Introduction
Prognosis of glioblastoma (GBM) is still dismal
despite multimodality treatments applied such as aggressive
surgical resection and adjuvant chemo-radiotherapy (1–3). A
major advance in GBM therapy as an advantageous treatment is the
involvement of DNA alkylating agent temozolomide (TMZ) (4). TMZ, an orally administered alkylating
agent with relatively low toxicity, exerts the antitumor activity
by interfering with DNA replication. Many randomized clinical
trials evidenced that TMZ-based chemo-therapy significantly
improved quality of life and prolonged survival of GBM patients
(5,6). However, TMZ resistance of GBM is a
key factor involved in poor responses and dismal prognosis.
Previous studies have evidenced that the resistance to TMZ of
molecular pathogenes in GBM is related to O6-methylguanine-DNA
methyltransferase (MGMT), which can lead to the replication of DNA
and the growth of GBM cells (7,8).
The mechanism of action of MGMT involves the removal
of alkyl groups from damaged DNA in a one-way suicide reaction by
transferring them to an internal cysteine residue (9). The expression of MGMT varies widely
in different kinds of tumor cells. It has been suggested that MGMT
is the most important determinant of resistance to TMZ (10). Increasing data indicated prognosis
of patients with high MGMT expression is much poorer than those
with low expression. Moreover, MGMT overexpression was able to
resist cell death induced by TMZ in both experimental and clinical
settings (11,12). Importantly, MGMT promoter
methylation is an essential predictor which is associated with
better clinical responses to chemoradiation and overall survival
(13,14).
Several transcription factors have been suggested to
be involved in the complex process of MGMT transcriptional
regulation, including p53, NF-κB, HIF-1α, and AP-1 (15–17).
NF-κB is a family of direct transcription factors which can bind to
specific DNA sequences in target genes involving in
immunoregulation, inflammation, growth, carcinogenesis and
apoptosis (18,19). Activated NF-κB can function as an
oncogene in a variety of tumors to promote tumor cell proliferation
and invasion, induce angiogenesis and metastasis, and prevent
apoptosis (20,21). It was also suggested that
inhibition of NF-κB activity by suppressors could strongly enhance
the apoptotic potential of the alkylating agent. A previous study
showed that NF-κB subunit p65 induced the increase of MGMT
expression, whereas NF-κB inhibitor abrogates the augmented
expression of MGMT in HEK293 cells. MGMT is a target gene for NF-κB
and plays an important role in NF-κB-mediated chemo-resistance to
alkylating agents (16).
Therefore, inhibiting the NF-κB-MGMT pathway is a significant
strategy to overcome TMZ-resistance.
Sulforaphane (SFN), a naturally-occurring member of
the isothiocyanate (ITC) family, is ample in normally consumed
cruciferous vegetables (22,23).
Recently, evidence from numerous epidemiological investigations
demonstrated that the higher dietary intakes of cruciferous
vegetables were associated with reduced risk of various cancers,
such as mammary gland tumorigenesis, colonic aberrant crypt foci,
stomach tumors, prostate, bladder and lung cancer (24). The underlying mechanisms of SFN in
anticancer activity have been reported in many respects including
detoxification enzymes, oxidative stress induction, checkpoint
activation in DNA damage, and inhibition of histone deacetylase
(HDAC) direct binding to cellular proteins (25). Previous reports suggested many
survival signaling pathways could be blocked by SFN in various
tumors. For example, SFN significantly suppressed the expression of
phosphorylated c-Jun N-terminal kinase (p-JNK), phosphorylated
extracellular signal-regulated kinases (p-ERK), p-Akt and
β-catenin, and then interrupting the MAPK, PI3K/Akt and Wnt
signaling pathway (26,27).
Increasing evidence shows an essential effect of SFN
on growth inhibition and apoptosis induction in human cancer cells,
and this research was performed to explore the sensitization of SFN
to TMZ in TMZ-resistant GBM cells.
Materials and methods
Cell culture and reagents
Human GBM cell lines (T98G, U87, LN229, U373 and
U251) were obtained from the China Academia Sinica Cell Repository,
Shanghai, China. Human cells U373 and U87 were exposed to TMZ
(Temodal, Schering-Plough, Whitehouse Station, NJ, USA) until
stable TMZ-resistant subclones (U373-R and U87-R) were derived from
the parental TMZ-sensitive cell lines. Both cells were exposed to
stepwise increasing concentrations of TMZ (2–100 μM) over a period
of 6 months.
Antibodies against MGMT, p65, Ki-67, caspase-3 and
MMP2/9 were obtained from Cell Signaling Technology (Danvers, MA,
USA). SFN (St. Louis, MO, USA) was prepared in DMSO at the stock
solution of 100 mM and further diluted to appropriate concentration
with cell culture medium immediately before use.
Western blot analysis and reverse
transcription-polymerase chain reaction (RT-PCR) analysis
Cell proteins were separated by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then
transferred onto polyvinylidene difluoride membranes (Millipore).
Protein binding to the MGMT, Ki-67, MMP2/9, p65 and caspase-3
antibody (Santa Cruz Biotechnology, Inc.) was assessed by enhanced
chemiluminescence and exposed to a chemiluminescent film.
Total RNA was isolated from GBM cells by the TRIzol
method. Reverse transcription was performed using a reverse
transcription kit (Qiagen), and the primers were synthesized by
GenePharma (Shanghai, China) (28).
Cell viability assay
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT) (Sigma) was used to determine the cell viability and
the 50% growth inhibitory concentration (IC50) of TMZ or
SFN in both parental and TMZ-resistant cell lines. Cells were
cultured in 96-well plate at a density of 5×103
cells/well in medium containing 10% fetal bovine serum (FBS) and
were incubated for 72 h. Subsequently, TMZ was added to culture
medium at final concentrations of 0, 500, 1000, 2000, 3000, 4000,
5000 or 6000 μM, SFN was added at a final concentration of 0, 5,
10, 20, 30, 40 or 50 μM. After incubation for 48 h, cells were
detached by trypsinization and the viable cell population was
determined using MTT. All experiments were done in triplicate.
Cell colony formation assay
U87-R, U373-R and T98G cells were seeded into 6-well
plates and allowed to attach overnight. Cells were then treated
with different concentration of SFN (0, 10 or 25 μM) and colony
formation assay was done as described (29). Only colonies with 50 or more cells
were counted as surviving colonies under an inverted
microscope.
Cell invasion assay
Cells were treated with different concentrations of
SFN (10, 20 or 30 μM), and after 8 h TMZ (250 μM) was added. After
48 h, the invasion ability of GBM cells was determined using
Transwell plates (BD Biosciences, Bedford, MA, USA) according to
the manufacturer's protocol.
Cell apoptosis assay
Cells cultured in 6-well plates for 24 h were
treated with SFN (10, 20 or 30 μM) and TMZ (250 μM, after 8 h) and
cultured for additional 48 h. For detection of apoptosis, the FITC
Annexin V Apoptosis Detection kit (BD Pharmingen, San Jose, CA,
USA) was used according to the manufacturer's protocol, and
quantification of apoptosis was determined through measurement of
caspase-3/7 activation using caspase-3/7 Detection in Living Cells
kit (Biotium Inc., Hayward, CA, USA).
NF-κB transcription reporter assay
The Luciferase reporter (Stratagene, La Jolla, CA,
USA) contains the NF-κB enhancer consensus sequences
[(TGGGGACTTTCCGC) ×5] and NF-κB-dependent firefly luciferase gene.
Cells were seeded in 24-wells plate, and then transiently
transfected with pNF-κB-luc plasmids using Lipofectamine 2000 after
24 h. Luciferase activity was measured with the Dual-Luciferase
assay system kit (Promega Corp.).
Establishment of subcutaneous tumors in a
nude mouse model
All animal experiment procedures were carried out
according to the regulations and internal biosafety and bioethics
guidelines of Tianjin Hospital and Tianjin Huanhu Hospital Medical
Ethics Committee. Nude mouse models were established with U373-R
GBM cells inoculated subcutaneously into the right flanks of
4–6-week-old female mice. Twenty-four mice were randomized in 4
groups (control, TMZ, SFN alone and the combination of TMZ and SFN)
when tumors reached a mass of ~200 mm3 in size. TMZ was
given at 50 mg/kg/day for 5 days/week for 4 cycles. SFN was
injected subcutaneously at 50 mg/kg/day for 28 consecutive days.
