Introduction
The epithelial-to-mesenchymal transition (EMT)
enables cancer to invade and metastasize (1,2). In
addition, accumulating evidence strongly suggests that the EMT
induces cancer chemoresistance (3)
and radioresistance (4,5) and has an immunoprotective effect
(6). Therefore, the EMT
constitutes a major malignant propensity to cancer development and
is a major obstacle to curing cancer. TGF-β is a well-known driving
factor of the EMT in many cancers (7). Activations of the Smad3,
PI3K/Akt/mTOR, and MEK/Erk cascades are thought to be key
mechanisms by which TGF-β induces EMT (8–11).
FGF-2 also induces EMT through the activation of the MEK/Erk
signaling pathway. Moreover, a combination of TGF-β and FGF-2
strongly induces EMT because TGF-β induces isoform switching of the
FGF-receptor and increases cellular sensitivity to FGF-2 (12). All these signaling pathways
ultimately upregulate the transcriptional factors slug, snail, and
ZEB1, to induce EMT (13–15).
Recent advances in cancer biology have expanded
therapeutic modalities to include cytotoxic chemotherapy, targeted
therapy, and immune-checkpoint therapy. For example, the survival
time of patients with advanced non-small cell lung cancer (NSCLC)
harboring a mutation in either the epidermal growth factor receptor
(EGFR) gene or the anaplastic lymphoma kinase (ALK)
gene has been greatly prolonged using tyrosine kinase inhibitors
(16) for the corresponding
mutation. Nevertheless, cancer still recurs even after the drastic
effects of targeted therapy. Although the mechanisms underlying
drug resistance, either to cytotoxic or targeted agents, are not
fully understood, they consist of mutational and non-mutational
mechanisms. EMT is one of the non-mutational mechanisms of drug
resistance (3). For example, EMT
markers were reportedly upregulated in patients with NSCLC showing
resistance to cisplatin-based chemoradiotherapy (17). EMT is also reportedly related to
resistance to docetaxel in patients with prostate cancer (18). The results of several in
vitro experiments have supported these clinical observations:
EMT markers were upregulated in ovarian cancer cell lines resistant
to paclitaxel (19), morphological
changes suggesting EMT were observed in colon cancer cells
resistant to oxaliplatin (20),
and the knockdown of snail increased the sensitivity of NSCLC cells
to cisplatin (21). Therefore, the
EMT appears to be closely related to resistance to cytotoxic
agents. In addition, EMT is a candidate for the application of a
non-mutational resistance mechanism to targeted therapy. The in
vitro induction of EMT rendered human NSCLC cells harboring
EGFR mutations less sensitive to EGFR-TKIs (22–24).
Therefore, EMT reversion to the original epithelial
phenotype might be a new therapeutic strategy for overcoming
resistance to chemotherapy and/or targeted therapy. Although some
agents, including metformin and mTOR inhibitors, can reportedly
revert the EMT phenotype, the relationship between this phenomenon
and changes in drug sensitivity is not fully understood. This study
examined the molecular mechanisms underlying EMT induction and
reversion by investigating changes in drug sensitivity and
immune-protectiveness according to EMT induction and reversion in
human lung adenocarcinoma cell lines harboring an EGFR
mutation.
Materials and methods
Cells and reagents
Human lung adenocarcinoma cell lines, PC-9 and
HCC-827, were used throughout the study. PC-9 was purchased from
Riken Cell Bank (accession no. RCB4455; Tsukuba, Japan). HCC-827
was obtained from the American Type Culture Collection (accession
no. CRL-2868; Manassas, VA, USA). The cell lines have an identical
activating mutation in EGFR: a deletion (del E746-A750) in exon 19.
The cells were cultured as a monolayer in RPMI-1640 medium (cat.
no. R8758; Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10%
fetal bovine serum (FBS), 100 U/ml penicillin and 100 mg/ml
streptomycin (cat. no. 10378-016; Life Technologies, Carlsbad, CA,
USA) in a 37°C humidified atmosphere containing 5%
CO2.
The EGFR-TKI gefitinib (cat. no. 3000; Tocris
Bioscience, Ellisville, MO, USA) was dissolved in DMSO (cat. no.
D2650; Sigma-Aldrich) and stored at −20°C until use. Cisplatin at a
concentration of 0.5 mg/ml (pH 2.5–5.5) was purchased from Nihon
Kayaku (Tokyo, Japan) and stored at 4°C. Metformin
(1,1-dimethylbiguanide hydrochloride, cat. no. D150959-5G;
Sigma-Aldrich) was dissolved in phosphate-buffered saline (PBS) at
a concentration of 100 mM and stored at 4°C. PP242 (cat. no.
165-24441; Wako, Osaka, Japan), a potent inhibitor of mTOR complex
1 and C2 (mTOR-C1 and -C2), was dissolved in DMSO at concentrations
of 10 mM and stored in aliquots at −80°C. Each agent was diluted in
complete medium, and the final concentration of DMSO was
<0.001%. Recombinant human TGF-β1 was purchased from PeproTech
(cat. no. 100-21C; Rock Hill, NJ, USA), and recombinant human FGF-2
was purchased from Cell Signaling Technology (cat. no. 8910LC;
Beverly, MA, USA).
Mouse monoclonal anti-β-actin (cat. no. A2228) and
anti-fibronectin (cat. no. F3648) were purchased from
Sigma-Aldrich. Rabbit monoclonal anti-vimentin (cat. no. 5741P),
anti-slug (cat. no. 9585S), anti-p70S6K (cat. no. 2708S),
anti-phospho-p70S6K (cat. no. 9205S), anti-Erk1/2 (cat. no. 9102S),
anti-phospho-Erk1/2 (cat. no. 9101S), anti-Akt (cat. no. 9272S),
anti-phospho-Akt on Ser473 (cat. no. 9271S), anti-Smad3 (cat. no.
9523S) and anti-phospho-Smad3 antibodies (cat. no. 9516S) were
purchased from Cell Signaling Technology. The anti-E cadherin
antibody was purchased from BD Transduction Laboratory (cat. no.
610181; Lexington, KY, USA). Anti-PD-L1/CD274 monoclonal antibody
was purchased from Spring Bioscience (cat. no. M4420; Pleasanton,
CA, USA). Anti-rabbit IgG-horseradish peroxidase-linked antibody
was purchased from Medical and Biological Laboratories (cat. no.
458; Nagoya, Japan). ECL™ horseradish peroxidase linked anti-mouse
antibody was purchased from GE Healthcare (cat. no. NA931; Uppsala,
Sweden). Anti-mouse IgG Fab2 Alexa Fluor® 488 molecular
probe (cat. no. 4408S) and anti-rabbit IgG Fab2 Alexa
Fluor® 555 molecular probe (cat. no. 4413S) was
purchased from Cell Signaling Technology.
In vitro induction and reversion of
EMT
EMT was induced by treating cells with TGF-β and/or
FGF-2. Based on a series of preliminary experiments, a combination
of TGF-β at a concentration of 10 ng/ml and FGF-2 at a
concentration of 10 ng/ml was admixed into the complete medium for
inducing EMT after 24 h of serum starvation. To evaluate the EMT
phenotypes, cells harvested at 48 h after the admixture of the
agents were used. For EMT reversion, PP242 at a concentration of
1.0, 10 or 100 nM, metformin at a concentration of 0.1, 1.0 or 10
mM, or DMSO at a concentration of 0.1 or 1.0% (vol/vol) was added
to cells in which the EMT had already been induced, with
TGF-β/FGF-2 being supplemented continuously. The phenotypes were
evaluated after 72 h of culture with the reverting agent.
Evaluation of cell viability
Cell viability was determined using the MTT dye
reduction method. A total of 500 cells/well were plated in 96-well
culture plates. After 24 h of serum-free culture, the cells were
treated to induce EMT and to revert EMT, as previously described.
Then, the cells were treated with various concentrations of
gefitinib or cisplatin for an additional 72 h of culture. To each
well, 15 μl of dye solution (cat. no. G402A; Promega, Madison, WI,
USA) was added for further incubation at 37°C for 4 h, followed by
the addition of 100 μl of stop solution (cat. no. G4001; Promega)
for a further 1 h of incubation. The absorbance at 570 nm in the
resulting solution was measured using Infinite® 200 PRO
(FPRO-T; Tecan, Seestrasse, Switzerland). Cell viability was
determined by dividing the absorbance value of the treated cells by
that of the untreated cells.
