Introduction
The fate of a cell upon exposure to ionizing
radiation depends in great part on their capacity to repair the
damage and maintain genomic integrity (1). For this purpose, the repair of
radiation-induced DNA double strand breaks (DSBs), being the most
devastating DNA lesions, and cell cycling tightly interrelate in a
well-defined manner (1–3).
Although the general coordination and interrelation
between DNA damage repair and cell cycling has intensively been
documented (4), the detailed
interplay of the molecular players is still not well understood.
Cyclin-dependent kinases (CDKs), for example, are a family of over
11 members whose activity is controlled by post-translational
modifications occurring in CDK-specific manner in the different
phases of the cell cycle (5).
Owing to their crucial function in cell cycling and DNA damage
repair and frequent aberrations of their activities in cancer
encouraged an intensive screening for small-molecule CDK
pharmacological inhibitors that block CDK activity (6–8).
Despite the intensive research regarding CDKs as target for cancer
therapy, studies evaluating their role in cancer cell response to
irradiation are rare.
CDK9, unlike most other CDKs functioning in cell
cycling, is particularly involved in gene transcription (9,10).
It is activated by forming a heterodimeric complex with either
cyclin T or cyclin K family members and inhibited by both 7SK and
HEXIM1 proteins (11). In
mammalian cells, two CDK9 isoforms of different molecular weight
have been identified. The functional difference between these
isoforms remains to be clarified (10). CDK9 together with its cyclin
partner (cyclin T or cyclin K) forms a complex known as positive
transcription elongation factor b (pTEFb) (12,13).
This complex promotes transcription elongation by phosphorylating
the CTD of the large subunit of RNA polymerase II (Rpb1-CTD) on S2
(10) or on S5 (14). In turn, pTEFb modulates other
cellular functions such as co-transcriptional histone modification,
mRNA processing and mRNA export (15,16).
A recent study showed that CDK9 together with cyclin K, but not T
is involved in maintaining genome integrity after replication
stress (17). The connection
between CDK9 and cell division has recently been reported in
mammalian cells suggesting a direct role for the CDK9/cyclin K
complex in checkpoint pathway regulation (17,18).
Moreover, coprecipitation studies showed that CDK9/cyclin K complex
interacts with ATR, ATRIP and other DNA repair and checkpoint
proteins (17).
Based on its function in cell cycle regulation and
DNA damage repair, we hypothesized targeting of CDK9 to potently
modify the cancer cell response to radiotherapy. Therefore, we
evaluated the significance of CDK9 inhibition using siRNA
technology and a pharmacological inhibitor (ZK304709) (19) for the radioresponse in a panel of
human head and neck squamous cell carcinoma (HNSCC) cell lines.
Materials and methods
Antibodies and reagents
Antibodies against GFP (Abcam, Cambridge, UK); BrdU
(BD, Heidelberg, Germany); ATM, phospho-ATM (S1981), CDK9, CHK2,
phospho-CHK2 (T86), cyclin E, DNA-PK, NBS1, phospho-Rb (S795),
phospho-Rpb1 (S2/5) and Rpb1 (Cell Signaling, Frankfurt, Germany);
cyclin A, PCNA, RAD50 and Rb (Santa Cruz, Heidelberg, Germany);
phospho-histone γH2AX (S139) (Millipore, Darmstadt, Germany); p53
binding protein 1 (53BP1) (Novus Biologicals, Littelton, CO, USA);
cyclin D1 (Zymed Laboratories Inc.); β-actin and anti-mouse IgG
FITC (Sigma-Aldrich, Taufkirchen, Germany); HRP-conjugated goat
anti-rabbit and anti-mouse secondary antibodies (Amersham,
Freiburg, Germany); Alexa 594 anti-mouse and Alexa 488 anti-rabbit
(Invitrogen, Cyclin T, Germany) were purchased as indicated. ECL
SuperSignal® West Dura Extended Substrate was from
Pierce (Bonn, Germany); complete protease inhibitor cocktail from
Roche (Mannheim, Germany); propidium iodide (PI) from Serva
(Heidelberg, Germany); the phosphatase inhibitors
Na3VO4 and NaF from Sigma-Aldrich,
Vectashield/DAPI mounting medium was from Alexis (Grünberg,
Germany) and oligofectamine from Invitrogen.
CDK inhibitor
ZK304709 is a pharmacological pan-CDK inhibitor
kindly provided by Bayer Pharma AG, Germany (19).
Cell culture
Human SAS, FaDu, HSC4, Cal33, UTSCC5 HNSCC cell
lines (a kind gift from R. Grenman, Turku University Central
Hospital, Finland, and M. Baumann, Dresden University of
Technology, Germany) were cultured in Dulbecco's modified Eagle's
medium (DMEM, PAA, Cölbe, Germany) containing 10% fetal bovine
serum (FBS, PAA) and 1% non-essential amino acids (NEAA, PAA) at
37°C in a humidified atmosphere containing 7% CO2. In
all experiments, asynchronously and exponentially growing cells
were used.
Generation of CDK9-EGFP
transfectants
CDK9 PCR fragment (hCDK9-NheI forward:
5′-ctaGCTAGCgccgccATG GCAAAGCAGTACGACTCGG-3′; hCDK9-BamHI
reverse: 5′-cgGGATCCcgGAAGACGCGCTCAAACTCCG-3′) was amplified using
placental DNA (UKD, Dresden, Germany), followed by ligation of CDK9
in frame into NheI/BamHI restriction sites of
pEGFP-N1 expression vector (CDK9-EGFP-N1). Stable transfection and
selection was performed as described (20).
Colony formation assay
The colony formation assay was applied for
measurement of clonogenic cell survival as published (20). In brief, single cells were grown on
polystyrene 6-well plates (BD) 24 h prior to irradiation with 0–8
Gy. After seven days (SAS), 11 days (FaDu), eight days (HSC4), 13
days (Cal33) or 14 days (UTSCC5), cells were fixed with 80%
ethanol, stained with Coomassie blue and cell colonies (>50
cells) were counted. Images of representative colonies were scanned
using Perfection 4490 Photo scanner (Epson, Meerbusch, Germany).