Mice were observed daily and euthanized after 5 days of treatment.
At the end, all the mice were sacrificed and tumors were collected
for further experiments.
Terminal
deoxynucleotidyltransferase-mediated dUTP nick end labelling
(TUNEL) assay
The apoptosis in the tumor specimens of mouse models
from the in vivo study was examined by TUNEL (Roche,
Indianapolis, IN, USA) according to the manufacturer's
protocol.
Statistical analysis
All of the statistical analyses were evaluated by
commercially available software SPSS version 13.0. Differences
between the groups were analyzed by using Student's t-test and
statistical significance was determined as P<0.05 or
P<0.01.
Results
Expression of MGMT in different GBM
cells
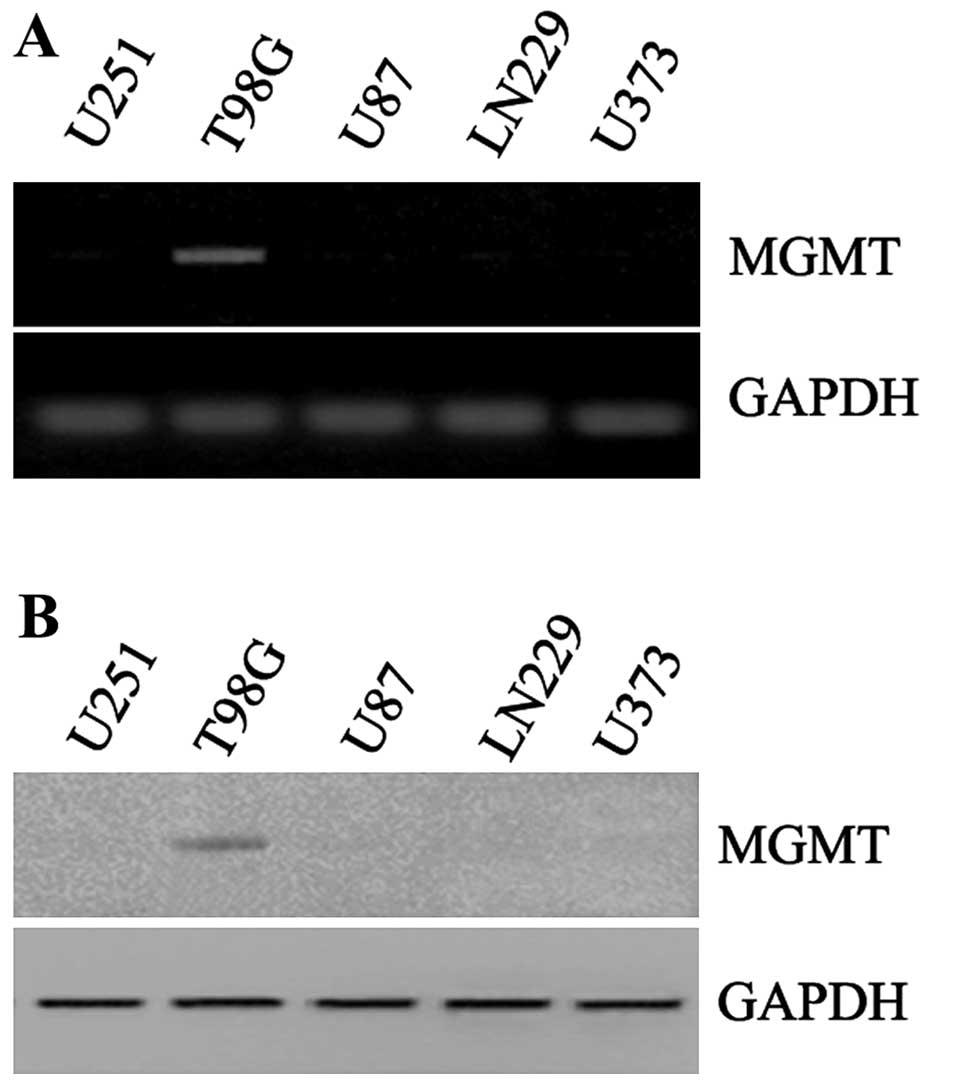
To explore the different conditions of MGMT in
various types of GBM cells, we firstly detected expression of MGMT
mRNA and protein in U251, T98G, U87, LN229 and U373 cells. The
expression of MGMT mRNA was high in T98G cells, and very low or
hardly detected in other GBM cells (Fig. 1A). Furthermore, the detection of
MGMT protein was consistent with mRNA status and much higher in
T98G cells (Fig. 1B). As reported,
MGMT overexpression was able to resist cell death induced by TMZ.
Cellular sensitivity to TMZ in five cell lines was also evaluated
after 48 h of treatment. IC50 values varied between the
different cell lines, and the IC50 of T98G was much high
than other cell lines (Table I).
Based on the above data, T98G cell line was considered as resistant
to TMZ and chosen to carry out the following experiments.
 | Table IDoses required inducing 50%
inhibition of cell growth (IC50) in GBM cell lines
treated with TMZ. |
Table I
Doses required inducing 50%
inhibition of cell growth (IC50) in GBM cell lines
treated with TMZ.
| Cell line | IC50 TMZ
(μM) |
|---|
| U251 | 851.3 |
| T98G | 3457.8 |
| U87 | 702.4 |
| LN229 | 954.2 |
| U373 | 483.5 |
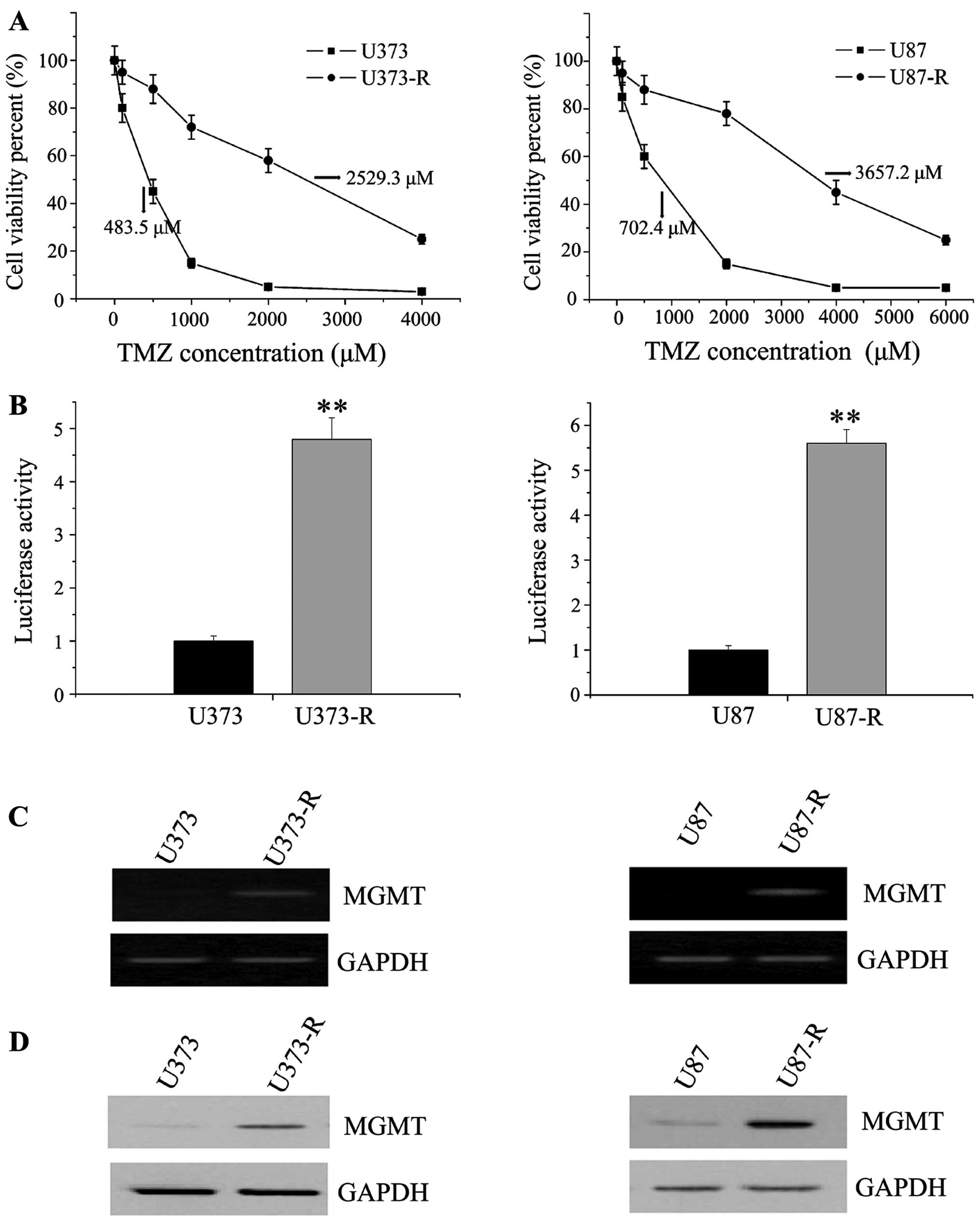
Establishment of TMZ-resistant U373 and
U87 GBM cells by chronic exposure to TMZ
We then successfully established TMZ-resistant GBM
cell models to search the methods of TMZ sensitization. Two
TMZ-resistant human glioma cell sublines, U373-R and U87-R, were
generated by a stepwise exposure to increasing TMZ concentrations
for 6 months. The two established variants showed effective
resistance to further TMZ treatment. The IC50 of U373-R
and U87-R showed more than 5-fold increase when compared with their
parental cell lines (2529.3 vs. 483.5 μM and 3657.2 vs. 702.4 μM)
(Fig. 2A). Previous it was
reported that most DNA-damaging chemotherapeutic agents including
TMZ could activate the transcription of NF-κB (30). We then detected the activity of
NF-κB in TMZ-inducing cells by transiently transfecting with
pNF-κB-luc reporter plasmids. When compared with control
counterparts, NF-κB transcription was significant activated in
U373-R and U87-R cells (Fig. 2B).
