Introduction
Approximately 40,000 brain tumor cases were reported
between 1973 and 2001 (1).
Astrocytomas, known as grade II astrocytic tumors (2), have a peak incidence in early
adulthood, and are the most common form of primary human brain
tumors that are usually associated with a poor prognosis (3). Systemic metastasis of malignant
astrocytomas rarely occurs. These tumors; however, are highly
invasive into adjacent and distant tissues of the normal brain,
which makes them surgically and medically unmanageable (4). Tumor invasion and metastasis are
direct consequences of uncontrolled cell migration (5). The key to understanding these
phenomena is to uncover altered cellular signaling underlying tumor
aggressiveness.
Motility is a physiological process needed for
immune responses, embryonic development and wound healing. Cancer
cells use this process to invade neighboring tissues and to
metastasize to distant organs ensuring cancer progression (6). Cell motility is a complicated process
that is tightly coordinated by the cell itself with the help of
various proteins (7). Rho GTPases
including RhoA, Rac and Cdc42 are considered key players and chief
regulators of cell migration, among many other previously described
cellular events (8,9). These small monomeric G proteins act
as molecular switches, their spatio-temporal regulation being
tightly regulated (10). Upstream
regulators include GTPase-activating proteins (GAPs), which
catalyze the conversion of the active GTP-bound Rho-family GTPases
to the inactive GDP-bound form, and Guanine-nucleotide exchange
factors (GEFs) that activate Rho GTPases by catalyzing the exchange
of bound GDP to GTP (11). Upon
stimulation with growth factors, intracellular Rho family small
GTPases substitute their bound GDP to GTP. Once active, they
interact with and activate specific downstream effectors mediating
a multitude of intracellular responses including the reorganization
of the actin cytoskeleton leading to cell migration and invasion
(12–14). Altered levels of
expression/activation of Rho GTPases have been implicated in the
invasion and progression of many cancers (15–17).
A previous study conducted in our laboratory showed that
overexpression or knockdown of RhoA leads to the decrease in 2D
motility though altered focal complex formation and maturation into
focal adhesions. The same study concluded that RhoA undergoes an
activation/inactivation cycle at the cell edges mediating
protrusion formation (18). This
mechanism plays a central role in tumor progression resulting in
cancer spreading and invasion (19).
The MAPK cascade is an important signaling pathway
that conveys extracellular signals into the nucleus. It is
activated by various extracellular stimuli contributing to the
regulation of cellular responses such as apoptosis, survival,
proliferation and differentiation (20). After detecting an extracellular
mitogen, Ras, a small GTPase, switches its GDP to GTP. This allows
a subsequent activation of MAP3K (Raf), MAP2K (MEK) and MAPK
(ERK1/2) respectively, activating downstream transcription factors
(21). ERK/MAPK pathway seems to
play an elaborate role in tumor progression since its inhibition
abolishes the growth of BRAF-mutated HT29 colon cancer cells in
mice (22). In ER-negative
MDA-MB-468 and MDA-MB-231 cells, TGFα-induced MAPK activation was
correlated to an ~5-fold increase in cellular motility. Similar
results were observed in ER-positive TGFα-stimulated MCF-7 breast
cancer cells with an ~2-fold increase in cell motility (23).
The relationship between ERK-MAPK and Rho GTPases in
cell motility is far from being uncovered. Studies conducted on
LN-18 cells revealed a gradual decrease in MAPK activation the
longer the cells are treated with ROCK-inhibitor Y27632 (24). A recent study described two
signaling pathways downstream from MAPK that regulate cell motility
in colon carcinoma cells. Fra-1, member of theFos family, was shown
to downregulate RhoA activity by inactivating β1-integrin. The
former step being necessary for urokinase-type plasminogen
activator receptor (uPAR)-dependent activation of Rac, downstream
MAPK-ERK, resulting in lamellipodial formation (21).
Anthrax lethal toxin (LeTx) is an exotoxin produced
by the gram-positive bacterium Bacillus Anthasis. LeTx is a
binary toxin that consists of two distinct proteins, the catalytic
lethal factor (LF) and the cell binding and internalization
protective antigen (PrAg) (25,26).
These two protein subunits act together to impart their
physiological effects. PrAg binds to cells though its cell surface
receptors capillary morphogenesis gene-2 (CMG2) and tumor
endothelial marker-8 (TEM8) and is cleaved by furin-like proteases
releasing a 20-kDa fragment and generating PrAg63, a
63-kDa active fragment (27,28).
Three or four LF molecules bound to PrAg63 undergo
endocytosis. The latter, upon endosome acidification, undergoes a
conformational change leading to the formation of pores allowing LF
translocation into the cytosol (29). Once in the cytosol, the enzymatic
subunit LF disrupts various cell processes notably cellular
signaling. Previous studies revealed that the LF/PA mixture
significantly impaired chemotaxis among polymorphonuclear
neutrophils (PMNs) and abolished cellular polarity (30,31).
Upon screening for novel inhibitors of MAPK signal transduction
pathways, activity profile of LeTx was similar to PD09859, a
selective inhibitor of the MAPK pathway (32). Later on, lethal factor of LeTx (LF)
was identified as a endoprotease that inhibits the MAPK pathway by
cleaving the amino terminus and inactivating MEKs subsequently
leading to growth inhibition and death (33,34).