Each point on the survival curves represents the mean surviving
fraction from three independent experiments.
Radiation exposure
Cells were irradiated at room temperature using
single doses of 200 kV X-rays (2, 4, 6 or 8 Gy; Yxlon Y.TU 320;
Yxlon; 0.5 mm copper filter; ~1.3 Gy/min, 20 mA) filtered with 0.5
mm Cu. The absorbed dose was measured using a Duplex dosimeter
(PTW, Freiburg, Germany).
Small interfering RNA-mediated knockdown
of CDK9
Human CDK9 small interfering ribonucleic acid
(siRNA) (#1 sequence: 5′-GGAGAAUUUUACUGUGUUUtt-3′; #2 sequence:
5′-GGU GCUGAUGGAAAACGAGtt-3′) were obtained from Applied Biosystems
(Darmstadt, Germany). Non-specific control siRNA (sequence:
5′-GCAGCUAUAUGAAUGUUGUtt-3′) was from MWG (Martinsried, Germany).
Cells were prepared for siRNA transfection (20 nmol/l) with
oligofectamine and were subjected 24 h after transfection to colony
formation assay and western blotting as previously published
(20).
Immunofluorescence staining
For detection of DSBs, the phosphorylated H2AX-S139
(γH2AX)/p53 binding protein 1 (p53BP1) foci assay was performed as
published (20).
γH2AX/p53BP1-positive nuclear foci of 50 cells were counted
microscopically with an Axioscope 2 plus fluorescence microscope
(Zeiss) and defined as DSBs. Fluorescence images were obtained
using an LSM 510 Meta equipped with Zeiss LSM 510 Software
(Zeiss).
DAPI staining
For apoptosis analysis, cells were trypsinized and
fixed with 80% ethanol for ≥24 h as published (21). Typical apoptotic nuclear shape was
analyzed using Vectashield/DAPI mounting medium. Apoptotic nuclei
of ≥100 cells from 3 independent experiments were counted
microscopically using an Axioscope 2 microscope (Zeiss).
Total protein extracts and western
blotting
Total protein extracts were isolated as previously
described (20). Therefore, cells
were lyzed using modified RIPA buffer [50 mM Tris-HCl (Carl Roth,
Karlsruhe, Germany), pH 7.4], 1% Nonidet-P40 (Sigma-Aldrich,
Taufkirchen, Germany), 0.25% sodium deoxycholate (AppliChem,
Darmstadt, Germany), 150 mM NaCl (VWR International, Darmstadt,
Germany), 1 mM ethylenediaminetetraacetic acid (Merck), complete
protease inhibitor cocktail (Roche, Mannheim, Germany), 1 mM
NaVO4 (AppliChem), 2 mM NaF (AppliChem)]. Samples were