At the end of this section, we investigated whether the increase in
TMZ resistance in GBM cells was associated with high MGMT
expression. PCR and western blot analyses revealed that MGMT
content increased significantly at both mRNA and protein levels in
the TMZ-resistant cells (Fig. 2C and
D). Taken together, these results suggested a link between the
onset of TMZ resistance and activity of NF-κB signaling
pathway.
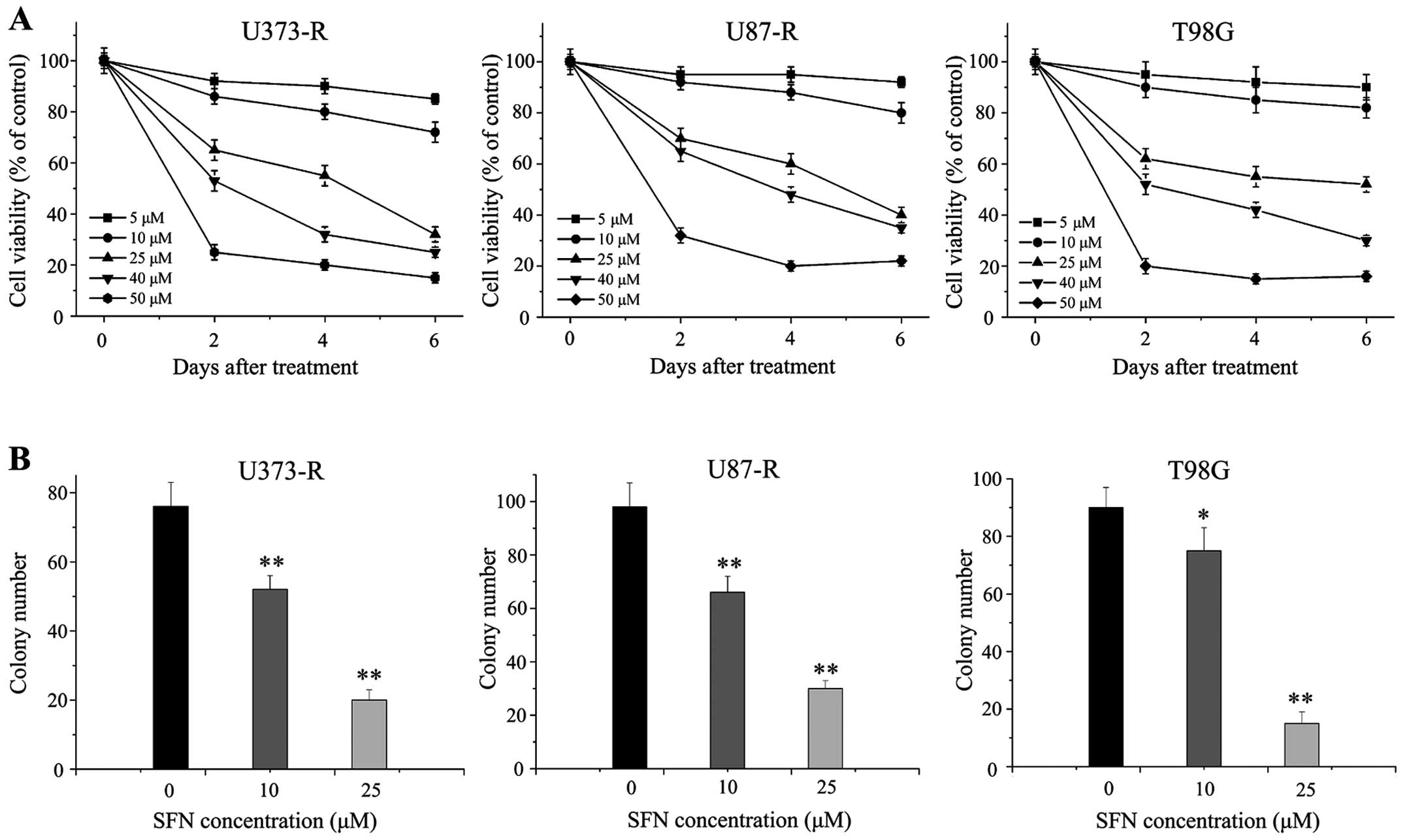
SFN inhibits proliferation of
TMZ-resistant GBM cells
SFN has been indicated for the prevention and
suppression of tumorigenesis in various solid tumors. Our previous
study demonstrated that SFN could effectively enhance TMZ-induced
apoptosis by inhibiting miR-21 via Wnt/β-catenin signaling in GBM
cells (31). Herein, we aimed to
examine the inhibition of growth of SFN in TMZ-resistant GBM cells.
The role of SFN in TMZ-resistant cell proliferation was
investigated by conducting MTT and colony formation assays. We
treated U373-R, U87-R and T98G cells with 5–50 μM for 48 h and
assessed the number of viable cells. The MTT assay showed that SFN
had a growth inhibitory effect on all the tumor cell lines in a
dose-dependent manner (Fig. 3A).
Next, colony formation assay suggested that cells treated with SFN
(10 and 25 μM) for 48 h formed significantly less colonies than
control cells (P<0.01) (Fig.
3B), suggesting an inhibitory effect of SFN on
anchorage-dependent growth of TMZ-resistant cells.