Real-time PCR (RT-PCR)
The expression of mRNA was semi-quantified using
RT-PCR with the TaqMan® Gene Expression Assays, Step One
Plus Real-Time PCR system, and Step One Software (Life
Technologies). After culturing cells at 80% confluence in 6-well
culture plates, the total RNA was extracted using the
RNeasy® Mini kit (cat. no. 74104; Qiagen, Venlo,
Limburg, The Netherlands) and the cDNA was immediately synthesized
using SuperScript® First-Strand Synthesis for RT-PCR
(cat. no. 11904-018; Life Technologies) for RT-PCR according to the
manufacturer's instructions. The gene expression data were
calculated relative to the glyceraldehyde-3-phosphate dehydrogenase
(GAPDH) level. TaqMan probes for GAPDH, E-cadherin, vimentin,
fibronectin, slug, and PD-L1 were obtained by ordering from Applied
Biosystems through Life Technologies (assay identification numbers:
Hs02758991_g1, Hs01023894_m1, Hs00185584_m1, Hs00365052_m1,
Hs00950344_m1 and Hs01125301_m1).
Western blot analysis
Cells prepared in a manner similar to that used for
the mRNA isolation studies mentioned above were lysed in RIPA
buffer (cat. no. 89900; Thermo Fisher Scientific) with protease and
phosphatase inhibitor (cat. no. 1861281; Thermo Fisher Scientific)
on ice to extract the total protein. After measurement with a BCA
assay (Pierce® BCA Protein assay kit, cat. no. 23225;
Thermo Fisher Scientific, Rockford, IL, USA), 10 μg of protein in
each lane in a 10% precast gel (Mini-PROTEAN®
TGXTM, cat. no. 456-1025, Bio-Rad, Hercules, CA, USA)
was electrophoresed using the Powger Pac Basic, Mini
PROTEAN® Tetra System (Bio-Rad), followed by transfer to
polyvinylidenedifluoride membranes (Trans-Blot®
TurboTM Transfer Pack 0.2 μm PVDF, cat. no. 170-4156;
Bio-Rad) using the Trans-Blot Turbo™ Transfer System (Bio-Rad). The
membranes were probed with antibodies using the iBind™ Western
Device, Cards and Solution kit (cat. no. SLF 1000, 1010 and 1020;
Novex by Life Technologies), followed by visualization using an
enhanced chemiluminescence substrate kit (Clarify™ Western ECL
Substrate, cat. no. 170-5060; Bio-Rad). The images were analyzed
using Molecular imager® ChemiDOC™ XRS+ (Bio-Rad) and
quantified by Image Lab® Software (Bio-Rad). The
expression of phosphorylated proteins was also determined as the
relative ratio per total protein.
Fluorescent immunohistochemistry
For the fluorescent immunohistochemical evaluation
of protein expression, cells were cultured up to 80% confluence in
a 4-well chamber slide (cat. no. 177399; Thermo Fisher Scientific)
and then washed with PBS and fixed with 4% paraformaldehyde at room
temperature for 15 min, followed by incubation with the primary
antibodies (diluted in PBS with 1% bovine serum albumin) at 4°C
overnight. Non-specific binding was blocked using 2% bovine serum
albumin. Then, the cells were washed with PBS and incubated with
secondary antibodies at room temperature for 30 min before being
mounted in ProLong® Gold Antifade Reagent with 4′,
6-diamidino-2-phenylindole (DAPI) (cat. no. 8961, Cell Signaling
Technology) for observation with EVOS FL (AMF4300; Life
Technologies).
Apoptosis assay
Apoptosis was examined using flow cytometry with
Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide
(PI) (cat. no. 556547; BD Biosciences, San Jose, CA, USA). Briefly,
cells were treated with various concentrations of gefitinib or
cisplatin for 24 h, trypsinized, washed with cold PBS, and
suspended in binding buffer (BD Biosciences) at a concentration of
1×107 cells/ml. Thereafter, they were stained with 5 μl
of Annexin V-FITC and 5 μl of PI in 100 μl of cell suspension and
incubated for 15 min at room temperature in the dark for processing
using flow cytometry. All the early apoptotic cells (Annexin
V-positive, PI-negative), necrotic/late apoptotic cells (double
positive), and living cells (double negative) were counted using
FACSCanto IITM (BD Biosciences).
Cell cycle
The cell cycle distributions were determined using a
PI single-color flow cytometry (FACSCanto II), according to the
manufacturer's instructions. Briefly, cells were trypsinized and
washed twice with cold PBS, then fixed with 70% ethanol. Before
analysis, the cell suspensions were washed with PBS, suspended in
100 μl of binding buffer containing 5 μl of PI at a concentration
of 1×107 cells/ml, followed by incubation for 15 min at
room temperature in the dark.
Wound healing assay
The cell migration ability was evaluated using a
wound healing assay. Cells were cultured until they became
confluent in 6-well plates, at which time the surfaces of the
plates were scratched with micropipette tips (cat. no. 2069-05;
Thermo Fisher Scientific) and the culture medium was immediately
exchanged, followed by culture for a further 9 h. The scratched
wounds were viewed using an inverted microscope (x10 objective)
(ECLIPSE TS100; Nikon, Tokyo, Japan). The scratched areas were
quantified using Image J (from National Institutes of Health), and
the wound closure rates were determined as a percentage of the
total repaired area per hour and were normalized to the
control.
Statistical analyses and ethical
considerations
Data are presented as the means ± standard errors,
and differences between groups were evaluated using the Student's
t-test. A P-value of <0.05 (2-tailed) was considered
statistically significant. All the experiments were conducted in
close adherence to the institutional regulations.
Results
In vitro induction of EMT
The EMT phenotype was confirmed by a decreased
expression of E-cadherin (an epithelial marker) and an increased
expression of vimentin, fibronectin (mesenchymal markers), and
slug. RT-PCR for the HCC-827 cells revealed that TGF-β upregulated
vimentin, fibronectin, and slug without downregulating E-cadherin;
that FGF-2 downregulated E-cadherin without upregulating vimentin,
fibronectin, and slug; and that the combination of TGF-β and FGF-2
downregulated E-cadherin and upregulated vimentin, fibronectin, and
slug, resulting in a perfect EMT phenotype. In the PC-9 cells, in
contrast, the combination of TGF-β and FGF-2 upregulated vimentin,
fibronectin, and slug without downregulating E-cadherin (Fig. 1A). The altered expressions of
E-cadherin at the cell junctions and of fibronectin in the
cytoplasm were further confirmed using fluorescent
immunohistochemistry, except that the PC-9 cells failed to show any
change in E-cadherin expression (Fig.
1B). The morphological changes in the cells were concordant
with these observations: that is, the HCC-827 cells became
elongated in morphology and less adhesive when they exhibited the
EMT phenotype, whereas the PC-9 cells were unaltered in morphology,
since the original cells were already ostensibly elongated and less
adhesive (Fig. 2A). A wound
healing assay disclosed increased cell-mobility for the EMT
phenotype in both cell lines (Fig.
2B). The accumulation of G0/G1 cells in the EMT phenotype was
observed in a cell cycle analysis (Fig. 2C). These findings clearly indicate
the successful induction of EMT in both cell lines. Based on this
fact, the combination of TGF-β and FGF-2 was used to induce EMT in
the remainder of this study.