stored at −80°C. Total protein amount was measured using the
bicinchoninic acid assay (Pierce, Bonn, Germany). After sodium
dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and
western blotting, protein detection was performed as published
(20).
Cell cycle analysis
Cells were incubated with 10 mM bromodeoxyuridine
(BrdU; Serva) for 10 min, trypsinized and fixed in 80% ice cold
ethanol. Then, cells were prepared for cell cycle analysis as
published (22). Detection of BrdU
was accomplished with anti-BrdU and anti-mouse IgG FITC antibodies
and total DNA staining with propidium iodide (PI) solution.
Acquisition of data was performed with a CyFlow (Partec, Münster,
Germany). The distribution of cells in the different phases of the
cell cycle was analyzed from the DNA dot-blots and histograms using
FloMax software.
Statistical analysis
Data were expressed as means ± SD of at least three
independent experiments. P-values are based on the Student's t-test
(Microsoft® Excel 2003). Results were considered
statistically significant when a P-value of <0.05 was
reached.
Results
CDK9 overexpression enhances the
clonogenic survival of HNSCC cells
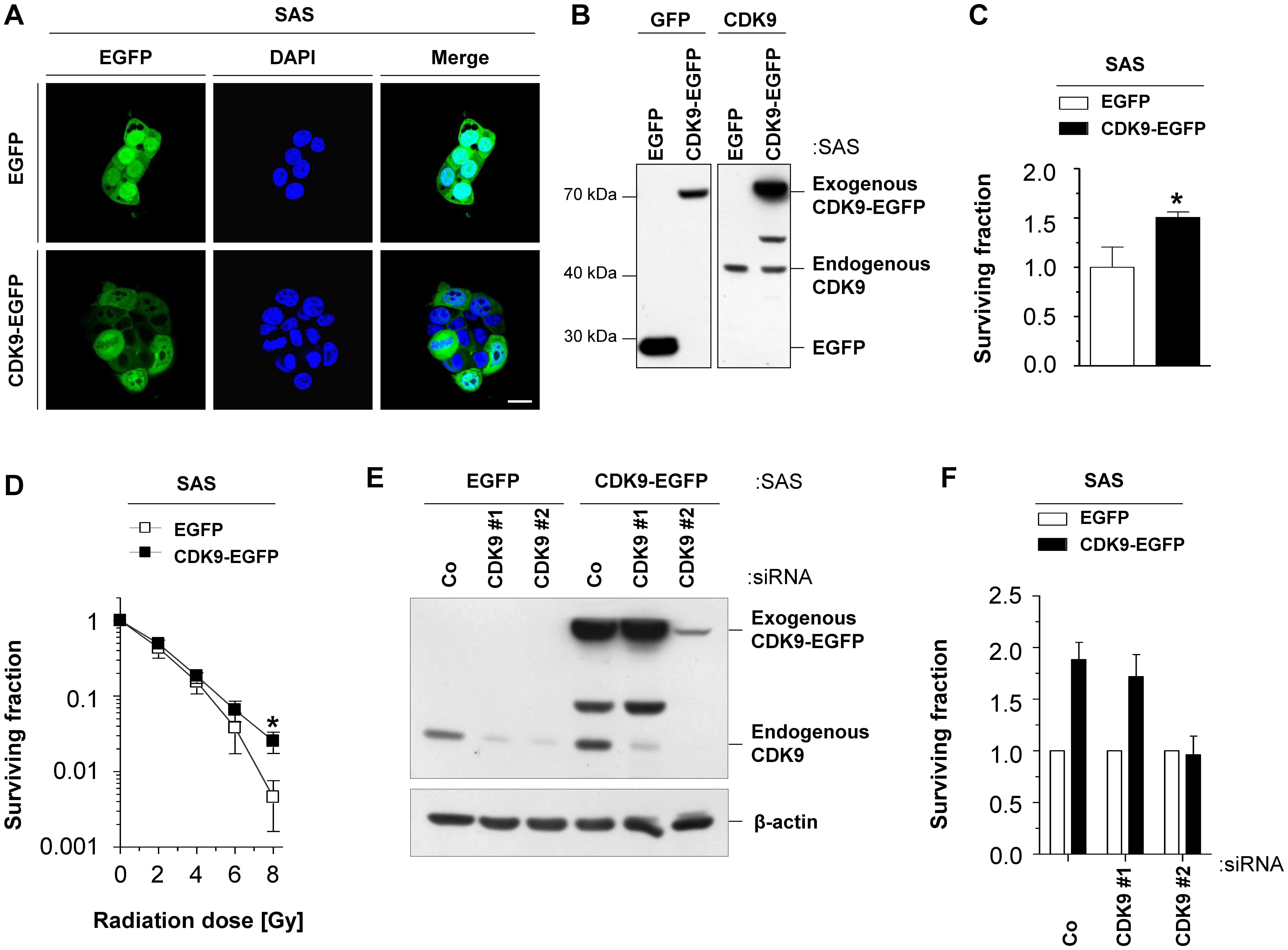
To better understand the role of CDK9 in clonogenic
survival of unirradiated and irradiated cells, we first stably
transfected SAS cells with CDK9-EGFP construct and characterized
its cytoplasmic and nuclear localization and expression in whole
cell lysates (Fig. 1A and B). We
further found that CDK9 overexpression significantly (P<0.05)
increased plating efficiency (Fig.
1C) and clonogenic radiation survival (Fig. 1D). To prove CDK9 dependence of
these results, we depleted CDK9 expression using siRNA, which
resulted in a plating efficiency similar to controls (Fig. 1E and F).
CDK9 knockdown radiosensitizes HNSCC cell
lines
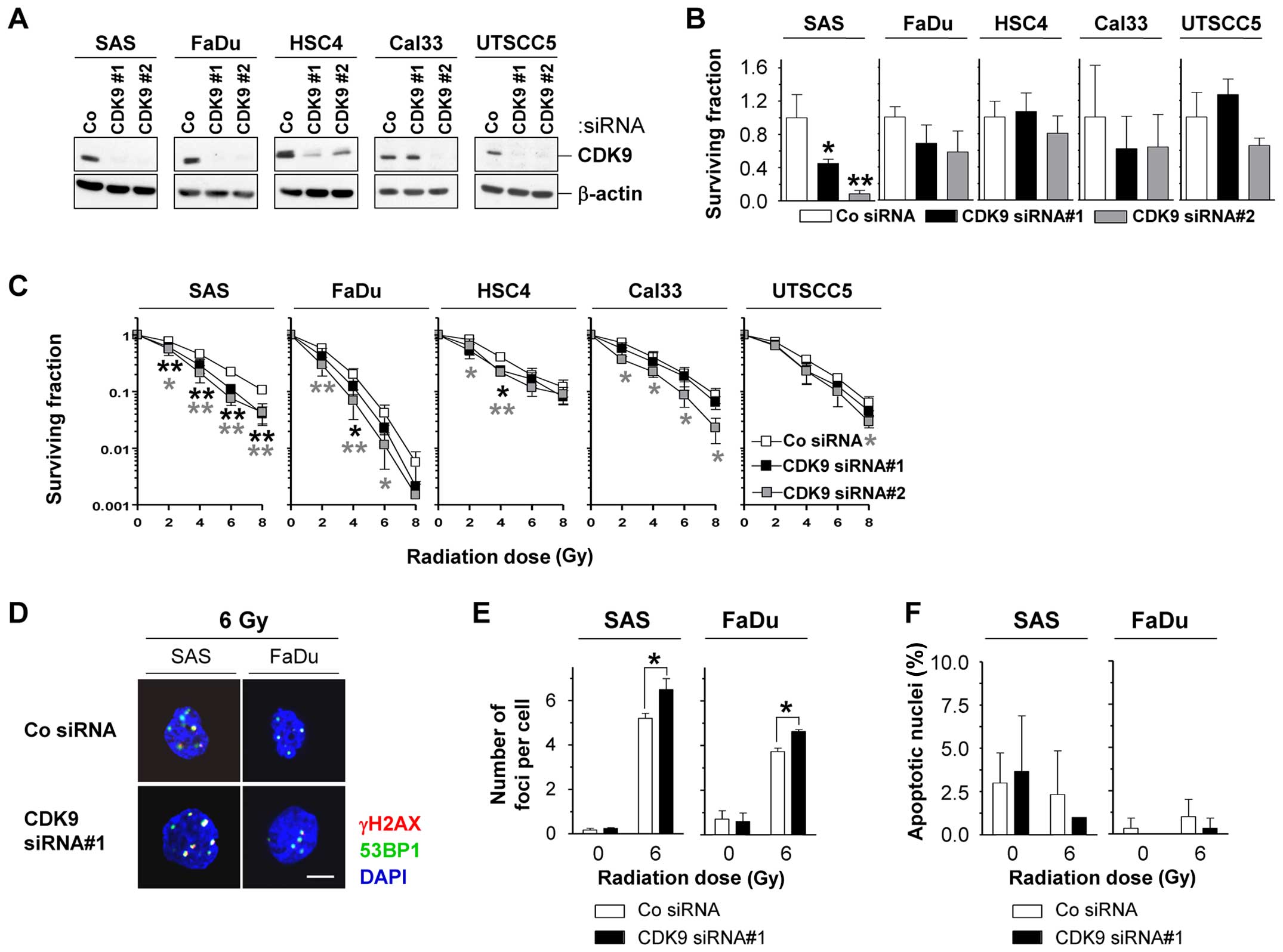
To investigate the potential of CDK9 as cancer
target in more detail, we next assessed the effects of CDK9
knockdown on plating efficiency and radiation survival in a panel
of five HNSCC cell lines. The efficient CDK9 knockdown (Fig. 2A) modified the plating efficiencies
rather moderately, except in SAS cells, which showed significantly
(P<0.05) reduced survival (Fig.
