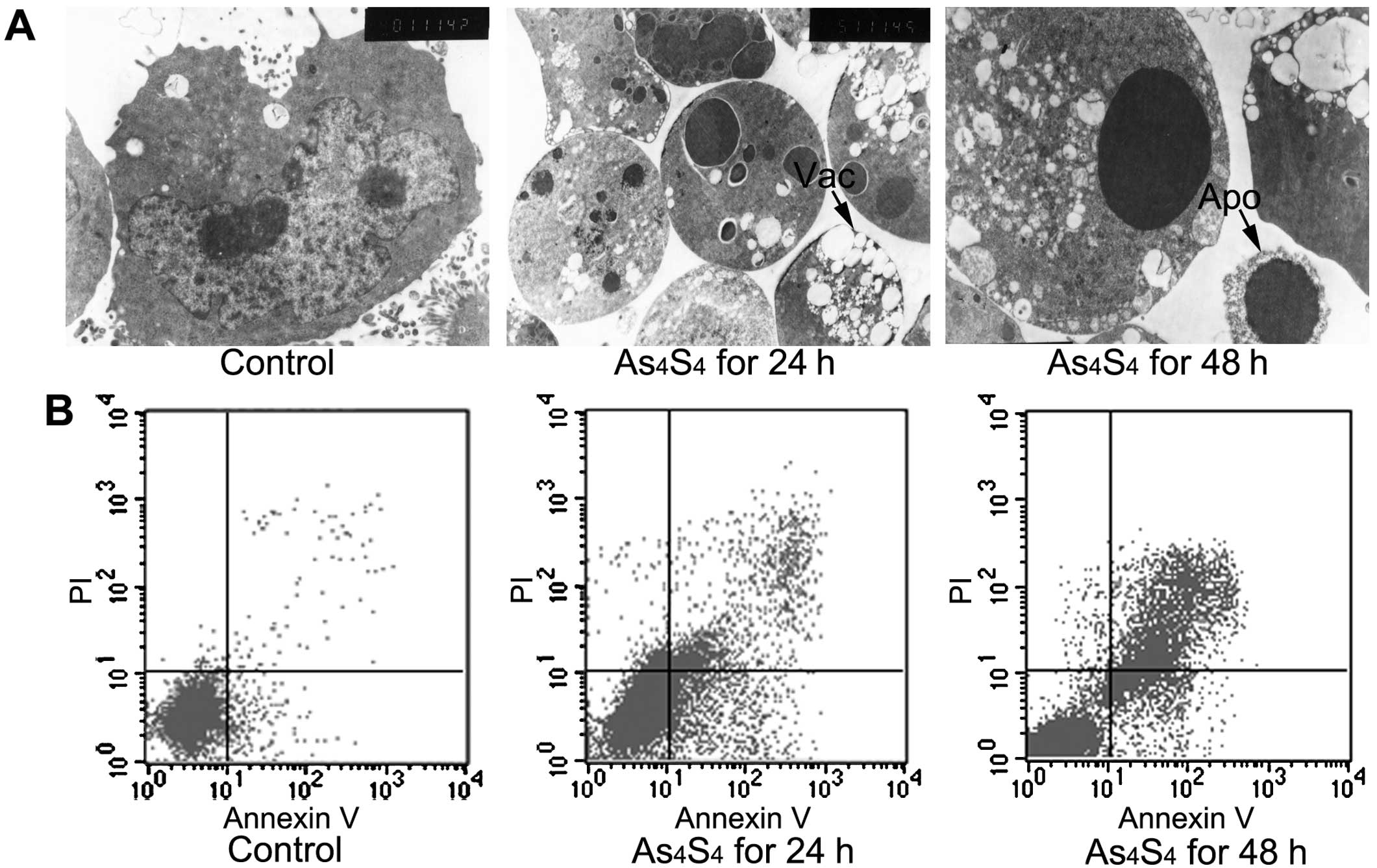
As4S4 induces
apoptosis of NB4-R1 cells
As4S4-induced apoptosis was
assessed by using TEM and FCM analysis. The NB4-R1 cells treated
with As4S4 showed morphological features of
cytoplasmic vacuolization, chromatin condensation, nuclear
fragmentation and formation of apoptotic bodies (Fig. 2A). The apoptotic cells were
quantified by FCM assay for Annexin V+ cells. The
percentage of apoptotic cells was significantly increased with
As4S4 treatment for 24 and 48 h (Fig. 2B).