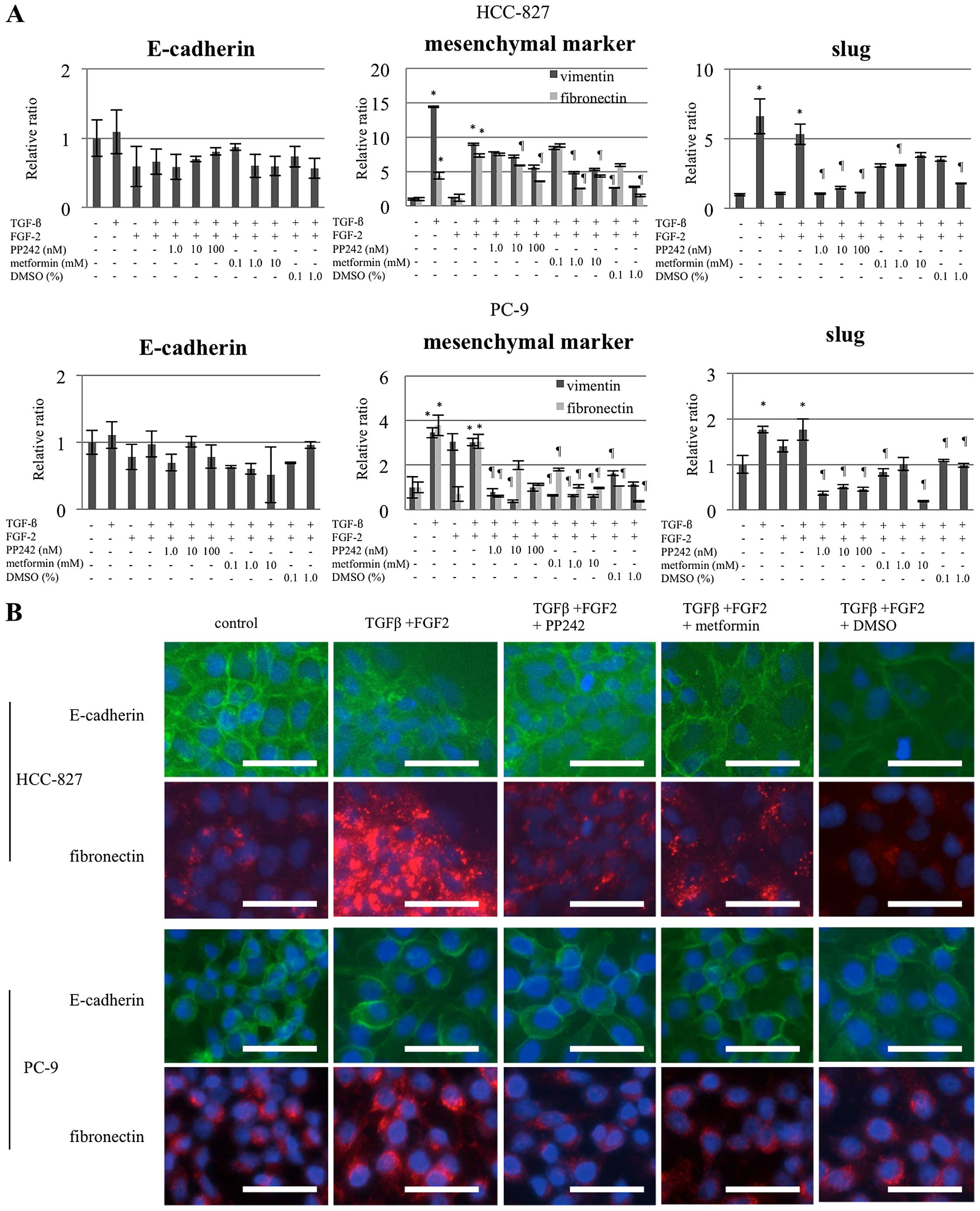 | Figure 1Evidence of EMT induction and
reversion as assessed by changes in the expressions of E-cadherin,
vimentin, fibronectin, and slug in the human adenocarcinoma lung
cancer cell lines, HCC827 and PC9. (A) The relative ratio of mRNA
normalized to that in the control was evaluated using RT-PCR for
cells treated with/without TGF-β (10 ng/ml) and/or FGF-2 (10
ng/ml). The treatment duration was 48 h after a 24-h serum
starvation period. TGF-β alone and the combination of TGF-β/FGF-2
showed comparable effects on increasing the expression of the
mesenchymal markers and slug in both cell lines, whereas the
combination, but not TGF-β alone, was effective in decreasing
E-cadherin expression in HCC827. The combination was thereafter
used to induce EMT throughout the study. In addition, PP242 (1.0,
10 or 100 nM), metformin (0.1, 1.0 or 10 mM), and DMSO (0.1 or
1.0%) reverted the altered expression of these markers. Of note,
treatment with these 3 drugs was started in cells that were already
exhibiting EMT induction. The columns and bars represent the means
and SEs (n=6, in triplicate) of the relative ratios compared with
the controls, respectively. Statistically significant differences
(P<0.05) compared with the control (no TGF-β/FGF-2) and
TGF-β/FGF-2-treated cells are indicated by * and
¶, respectively. (B) The changes in E-cadherin
expression on the cell surface and the changes in fibronectin
expression in the cytoplasm were confirmed using fluorescent
immunohistochemical staining. The doses for PP242, metformin, and
DMSO were 100 nM, 1.0 mM, and 1.0%, respectively. Cells were
counterstained with DAPI. The bars represent 50 μm. |
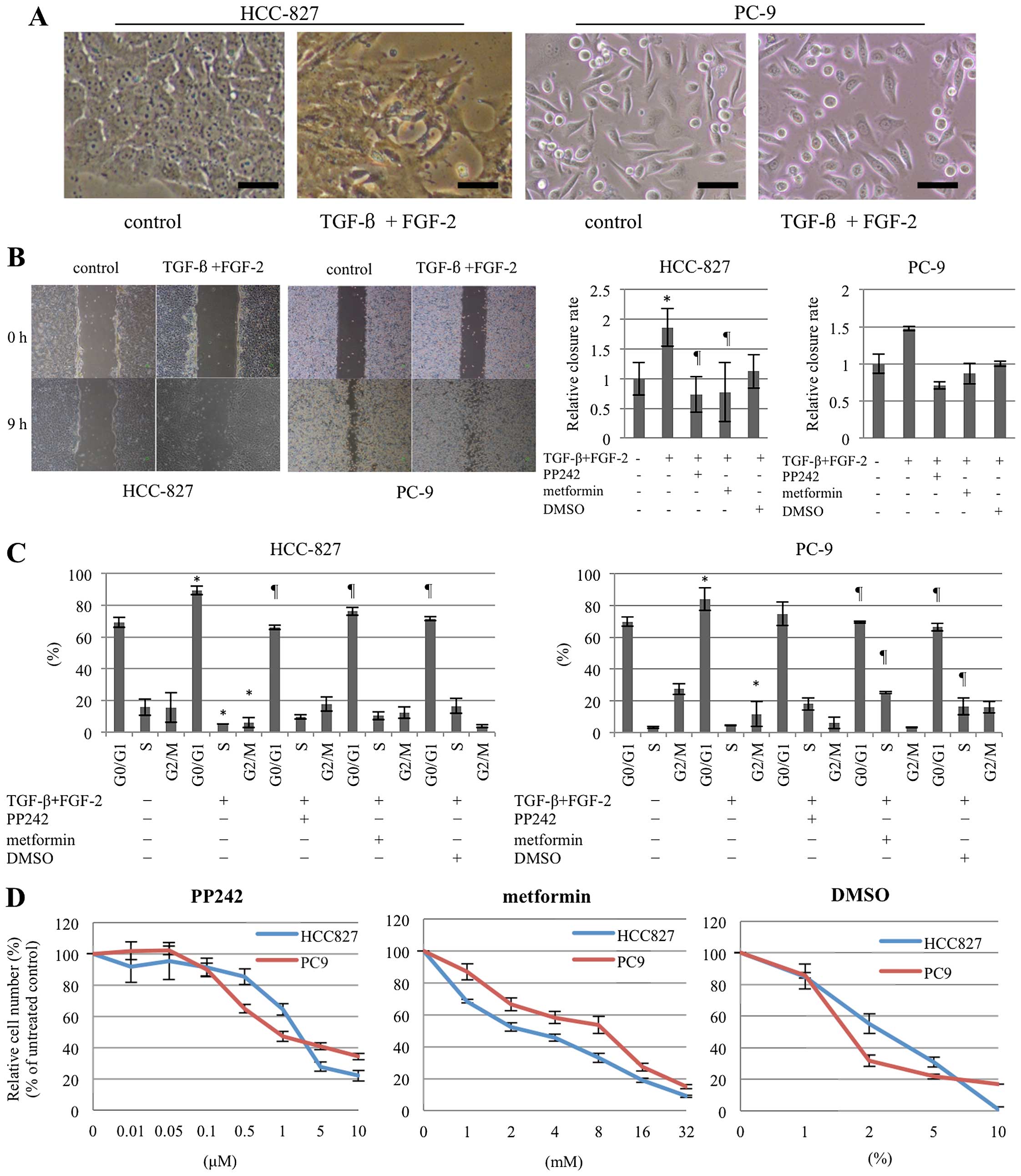 | Figure 2Phenotypic alterations according to
EMT induction and reversion. (A) The combination of TGF-β/FGF-2 (10
ng/ml, each) induced clear morphological changes in HCC-827,
resulting in an elongated and less-adhesive appearance; in
contrast, the morphology of the PC-9 cells was unaltered, since the
original cells already exhibited an elongated and less-adhesive
appearance. The bars represent 50 μm. (B) A wound healing assay
disclosed enhanced cell motility after treatment with TGF-β/FGF-2
in both cell lines, whereas PP242, metformin, and DMSO at the same
doses as those used in Fig. 1B
suppressed the phenomenon, as shown by the representative phase
contrast images on the left. For quantification, the distance of
cell migration was measured after 9 h in 3 fields (n=9, in
triplicate) for each group; the results are presented on the right.