2B). In combination with irradiation, HSC4, Cal33 and UTSCC5
cells were only slightly altered in the radiosensitivity upon CDK9
depletion, while SAS and FaDu cells were significantly (P<0.05)
sensitized to X-ray irradiation (Fig.
2C). In line with the CDK9-dependent radiosensitization
occurred a significantly (P<0.05) increased number of
γH2AX/53BP1-positive foci per cell in 6-Gy irradiated CDK9
knockdown SAS and FaDu cell cultures relative to controls (Fig. 2D and E). Enhanced rates of
apoptosis were not found after CDK9 knockdown alone or in
combination with irradiation (Fig.
2F).
CDK9 depletion fails to modulate DNA
repair proteins
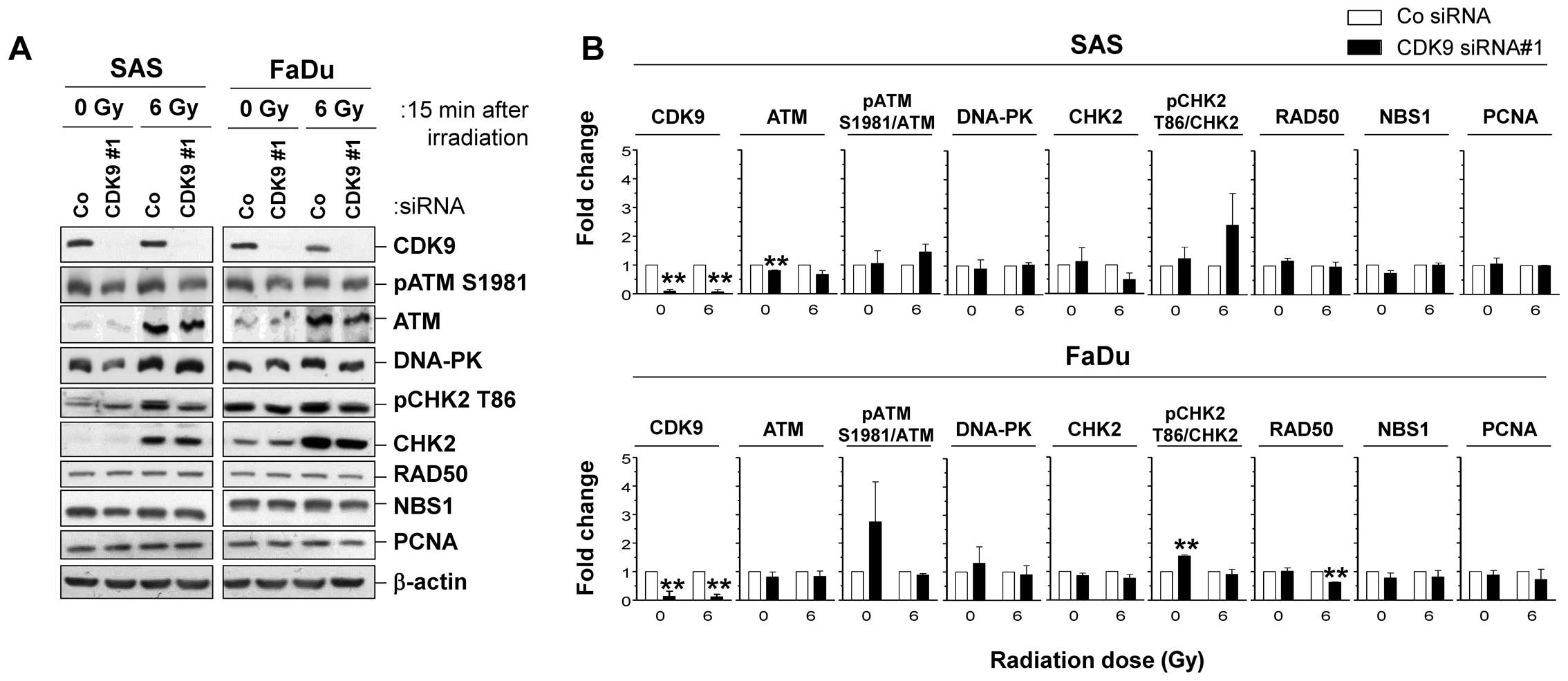
In SAS and FaDu cells showing the strongest
radiosensitization by CDK9 silencing, we analyzed the expression
and phosphorylation patterns of various proteins involved in DNA
repair 15 min after 6 Gy X-rays (Fig.
3A). Surprisingly, we found no significant alterations in
expression and phosphorylation of DNA repair proteins upon CDK9
knockdown in SAS and FaDu cells (Fig.
3B).