The aim of the present study was to investigate the
effect of LeTx on motility and invasion of astrocytomas cancer cell
lines. First, we studied its effect on cellular proliferation and
viability upon treating cells with recombinant toxin. Then, we
examined its effect on 2D motility, adhesion and invasion of
astrocytoma cells, in addition to identify its effect on Rho
GTPases. Our results revealed MAPK-mediated inhibition of random
2D-motility and 3D-motility in collagen. Decrease in the motile and
invasive profile of astrocytoma cells was opposed to an increase in
RhoA activity previously shown to cause cell stabilization that
contradicts free invasion. LeTx did not seem have an effect on the
activation of PI3K, a signal transduction pathway involved in cell
survival and proliferation.
Materials and methods
Cell culture
Human astrocytoma cell line SF268, obtained from Dr
Marc Symons, was cultured in Dulbecco's modified Eagle's medium
(DMEM) supplemented with 10% fetal bovine serum (FBS) and 100 U
penicillin/streptomycin at 37°C and 5% CO2 in a
humidified chamber.
Proliferation inhibition assay
(cytotoxicity)
Sensitivity of astrocytoma cell line to LeTx
(PrAg/LF) was determined using a proliferation inhibition assay as
previously described. Briefly, aliquots of 1,000 cells/well, in 100
μl of cell culture medium, containing a fixed concentration of
10−9 M LF, were plated onto a flat-bottom 96-well plate
(Corning Inc., Corning, NY, USA). Then, 50 μl of PrAg in media were
added to each well to yield concentrations ranging from
10−8 to 10−13 M. Following a 72-h incubation
at 37°C/5% CO2, 50 μl of XTT cell proliferation reagent
(Roche, Basel, Switzerland) was added to each well and the plates
incubated for another 4 h. Absorbance was then read at 450 nm using
a microplate reader (Thermo Fisher Scientific, Waltham, MA, USA).
Absorbance was plotted against the log of concentration, and a
non-linear regression with a variable slope sigmoidal dose-response
curve was generated along with inhibitory concentration 50
(IC50) using GraphPad Prism 5 software (Graphpad
Software Inc., San Diego, CA, USA).
Antibodies and reagents
Mouse monoclonal anti-ERK antibody, mouse monoclonal
anti-vinculin antibody, rabbit polyclonal anti-PERK antibody was
obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Mouse
monoclonal anti-RhoA, rabbit polyclonal antibody to pan-AKT, rabbit
polyclonal antibody to pan-AKT (phospho T308) from Abcam.
Fluorescent secondary antibodies (Alexa Fluor 488)
were obtained from Invitrogen. To visualize the actin cytoskeleton,
cells were stained with rhodamin phalloidin (Invitrogen).
Treatment with toxin
Sublethal anthrax lethal toxin concentration range
was determined after a literature review. A 2.84 mg/ml lethal
factor (LF) and a 5.23 mg/ml protective antigen (PA) were diluted
separately in DMEM to reach a concentration ratio of 1:3,
respectively. Cells were treated by adding equal volumes of each of
the two dilutions for 2 h. Following treatment cells were left for
24 h in complete medium before running the desired experiment.
Wound healing assay
Cells were grown to confluence on culture plates and
a wound was made in the monolayer with a sterile pipette tip. Cells
were then washed twice with PBS to remove debris and new medium was
added. Phase-contrast images of the wounded area were captured at 0
and 24 h after wounding. Wound widths were measured at 11 different
points for each wound, and the average rate of wound closure was
calculated (in μm/h).
Motility assay/analyzing 2D motility
For motility analysis, images of cells moving
randomly in serum were collected every 60 sec for 2 h using a 20x
objective. During imaging, the temperature was controlled using a
Nikon heating stage which was set at 37°C. The medium was buffered
using HEPES and overlayed with mineral oil. The speed of cell
movement was quantified using the ROI tracker plugin in ImageJ
software, which was used to calculate the total distance travelled
by individual cells. The speed is then calculated by dividing this
distance by the time (120 min) and reported in μm/min. The speed of
at least 15 cells for each condition was calculated. The net
distance travelled by the cell was calculated by measuring the
distance travelled between the first and the last frames.
Adhesion assay
Plates (96-well) were coated with collagen using
collagen solution, type I from rat tail (Sigma) overnight at 37°C
then washed with washing buffer (0.1% BSA in DMEM). The plates were
then blocked with 0.5% BSA in DMEM at 37°C in a CO2
incubator for 1 h. Then washing the plates and chilling them on ice
followed. The cells were trypsinized and counted to
4×105 cell/ml. A total of 50 ml of cells were added in
each well and incubated at 37°C in a CO2 incubator for
30 min. The plates were then shaken and washed 3 times. Cells were
then fixed with 4% paraformaldehyde at room temperature for 10 min,
washed, and stained with crystal violet (5 mg/ml in 2% ethanol) for
10 min. Following the staining with crystal violet, the plates were
washed extensively with water, and left to dry completely. Crystal
violet was solubilized by incubating the cells with 2% SDS for 30
min. The absorption of the plates was read at 550 mm using a plate
reader.
Immunostaining
The cells were plated on coverslips, and the
appropriate treatment was applied. Cells were fixed with 4%
paraformaldehyde for 10 min, and permeabilized with 0.5% Triton
X-100 for 10 min. To decrease background fluorescence, cells were
rinsed with 0.1 M glycine then incubated with 0.1 M glycine for 10
min. For blocking, cells were incubated 4 times with 1% BSA, 1% FBS
in PBS for 5 min. Samples were stained with primary antibodies for
2 h and with fluorophore-conjugated secondary antibodies for 2 h.
Fluorescent images were taken using a x60 objective on a
fluorescent microscope.