Statistically significant differences (P<0.05) compared with the
control (no TGF-β/FGF-2) and TGF-β/FGF-2-treated cells are
indicated by * and ¶, respectively. (C) FACS
analyses demonstrated a significant accumulation at the G0/G1
phases together with a significant decrease in the S and G2/M
phases in TGF-β/FGF-2-treated cells in both cell lines. PP242,
metformin, and DMSO at the same doses as those used in Fig. 1B reverted these alterations.
Statistically significant differences (P<0.05) compared with the
control (no TGF-β/FGF-2) and TGF-β/FGF-2-treated cells are
indicated by * and ¶, respectively. (D) MTT
assays for PP242, metformin, and DMSO revealed dose-dependent
growth inhibition of the cells. Based on the results, the dose
ranges for these 3 agents were determined so that growth inhibition
was not so significant within these dose ranges. The dots and bars
represent the means and SEs (n=3, in triplicate), respectively. |
Reversion of EMT induction
For the EMT reversion that was induced as described
above, PP242, metformin, and DMSO were used. Because these agents
are known to be cytotoxic, the cell survival curves resulting from
the use of these agents were determined using an MTT assay
(Fig. 2D). Based on the resulting
data, doses of 1.0, 10, or 100 nM for PP242; 0.1, 1.0, or 10 mM for
metformin; and 0.1 or 1.0% (vol/vol) for DMSO were chosen because
their cytotoxicity was considered not to be very high at these
doses. Although none of the agents significantly increased the
expression of E-cadherin through EMT reversion in the PC-9 cells,
PP242 and metformin administered at the lowest concentrations
tended to increase E-cadherin expression in the HCC-827 cells. The
three agents significantly decreased the expressions of vimentin,
fibronectin, and slug in both cell lines (Fig. 1A). Fluorescent immunohistochemistry
studies also supported the findings in HCC-827 cells: treatment
with PP242, metformin, or DMSO suppressed the expression of
fibronectin in the cytoplasm, with E-cadherin expression on the
surface being unaltered (Fig. 1B).
A wound healing assay disclosed the reversion of cell-migration
activity with each of the agents in both cell lines (Fig. 2B). All three reverting agents
suppressed the G0/G1 accumulation resulting from EMT induction and
reverted the cell cycle to the pattern of the original cells in
both cell lines (Fig. 2C). These
observations indicate that the agents are capable of reverting the
TGF-β/FGF-2-induced EMT phenotype.
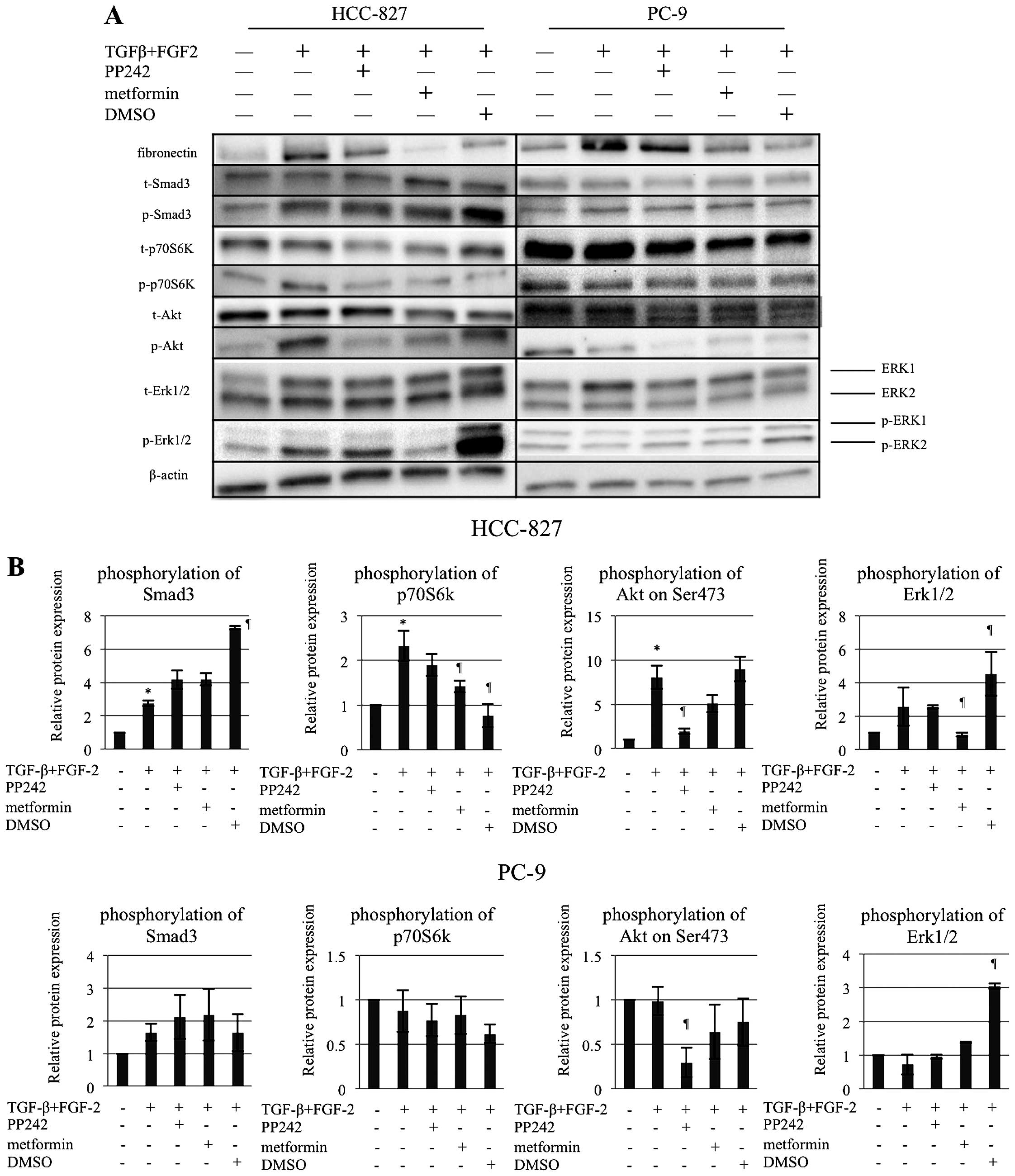
Alterations of Smad3, MEK/Erk,
mTOR-C1/p70S6K, and mTOR-C2/Akt (Ser473) pathways according to EMT
induction and reversion
The possible mechanisms underlying
TGF-β/FGF-2-induced EMT and its reversion by PP242, metformin, and
DMSO were scrutinized by immunoblotting for phosphorylated Smad3,
Erk1/2, p70S6K, and Akt (Ser473) (Fig.
3). In the PC-9 cells, the activation of the Smad3 pathway
alone was related to EMT induction, whereas none of the 3 reverting
agents suppressed the activated Smad3. All the agents showed a
tendency to suppress the phosphorylation of Akt (Ser473). DMSO
drastically and very paradoxically activated the phosphorylation of
Erk1/2 (Fig. 3). On the contrary,
in HCC-827, all the four pathways were related to the induction of
EMT. As to the reverting agents, PP242 significantly suppressed the
activated phosphorylation of Akt (Ser473) and showed a tendency to
inhibit the activated phosphorylation of p70S6K; metformin
significantly suppressed the activated phosphorylation of Erk1/2
and p70S6K and showed a tendency to inhibit the activated
phosphorylation of Akt (Ser473); and DMSO significantly suppressed
the activated phosphorylation of p70S6K, whereas it again
significantly and paradoxically further activated the
phosphorylation of Smad 3 and Erk1/2 (Fig. 3).