CDK9 knockdown modulates cell cycling in
SAS and FaDu cells
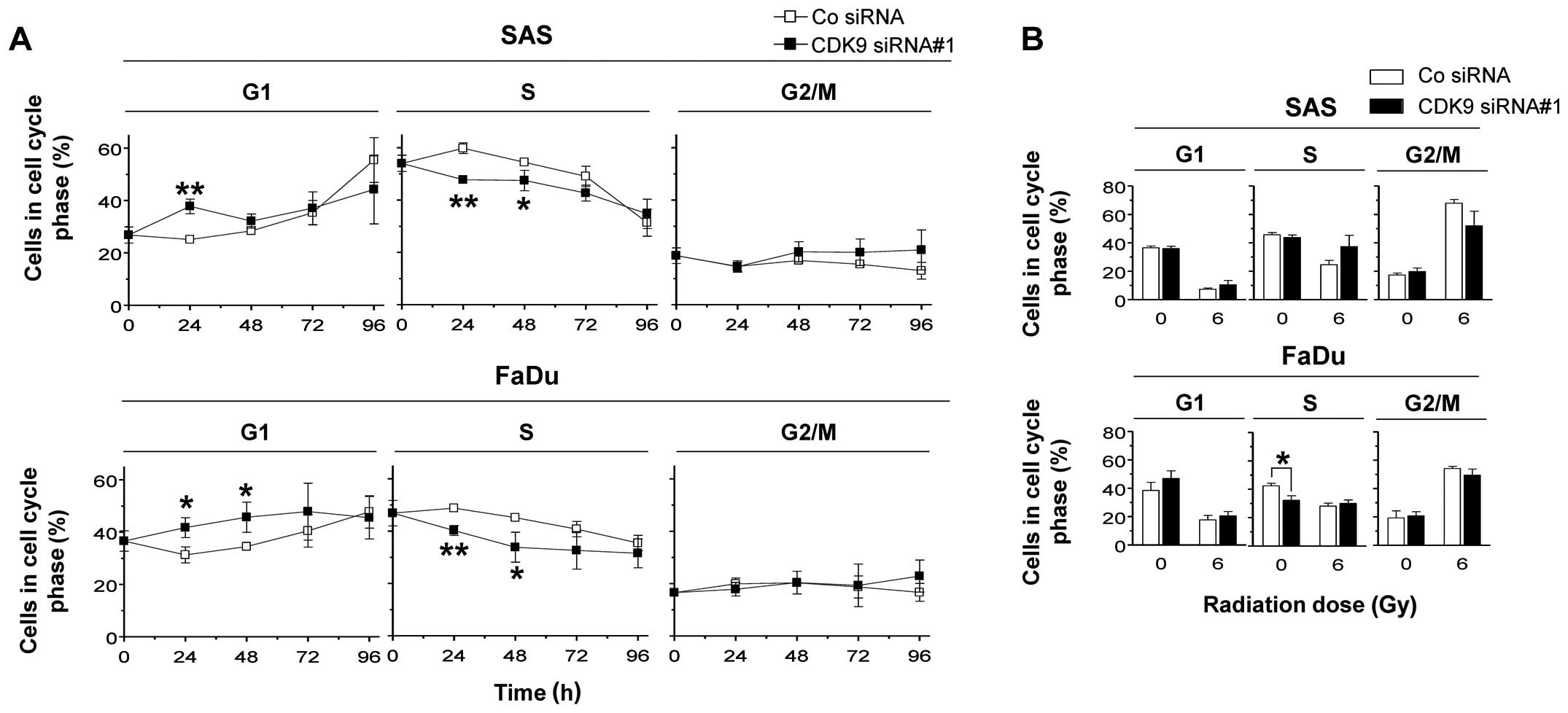
Next, we sought to investigate whether CDK9 play a
role in cell cycling and found that CDK9 knockdown in SAS and FaDu
cells, respectively, results in a significant (P<0.05) increase
in G1 phase cells and a significant (P<0.05) decrease in the S
phase cell population (Fig. 4A).
No changes were observed in the G2/M phase cell population
(Fig. 4A). In combination with
irradiation, CDK9-depleted SAS and FaDu cells showed no significant
alterations in their cell cycle distribution (Fig. 4B).
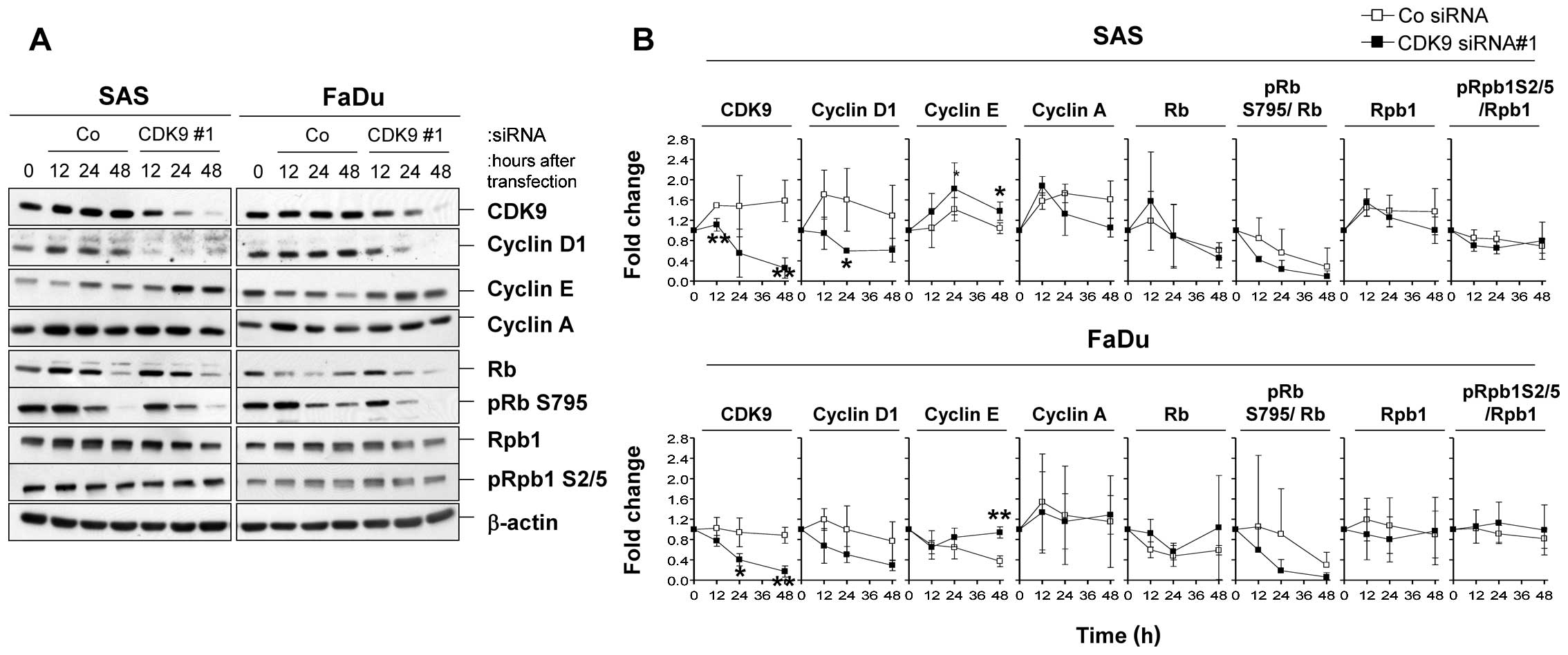
Silencing of CDK9 impacts on cell cycle
protein expression
Next we analyzed the expression and phosphorylation
of a panel of cell cycle regulatory proteins and of Rpb1 upon CDK9
knockdown (Fig. 5A).