Boyden chamber/invasion assay
Cells were treated with toxin or left untreated as
control, and the invasion assay was performed following the
treatment period using the collagen-based invasion assay
(Millipore) according to the manufacturer's instructions. Briefly,
24 h prior to the assay, cells were starved with serum-free medium.
Cells were harvested, centrifuged and then resuspended in quenching
medium (without serum). Cells were then brought to a concentration
of 1×106 cells/ml. In the meantime, inserts were
prewarmed with 300 μl of serum-free medium for 30 min at room
temperature. After rehydration, 250 μl of medium was removed from
the inserts, and 250 μl of cell suspension was added. Inserts were
then placed in a 24-well plate, and 500 μl of complete medium (with
10% serum) was added to the lower wells. Plates were incubated for
24 h at 37°C in a CO2 incubator. Following the
incubation period, inserts were stained for 20 min at room
temperature with 400 μl of cell stain provided with the kit. The
stain was then extracted with extraction buffer (also provided).
The extracted stain (100 μl) was then transferred to a 96-well
plate suitable for colorimetric measurement using a plate reader.
Optical density was then measured at 560 nm.
Western blotting
Cell lysates were prepared by scraping the cells in
a sample buffer (4% SDS, 10% β-mercaptoethanol, 20% glycerol,
0.004% bromophenol blue, and 0.125 M Tris-HCl at a pH of 6.8). The
resulting lysates were boiled for 5 min. Protein samples were
separated by SDS-PAGE on 10% (for ERK and P-ERK) or 15% (for RhoA)
gels and transferred to PVDF membranes overnight at 30 V. The
membranes were then blocked with 5% non-fat dry milk in PBS
containing 0.1% Tween-20 for 1 h at room temperature and incubated
with primary antibody at a concentration of 1:100 for 2 h at room
temperature. After the incubation with the primary antibody, the
membranes were washed and incubated with secondary antibody at a
concentration of 1:1,000 for 1 h at room temperature. The membranes
were then washed, and the bands visualized by treating the
membranes with western blotting chemiluminescent reagent ECL (GE
Healthcare). The results were obtained on X-ray film (Agfa
Healthcare).
Pull-down assay
Cells were lysed and incubated with GST-RBD or
GST-CRIB and the pull-down assay was performed using the
RhoA/Rac1/Cdc42 Activation Assay Combo kit (Cell Biolabs Inc., San
Diego, CA, USA) following the manufacturer's instructions. Lysates
were incubated with GST-RBD (for RhoA) or GST-CRIB for Rac for 1 h
at 4°C. GTP-RhoA and GTP-Rac were detected by western blotting
using the anti-RhoA and the anti-Rac antibodies provided in the
kit. Total proteins were collected prior to the incubation with GST
beads and used as a loading control.
Results
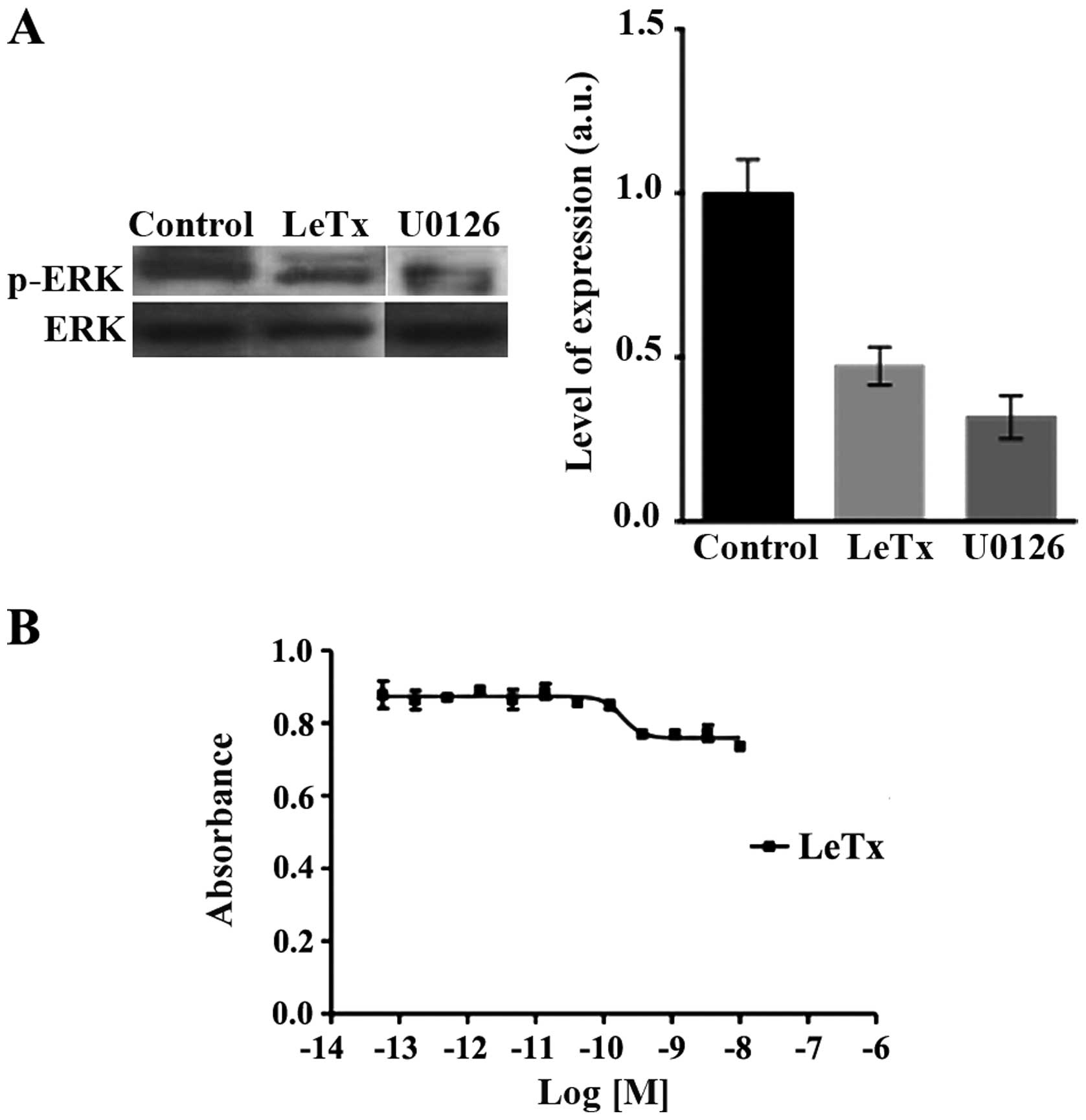
LeTx acts though the MAPK pathway
LeTx is known to inhibit the MAPK pathway in part by
inducing the cleavage of MEK upstream of ERK (20,21).