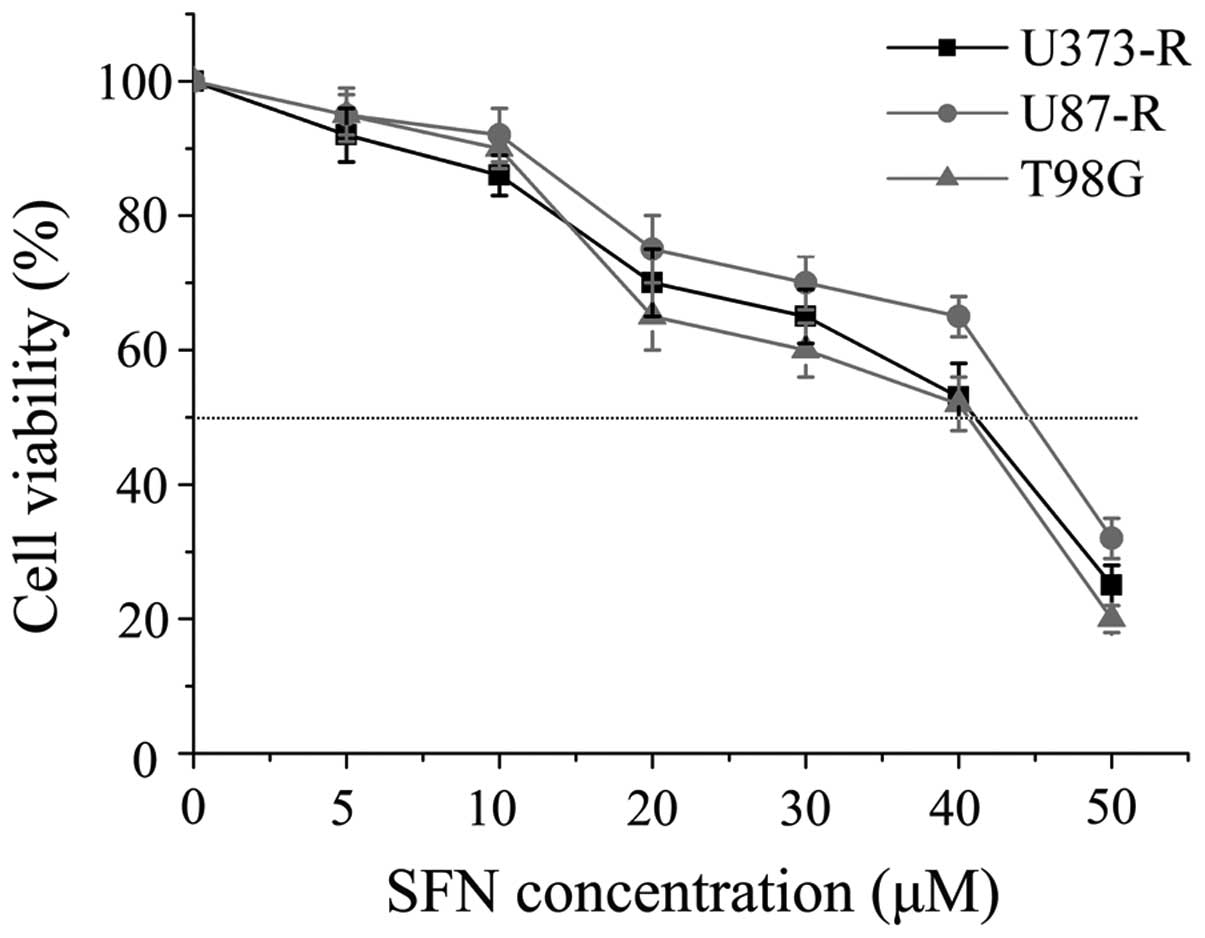
SFN downregulates the expression of MGMT
through inhibiting NF-κB in TMZ-resistant GBM cells
To choose the correct dose for the following in
vitro combination experiments, the IC50 value of SFN
was investigated in these three cell lines. The IC50 of
SFN for U373-R, U87-R and T98G was 41.7, 44.8 and 41.5 μM,
respectively (Fig. 4). Based on
the above results, concentrations lower than the respective
IC50 value of SFN was identified for further study in
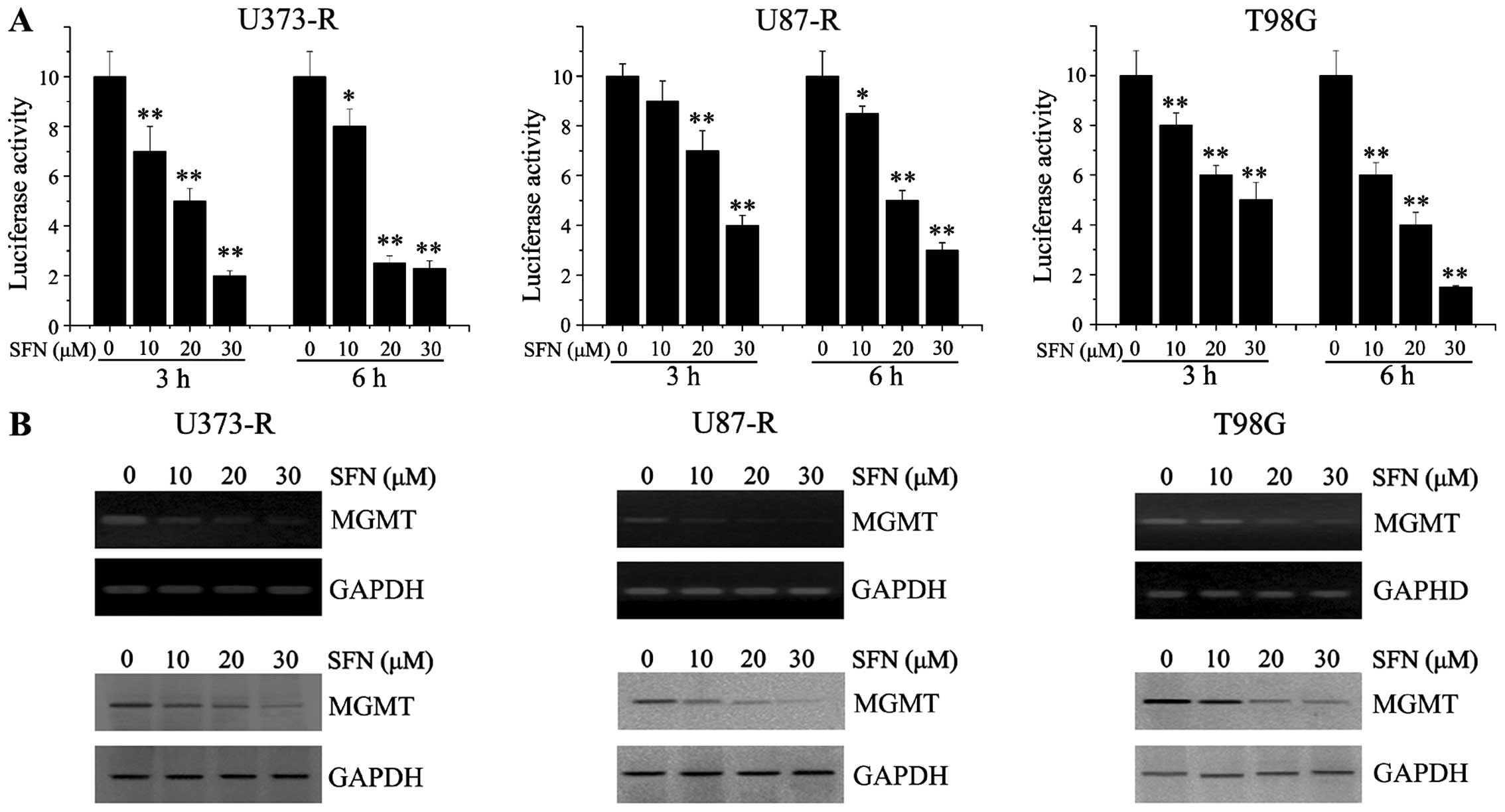
TMZ-resistant cells. To determine whether SFN as a chemosensitizer
in TMZ-resistant GBM was mediated by decreasing the expression of
MGMT through inhibiting NF-κB, we examined the effects of SFN on
NF-κB transcription activity. The U373-R, U87-R and T98G cells were
transfected with the NF-κB reporter plasmids. A range of
concentrations of SFN was added to the transfected cells, as
observed in Fig 5A, the
transcriptional activity of NF-κB was significantly attenuated by
SFN in a concentration-dependent manner. A previous study provided
evidence that MGMT is a target gene for NF-κB (16). We then analyzed the expression of
MGMT following SFN treatment in TMZ-resistant GBM cells. The PCR
and western blot analyses revealed that SFN resulted in decreased
expression of MGMT in a dose-dependent manner (Fig. 5B).
SFN reverses TMZ-resistant GBM cells to
TMZ-induced cytotoxicity in vitro
In order to examine whether SFN can enhance the
cytotoxity of TMZ, a series of experiments were conducted, and the
dose of 250 μM TMZ was selected, which has been evidenced as no
growth inhibitory effects on TMZ-resistant cells. Initially,
nonlinear regression of a sigmoid dose response model and
combination index (CI) approaches were performed to test drug
interaction between SFN and TMZ. The results revealed antagonistic
effects (CI>1) when cells were simultaneously treated with TMZ
and each concentration of SFN for T98G, U87-R and U373-R.