Interestingly, in line with CDK9 depletion, we found a rapid
decline in the level of cyclin D1, an induction of cyclin E, and a
slight reduction in Rb S795 phosphorylation in both SAS and FaDu
cell lines as compared to controls (Fig. 5B). Moreover, levels of RB,
Rpb1-CTD, or phospho-Rpb1-CTD(S2/5) remained stable. These results
suggest that CDK9 plays a role in cell cycle regulation.
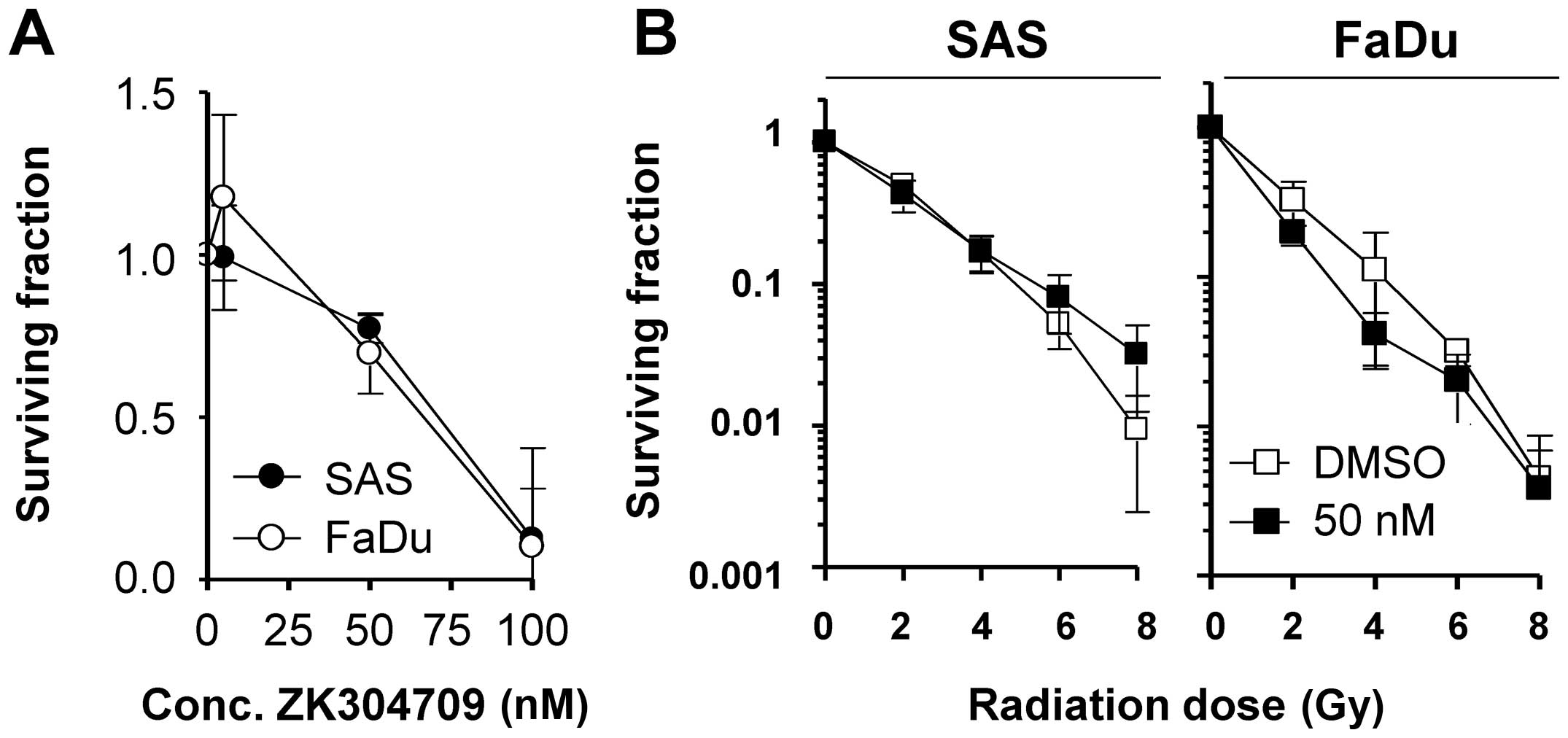
Pharmacological inhibition of CDK9
mediates cytotoxicity but not radiosensitization
In addition to siRNA-mediated CDK9 inhibition, we
finally explored how plating efficiency and radiation survival are
affected by the pharmacological CDK9/CDK2/CDK1 inhibitor ZK304709
in SAS and FaDu cells (Fig. 6A).
While ZK304709 reduced the plating efficiency of SAS and FaDu cells
concentration-dependently, neither cell line could be
radiosensitized when pretreated with 50 nM ZK304709 (Fig. 6B).
Discussion
Optimization of multimodal therapy concepts is
essential to improve cancer patient outcome. Therefore, the
identification of new and promising molecular targets is necessary.
Among the hallmarks of cancer (1),
the unlimited proliferation capability of cancer cells is an
important characteristic and cell cycle regulators are promising
candidates for targeted therapy. In this study, we investigated the
role of CDK9 for the cellular radiation response of HNSCC
cells.
By means of overexpression, siRNA and
pharmacological inhibition of CDK9, we found radioprotection by
CDK9 overexpression as well as cell line-dependent
radiosensitization which could not be recapitulated with the
CDK9/CDK2/CDK1 inhibitor ZK304709. Accordingly, we observed that
silencing of CDK9 perturbed the repair of irradiation-induced
residual DSBs in SAS and FaDu cells. These findings coincide with
our own data showing a strong correlation between clonogenic
radiation survival and the number of residual DSBs (20,23).
Regarding apoptosis, several studies suggested that CDK9 inhibition
induces apoptosis in cancer cells (24,25).