Western blots revealed a decrease in the activation of ERK as seen
in the decrease in p-ERK levels compared to total ERK levels upon
LeTx or U0126 treatment as expected (Fig. 1A). Paradoxically, LeTx has no net
effect on astrocytoma cell viability. Using an increasing
concentration of recombinant anthrax lethal toxin followed by the
evaluation of cell viability via XTT, revealed no significant
effect of the toxin on astrocytoma cells viability (Fig. 1B). Cells are slightly sensitive to
PrAg/LF, indicating a partial resistance to the LF-mediated
inhibition of the MAPK pathway.
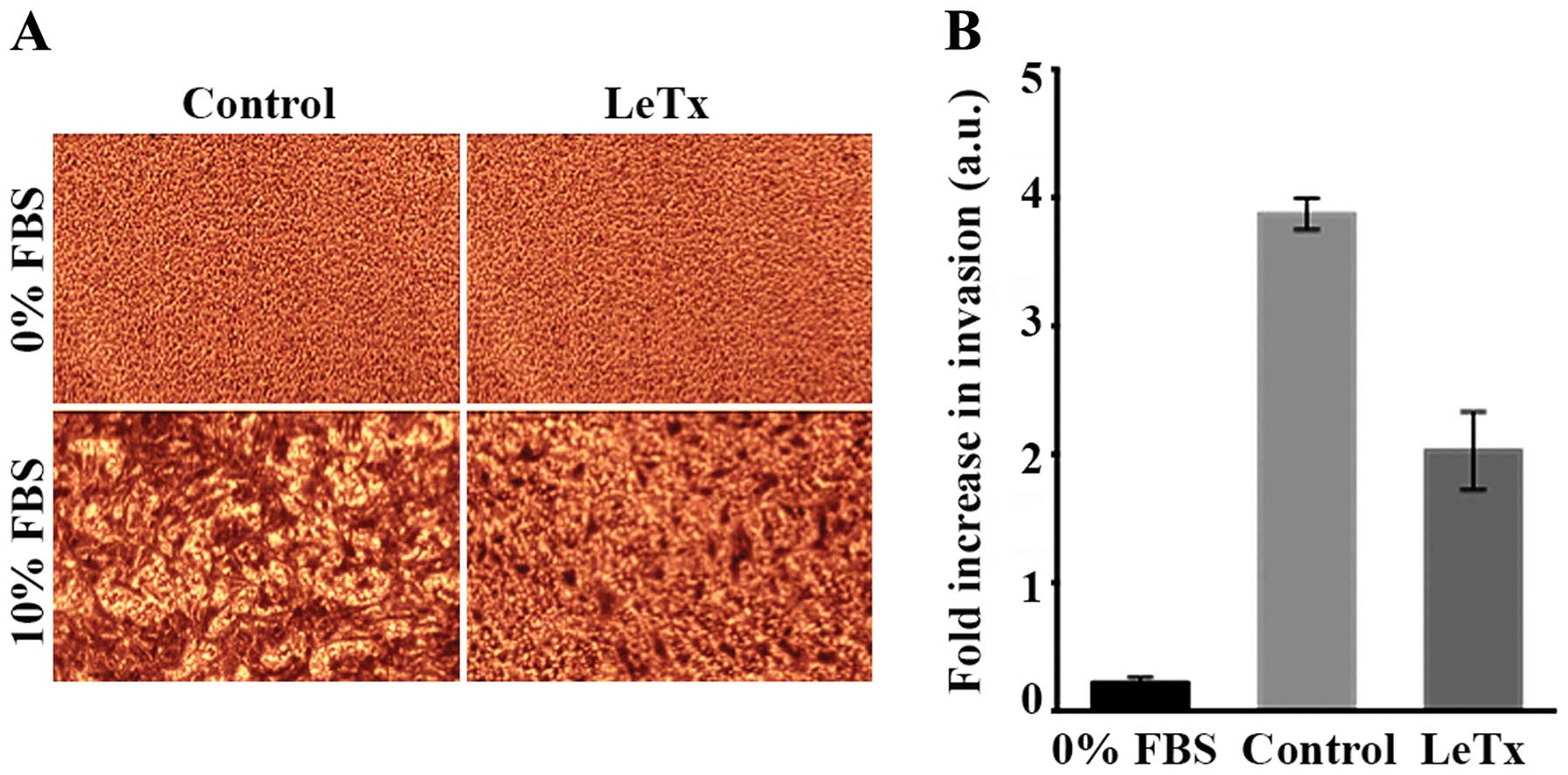
LeTx decreases astrocytoma invasion
Since LeTx had no effect on astrocytoma viability,
we wanted to test its potential effect on astrocytoma cell
invasion. In order to do so, we performed an in vitro
Transwell migration assay using FBS as a chemoattractant. A
negative control was run in parallel whereby serum-free media was
introduced into the well and the corresponding insert. The results
showed a 2-fold decrease in cellular invasion in treated cells as
compared to control (Fig. 2).