Nevertheless, the response to concomitant treatment was drastically
reversed when the administration order was changed. Synergistic
effects (CI<1) were observed when different concentrations of
SFN were used as pretreatment in TMZ-resistant cells for 8 h before
exposure to TMZ. Sequential exposure also resulted in high- dose
reduction index (DRI) values suggesting that TMZ doses could be
signicantly reduced to achieve comparable cytotoxicity (Table II). The three cell lines were then
treated in schedule dependency with the previous (8 h) exposure to
SFN. To identify the synergistic effects of SFN and TMZ on the
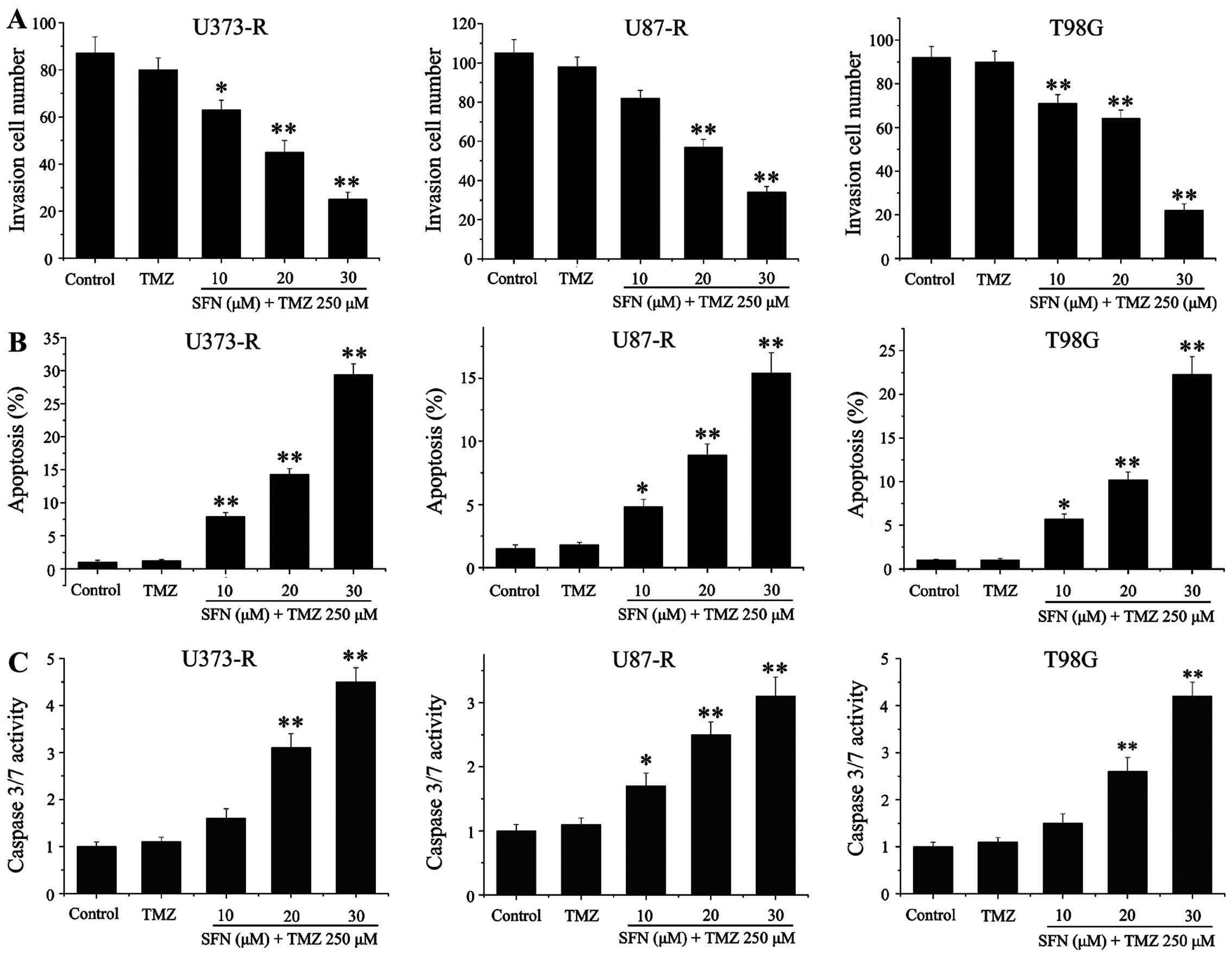
invasive ability, Transwell Matrigel invasion assay was used in
T98G, U87-R and U373-R cell lines. The results suggested that TMZ
alone could not inhibit cell invasion, but cells pretreated with
different concentrations of SFN combined with TMZ significantly
reduced GBM cell invasive capacity (Fig. 6A). We further investigated the
effect of SFN on TMZ-inducing apoptosis, TMZ alone did not
demonstrate apoptosis induction in the cell lines compared with
control, however, the sequential therapy of the SFN and TMZ caused
a significant increase of apoptotic death even at lower doses of
SFN, suggesting that a synergistic induction of apoptosis developed
in the cells co-treated with SFN and TMZ (Fig. 6B). Moreover, caspase-3/7 activity
was also considerably elevated after synergism with SFN in
TMZ-resistant cells (Fig. 6C).
Collectively, these observations provide strong evidence for the
role of pretreatment of SFN in reversing TMZ-resistant GBM cells to
TMZ.
| Table IIMedian dose effect analysis to
characterize the interactions between SFN and TMZ. |
Table II
Median dose effect analysis to
characterize the interactions between SFN and TMZ.
| Concomitant TMZ
(250 μm) | Sequential TMZ (250
μm) |
|---|
|
|
|
|---|
| SFN (μm) | AF | AF | CI | DRI | AF | CI | DRI |
|---|
| U373-R |
| 5 | 0.02 | 0.01 | 28.12 | 0.29 | 0.27 | 0.93 | 2.63 |
| 10 | 0.07 | 0.05 | 15.73 | 0.37 | 0.41 | 0.78 | 4.67 |
| 20 | 0.13 | 0.09 | 6.54 | 0.64 | 0.79 | 0.54 | 7.85 |
| 30 | 0.25 | 0.14 | 2.45 | 0.98 | 0.85 | 0.15 | 26.79 |
| U87-R |
| 5 | 0.01 | 0.01 | 12.57 | 0.57 | 0.29 | 0.98 | 3.45 |
| 10 | 0.02 | 0.02 | 8.65 | 0.98 | 0.37 | 0.73 | 4.13 |
| 20 | 0.03 | 0.02 | 6.42 | 1.21 | 0.45 | 0.65 | 6.27 |
| 30 | 0.05 | 0.04 | 3.96 | 1.45 | 0.67 | 0.27 | 17.89 |
| T98G |
| 5 | 0.01 | 0.02 | 57.52 | 0.09 | 0.32 | 0.87 | 1.75 |
| 10 | 0.02 | 0.02 | 21.68 | 0.17 | 0.46 | 0.63 | 3.27 |
| 20 | 0.10 | 0.13 | 11.21 | 0.46 | 0.67 | 0.45 | 5.69 |
| 30 | 0.35 | 0.41 | 7.49 | 0.83 | 0.82 | 0.20 | 18.53 |
SFN enhances TMZ chemosensitivity in
vivo
To facilitate the in vitro experiments,
stable chemo-resistant cell line U373-R was used to establish
subcutaneous tumors. We subsequently investigated the synergistic
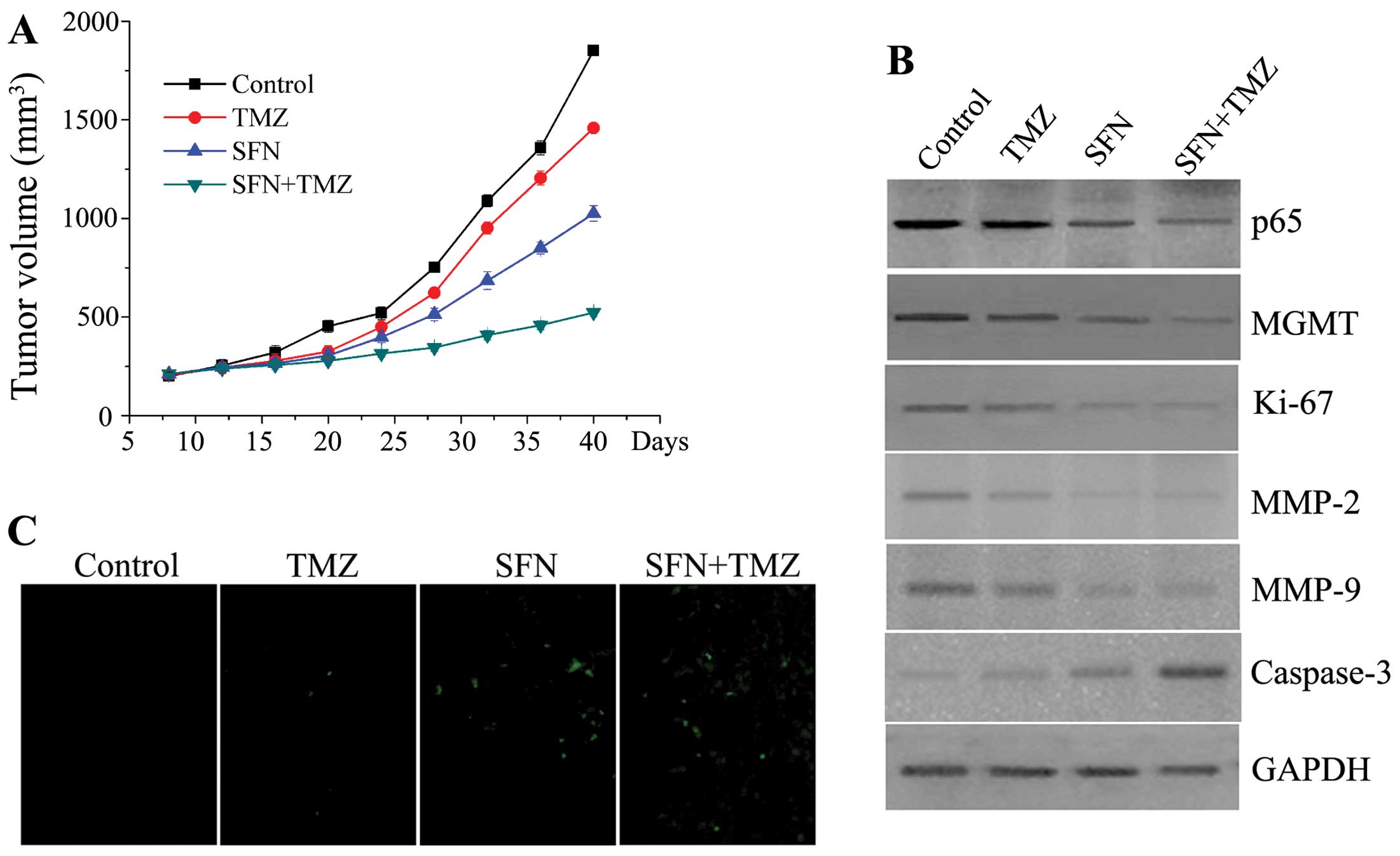
effects of a combination of SFN and TMZ in glioma xenograft nude
mouse models. In xenograft growth assay, TMZ alone did not
effectively suppress growth of the U373-R tumor (P>0.05).
However, SFN alone suppressed tumor growth more significantly than
control groups from day 28 (P<0.01), and the combination of SFN
and TMZ decreased tumor growth to a more significant extent,
moreover, the differences in tumor mass of the combined groups were
still marked compared with control and TMZ alone group at the
termination of the study (P<0.01) (Fig. 7A). Therefore, the SFN plus TMZ
treatment inhibited tumor growth significantly in the TMZ-resistant
glioma xenografts compared with the TMZ alone. To extend these
observations, western blot analysis and TUNEL analysis were used to
examine solid tumors after 40-day observation. As demonstrated in
Fig. 7B, the expression of NF-κB
p65 and MGMT protein were significant decreased in combined groups.
The protein levels of Ki-67, MMP2/9 and caspase-3 related with the
cell proliferation, invasion, and apoptosis in combined group were
prominently changed which is similar to results obtained from the
in vitro study. The TUNEL assay analysis verified the
synergistic pro-apoptotic effects of the combination of SFN and TMZ
in vivo (Fig. 7C). In
conclusion, these data demonstrated that SFN could effectively
inhibit TMZ-mediated cell growth and enhance TMZ-mediated cell