Alterations of drug sensitivity according
to EMT induction and reversion
Both PC-9 and HCC-827 cells exhibiting
TGF-β/FGF-2-induced EMT were significantly resistant to gefitinib,
compared with untreated cells, whereas treatment with TGF-β alone
or FGF-2 alone had no effect or only a modest effect on sensitivity
to gefitinib (Fig. 4A). On the
contrary, PC-9 cells exhibiting TGF-β/FGF-2-induced EMT showed an
unaltered sensitivity to cisplatin, whereas HCC-827 cells
exhibiting TGF-β/FGF-2-induced EMT were significantly resistant to
cisplatin (Fig. 4A). PP242 and
metformin had modest re-sensitizing effects for gefitinib in cells
exhibiting EMT induction in both cell lines, with the effects of
DMSO being unequivocal. Although none of the agents had any effect
on cisplatin sensitivity in the PC-9 cells, PP242 and DMSO had a
modest re-sensitizing effect on cisplatin in the HCC-827 cells,
whereas metformin seemingly had no effect on the sensitivity of the
cells to cisplatin (Fig. 4A).
These phenomena were precisely supported by the apoptosis assay.
Apoptosis induced by gefitinib in the PC-9 and HCC-827 cells and
apoptosis induced by cisplatin in the HCC-827 cells, but not in the
PC-9 cells, was significantly suppressed in cells exhibiting the
EMT phenotype, compared with the control cells (Fig. 4B). Nevertheless, PP242, metformin,
and DMSO restored apoptosis with gefitinib, but not with cisplatin,
in the PC-9 cells, whereas all three of the reverting agents
restored apoptosis caused by either gefitinib or cisplatin that was
suppressed in the HCC-827 cells exhibiting EMT induction (Fig. 4B).
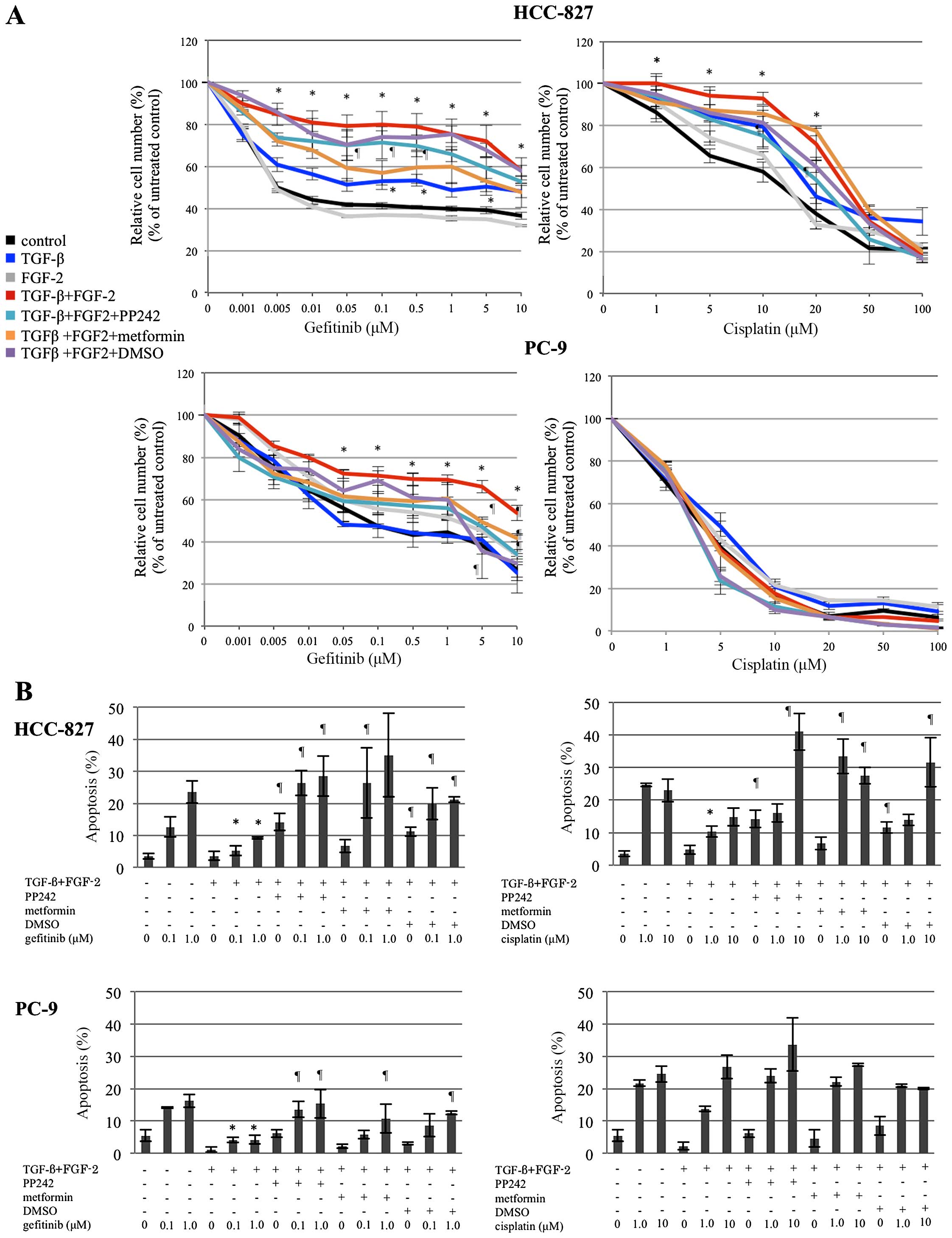 | Figure 4Alterations of sensitivity to
gefitinib and cisplatin according to EMT induction and reversion.
(A) MTT assay after 72 h of treatment with gefitinib or cisplatin
showed that sensitivity to gefitinib was not significantly altered
when the cells were treated with TGF-β or FGF-2 alone, whereas the
combination of these 2 agents induced significant resistance to
gefitinib. PP242 and metformin partly restored the sensitivity to
gefitinib in both cell lines. As to the sensitivity to cisplatin,
similar effects were only observed in the HCC-827 cells. The dots
and bars represent the means and SEs (n=6, in triplicate),
respectively. (B) Apoptotic cells were counted using Annexin V
staining at 24 h after exposure to gefitinib or cisplatin, then
assessed using FACS. The percentages for all the early apoptotic
cells and for the necrotic/late apoptotic cells were then
calculated. Apoptosis increased with gefitinib or cisplatin
treatment in a dose-dependent manner in the control (no
TGF-β/FGF-2). In both cell lines with TGF-β/FGF-2-induced EMT,
however, apoptosis was significantly suppressed after treatment
with gefitinib. Treatment with PP242, metformin, and DMSO
significantly restored the sensitivity to gefitinib, as assessed by
evaluating apoptosis. Similar effects were also observed for the
sensitivity to cisplatin in HCC-827 cells, but not in PC-9 cells.