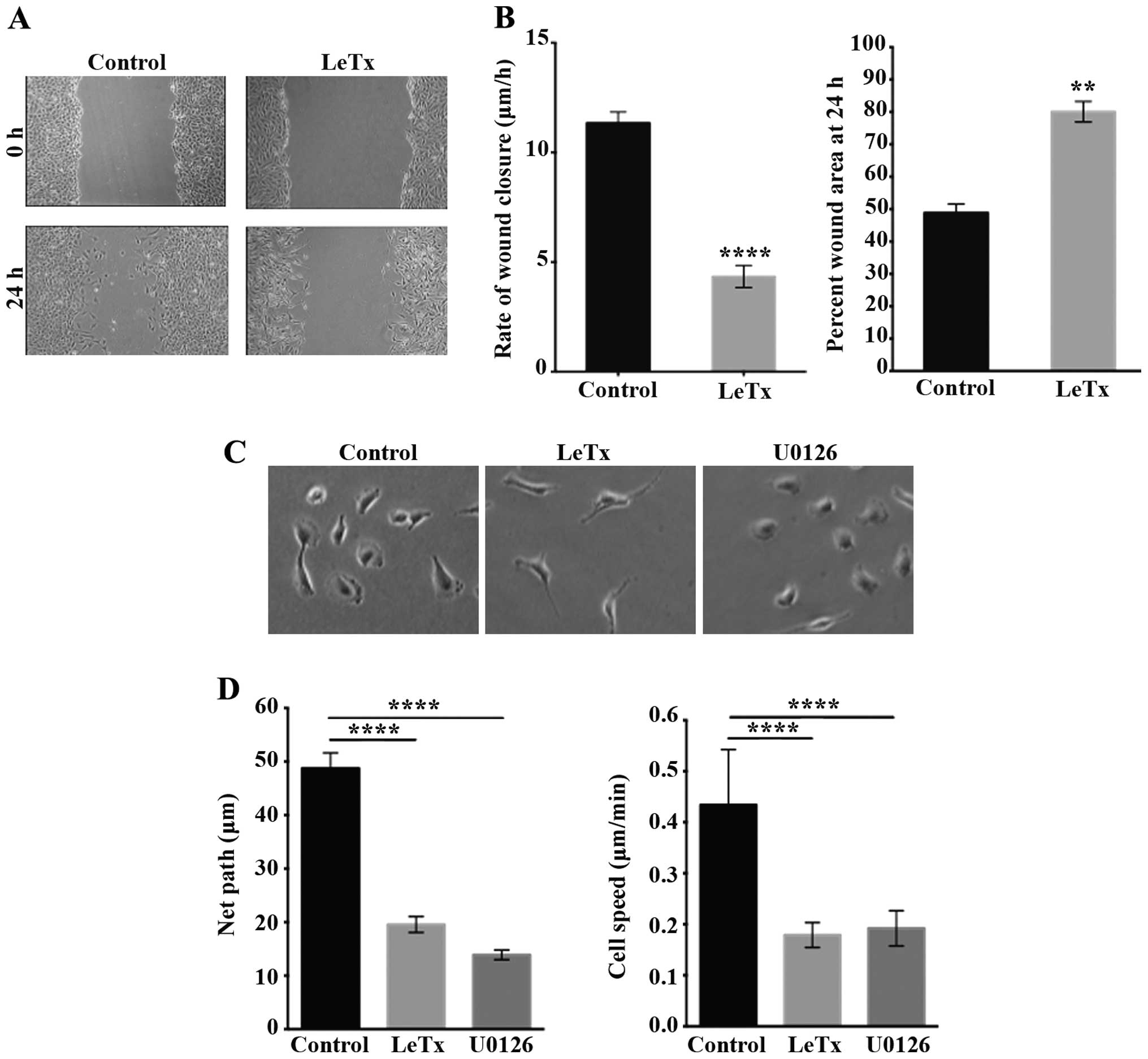
LeTx decreases astrocytoma cell
motility
In order to further study the effect of LeTx on
astrocytoma invasion we looked at the behavior of the cells in 2D
in order to observe their phenotype. First the 2D migration was
examined by performing a wound closure assay. Treatment with LeTx
caused decrease in the rate of wound closure from 11 to 4 μm/h
(Fig. 3A and B). The area of the
wounds we calculated both at time 0 and 24 h after inflicting the
wound (Fig. 3B). The results
reveal that control cells were able to close >50% of the wound
after 24 h, as opposed to treated cells where only 20% of the wound
was closed (Fig. 3B). The net path
taken by individual cells significantly decreased >2.5-fold in
cells treated with LeTx or with the MEK1/2 inhibitor U0126 as
determined by time-lapse imaging to detect random 2D cell migration
rates and profiles (Fig. 3D).