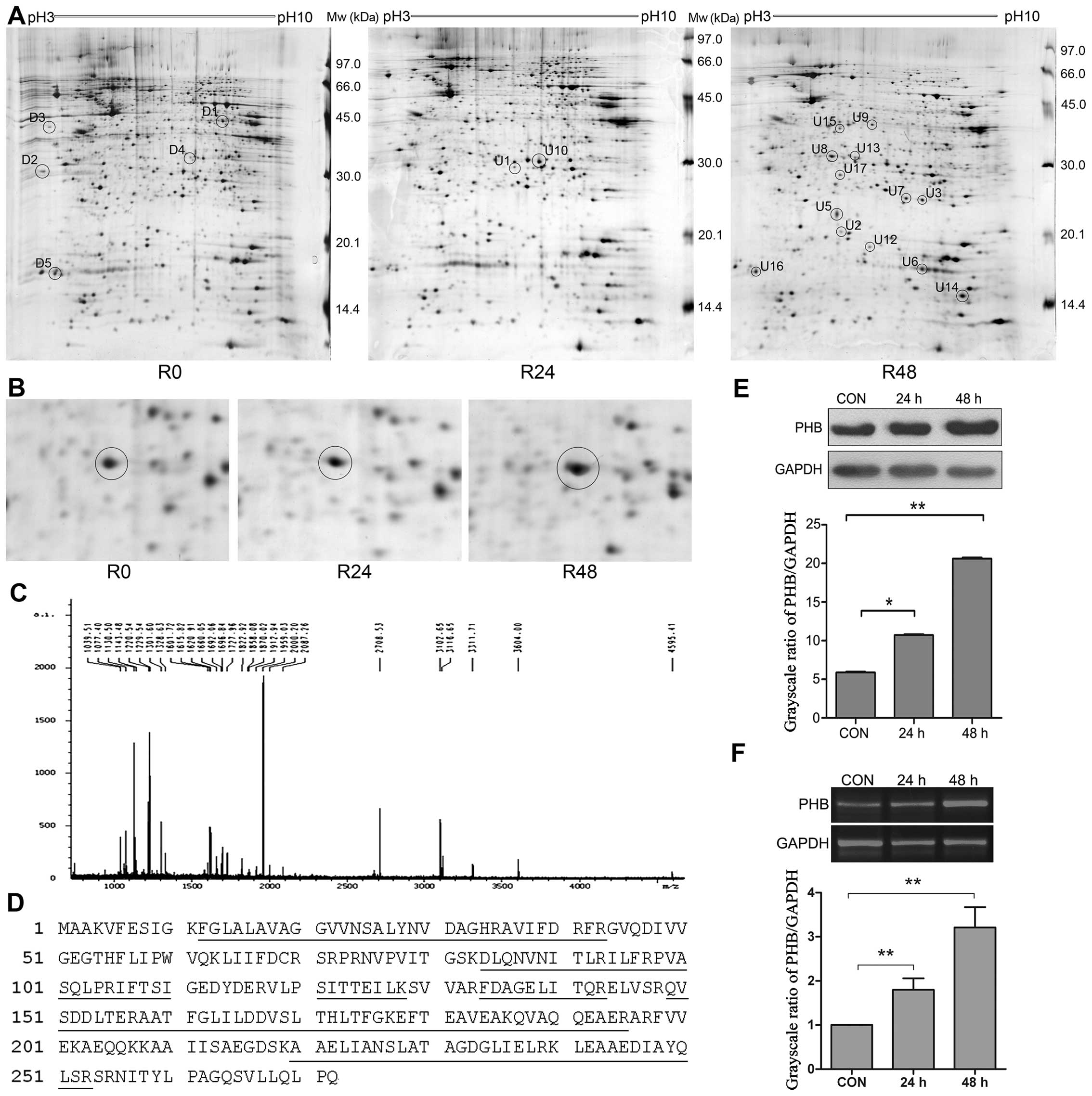
PHB is an upregulated protein induced by
As4S4
We next used proteomic approaches to screen and
identify proteins that were differentially expressed following
As4S4 treatment. The comparison of 2-DE
protein profiles of NB4-R1 cells at 0 h with that at 24 and 48 h
As4S4 treatment were performed, and 22
protein spots with at least a 2-fold increase or decrease in
density were selected for further analysis (Fig. 3A and B).
These spots were cut out, followed by in-gel trypsin
digestion and MALDI-TOF MS analysis. The protein spots which were
not identified by MALDI-TOF-MS were further analyzed by MALDI-TOF
MS/MS. PMF and peptide amino acid sequence were analyzed for
protein identification using the Mascot search program. Fig. 3C showed the PMF of spot U8 analyzed
by MALDI-TOF-MS. spot U8 was identified as prohibitin (PHB) and the
corresponding protein sequence is shown in Fig. 3D. The annotation of the 22
identified proteins is shown in Table
I.
 | Table IIdentification of differentially
expressed protein spots by MALDI-TOF-MS and MALDI-TOF-MS/MS. |
Table I
Identification of differentially
expressed protein spots by MALDI-TOF-MS and MALDI-TOF-MS/MS.