death in chemo-resistant xenograft nude mouse models.
Discussion
The inherent chemo- and radio-resistance of GBM are
the essential contributing factors that lead to an aggressive
clinical course and poor patient outcome. TMZ is widely recognized
as the first-line agent in patients with GBM, but the rapid
emergence of resistance to majority of GBM is still an urgent issue
to tackle (32,33). Many studies have been devoted to
sensitize TMZ to improve overall survival of GBM patients.
Bevacizumab in combination with TMZ could effectively inhibit tumor
cell proliferation, reduce tumor associated inflammation, and
induce cancer cell death (34).
P4HB inhibition was identified as an effective method in
combination with TMZ for the treatment of TMZ-resistant GBM
(35). Valparaiso acid can
regulate the effectiveness of TMZ-radio-chemotherapy and prolong
overall survival in patients with newly diagnosed GBM (36).
GBM is considered as a heterogeneous group of tumors
with differing cellular lineages, genetic alteration, biological
behavior and response to chemotherapeutics. TMZ resistance in GBM
also involves different cellular pathways resulting in large number
of gene changes (37,38). Although some of the potential
mechanisms have not been revealed, persistent NF-κB activity can be
a result of tumor amplification, invasion and chemo-resistance by
targeting transcription of various genes involved in
immunoregulation, inflammation, growth, carcinogenesis and
apoptosis (39). It is known that
NF-κB is a significant factor involved in MGMT transcription
independent of MGMT methylation status and aberrantly activates as
part of the DNA damage response. It was previously shown that high
NF-κB promotes GBM tumor growth and is associated with resistance
to alkylating agent-based chemotherapy through the transcriptional
activation of genes (16). Several
therapeutic inhibitors which can block NF-κB activation are in
development. Dehydroxymethylepoxyquinomicin (DHMEQ) has been
reported to be an effective NF-κB inhibitor with antiproliferative
properties in GBM (40).
Triptolide was able to inhibit NF-κB signaling in glioma initiating
cells, and then synergistically enhances TMZ-induced apoptosis
(41). Smac mimetic is reported to
sensitize glioblastoma cells to TMZ-induced apoptosis, and NF-κB
signaling pathway is identified as a critical mediator (42). Accordingly, inhibiting NF-κB
activity can increase sensitivity of GBM cells to alkylating
chemotherapeutic treatment and may assist in overcoming
treatment-induced chemo-resistance.
Despite these gains, the development of effective
therapies to improve the efficacy of TMZ is currently under
investigation to achieve durable clinical responses. A new strategy
for GBM chemotherapy which is aimed to increase antitumor responses
is the combination of natural compound with TMZ (43). Therefore, the present study was
performed to detect the role of SFN in sensitizing chemotherapeutic
agents of TMZ in TMZ-resistant glioma cells. We examined different
malignant glioma cell lines and exhibited differential expression
of the MGMT gene. Moreover, IC50 values varied between
the different cell lines, and the IC50 of T98G was much
high than other cell lines. We then successfully established
TMZ-resistant glioma cell models to confirm the sensitization of
SFN to TMZ.
An extensive amount of studies have shown that
chemo-prevention property of cruciferous vegetables can directly or
indirectly affect survival signaling pathways in cancer cells
(44). SFN inhibits TPA-induced
NF-κB activation and COX-2 expression by blocking two distinct
signaling pathways mediated by ERK1/2-IKKa and NAK-IKKb in MCF-10A
cells (45). The incidence of
metastatic nodules in the lung with SFN treatment is decreased in
transgenic adenocarcinoma of the mouse prostate (TRAMP) (46). SFN can downregulate the expression
of β-catenin through activation of caspase-3 in human cervical
carcinoma HeLa and hepatocarcinoma HepG2 cells. The combination of
SFN and chloroquine significantly reduce the incidence as well as
size of lymph node metastasis by inhibiting EMT and suppressing
proangiogenic cytokine VEGF in prostate cancer compared with
control group (22). SFN
counteracts aggressiveness of pancreatic cancer driven by
dysregulated Cx43-mediated gap junctional intercellular
communication (47).