However, we could not observe any induction in apoptosis following
CDK9 depletion in either SAS or FaDu cells.
CDK9 is the catalytic part of pTEFb which stimulates
transcription elongation by phosphorylating the CTD of the large
subunit (Rpb1-CTD) of RNA polymerase II (RNAPII) (14,16,26).
Previous studies reported a possible selective regulatory role of
CDK9 on the expression of a restricted subset of genes instead of
all genes controlled by RNA polymerase II (9). Therefore, it is possible that CDK9
may specifically regulate one or more DNA damage response proteins.
Similar to our study showing no obvious DNA damage repair protein
modification upon CDK9 silencing, Yu and Cortez demonstrated that
depletion of CDK9 lacked significantly up or down regulation of DNA
damage response genes using genome-wide expression analysis
(10). Although these results
cannot explain how CDK9 modulates the DNA damage repair of DSBs,
one might speculate that CDK9 plays a direct role in maintaining
genomic integrity via yet to be determined mechanisms (10).
In contrast to DNA repair, our results suggest a
function of CDK9 in cell cycling. CDK9 depletion delayed cell cycle
transition based on elevated G1 phase and declined S phase cell
populations. Similarly, Cai and colleagues reported that reduction
of CDK9 was associated with changes in cell cycle distribution that
are consistent with cell cycle delay (24). The S phase retardation after CDK9
silencing seems to be a consequence of the accumulation of cells in
the G1 phase. Mammalian cells exhibit variation in their response
to irradiation as they move through the cell cycle. Cells in the
G2-M phase are the most radiosensitive and S phase cells are the
most radioresistant ones while G1 phase cells show moderate
radiosensitivity (27).
Accordingly, the retardation of S phase population may contribute
to the enhanced radiosensitivity of cancer cells upon CDK9
knockdown.
On the molecular level, Rb is one of the known
substrates for CDK9 (28).
Therefore, it is not surprising that Rb was found to be
hypophosphorylated after silencing of CDK9. In addition, CDK9
depletion was associated with a remarkable decrease in cyclin D1
levels in SAS and FaDu. Together with its binding partners CDK4 and
CDK6, cyclin D1-dependent kinase activity promotes G1 phase
progression by phosphorylating and inactivating Rb (29,30).
Because of the central role of Rb in cell cycle progression,
especially during the G1 phase, the observed cell cycle changes
after CDK9 knockdown seem to be attributed to Rb
hypophosphorylation (24). On the
contrary, the level of cyclin E, another G1 phase cyclin, was
elevated after CDK9 depletion which might indicate an attempt to
tune the cell cycle disturbance due to suppression of cyclin
D1-dependent kinase activity in the G1 phase (31).
Despite the well-known role of CDK9 in
phosphorylating Rpb1-CTD of RNA polymerase II on S2 and S5 residues
(8,14,16,26)
we unexpectedly observed unaffected phosphorylation of Rpb1-CTD
(S2/5) after depletion of CDK9. One possible explanation is that
other CDKs such as CDK12 and CDK13 also regulate, like CDK9,
transcription elongation by phosphorylating Rpb1-CTD (32,33).
This may explain why CDK9 was dispensable for Rpb1-CTD
phosphorylation but cannot reveal the CDK9-related changes in
cyclin D1 and cyclin E expression.
Finally, the pharmacological multi-target inhibitor
ZK304709 was used in combination with radiotherapy to stress the
need of CDK9 for clonogenic radiation survival in a more clinically
relevant approach (19). Although
ZK304709 showed inhibitory efficacy in human tumor xenografts as
well as in human pancreatic carcinoma models (19,34),
ZK304709 mediated cytotoxicity without enhanced radiosensitivity in
the tested HNSCC cells.
These findings suggest a critical role of CDK9 in
the cellular response to ionizing radiation. However, further
examinations are required to better understand the molecular
circuitry of CDK9 involvement level in DNA damage repair and cell
cycle regulation and explore the potential of CDK9 targeting for
the clinic.
Acknowledgements
This study was supported in part by the
Bundesministerium für Bildung und Forschung (BMBF Contract 03ZIK041
to N.C.), the Saxon Ministry of Science and Arts and the EFRE
Europäische Fonds für regionale Entwicklung, Europa fördert Sachsen
(100066308). A. Soffar was awarded with a stipend from the Ministry
of Higher Education and Scientific Research of the Arab Republic of
Egypt (MHESR) and the Deutscher Akademischer Austauschdienst
(DAAD). We thank A. Soffar for excellent technical assistance and
thank R. Grenman and M. Baumann for kindly providing the cell
lines. The pharmacological pan-CDK inhibitor ZK304709 was kindly
provided by Bayer Pharma AG, Germany.
References
|
1
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dikomey E, Brammer I, Johansen J, Bentzen
SM and Overgaard J: Relationship between DNA double-strand breaks,
cell killing, and fibrosis studied in confluent skin fibroblasts
derived from breast cancer patients. Int J Radiat Oncol Biol Phys.
46:481–490. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jeggo PA and Löbrich M: Contribution of
DNA repair and cell cycle checkpoint arrest to the maintenance of
genomic stability. DNA Repair (Amst). 5:1192–1198. 2006. View Article : Google Scholar
|
|
4
|
Cann KL and Hicks GG: Regulation of the
cellular DNA double-strand break response. Biochem Cell Biol.
85:663–674. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Endicott JA and Noble ME: Structural
characterization of the cyclin-dependent protein kinase family.
Biochem Soc Trans. 41:1008–1016. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Canavese M, Santo L and Raje N: Cyclin
dependent kinases in cancer: Potential for therapeutic
intervention. Cancer Biol Ther. 13:451–457. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Diaz-Padilla I, Siu LL and Duran I:
Cyclin-dependent kinase inhibitors as potential targeted anticancer
agents. Invest New Drugs. 27:586–594. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Malumbres M, Pevarello P, Barbacid M and
Bischoff JR: CDK inhibitors in cancer therapy: What is next? Trends
Pharmacol Sci. 29:16–21. 2008. View Article : Google Scholar
|
|
9
|
Garriga J, Bhattacharya S, Calbó J,
Marshall RM, Truongcao M, Haines DS and Graña X: CDK9 is
constitutively expressed throughout the cell cycle, and its
steady-state expression is independent of SKP2. Mol Cell Biol.