| Spot | Protein name | NCBInr ID no. | Function
classification | Mr (Da) | pI | Peptides
(MALDI/MS) | Sequence coverage
(%) | Protein
expressionb R24/R48 |
|---|
|
|
|
|---|
| Theor. | Observ. | Theor. | Observ. | Match | Total |
|---|
| D1 | Poly C binding
protein 1 (PCBP1) | gi|6754994 | Regulates gene
expression | 37474 | 43062 | 6.66 | 7.83 | 17 | 28 | 52 | 0.57/0.19 |
| D2 | Acidic leucine-rich
nuclear phosphoprotein 32 family member A (ANP32A) | gi|5453880 | Cell proliferation,
differentiation, apoptosis | 28568 | 30123 | 3.99 | 3.88 | 8 | 14 | 31 | 0.70/0.42 |
| D3a | SET/protein
phosphatase 2A inhibitor (SET/I2PP2A) | gi|170763500 | Multitasking
protein | 33469 | 41249 | 4.23 | 4.01 | 7 | 13 | 27 | 0.34/0.10 |
| D4 | Eukaryotic
translation initiation factor 4H isoform 1 (eIF4H-1) | gi|11559923 | Protein
synthesis | 27368 | 32661 | 6.67 | 7.16 | 14 | 29 | 48 | 0.64/0.20 |
| D5 | 60S acidic
ribosomal protein P2 (RPP2) | gi|4506671 | Protein
synthesis | 11658 | 16831 | 4.42 | 4.13 | 7 | 20 | 77 | 0.40/0.30 |
| U1 | High mobility group
protein B1 (HMGB1) | gi|4504425 | Signal
transduction | 24878 | 29744 | 5.62 | 6.88 | 11 | 20 | 48 | 4.58/2.95 |
| U2 | Transgelin-2
(TAGLN2) | gi|4507357 | Not be
determined | 22377 | 20417 | 8.41 | 5.58 | 15 | 19 | 56 | 2.50/6.07 |
| U3 | Eukaryotic
translation initiation factor5A) (eIF5A-1 | gi|183448388 | Protein synthesis,
cellular growth, differentiation and proliferation | 16821 | 16949 | 5.08 | 7.37 | 9 | 31 | 52 | 2.46/10.14 |
| U4 | Transcription
factor(TF) | gi|388307 | Transcription | 20700 | 22567 | 6.28 | 5.49 | 2 | 43 | 12 | 6.18/19.98 |
| U5 | α-tubulin | gi|37492 | Cellular motility
and transportation | 50126 | 22567 | 5.02 | 5.49 | 3 | 26 | 9 | 6.18/19.98 |
| U6 | Histone H2B type
1-M (H2B1M) | gi|4504263 | Transcription, DNA
repair | 13981 | 16949 | 10.31 | 7.37 | 12 | 31 | 67 | 2.12/15.87 |
| U7 | Rho GDP
dissociation inhibitor β 2 (RhoGDI2) | gi|56676393 | Signal transduction
and regulates Rho GTPases | 22974 | 24685 | 5.10 | 7.01 | 8 | 33 | 54 | 5.31/16.83 |
| U8 | Prohibitin
(PHB) | gi|4505773 | Cell proliferation,
tumor suppressor | 29786 | 31560 | 5.57 | 5.37 | 13 | 14 | 61 | 2.18/3.68 |
| U9 | Ribosomal
phosphoprotein P0 (RPP0) | gi|4506667 | Protein synthesis
and apoptosis | 34252 | 39054 | 5.71 | 6.27 | 14 | 19 | 46 | 16.16/22.4 |
| U10 | Heat shock 27 kDa
protein (HSP27) | gi|4504517 | Stress
resistance | 22768 | 28891 | 5.98 | 6.34 | 11 | 18 | 46 | 2.77/1.79 |
| U11 | Elongation factor
1-β (EF-1-β) | gi|18203449 | Protein
synthesis | 24748 | 32071 | 4.50 | 4.38 | 6 | 13 | 37 | 1.53/2.84 |
| U12 | Keratin-2 | gi|47132620 | Proliferation and
keratinization | 65393 | 18903 | 8.07 | 6.21 | 11 | 38 | 25 | 4.23/14.82 |
| U13 | ERP29 | gi|5803013 | Protein
processing | 28975 | 31332 | 6.77 | 5.89 | 12 | 28 | 42 | 1.30/5.06 |
| U14 | β-actin (ACTB) | gi|4501885 | Cellular
motility | 41710 | 14843 | 5.29 | 8.26 | 10 | 20 | 23 | 1.90/13.48 |
| U15 | GTPase-activating
protein | gi|62911375 | Increase GTP
hydrolysis | 23439 | 27647 | 5.21 | 5.25 | 6 | 17 | 30 | 1.70/3.27 |
| U16a | Neuropolypeptide
h3 | gi|913159 | Serine protease
inhibitor | 20913 | 66684 | 7.42 | 5.88 | - | - | 31 | 0.95/3.18 |
| U17 | Proteasome β 4
subunit (PSMB4) | gi|22538467 | Proteolysis | 29185 | 28177 | 5.72 | 5.56 | 13 | 25 | 35 | 1.32/3.42 |
PHB was identified from the spot U8, which was
upregulated induced by As4S4. The increase in
PHB protein was confirmed by western blot analysis. As shown in
Fig. 3E, there was a 2.0- and
3.9-fold increase in PHB protein with As4S4
for 24 and 48 h, respectively. At mRNA level, PHB expression was
increased by 1.8- and 3.2-fold with As4S4 for
24 and 48 h, respectively (Fig.
3F). The results indicate an upregulation of PHB gene
expression at both mRNA and protein levels.