Average speed of individual cells also significantly decreased upon
treatment from ~0.45 to ~0.2 μm/min (Fig. 3D). Time lapse movies allowed us to
examine the migration profile and the phenotype of individual cells
in response to treatment with LeTx. Treated cells displayed an
extended shape with thin elongated protrusions. Cells treated with
LeTx seemed to lack de-adhesion and to be unable to retract their
tail at times (Fig. 3C) which
might explain the decrease in cell migration. This phenotype was
not, however, seen in cells treated with U0126 which suggest
another mechanism through which this drug is affecting migration in
these cells.
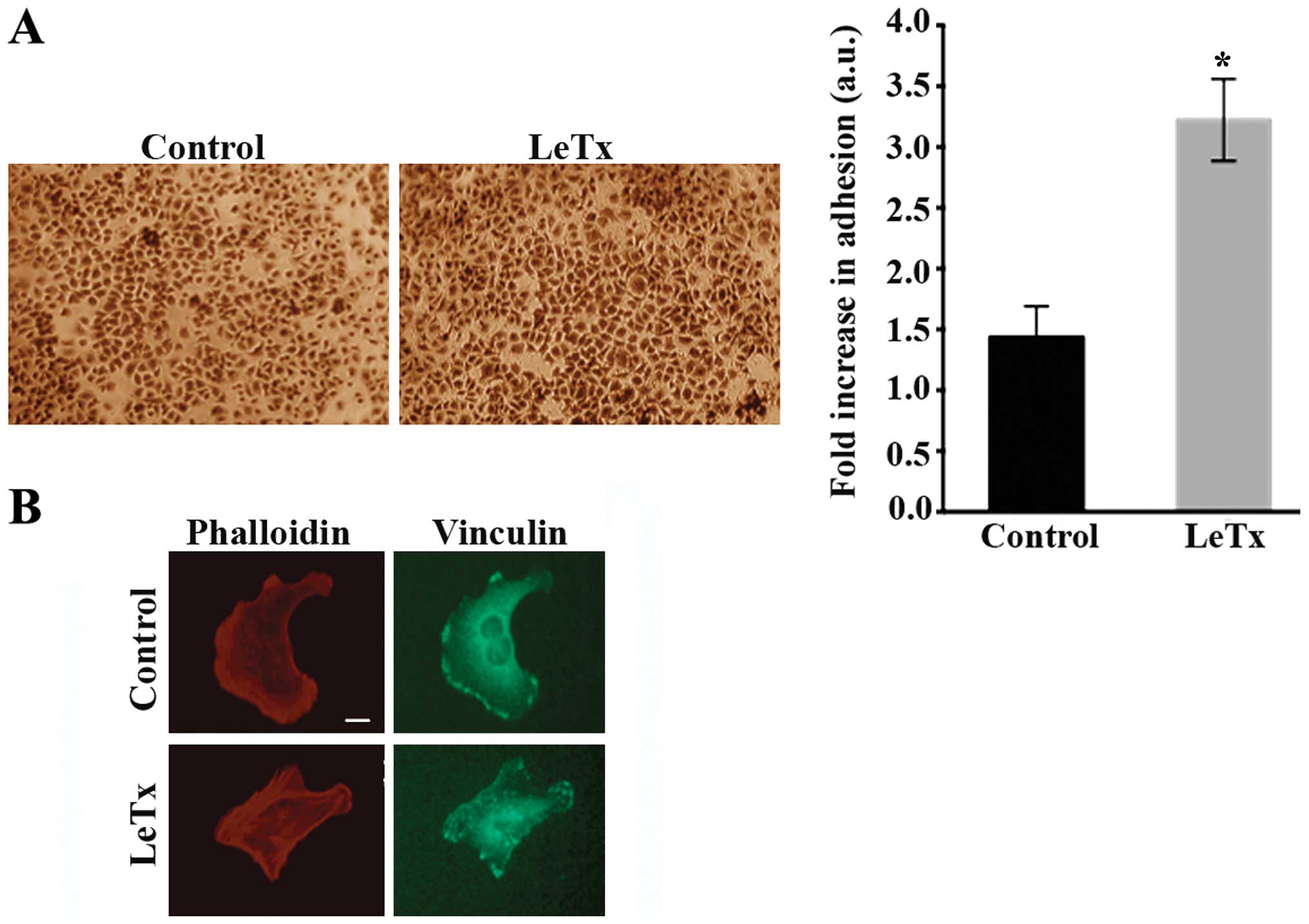
LeTx increases cell adhesion
Having suspected an inability of the cells to detach
during migration in the previous experiment which might indicate
exaggerated adhesions, we were interested to look at the effect of
LeTx on cell adhesion. Cells treated with LeTx displayed a 2-fold
increase in cellular adhesion as compared to control cells
(Fig. 4A) which is consistent with
the elongated phenotype. The elongated phenotype was also
reminiscent of a phenotype we have previously reported in
astrocytoma cells where the RhoA GAP StarD13 was knocked down which
leads to an increase in RhoA activity and adhesion structures
resulting in the inability of cells to migrate (18).
Indeed, cells treated with LeTx treated cells
exhibited a higher density of intracellular stress fibers as
reflected with phalloidin staining which might indicate an increase
in RhoA activity. In addition, immunostaining the cells with
anti-vinculin antibody revealed focal adhesions that were more
prevalent in treated cells. Large punctate structures were present
at the leading edge and the tail as well as the cell body, unlike
control cells where focal adhesions were mostly limited to the
lamellipodium (Fig. 4B).