23:5165–5173. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yu DS and Cortez D: A role for CDK9-cyclin
K in maintaining genome integrity. Cell Cycle. 10:28–32. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Michels AA, Fraldi A, Li Q, Adamson TE,
Bonnet F, Nguyen VT, Sedore SC, Price JP, Price DH, Lania L, et al:
Binding of the 7SK snRNA turns the HEXIM1 protein into a P-TEFb
(CDK9/cyclin T) inhibitor. EMBO J. 23:2608–2619. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Egloff S and Murphy S: Cracking the RNA
polymerase II CTD code. Trends Genet. 24:280–288. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Peng J, Marshall NF and Price DH:
Identification of a cyclin subunit required for the function of
Drosophila P-TEFb. J Biol Chem. 273:13855–13860. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhou M, Halanski MA, Radonovich MF,
Kashanchi F, Peng J, Price DH and Brady JN: Tat modifies the
activity of CDK9 to phosphorylate serine 5 of the RNA polymerase II
carboxyl-terminal domain during human immunodeficiency virus type 1
transcription. Mol Cell Biol. 20:5077–5086. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pirngruber J, Shchebet A, Schreiber L,
Shema E, Minsky N, Chapman RD, Eick D, Aylon Y, Oren M and Johnsen
SA: CDK9 directs H2B monoubiquitination and controls
replication-dependent histone mRNA 3′-end processing. EMBO Rep.
10:894–900. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Romano G and Giordano A: Role of the
cyclin-dependent kinase 9-related pathway in mammalian gene
expression and human diseases. Cell Cycle. 7:3664–3668. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yu DS, Zhao R, Hsu EL, Cayer J, Ye F, Guo
Y, Shyr Y and Cortez D: Cyclin-dependent kinase 9-cyclin K
functions in the replication stress response. EMBO Rep. 11:876–882.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu H and Herrmann CH: Differential
localization and expression of the Cdk9 42k and 55k isoforms. J
Cell Physiol. 203:251–260. 2005. View Article : Google Scholar
|
|
19
|
Siemeister G, Luecking U, Wagner C, Detjen
K, Mc Coy C and Bosslet K: Molecular and pharmacodynamic
characteristics of the novel multi-target tumor growth inhibitor ZK
304709. Biomed Pharmacother. 60:269–272. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Storch K, Eke I, Borgmann K, Krause M,
Richter C, Becker K, Schröck E and Cordes N: Three-dimensional cell
growth confers radioresistance by chromatin density modification.
Cancer Res. 70:3925–3934. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mazzeo E, Hehlgans S, Valentini V, Baumann
M and Cordes N: The impact of cell-cell contact, E-cadherin and EGF
receptor on the cellular radiosensitivity of A431 cancer cells.
Radiat Res. 178:224–233. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cordes N, Frick S, Brunner TB, Pilarsky C,
Grützmann R, Sipos B, Klöppel G, McKenna WG and Bernhard EJ: Human
pancreatic tumor cells are sensitized to ionizing radiation by
knockdown of caveolin-1. Oncogene. 26:6851–6862. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Eke I, Sandfort V, Storch K, Baumann M,
Röper B and Cordes N: Pharmacological inhibition of EGFR tyrosine
kinase affects ILK-mediated cellular radiosensitization in vitro.
Int J Radiat Biol. 83:793–802. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cai D, Latham VM Jr, Zhang X and Shapiro
GI: Combined depletion of cell cycle and transcriptional
cyclin-dependent kinase activities induces apoptosis in cancer
cells. Cancer Res. 66:9270–9280. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gojo I, Zhang B and Fenton RG: The
cyclin-dependent kinase inhibitor flavopiridol induces apoptosis in
multiple myeloma cells through transcriptional repression and
down-regulation of Mcl-1. Clin Cancer Res. 8:3527–3538.
2002.PubMed/NCBI
|
|
26
|
Ramanathan Y, Rajpara SM, Reza SM, Lees E,
Shuman S, Mathews MB and Pe'ery T: Three RNA polymerase II
carboxyl-terminal domain kinases display distinct substrate
preferences. J Biol Chem. 276:10913–10920. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sinclair WK and Morton RA: X-ray
sensitivity during the cell generation cycle of cultured Chinese
hamster cells. Radiat Res. 29:450–474. 1966. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Simone C, Bagella L, Bellan C and Giordano
A: Physical interaction between pRb and cdk9/cyclinT2 complex.
Oncogene. 21:4158–4165. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lundberg AS and Weinberg RA: Functional
inactivation of the retinoblastoma protein requires sequential
modification by at least two distinct cyclin-cdk complexes. Mol
Cell Biol. 18:753–761. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Weinberg RA: The retinoblastoma protein
and cell cycle control. Cell. 81:323–330. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bowe DB, Kenney NJ, Adereth Y and
Maroulakou IG: Suppression of Neu-induced mammary tumor growth in
cyclin D1 deficient mice is compensated for by cyclin E. Oncogene.
21:291–298. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bartkowiak B, Liu P, Phatnani HP, Fuda NJ,
Cooper JJ, Price DH, Adelman K, Lis JT and Greenleaf AL: CDK12 is a
transcription elongation-associated CTD kinase, the metazoan
ortholog of yeast Ctk1. Genes Dev. 24:2303–2316. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Blazek D, Kohoutek J, Bartholomeeusen K,
Johansen E, Hulinkova P, Luo Z, Cimermancic P, Ule J and Peterlin
BM: The Cyclin K/Cdk12 complex maintains genomic stability via
regulation of expression of DNA damage response genes. Genes Dev.
25:2158–2172. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Scholz A, Wagner K, Welzel M, Remlinger F,
Wiedenmann B, Siemeister G, Rosewicz S and Detjen KM: The oral
multitarget tumour growth inhibitor, ZK 304709, inhibits growth of
pancreatic neuroendocrine tumours in an orthotopic mouse model.
Gut. 58:261–270. 2009. View Article : Google Scholar
|