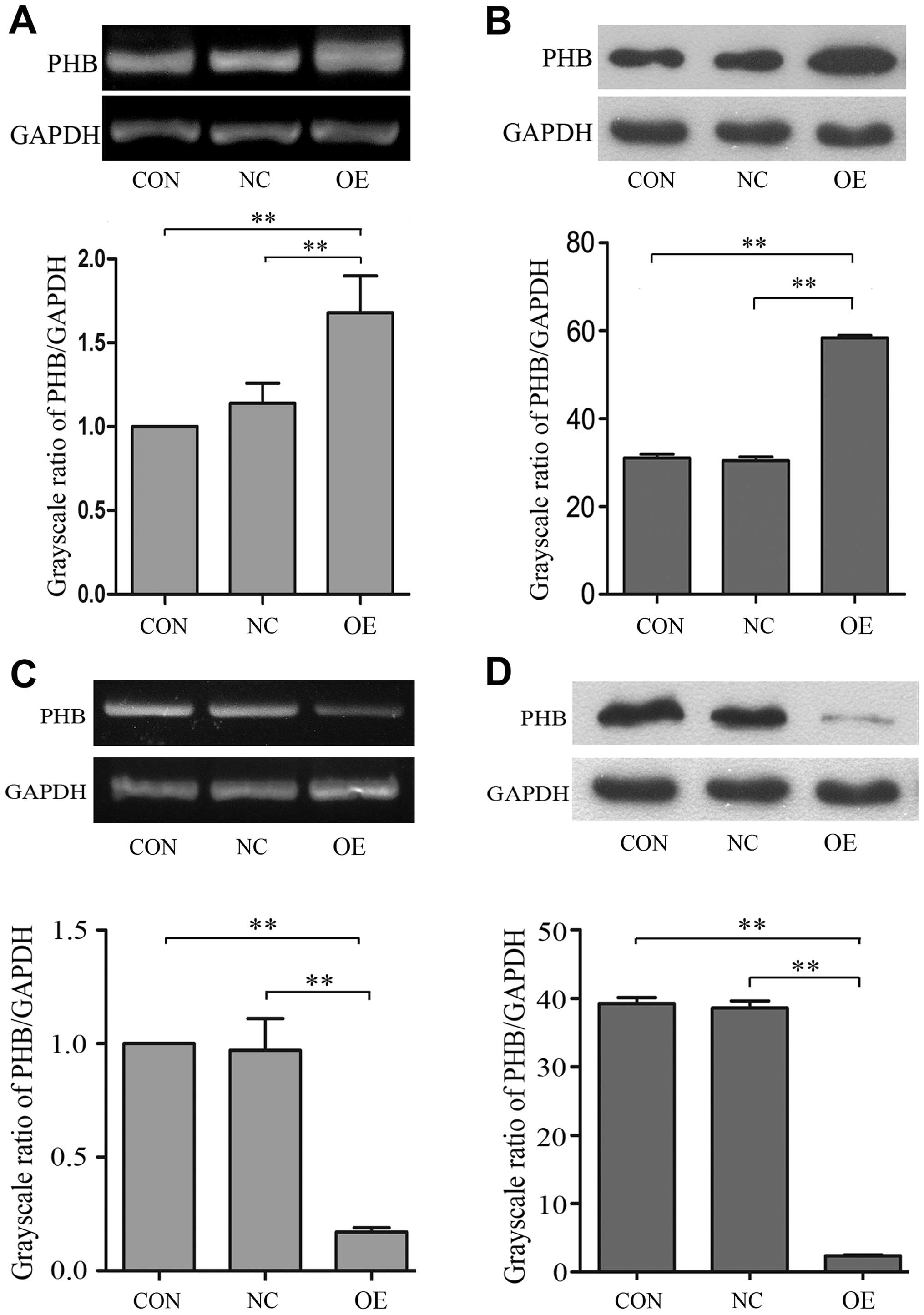
Generation of PHB-overexpression and
PHB-knockdown NB4-R1 cells
To investigate whether PHB plays a functional role
in NB4-R1 cell apoptosis, we used the PHB gene overexpressing
vector (pEGFP-N1-3FLAG-PHB) to generate PHB-overexpression NB4-R1
cells (OE group). The PHB-overexpression efficiency was then
validated by qRT-PCR and western blot analysis, respectively. Our
results showed that PHB expression in OE group was increased by
67.8% at mRNA level and 45.8% at protein level (Fig. 4A and B). Similarly, the RNA
interference vector (pGCSIL-GFP-PHB) of PHB gene was used to
generate PHB-knockdown NB4-R1 cells (KD group). Our results showed
that PHB expression was reduced by 83.5% at mRNA level and 89.7% at
protein level, respectively (Fig. 4C
and D).
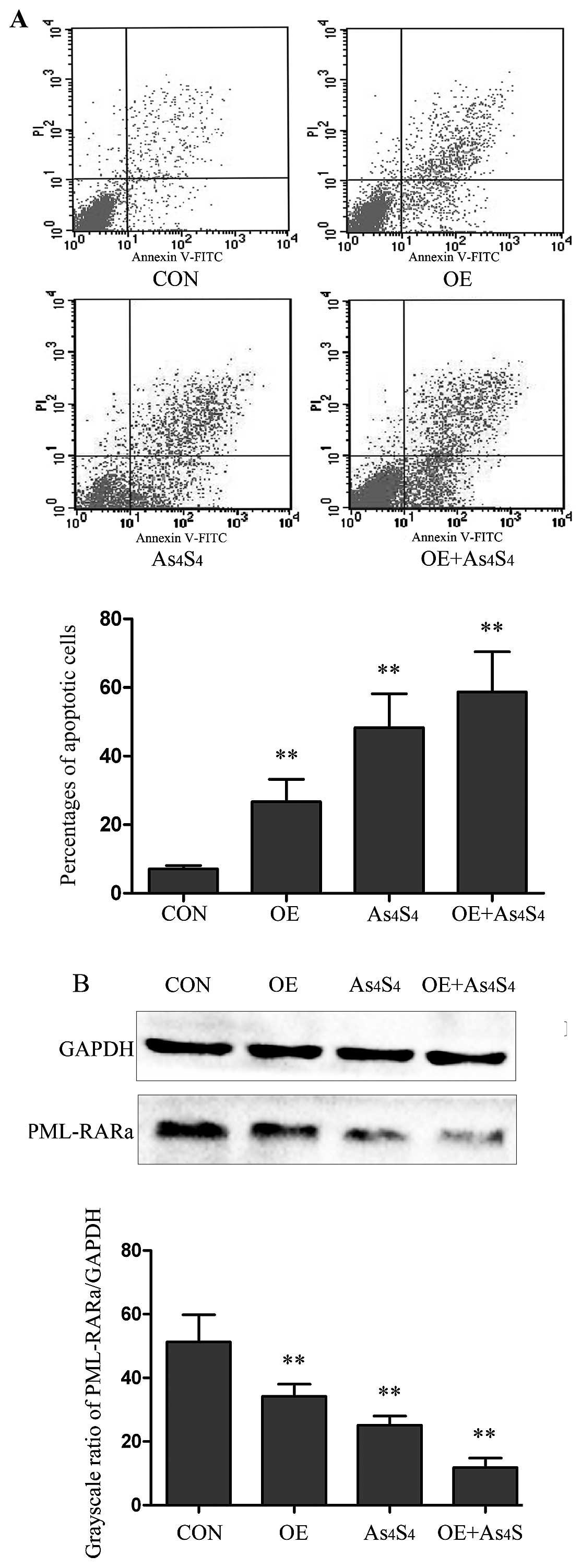
PHB-overexpression promotes NB4-R1
apoptosis and PML-RARα fusion protein degradation
Our results showed after 48 h of transfection, the
percentages of apoptotic cells in OE group was increased by
3.8-fold in comparison with the parental NB4-R1 cells (26.73±6.53
vs. 7.11±1.02%, P<0.01) (Fig.