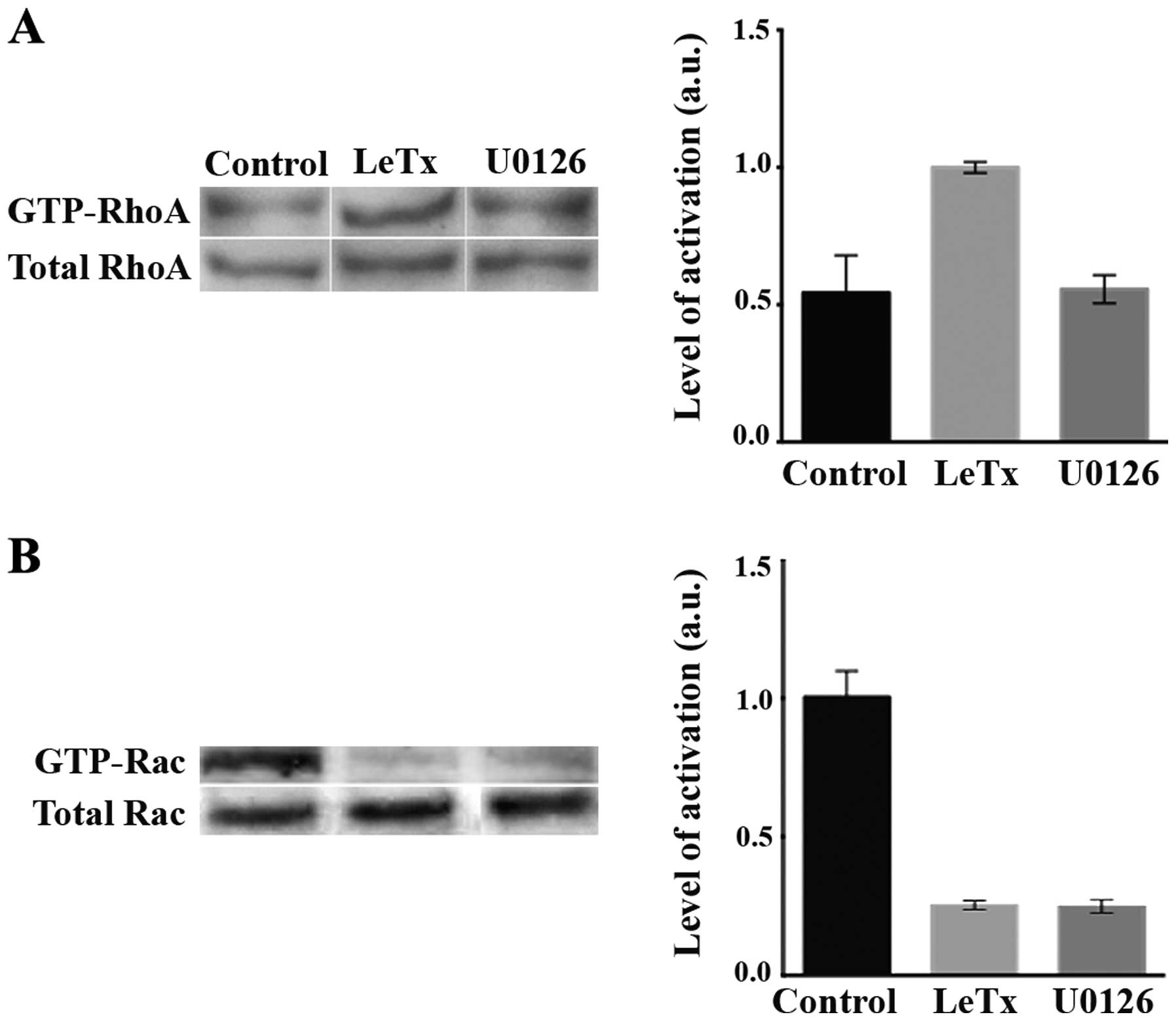
Effect of LeTx and U0126 on RhoA and Rac
activation
It is well established in the literature that RhoA
is a major contributor to the formation and maturation of focal
complexes into focal adhesions. After being shown to increase cell
adhesion, we aimed to check for the effect of LeTx on the
activation of RhoA. Treated cells revealed a higher RhoA activity
as compare to control (Fig. 5A).
Treating the cells with U0126, however, did not lead to an increase
in RhoA activation. This is in accordance with the phenotype seen
in Fig. 3C where the cells treated
with U0126 lacked the elongated phenotype seen in the cells treated
with LeTx.
We also looked at the activation of Rac which is a
major regulator of cell migration. In response to both LeTx and
U0126 treatment, the activation of Rac was substantially
decreased.