Here, we provide evidence that SFN could exert
antitumor effect on TMZ-resistant cells, and finally reverse
chemo-resistance to TMZ by downregulating MGMT expression through
NF-κB signaling. Initially, we investigated proliferation of the
SFN inhibition of TMZ-resistant GBM cells. Next, we revealed
function of SFN sensitizing TMZ by blocking NF-κB signaling in
resistant GBM cells, and then, we focused on the synergistic
effects of SFN and TMZ in inhibiting proliferation, invasion and
inducing apoptosis in vitro and in vivo. In this
section, we demonstrated that the key point of synergism is
administration of SFN before cells exposure to TMZ. Only sequential
schedule of drug administration was efficient in TMZ-resistance
cell lines. Pre-existing levels of MGMT protein may be in part a
result of response to TMZ which is an important observation in the
following in vivo preclinical models or clinical trials.
The novelty of the present study resides in the
demonstration that SFN confers increased sensitivity toward TMZ in
an NF-κB-dependent manner in TMZ-resistant cells. Our data suggest
that long-term exposure to TMZ may result in the development of TMZ
resistance and promote malignant phenotypes in human malignant
glioma cells. SFN as a potent antitumor agent may reverse TMZ
resistance in GBM treatment and successfully translate the
experimental knowledge into robust clinical trials, what could be
clinically advantageous for GBM patients with intrinsic or acquired
drug resistance. This study provides evidence that SFN may be of
great clinical value for sensitizing NF-κB-MGMT-activity cells to
TMZ resistance.
Acknowledgements
This study was supported by the China National
Natural Scientific Fund (nos. 81201973, 81502654, 81172596 and
81482814) and Beijing Health System High Level Health Technology
Personnel Training Project Foundation.
References
|
1
|
Wakimoto H, Tanaka S, Curry WT, Loebel F,
Zhao D, Tateishi K, Chen J, Klofas LK, Lelic N, Kim JC, et al:
Targetable signaling pathway mutations are associated with
malignant phenotype in IDH-mutant gliomas. Clin Cancer Res.
20:2898–2909. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yiin JJ, Hu B, Schornack PA, Sengar RS,
Liu KW, Feng H, Lieberman FS, Chiou SH, Sarkaria JN, Wiener EC, et
al: ZD6474, a multitargeted inhibitor for receptor tyrosine
kinases, suppresses growth of gliomas expressing an epidermal
growth factor receptor mutant, EGFRvIII, in the brain. Mol Cancer
Ther. 9:929–941. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yue X, Lan F, Hu M, Pan Q, Wang Q and Wang
J: Downregulation of serum microRNA-205 as a potential diagnostic
and prognostic biomarker for human glioma. J Neurosurg. Jul
31–2015.(Epub ahead of print). PubMed/NCBI
|
|
4
|
Yeom SY, Nam DH and Park C: RRAD promotes
EGFR-mediated STAT3 activation and induces temozolomide resistance
of malignant glioblastoma. Mol Cancer Ther. 13:3049–3061. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Loftus JC, Dhruv H, Tuncali S, Kloss J,
Yang Z, Schumacher CA, Cao B, Williams BO, Eschbacher JM, Ross JT,
et al: TROY (TNFRSF19) promotes glioblastoma survival signaling and
therapeutic resistance. Mol Cancer Res. 11:865–874. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Oliva CR, Nozell SE, Diers A, McClugage SG
III, Sarkaria JN, Markert JM, Darley-Usmar VM, Bailey SM, Gillespie
GY, Landar A, et al: Acquisition of temozolomide chemoresistance in
gliomas leads to remodeling of mitochondrial electron transport
chain. J Biol Chem. 285:39759–39767. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ramirez YP, Mladek AC, Phillips RM,
Gynther M, Rautio J, Ross AH, Wheelhouse RT and Sakaria JN:
Evaluation of novel imidazotetrazine analogues designed to overcome
temozolomide resistance and glioblastoma regrowth. Mol Cancer Ther.
14:111–119. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Park I, Mukherjee J, Ito M, Chaumeil MM,
Jalbert LE, Gaensler K, Ronen SM, Nelson SJ and Pieper RO: Changes
in pyruvate metabolism detected by magnetic resonance imaging are
linked to DNA damage and serve as a sensor of temozolomide response
in glioblastoma cells. Cancer Res. 74:7115–7124. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Smalley S, Chalmers AJ and Morley SJ: mTOR
inhibition and levels of the DNA repair protein MGMT in T98G
glioblastoma cells. Mol Cancer. 13:1442014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gupta SK, Mladek AC, Carlson BL,
Boakye-Agyeman F, Bakken KK, Kizilbash SH, Schroeder MA, Reid J and
Sarkaria JN: Discordant in vitro and in vivo chemopotentiating
effects of the PARP inhibitor veliparib in temozolomide-sensitive
versus-resistant glioblastoma multiforme xenografts. Clin Cancer
Res. 20:3730–3741. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cen L, Carlson BL, Pokorny JL, Mladek AC,
Grogan PT, Schroeder MA, Decker PA, Anderson SK, Giannini C, Wu W,
et al: Efficacy of protracted temozolomide dosing is limited in
MGMT unmethylated GBM xenograft models. Neuro Oncol. 15:735–746.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Melguizo C, Prados J, González B, Ortiz R,
Concha A, Alvarez PJ, Madeddu R, Perazzoli G, Oliver JA, López R,
et al: MGMT promoter methylation status and MGMT and CD133
immunohistochemical expression as prognostic markers in
glioblastoma patients treated with temozolomide plus radiotherapy.
J Transl Med. 10:2502012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Etcheverry A, Aubry M, Idbaih A, Vauleon
E, Marie Y, Menei P, Boniface R, Figarella-Branger D, Karayan-Tapon
L, Quillien V, et al: DGKI methylation status modulates the
prognostic value of MGMT in glioblastoma patients treated with
combined radio-chemotherapy with temozolomide. PLoS One.
9:e1044552014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nguyen SA, Stechishin OD, Luchman HA, Lun
XQ, Senger DL, Robbins SM, Cairncross JG and Weiss S: Novel MSH6
mutations in treatment-naïve glioblastoma and anaplastic
oligodendroglioma contribute to temozolomide resistance
independently of MGMT promoter methylation. Clin Cancer Res.
20:4894–4903. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lee KE: Immunohistochemical assessment of
O(6)-methylguanine-DNA methyltransferase (MGMT) and its
relationship with p53 expression in endometrial cancers. J Cancer
Prev. 18:351–354. 2013. View Article : Google Scholar
|
|
16
|
Lavon I, Fuchs D, Zrihan D, Efroni G,
Zelikovitch B, Fellig Y and Siegal T: Novel mechanism whereby
nuclear factor kappaB mediates DNA damage repair through regulation
of O(6)-methylguanine-DNA-methyltransferase. Cancer Res.
67:8952–8959. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Persano L, Pistollato F, Rampazzo E, Della
Puppa A, Abbadi S, Frasson C, Volpin F, Indraccolo S, Scienza R and
Basso G: BMP2 sensitizes glioblastoma stem-like cells to
Temozolomide by affecting HIF-1α stability and MGMT expression.
Cell Death Dis. 3:e4122012. View Article : Google Scholar
|
|
18
|
Janssens S, Tinel A, Lippens S and Tschopp
J: PIDD mediates NF-kappaB activation in response to DNA damage.
Cell. 123:1079–1092. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rawat N, Alhamdani A, McAdam E, Cronin J,
Eltahir Z, Lewis P, Griffiths P, Baxter JN and Jenkins GJ: Curcumin
abrogates bile-induced NF-κB activity and DNA damage in vitro and
suppresses NF-κB activity whilst promoting apoptosis in vivo,
suggesting chemopreventative potential in Barrett's oesophagus.
Clin Transl Oncol. 14:302–311. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dong QG, Sclabas GM, Fujioka S, Schmidt C,
Peng B, Wu T, Tsao MS, Evans DB, Abbruzzese JL, McDonnell TJ, et
al: The function of multiple IkappaB: NF-kappaB complexes in the
resistance of cancer cells to Taxol-induced apoptosis. Oncogene.