5A), and the PML-RARα fusion protein was reduced by 1.5-fold in
comparison with the control (34.21±3.81 vs. 51.31±8.55%, P<0.01)
(Fig. 5B).
The response of the OE cells to
As4S4 was evaluated in comparison with
parental NB4-R1 cells. OE cells showed an increase in
As4S4-induced apoptosis. With
As4S4 at the concentration of 25 μM for 48 h,
the apoptotic cells in NB4-R1 and OE cells were 48.33±9.84 and
58.71±11.74%, respectively (Fig.
5A). PML-RARα fusion protein was assessed by western blot
analysis, and the results showed that As4S4
treatment led to greater reduction of PML-RARα protein in OE cells
than that in NB4-R1 cells. In comparison with untreated NB4-R1
cells, As4S4 treatment reduced PML-RARα
protein by 51.0 and 76.9% in NB4-R1 and OE cells, respectively (the
grayscale ratios of PML-RARα/GAPDH: 25.14±2.87 and 11.86±2.99%,
P<0.05) (Fig. 5B).
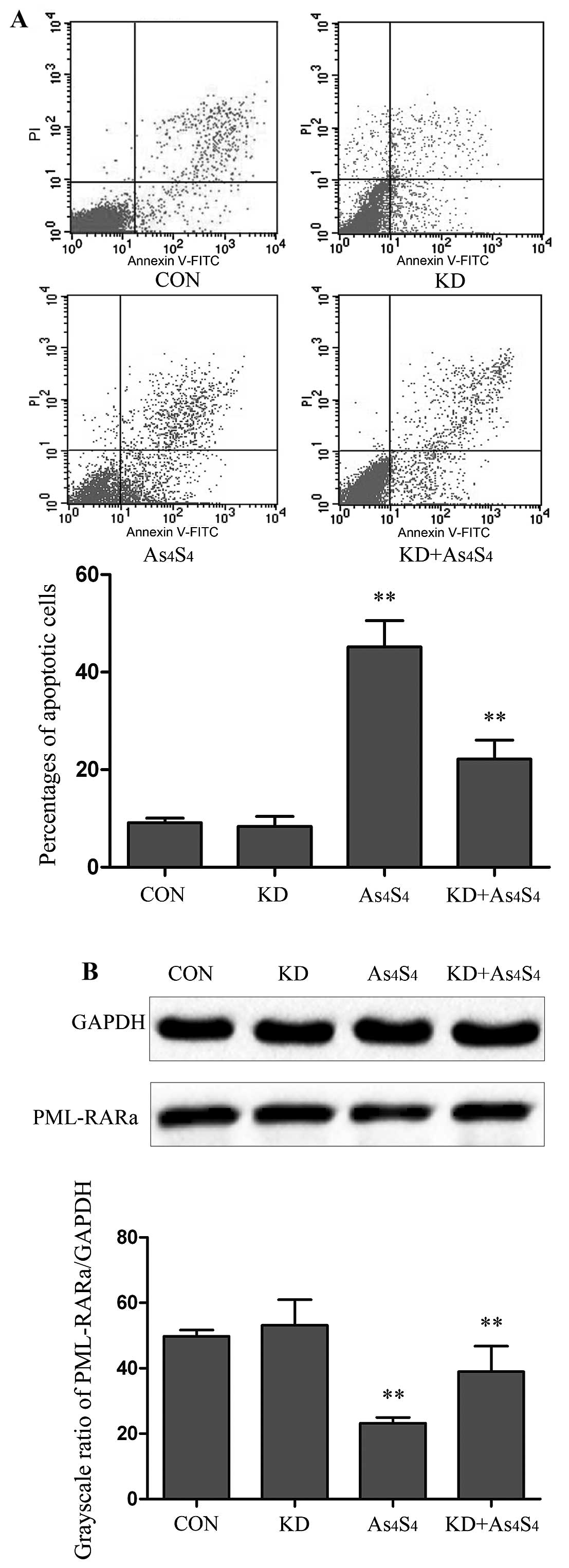
PHB-knockdown reduces
As4S4-induced apoptosis and degradation of
PML-RARα protein
PHB-knockdown NB4-R1 cells (KD) was evaluated in
comparison with parental NB4-R1 cells. With no
As4S4 treatment, there was no significant
difference in the baseline apoptotic cells between KD and NB4-R1
cells. Similarly, no significant difference was seen between KD and
the NB4-R1 in the expression of PML-RARα fusion proteins, as
determined by PML-RARα/GAPDH (53.16±7.83 vs. 49.78±1.89%) (Fig. 6A and B).