The columns and bars represent the means and SEs (n=3, in
triplicate), respectively. Statistically significant differences
(P<0.05) compared with the control (no TGF-β/FGF-2) and
TGF-β/FGF-2-treated cells are indicated by * and
¶, respectively, in (A) and (B). |
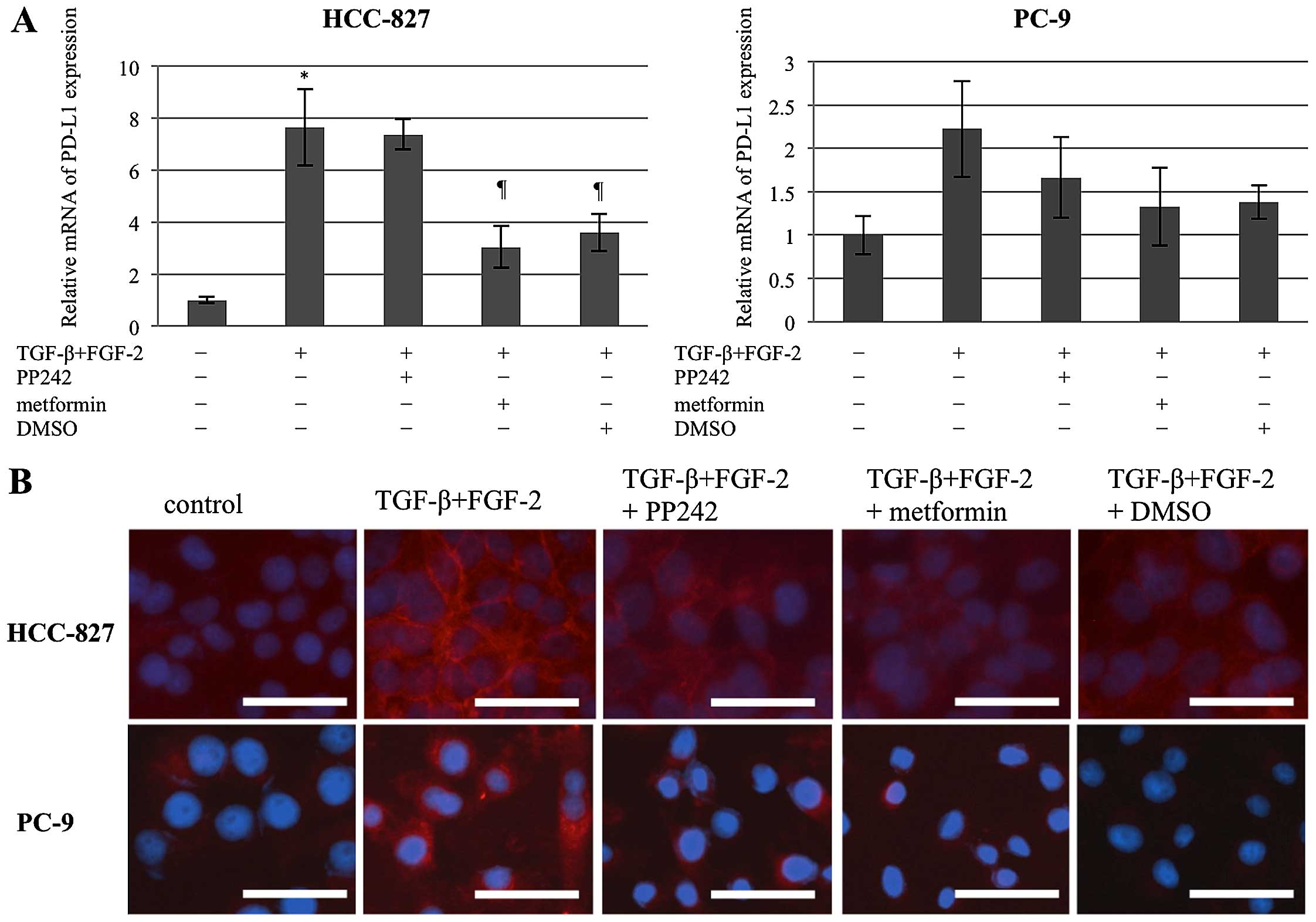
Alteration of PD-L1 expression according
to EMT induction and reversion
In the HCC-827 cells, TGF-β/FGF-2-induced EMT cells
were related to a strong increase in PD-L1 expression, and
metformin and DMSO, but not PP242, suppressed this phenomenon. The
PC-9 cells also displayed a similar phenomenon, although the
difference was not statistically significant (Fig. 5A). Fluorescent immunohistochemistry
for PD-L1 expression on the cellular membranes supported these
findings, except that PP242 significantly suppressed EMT-induced
PD-L1 overexpression (Fig. 5B), in
contrast to the absence of any effect when assessed using RT-PCR
(Fig. 5A).
Discussion
This study, which was performed using two NSCLC cell
lines harboring an EGFR mutation, confirmed the induction of
the EMT using a combination of TGF-β and FGF-2. It also
demonstrated the reversion of EMT induction using any of the
agents, PP242, metformin, or DMSO. These phenotypic changes were
supported by morphological changes; the altered expression of an
epithelial marker, mesenchymal markers, and slug; altered cell
mobility; and an altered cell cycle, with the slight exception that
the expression of E-cadherin was unaltered among the original
phenotype, induced EMT phenotype, and reverted phenotype in PC-9
cells. This exception, however, is concordant with a previously
reported finding that the morphology and expression of E-cadherin
were not significantly altered in PC-9 cells exhibiting EMT
(25). To elucidate the mechanisms
underlying this phenomenon, the known important pathways for EMT,
Smad3 (2,7,9,10),
MEK/Erk (2,7,8,11),
mTOR-C1/p70S6K (2,7,11)
and mTOR-C2/Akt (Ser473), (2,7,26),
were examined. The results showed that all the pathways in the
HCC-827 cells and the Smad3 pathway in the PC-9 cells were
correlated with EMT induction. Whereas, the mechanisms involved in
reverting the EMT seemed slightly more complex. PP242 significantly
inhibited mTOR-C2/Akt in both cell lines and tended to inhibit
mTOR-C1/p70S6K in HCC-827 cells; metformin significantly inhibited
MEK/Erk and mTOR-C1/p70S6K in HCC-827 cells and tended to inhibit
mTOR-C2/Akt in both cell lines; and DMSO inhibited mTOR-C1/p70S6K
in both cell lines but had some paradoxical effects on the MEK/Erk
pathway. This paradoxical effect of DMSO suggests the possible
involvement of other pathways in EMT, although no evidence to
support this hypothesis is presently available. In addition, as
also supported by the apoptosis analyses, the induction and
reversion of EMT were correlated with drug resistance to an
EGFR-TKI and the ability to overcome resistance to an EGFR-TKI,
respectively, in both cell lines, and an altered sensitivity to
cisplatin was observed solely in HCC-827 cells, again with some
differences in the effects of the three reverting agents.
Interestingly, the combination of TGF-β and FGF-2 had significant
effects on drug resistance in this study, whereas TGF-β or FGF-2
alone showed absent or equivocal effects, suggesting a synergistic
effect of these cytokines.
The changes in drug sensitivity according to the
acquisition and reversion of the EMT phenotype were consistent with
those of previous reports, that is, a decreased sensitivity to
gefitinib was observed in EGFR mutant NSCLC cells exhibiting
an EMT phenotype induced by TGF (27), HGF (28), and IL-6 (29). A decreased sensitivity to cisplatin
in NSCLC cells (30) and to
trastuzumab in breast cancer cells (31) as a result of EMT induction with
TGF, and to etoposide in small cell lung cancer cells with an
HGF-induced EMT phenotype (32)
were also reported. Previous studies have disclosed that an Erk
inhibitor (27) and mTOR
inhibitors (26,33–35)
restored drug sensitivity affected by the EMT. In addition,
metformin was shown to cause EMT reversion through the inhibition
of the mTOR, Erk, and JAK/STAT3 pathways (29,31).
These findings suggest the involvement of the Erk, mTOR, and
JAK/STAT3 pathways in EMT reversion and its relation to drug
resistance. Although the mechanisms involved have not yet been
defined, other agents such as crizotinib (32), resveratrol (36), tranilast (37), the eukaryotic initiation factor
inhibitor GC7 (N1-guanyl-1,7-diaminoheptane) (38), propolis (39), eribulin (40) and TTF-1 (41) have been reported to decrease
EMT-induced drug resistance. Therefore, this study, in addition to
confirming the restoration of drug sensitivity and EMT reversion by
an mTOR inhibitor and metformin, provided the new findings that
DMSO reverted the EMT and restored drug sensitivity through the
inhibition of the mTOR-C1/p70S6K pathway and had some paradoxical
effects on other pathways. Although DMSO is known to have multiple
effects on cellular functions, such as an increased intracellular
calcium concentration and the differentiation of embryonal cells
and leukemia cells (42–44), its role on modifying the EMT
phenotype has not been reported. Furthermore, since DMSO has also
been reported to reduce vimentin expression and to promote a
fibroblast-like morphology in cultured rat neonatal hepatocytes
(45,46), these observations may be reflected
by the reversion of the EMT. DMSO also reportedly induces the
differentiation of a human promyelocytic leukemia cell line through
the activation of the MEK/Erk pathway (47). The activation of the MEK/Erk
pathway was also demonstrated in this study, although the
activation of the pathway seems to be in the opposite direction for
EMT reversion. As the effects of DMSO on EMT reversion appeared to
be paradoxical in this study, the biological functions of DMSO
should be further clarified.