21:6510–6519. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li F and Sethi G: Targeting transcription
factor NF-kappaB to overcome chemoresistance and radioresistance in
cancer therapy. Biochim Biophys Acta. 1805:167–180. 2010.PubMed/NCBI
|
|
22
|
Vyas AR, Hahm ER, Arlotti JA, Watkins S,
Stolz DB, Desai D, Amin S and Singh SV: Chemoprevention of prostate
cancer by d,l-sulforaphane is augmented by pharmacological
inhibition of autophagy. Cancer Res. 73:5985–5995. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cornblatt BS, Ye L, Dinkova-Kostova AT,
Erb M, Fahey JW, Singh NK, Chen MS, Stierer T, Garrett-Mayer E,
Argani P, et al: Preclinical and clinical evaluation of
sulforaphane for chemoprevention in the breast. Carcinogenesis.
28:1485–1490. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lenzi M, Fimognari C and Hrelia P:
Sulforaphane as a promising molecule for fighting cancer. Cancer
Treat Res. 159:207–223. 2014. View Article : Google Scholar
|
|
25
|
Pledgie-Tracy A, Sobolewski MD and
Davidson NE: Sulforaphane induces cell type-specific apoptosis in
human breast cancer cell lines. Mol Cancer Ther. 6:1013–1021. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lin LC, Yeh CT, Kuo CC, Lee CM, Yen GC,
Wang LS, Wu CH, Yang WC and Wu AT: Sulforaphane potentiates the
efficacy of imatinib against chronic leukemia cancer stem cells
through enhanced abrogation of Wnt/β-catenin function. J Agric Food
Chem. 60:7031–7039. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chaudhuri D, Orsulic S and Ashok BT:
Antiproliferative activity of sulforaphane in Akt-overexpressing
ovarian cancer cells. Mol Cancer Ther. 6:334–345. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lan F, Yu H, Hu M, Xia T and Yue X:
miR-144-3p exerts anti-tumor effects in glioblastoma by targeting
c-Met. J Neurochem. 135:274–286. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yamini B, Yu X, Dolan ME, Wu MH, Darga TE,
Kufe DW and Weichselbaum RR: Inhibition of nuclear factor-kappaB
activity by temozolomide involves O6-methylguanine induced
inhibition of p65 DNA binding. Cancer Res. 67:6889–6898. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Caporali S, Levati L, Graziani G, Muzi A,
Atzori MG, Bonmassar E, Palmieri G, Ascierto PA and D‘Atri S: NF-κB
is activated in response to temozolomide in an AKT-dependent manner
and confers protection against the growth suppressive effect of the
drug. J Transl Med. 10:2522012. View Article : Google Scholar
|
|
31
|
Lan F, Pan Q, Yu H and Yue X: Sulforaphane
enhances temozolomide-induced apoptosis because of down-regulation
of miR-21 via Wnt/β-catenin signaling in glioblastoma. J Neurochem.
134:811–818. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li R, Tang D, Zhang J, Wu J, Wang L and
Dong J: The temozolomide derivative 2T-P400 inhibits glioma growth
via administration route of intravenous injection. J Neurooncol.
116:25–30. 2014. View Article : Google Scholar
|
|
33
|
Grossman SA, Ye X, Piantadosi S, Desideri
S, Nabors LB, Rosenfeld M and Fisher J; NABTT CNS Consortium.
Survival of patients with newly diagnosed glioblastoma treated with
radiation and temozolomide in research studies in the United
States. Clin Cancer Res. 16:2443–2449. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Grossman R, Rudek MA, Brastianos H, Zadnik
P, Brem H, Tyler B and Blakeley JO: The impact of bevacizumab on
temozolomide concentrations in intracranial U87 gliomas. Cancer
Chemother Pharmacol. 70:129–139. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sun S, Lee D, Ho AS, Pu JK, Zhang XQ, Lee
NP, Day PJ, Lui WM, Fung CF and Leung GK: Inhibition of prolyl
4-hydroxylase, beta polypeptide (P4HB) attenuates temozolomide
resistance in malignant glioma via the endoplasmic reticulum stress
response (ERSR) pathways. Neuro Oncol. 15:562–577. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Weller M, Gorlia T, Cairncross JG, van den
Bent MJ, Mason W, Belanger K, Brandes AA, Bogdahn U, MacDonald DR,
Forsyth P, et al: Prolonged survival with valproic acid use in the
EORTC/NCIC temozolomide trial for glioblastoma. Neurology.
77:1156–1164. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liu X, Han EK, Anderson M, Shi Y,
Semizarov D, Wang G, McGonigal T, Roberts L, Lasko L, Palma J, et
al: Acquired resistance to combination treatment with temozolomide
and ABT-888 is mediated by both base excision repair and homologous
recombination DNA repair pathways. Mol Cancer Res. 7:1686–1692.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ciechomska IA, Gabrusiewicz K,
Szczepankiewicz AA and Kaminska B: Endoplasmic reticulum stress
triggers autophagy in malignant glioma cells undergoing
cyclosporine a-induced cell death. Oncogene. 32:1518–1529. 2013.
View Article : Google Scholar
|
|
39
|
Tergaonkar V, Pando M, Vafa O, Wahl G and
Verma I: p53 stabilization is decreased upon NFkappaB activation: A
role for NFkappaB in acquisition of resistance to chemotherapy.
Cancer Cell. 1:493–503. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Brassesco MS, Roberto GM, Morales AG,
Oliveira JC, Delsin LE, Pezuk JA, Valera ET, Carlotti CG Jr, Rego
EM, de Oliveira HF, et al: Inhibition of NF-κB by
dehydroxymethylepoxyquinomicin suppresses invasion and
synergistically potentiates temozolomide and γ-radiation
cytotoxicity in glioblastoma cells. Chemother Res Pract.
2013:5930202013.
|
|
41
|
Sai K, Li WY, Chen YS, Wang J, Guan S,
Yang QY, Guo CC, Mou YG, Li WP and Chen ZP: Triptolide
synergistically enhances temozolomide-induced apoptosis and
potentiates inhibition of NF-κB signaling in glioma initiating
cells. Am J Chin Med. 42:485–503. 2014. View Article : Google Scholar
|
|
42
|
Wagner L, Marschall V, Karl S, Cristofanon
S, Zobel K, Deshayes K, Vucic D, Debatin KM and Fulda S: Smac
mimetic sensitizes glioblastoma cells to Temozolomide-induced
apoptosis in a RIP1- and NF-κB-dependent manner. Oncogene.
32:988–997. 2013. View Article : Google Scholar
|
|
43
|
Turrini E, Ferruzzi L and Fimognari C:
Natural compounds to overcome cancer chemoresistance: Toxicological
and clinical issues. Expert Opin Drug Metab Toxicol. 10:1677–1690.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Asakage M, Tsuno NH, Kitayama J, Tsuchiya
T, Yoneyama S, Yamada J, Okaji Y, Kaisaki S, Osada T, Takahashi K,
et al: Sulforaphane induces inhibition of human umbilical vein
endothelial cells proliferation by apoptosis. Angiogenesis.
9:83–91. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kim HN, Kim DH, Kim EH, Lee MH, Kundu JK,
Na HK, Cha YN and Surh YJ: Sulforaphane inhibits phorbol
ester-stimulated IKK-NF-κB signaling and COX-2 expression in human
mammary epithelial cells by targeting NF-κB activating kinase and
ERK. Cancer Lett. 351:41–49. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Singh SV, Warin R, Xiao D, Powolny AA,
Stan SD, Arlotti JA, Zeng Y, Hahm ER, Marynowski SW, Bommareddy A,
et al: Sulforaphane inhibits prostate carcinogenesis and pulmonary
metastasis in TRAMP mice in association with increased cytotoxicity
of natural killer cells. Cancer Res. 69:2117–2125. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Forster T, Rausch V, Zhang Y, Isayev O,
Heilmann K, Schoensiegel F, Liu L, Nessling M, Richter K, Labsch S,
et al: Sulforaphane counteracts aggressiveness of pancreatic cancer
driven by dysregulated Cx43-mediated gap junctional intercellular
communication. Oncotarget. 5:1621–1634. 2014. View Article : Google Scholar : PubMed/NCBI
|