The KD cells were then used to examine its response
to As4S4 treatment.
As4S4-induced apoptosis was evaluated with
As4S4 at the concentration of 25 μM for 48 h.
In comparison with parental NB4-R1 cells, the KD showed a lesser
degree of cellular apoptosis. The percentages of apoptotic cells in
NB4-R1 and KD were determined to be 45.17±5.43 and 22.16±3.92%,
respectively (Fig. 6A). Thus,
there was a 2.0-fold less As4S4-induced
apoptosis in KD than that in parental NB4-R1.
PML-RARα fusion protein of KD cells by
western blot analysis
By using the grayscale ratios of PML-RARα/GAPDH, the
levels of PML-RARα protein were determined to be 49.78±1.89% in the
untreated cells, and 24.21±1.73 and 37.95±7.79% in
As4S4-treated NB4-R1 and KD cells,
respectively. Using the untreated cells as the baseline,
As4S4 lowered PML-RARα protein by 51.3 and
23.7% in NB4-R1 and KD cells, respectively (Fig. 6B). The results indicate that KD
cells presented with a lesser degree of
As4S4-induced PML-RARα degradation, ~50% of
that in parental NB4-R1 cells.
Discussion
Arsenic agents have been proved highly effective in
the treatment of APL. It is particularly useful for
relapsed/refractory APL with ATRA-resistance (18). As4S4, is a
new and promising oral arsenic formulation. A multicenter study in
China has shown that a complete remission (CR) rate of 99.1% and a
disease-free survival (DFS) rate of 98.1% at 2 years were achieved
in 108 APL cases treated with an oral As4S4
combined with ATRA (19, 20). In the present study, we
demonstrated that As4S4 inhibited the growth
and induced apoptosis of ATRA-resistant NB4-R1 cells. The result is
consistent with previous findings (21,22).
By using comparative proteomic approach, we identified PHB was
significantly upregulated during
As4S4-induced NB4-R1 apoptosis. As PHB is of
particular interest, further experiments were performed to modulate
the gene expression, either PHB overexpression or PHB knockdown.
The results with modulation of PHB expression implicate its
activity in promoting As4S4-induced
apoptosis.
PHB was selected in this study for its diverse roles
in the regulation of proliferation, apoptosis and gene
transcription (23–27). PHB proteins have been found to
localize in the mitochondria, nucleus and plasma membrane of
mammalian cells. PHB is implicated in diverse cellular processes,
including mitochondrial biogenesis, cell death and replicative
senescence. A functional role for PHB as a regulator of
transcription has been shown for its interactions with p53, E2F and
Rb (28–30). PHB has been associated with various
types of cancer. The role of PHB in cancer cell proliferation or
tumor suppression is considered controversial. PHB was shown to be
necessary for the activation of C-Raf by the oncogene Ras in HeLa
cells (31). However, many reports
have shown evidence that PHB has antitumorigenic activity in
prostate, gastric and ovarian cancer (32–35).
PHB overexpression was shown to result in the inhibition of
prostate cancer cell growth and the knockdown of PHB by siRNA
accelerates tumor growth (33).
In the present study, stable clones of KD
(PHB-knockdown NB4-R1 cells) and OE (PHB-overexpression NB4-R1
cells) were established and used to determine the cellular response
to As4S4. The results showed that PHB
overexpression enhanced apoptosis of NB4-R1 cells, and reduction of
PML-RARα fusion protein. Although PHB knockdown had no significant
effect on baseline apoptosis and PML-RARα fusion protein, a
downregulation of PHB was associated with an attenuated apoptosis
and lesser reduction of PML-RARα protein in the cells treated with
As4S4. These results strongly support that
PHB has antitumorigenic activity.
The effects of PHB on cellular processes may be due
to its subcellular localization in different type cells. The
subcellular localization of PHB has been shown to affect cell fate,
specifically apoptosis (36). PHB
has been shown with an increased level on the cell membrane that
facilitates tumorigenesis through its interaction with c-Raf
induced by the Ras oncogene (37,38),
whereas increased levels of PHB in the nucleus induce apoptosis by
increasing the transcriptional activity of p53 and its
translocation to the cytoplasm (39). We have found the increased levels
of PHB, either modulated by As4S4 or by PHB
overexpression vectors, in the nucleus locations of APL cells.
The PML-RARα fusion protein is the key molecule that
drives APL cells. This fusion protein also serves as the
therapeutic target of ATRA and arsenic agents (40). While ATRA induces APL to undergo
differentiation by targeting the RARα moiety, arsenic agents induce
apoptosis through SUMO-1-mediated degradation of the PML moiety of
the fusion protein (41). However,
other molecules involved in the process remain to be identified. In
this study, we showed a close relationship of upregulation of PHB
with reduction of PML-RARα during
As4S4-induced apoptosis. Consistently, PHB
knockdown experiments showed a reduced degradation of PML-RARα
protein. These results indicate that PHB is involved in the APL
cell apoptosis. However, the biochemical pathway of PHB activity in
relation to PML-RARα remains the subject of investigations.
In conclusion, PHB was identified among the
upregulated proteins associated with
As4S4-induced apoptosis of NB4-R1 cells. The
experiments with modulation of PHB expression indicate that PHB
overexpression enhances apoptosis and degradation of PML-RARα
fusion protein, and consistently PHB knockdown attenuated the
cellular response to As4S4 treatment.