This study also demonstrated a close relationship
between PD-L1 expression and EMT induction/reversion. PD-L1
expression on the surface of cancer cells is known to be important
for escaping from host immunity. Although the mechanisms involved
in regulating PD-L1 expression are not fully understood, a recent
report also disclosed a link between the induction of EMT and the
overexpression of PD-L1. This study illustrated a potential
mechanism where an EMT activator, zinc-finger E-box-binding
homeobox 1, relieved the microRNA-200-induced suppression of PD-L1,
leading to CD8+ tumor-infiltrating lymphocyte-induced
immunosuppression (48). Another
recent study also demonstrated the overexpression of PD-L1 in the
TGF-β-driven EMT and the downregulation of PD-L1 expression through
the inhibition of PI3K or Erk in breast cancer cells (49). TGF-β is a well-known immune
suppresser related to the Smad3 and non-Smad3 pathways (6). In this study, although EMT induction
was related to an elevation in Smad3 expression, its reversion was
not related to a decrease in Smad3 expression, whereas PD-L1
expression was elevated in the process of EMT induction and was
suppressed during the process of reversion, suggesting the
involvements of the non-Smad3 pathways in EMT-driven immune
suppression, at least in the PC-9 and HCC-827 cells. The PI3K/Akt
and MEK/Erk pathways, which are both common downstream signal
pathways of ALK and EGFR, reportedly mediate the modulation of
PD-L1 expression in cancer cells with either an
ALK-translocation or EGFR mutation (50). Taken together, these results
suggest that EMT-related immune suppression and its reversion in
cancer cells with activated EGFR signaling may occur as a result of
changes in PD-L1 expression.
Many fragments of evidence regarding the EMT in
cancer cells as it relates to immune-protection, drug sensitivity,
and reversion have been provided by many studies. However, this
study provided a comprehensive demonstration of the entire story of
EMT induction and reversion, including the accompanying changes in
drug sensitivity and immune suppression. Moreover, this study
identified the mechanisms underlying these observations and
suggested that different mechanisms may be involved in different
cell lines. We believe that these points are the strengths of this
study. The addition of DMSO to the list of EMT-reverting agents
together with the disclosure of its unique roles in EMT reversion
gives further strength to this study. Nevertheless, this study did
have some limitations: i) the study was exclusively performed in
two NSCLC cell lines with the same EGFR mutation, ii) the
possibility that the observed findings might be limited to cells
with activated EGFR cannot be excluded because the change in
sensitivity to cisplatin was only modest, iii) the findings are
limited to the induction of the EMT through TGF-β and FGF-2, and
other growth factors and cytokines known to induce the EMT, such as
HGF, insulin-like growth factor, and IL-6, were not studied, and
iv) all the findings were derived from in vitro experiments
lacking interactions between the tumor and cancer
microenvironments, which are thought to be very important for some
epigenetic changes, including EMT. The last issue seems to be
especially crucial in the context of the relationship between the
EMT and immune suppression, where interactions between the cancer
cells and the host immune cells are critical. Obviously, further
elucidation and verification of the observations in other cell
lines, both in vitro and in vivo, are needed.
In conclusion, EMT reversion has the potential to
overcome drug resistance of cancer cells, especially for treatments
involving molecular targeted therapy and immune checkpoint therapy.
Further elucidation of the mechanisms underlying EMT reversion and
the identification of more potent agents capable of reverting EMT
are warranted for clinical applications.
Acknowledgements
This study was supported by grants from the Ministry
of Education, Culture, Sports, Science and Technology in Japan
(Kiban-C #26461182), Chugai Pharmaceutical Co., Ltd., Eli Lilly
Japan K.K., Bristol-Myers Squibb Co., Ltd., Japan, Kyowa Hakko
Kirin Co., Ltd., Mochida Pharmaceutical Co., Ltd., and Merck Serono
Co., Ltd., Japan to Y.T.
References
|
1
|
Prall F: Tumour budding in colorectal
carcinoma. Histopathology. 50:151–162. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Thomson S, Buck E, Petti F, Griffin G,
Brown E, Ramnarine N, Iwata KK, Gibson N and Haley JD: Epithelial
to mesenchymal transition is a determinant of sensitivity of
non-small-cell lung carcinoma cell lines and xenografts to
epidermal growth factor receptor inhibition. Cancer Res.
65:9455–9462. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kurrey NK, Jalgaonkar SP, Joglekar AV,
Ghanate AD, Chaskar PD, Doiphode RY and Bapat SA: Snail and slug
mediate radioresistance and chemoresistance by antagonizing
p53-mediated apoptosis and acquiring a stem-like phenotype in
ovarian cancer cells. Stem Cells. 27:2059–2068. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee JK, Joo KM, Lee J, Yoon Y and Nam DH:
Targeting the epithelial to mesenchymal transition in glioblastoma:
The emerging role of MET signaling. Onco Targets Ther. 7:1933–1944.
2014.PubMed/NCBI
|
|
6
|
Kudo-Saito C, Shirako H, Takeuchi T and
Kawakami Y: Cancer metastasis is accelerated through
immunosuppression during Snail-induced EMT of cancer cells. Cancer
Cell. 15:195–206. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gavert N and Ben-Ze'ev A:
Epithelial-mesenchymal transition and the invasive potential of
tumors. Trends Mol Med. 14:199–209. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Grände M, Franzen A, Karlsson JO, Ericson
LE, Heldin NE and Nilsson M: Transforming growth factor-beta and
epidermal growth factor synergistically stimulate epithelial to
mesenchymal transition (EMT) through a MEK-dependent mechanism in
primary cultured pig thyrocytes. J Cell Sci. 115:4227–4236. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shi Y and Massagué J: Mechanisms of
TGF-beta signaling from cell membrane to the nucleus. Cell.
113:685–700. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen XF, Zhang HJ, Wang HB, Zhu J, Zhou
WY, Zhang H, Zhao MC, Su JM, Gao W, Zhang L, et al: Transforming
growth factor-β1 induces epithelial-to-mesenchymal transition in
human lung cancer cells via PI3K/Akt and MEK/Erk1/2 signaling
pathways. Mol Biol Rep. 39:3549–3556. 2012. View Article : Google Scholar
|
|
12
|
Shirakihara T, Horiguchi K, Miyazawa K,
Ehata S, Shibata T, Morita I, Miyazono K and Saitoh M: TGF-β
regulates isoform switching of FGF receptors and
epithelial-mesenchymal transition. EMBO J. 30:783–795. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shih JY and Yang PC: The EMT regulator
slug and lung carcinogenesis. Carcinogenesis. 32:1299–1304. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen H, Zhu G, Li Y, Padia RN, Dong Z, Pan
ZK, Liu K and Huang S: Extracellular signal-regulated kinase
signaling pathway regulates breast cancer cell migration by
maintaining slug expression. Cancer Res. 69:9228–9235. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lau MT and Leung PC: The PI3K/Akt/mTOR
signaling pathway mediates insulin-like growth factor 1-induced
E-cadherin down-regulation and cell proliferation in ovarian cancer
cells. Cancer Lett. 326:191–198. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lyons JG, Patel V, Roue NC, Fok SY, Soon
LL, Halliday GM and Gutkind JS: Snail up-regulates proinflammatory
mediators and inhibits differentiation in oral keratinocytes.
Cancer Res. 68:4525–4530. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shintani Y, Okimura A, Sato K, Nakagiri T,
Kadota Y, Inoue M, Sawabata N, Minami M, Ikeda N, Kawahara K, et
al: Epithelial to mesenchymal transition is a determinant of
sensitivity to chemoradiotherapy in non-small cell lung cancer. Ann
Thorac Surg. 92:1794–1804. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Marín-Aguilera M, Codony-Servat J, Reig Ò,
Lozano JJ, Fernández PL, Pereira MV, Jiménez N, Donovan M, Puig P,
Mengual L, et al: Epithelial-to-mesenchymal transition mediates
docetaxel resistance and high risk of relapse in prostate cancer.
Mol Cancer Ther. 13:1270–1284. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kajiyama H, Shibata K, Terauchi M,
Yamashita M, Ino K, Nawa A and Kikkawa F: Chemoresistance to
paclitaxel induces epithelial-mesenchymal transition and enhances
metastatic potential for epithelial ovarian carcinoma cells. Int J
Oncol. 31:277–283. 2007.PubMed/NCBI
|
|
20
|
Yang AD, Fan F, Camp ER, van Buren G, Liu
W, Somcio R, Gray MJ, Cheng H, Hoff PM and Ellis LM: Chronic
oxaliplatin resistance induces epithelial-to-mesenchymal transition
in colorectal cancer cell lines. Clin Cancer Res. 12:4147–4153.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhuo W, Wang Y, Zhuo X, Zhang Y, Ao X and
Chen Z: Knockdown of Snail, a novel zinc finger transcription
factor, via RNA interference increases A549 cell sensitivity to
cisplatin via JNK/mitochondrial pathway. Lung Cancer. 62:8–14.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chang TH, Tsai MF, Su KY, Wu SG, Huang CP,
Yu SL, Yu YL, Lan CC, Yang CH, Lin SB, et al: Slug confers
resistance to the epidermal growth factor receptor tyrosine kinase
inhibitor. Am J Respir Crit Care Med. 183:1071–1079. 2011.