Acknowledgements
The present study is supported by a research grant
from the Natural Science Foundation of China (NSFC, grant no.
30701133), the Shaanxi Province Science and Technology Development
Fund (SPSTDF, grant no. 2012KTCL03-12). The authors thank Dr
Qunling Zhang from Shanghai Institute of Hematology for providing
the NB4-R1 cell line; Dr Xinyang Wang from the First Affiliated
Hospital, Xi'an Jiaotong University for their technological
assistance; and Dr Byron Song from Univerity of Tronto, Ontario,
Canada for critically reading the manuscript.
Abbreviations:
|
As4S4
|
arsenic sulfide
|
|
APL
|
acute promyelocytic leukemia
|
|
PHB
|
prohibitin
|
|
ATRA
|
all-trans retinoic acid
|
|
PML-RARα
|
promyelocytic leukemia-retinoic acid
receptor-α
|
References
|
1
|
Sahin U, Lallemand-Breitenbach V and de
Thé H: PML nuclear bodies: Regulation, function and therapeutic
perspectives. J Pathol. 234:289–291. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rabellino A, Carter B, Konstantinidou G,
Wu SY, Rimessi A, Byers LA, Heymach JV, Girard L, Chiang CM,
Teruya-Feldstein J, et al: The SUMO E3-ligase PIAS1 regulates the
tumor suppressor PML and its oncogenic counterpart PML-RARA. Cancer
Res. 72:2275–2284. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Guo Y, Dolinko AV, Chinyengetere F,
Stanton B, Bomberger JM, Demidenko E, Zhou DC, Gallagher R, Ma T,
Galimberti F, et al: Blockade of the ubiquitin protease UBP43
destabilizes transcription factor PML/RARα and inhibits the growth
of acute promyelocytic leukemia. Cancer Res. 70:9875–9885. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
de Thé H and Chen Z: Acute promyelocytic
leukaemia: Novel insights into the mechanisms of cure. Nat Rev
Cancer. 10:775–783. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang ZY and Chen Z: Acute promyelocytic
leukemia: From highly fatal to highly curable. Blood.
111:2505–2515. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tomita A, Kiyoi H and Naoe T: Mechanisms
of action and resistance to all-trans retinoic acid (ATRA) and
arsenic trioxide (As2O3) in acute
promyelocytic leukemia. Int J Hematol. 97:717–725. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mathews V, George B, Lakshmi KM,
Viswabandya A, Bajel A, Balasubramanian P, Shaji RV, Srivastava VM,
Srivastava A and Chandy M: Single-agent arsenic trioxide in the
treatment of newly diagnosed acute promyelocytic leukemia: Durable
remissions with minimal toxicity. Blood. 107:2627–2632. 2006.
View Article : Google Scholar
|
|
8
|
Lengfelder E, Hofmann WK and Nowak D:
Impact of arsenic trioxide in the treatment of acute promyelocytic
leukemia. Leukemia. 26:433–442. 2012. View Article : Google Scholar
|
|
9
|
Wu J, Shao Y, Liu J, Chen G and Ho PC: The
medicinal use of realgar (As4S4) and its
recent development as an anticancer agent. J Ethnopharmacol.
135:595–602. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lengfelder E, Hofmann WK and Nowak D:
Treatment of acute promyelocytic leukemia with arsenic trioxide:
Clinical results and open questions. Expert Rev Anticancer Ther.
13:1035–1043. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
van Meerloo J, Kaspers GJ and Cloos J:
Cell sensitivity assays: the MTT assay. Methods Mol Biol.
731:237–245. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Qian X, Dong H, Hu X, Tian H, Guo L, Shen
Q, Gao X and Yao W: Analysis of the interferences in quantitation
of a site-specifically PEGylated exendin-4 analog by the Bradford
method. Anal Biochem. 465:50–52. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rabilloud T and Lelong C: Two-dimensional
gel electrophoresis in proteomics: A tutorial. J Proteomics.
74:1829–1841. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dupont FM, Vensel WH, Tanaka CK, Hurkman
WJ and Altenbach SB: Deciphering the complexities of the wheat
flour proteome using quantitative two-dimensional electrophoresis,
three proteases and tandem mass spectrometry. Proteome Sci.
9:102011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lelong C, Chevallet M, Luche S and
Rabilloud T: Silver staining of proteins in 2DE gels. Methods Mol
Biol. 519:339–350. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Granvogl B, Plöscher M and Eichacker LA:
Sample preparation by in-gel digestion for mass spectrometry-based
proteomics. Anal Bioanal Chem. 389:991–1002. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu Y, He P, Zhang M and Wu D: Lentiviral
vector-mediated RNA interference targeted against prohibitin
inhibits apoptosis of the retinoic acid-resistant acute
promyelocytic leukemia cell line NB4-R1. Mol Med Rep. 6:1288–1292.
2012.PubMed/NCBI
|
|
18
|
Lu DP, Qiu JY, Jiang B, Wang Q, Liu KY,
Liu YR and Chen SS: Tetra-arsenic tetra-sulfide for the treatment
of acute promyelocytic leukemia: A pilot report. Blood.
99:3136–3143. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhu HH, Wu DP, Jin J, Li JY, Ma J, Wang
JX, Jiang H, Chen SJ and Huang XJ: Oral tetra-arsenic tetra-sulfide
formula versus intravenous arsenic trioxide as first-line treatment
of acute promyelocytic leukemia: A multicenter randomized
controlled trial. J Clin Oncol. 31:4215–4221. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhu HH and Huang XJ: Oral arsenic and
retinoic acid for non-high-risk acute promyelocytic leukemia. N
Engl J Med. 371:2239–2241. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang L, Zhou GB, Liu P, Song JH, Liang Y,
Yan XJ, Xu F, Wang BS, Mao JH, Shen ZX, et al: Dissection of
mechanisms of Chinese medicinal formula Realgar-Indigo naturalis as
an effective treatment for promyelocytic leukemia. Proc Natl Acad
Sci USA. 105:4826–4831. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen S, Fang Y, Ma L, Liu S and Li X:
Realgar-induced apoptosis and differentiation in all-trans retinoic
acid (ATRA)-sensitive NB4 and ATRA-resistant MR2 cells. Int J
Oncol. 40:1089–1096. 2012.
|
|
23
|
Zhou TB and Qin YH: Signaling pathways of
prohibitin and its role in diseases. J Recept Signal Transduct Res.
33:28–36. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rossi L, Bonuccelli L, Iacopetti P,
Evangelista M, Ghezzani C, Tana L and Salvetti A: Prohibitin 2
regulates cell proliferation and mitochondrial cristae
morphogenesis in planarian stem cells. Stem Cell Rev. 10:871–887.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu YH, Peck K and Lin JY: Involvement of
prohibitin upregulation in abrin-triggered apoptosis. Evid Based
Complement Alternat Med. 2012:6051542012. View Article : Google Scholar
|
|
26
|
Puppin C, Passon N, Franzoni A, Russo D
and Damante G: Histone deacetylase inhibitors control the
transcription and alternative splicing of prohibitin in thyroid
tumor cells. Oncol Rep. 25:393–397. 2011.
|
|
27
|
Joshi B, Ko D, Ordonez-Ercan D and
Chellappan SP: A putative coiled-coil domain of prohibitin is
sufficient to repress E2F1-mediated transcription and induce
apoptosis. Biochem Biophys Res Commun. 312:459–466. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chander H, Halpern M, Resnick-Silverman L,
Manfredi JJ and Germain D: Skp2B attenuates p53 function by
inhibiting prohibitin. EMBO Rep. 11:220–225. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Joshi B, Rastogi S, Morris M, Carastro LM,
DeCook C, Seto E and Chellappan SP: Differential regulation of
human YY1 and caspase 7 promoters by prohibitin through E2F1 and
p53 binding sites. Biochem J. 401:155–166. 2007. View Article : Google Scholar :
|
|
30
|
Wang S, Fusaro G, Padmanabhan J and
Chellappan SP: Prohibitin co-localizes with Rb in the nucleus and
recruits N-CoR and HDAC1 for transcriptional repression. Oncogene.
21:8388–8396. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rajalingam K, Wunder C, Brinkmann V,
Churin Y, Hekman M, Sievers C, Rapp UR and Rudel T: Prohibitin is
required for Ras-induced Raf-MEK-ERK activation and epithelial cell
migration. Nat Cell Biol. 7:837–843. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang S and Faller DV: Roles of prohibitin
in growth control and tumor suppression in human cancers. Transl
Oncogenomics. 3:23–37. 2008.PubMed/NCBI
|
|
33
|
Dart DA, Spencer-Dene B, Gamble SC, Waxman
J and Bevan CL: Manipulating prohibitin levels provides evidence
for an in vivo role in androgen regulation of prostate tumours.
Endocr Relat Cancer. 16:1157–1169. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang Y, Chen Y, Qu C, Zhou M, Ni Q and Xu
L: siRNA targeting prohibitins inhibits proliferation and promotes
apoptosis of gastric carcinoma cell line SGC7901 in vitro and in
vivo. Cell Mol Biol (Noisy-le-grand). 60:26–32. 2014.
|
|
35
|
Jia L, Ren JM, Wang YY, Zheng Y, Zhang H,
Zhang Q, Kong BH and Zheng WX: Inhibitory role of prohibitin in
human ovarian epithelial cancer. Int J Clin Exp Pathol.
7:2247–2255. 2014.PubMed/NCBI
|
|
36
|
Theiss AL and Sitaraman SV: The role and
therapeutic potential of prohibitin in disease. Biochim Biophys
Acta. 1813:1137–1143. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rajalingam K and Rudel T: Ras-Raf
signaling needs prohibitin. Cell Cycle. 4:1503–1505. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chowdhury I, Thompson WE and Thomas K:
Prohibitins role in cellular survival through Ras-Raf-MEK-ERK
pathway. J Cell Physiol. 229:998–1004. 2014. View Article : Google Scholar
|
|
39
|
Song W, Tian L, Li SS, Shen DY and Chen
QX: The aberrant expression and localization of prohibitin during
apoptosis of human cholangiocarcinoma Mz-ChA-1 cells. FEBS Lett.
588:422–428. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhou GB, Zhang J, Wang ZY, Chen SJ and
Chen Z: Treatment of acute promyelocytic leukaemia with all-trans
retinoic acid and arsenic trioxide: A paradigm of synergistic
molecular targeting therapy. Philos Trans R Soc Lond B Biol Sci.
362:959–971. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shinagawa K: All-trans retinoic acid and
arsenic trioxide: Their molecular mechanisms of action and updated
clinical progress in APL therapy. Rinsho Ketsueki. 52:469–483.
2011.(In Japanese). PubMed/NCBI
|