View Article : Google Scholar
|
|
23
|
Sharma SV, Lee DY, Li B, Quinlan MP,
Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach
MA, et al: A chromatin-mediated reversible drug-tolerant state in
cancer cell subpopulations. Cell. 141:69–80. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sato M, Shames DS and Hasegawa Y: Emerging
evidence of epithelial-to-mesenchymal transition in lung
carcinogenesis. Respirology. 17:1048–1059. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang HJ, Wang HY, Zhang HT, Su JM, Zhu J,
Wang HB, Zhou WY, Zhang H, Zhao MC, Zhang L, et al: Transforming
growth factor-β1 promotes lung adenocarcinoma invasion and
metastasis by epithelial-to-mesenchymal transition. Mol Cell
Biochem. 355:309–314. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lamouille S, Connolly E, Smyth JW, Akhurst
RJ and Derynck R: TGF-β-induced activation of mTOR complex 2 drives
epithelial-mesenchymal transition and cell invasion. J Cell Sci.
125:1259–1273. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Buonato JM and Lazzara MJ: ERK1/2 blockade
prevents epithelial-mesenchymal transition in lung cancer cells and
promotes their sensitivity to EGFR inhibition. Cancer Res.
74:309–319. 2014. View Article : Google Scholar :
|
|
28
|
Ishikawa D, Takeuchi S, Nakagawa T, Sano
T, Nakade J, Nanjo S, Yamada T, Ebi H, Zhao L, Yasumoto K, et al:
mTOR inhibitors control the growth of EGFR mutant lung cancer even
after acquiring resistance by HGF. PLoS One. 8:e621042013.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li L, Han R, Xiao H, Lin C, Wang Y, Liu H,
Li K, Chen H, Sun F, Yang Z, et al: Metformin sensitizes
EGFR-TKI-resistant human lung cancer cells in vitro and in vivo
through inhibition of IL-6 signaling and EMT reversal. Clin Cancer
Res. 20:2714–2726. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yu HG, Wei W, Xia LH, Han WL, Zhao P, Wu
SJ, Li WD and Chen W: FBW7 upregulation enhances cisplatin
cytotoxicity in non-small cell lung cancer cells. Asian Pac J
Cancer Prev. 14:6321–6326. 2013. View Article : Google Scholar
|
|
31
|
Cufí S, Vazquez-Martin A,
Oliveras-Ferraros C, Martin-Castillo B, Joven J and Menendez JA:
Metformin against TGFβ-induced epithelial-to-mesenchymal transition
(EMT): From cancer stem cells to aging-associated fibrosis. Cell
Cycle. 9:4461–4468. 2010. View Article : Google Scholar
|
|
32
|
Cañadas I, Rojo F, Taus Á, Arpí O,
Arumí-Uría M, Pijuan L, Menéndez S, Zazo S, Dómine M, Salido M, et
al: Targeting epithelial-to-mesenchymal transition with Met
inhibitors reverts chemoresistance in small cell lung cancer. Clin
Cancer Res. 20:938–950. 2014. View Article : Google Scholar
|
|
33
|
Lamouille S and Derynck R: Cell size and
invasion in TGF-beta-induced epithelial to mesenchymal transition
is regulated by activation of the mTOR pathway. J Cell Biol.
178:437–451. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Maru S, Ishigaki Y, Shinohara N, Takata T,
Tomosugi N and Nonomura K: Inhibition of mTORC2 but not mTORC1
up-regulates E-cadherin expression and inhibits cell motility by
blocking HIF-2α expression in human renal cell carcinoma. J Urol.
189:1921–1929. 2013. View Article : Google Scholar
|
|
35
|
Kim EY, Kim A, Kim SK, Kim HJ, Chang J,
Ahn CM and Chang YS: Inhibition of mTORC1 induces loss of
E-cadherin through AKT/GSK-3β signaling-mediated upregulation of
E-cadherin repressor complexes in non-small cell lung cancer cells.
Respir Res. 15:262014. View Article : Google Scholar
|
|
36
|
Shi XP, Miao S, Wu Y, Zhang W, Zhang XF,
Ma HZ, Xin HL, Feng J, Wen AD and Li Y: Resveratrol sensitizes
tamoxifen in antiestrogen-resistant breast cancer cells with
epithelial-mesenchymal transition features. Int J Mol Sci.
14:15655–15668. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Darakhshan S and Ghanbari A: Tranilast
enhances the anti-tumor effects of tamoxifen on human breast cancer
cells in vitro. J Biomed Sci. 20:762013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xu G, Yu H, Shi X, Sun L, Zhou Q, Zheng D,
Shi H, Li N, Zhang X and Shao G: Cisplatin sensitivity is enhanced
in non-small cell lung cancer cells by regulating
epithelial-mesenchymal transition through inhibition of eukaryotic
translation initiation factor 5A2. BMC Pulm Med. 14:1742014.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kao HF, Chang-Chien PW, Chang WT, Yeh TM
and Wang JY: Propolis inhibits TGF-β1-induced
epithelial-mesenchymal transition in human alveolar epithelial
cells via PPARγ activation. Int Immunopharmacol. 15:565–574. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yoshida T, Ozawa Y, Kimura T, Sato Y,
Kuznetsov G, Xu S, Uesugi M, Agoulnik S, Taylor N, Funahashi Y, et
al: Eribulin mesilate suppresses experimental metastasis of breast
cancer cells by reversing phenotype from epithelial-mesenchymal
transition (EMT) to mesenchymal-epithelial transition (MET) states.
Br J Cancer. 110:1497–1505. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Saito RA, Watabe T, Horiguchi K, Kohyama
T, Saitoh M, Nagase T and Miyazono K: Thyroid transcription
factor-1 inhibits transforming growth factor-beta-mediated
epithelial-to-mesenchymal transition in lung adenocarcinoma cells.
Cancer Res. 69:2783–2791. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Choi T: Dimethyl sulfoxide inhibits
spontaneous oocyte fragmentation and delays inactivation of
maturation promoting factor (MPF) during the prolonged culture of
ovulated murine oocytes in vitro. Cytotechnology. 63:279–284. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Santos NC, Figueira-Coelho J,
Martins-Silva J and Saldanha C: Multidisciplinary utilization of
dimethyl sulfoxide: Pharmacological, cellular, and molecular
aspects. Biochem Pharmacol. 65:1035–1041. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Szmant HH: Physical properties of dimethyl
sulfoxide and its function in biological systems. Ann NY Acad Sci.
243:20–23. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Pagan R, Sánchez A, Martin I, Llobera M,
Fabregat I and Vilaró S: Effects of growth and differentiation
factors on the epithelial-mesenchymal transition in cultured
neonatal rat hepatocytes. J Hepatol. 31:895–904. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Pagan R, Martín I, Llobera M and Vilaró S:
Epithelial-mesenchymal transition of cultured rat neonatal
hepatocytes is differentially regulated in response to epidermal
growth factor and dimethyl sulfoxide. Hepatology. 25:598–606. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yu HN, Lee YR, Noh EM, Lee KS, Song EK,
Han MK, Lee YC, Yim CY, Park J, Kim BS, et al: Tumor necrosis
factor-alpha enhances DMSO-induced differentiation of HL-60 cells
through the activation of ERK/MAPK pathway. Int J Hematol.
87:189–194. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chen L, Gibbons DL, Goswami S, Cortez MA,
Ahn YH, Byers LA, Zhang X, Yi X, Dwyer D, Lin W, et al: Metastasis
is regulated via microRNA-200/ZEB1 axis control of tumour cell
PD-L1 expression and intratumoral immunosuppression. Nat Commun.
5:52412014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Alsuliman A, Colak D, Al-Harazi O, Fitwi
H, Tulbah A, Al-Tweigeri T, Al-Alwan M and Ghebeh H: Bidirectional
crosstalk between PD-L1 expression and epithelial to mesenchymal
transition: Significance in claudin-low breast cancer cells. Mol
Cancer. 14:1492015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ota K, Azuma K, Kawahara A, Hattori S,
Iwama E, Tanizaki J, Harada T, Matsumoto K, Takayama K, Takamori S,
et al: Induction of PD-L1 expression by the EML4-ALK oncoprotein
and downstream signaling pathways in non-small cell lung cancer.
Clin Cancer Res. 21:4014–4021. 2015. View Article : Google Scholar : PubMed/NCBI
|