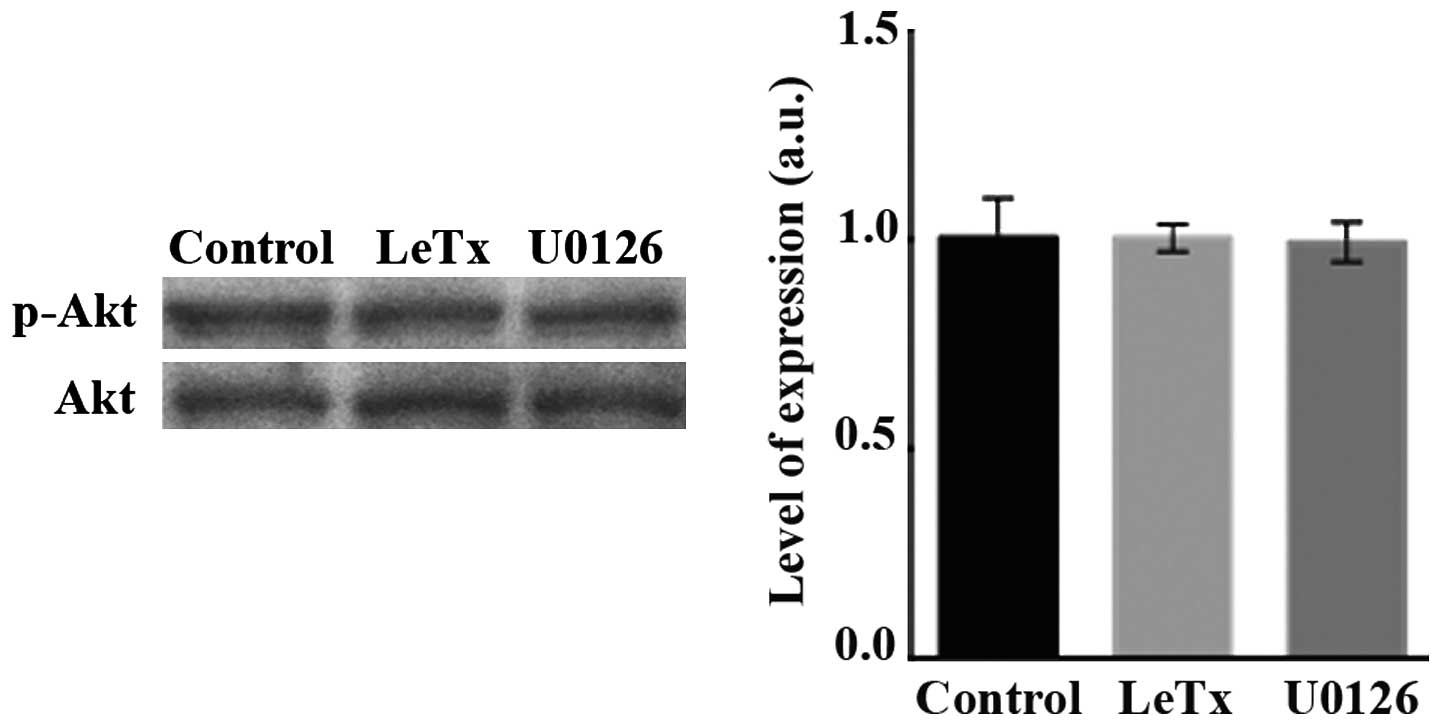
LeTx and U0126 treatments do not affect
the PI3K pathway
Finally, since PI3K is a known activator or Rho
GTPases (9), we examined whether
LeTx and U0126 are exerting their effects on Rac or RhoA through
affecting the PI3K pathway. Treating the cells with LeTx or U0126
did not affect the level of phosphorylation of Akt downstream of
PI3K (Fig. 6).
Discussion
Invasiveness of brain carcinoma and its infiltration
to the neighboring locations in brain limits the survival of
patients from several years to several months, depending on the
stage of this cancer. Previous studies were able to determine that
inactivation of MAPK pathway though proteolysis was enough to kill
different tumor types (35). A
study by Huang et al (30)
showed that renal cell carcinoma having high expression levels of
MKK1 and ERK2 were sensitive to the fusion anthrax lethal toxin due
to its capability of inhibiting the MAPK pathway leading to cell
death.
Based on the ability of toxin to inhibit MAPK
pathway resulting in cell death, astrocytoma cells were tested with
an increasing gradient of toxin concentration to assess their
sensitivity to the toxin. Surprisingly, no significant cytotoxic
effect of toxin on astrocytoma cells was observed. A previous study
aiming to unveil the mechanism of action of LeTx and its
pathological/physiological effect on peripheral polymorphonuclear
neutrophils (PMNs) showed that treating those cells with low
concentrations of LeTx caused paralysis of directed migration and
impaired chemokinesis and cell polarity, which was not accompanied
by significant apoptosis or necrosis (31). This former study was among the few
tackling the potential relationship between LeTx and impaired
cellular movement. Additionally, most of the studies were conducted
on immune cells and none on tumor cells. Hence, the significance of
the present study as a novel approach to treat highly motile and
invasive tumor cells with an agent that does not necessarily affect
cell viability.
In the present study, treating astrocytoma cells
with the toxin led to a decrease in 2D motility as shown by
time-lapse movies and their quantitation in addition to
wound-healing assay. It is important to note that upon treating the
cells with toxin a change in cellular phenotype was observed where
the cells gained an elongated morphology. Cells displayed an
unusual shape with the absence of defined leading edge and tail.
Over and above that, no matter how far the cell body migrated, the
‘tail region’ seemed to be stuck in place unable to detach and
retract. This phenotype was previously observed in astrocytoma
cells undergoing random 2D migration in serum whenever RhoA was
overexpressed or a RhoA GAP called StarD13 was knocked down
(18). We hypothesized that LeTx
could be impairing cell migration, in part, through the
deregulation of RhoA activation which leads to the phenotype
observed.
As we have previously established (18), the activation of RhoA needs to
cycle during the cell motility cycle. Initially, RhoA activation
increases which is needed for focal adhesions to form which enables
the protrusion to exert pressure on the ECM to pull the cell
forward. Following that, RhoA activation needs to decrease in order
for the focal adhesion to dissolve for the cell tail to detach and
for the cell to move forward. A persistent activation of RhoA was
shown to inhibit cell migration in astrocytoma through persistent
cell adhesions that disable the cells from moving. Consistently, in
the present study when we treated the cells with LeTx, we observed
an increase in cell adhesion and an exaggerated phenotype of focal
adhesions and an increase in stress fiber formation which is
indicative of an increase in RhoA activation.
For the above reason, a pulldown assay using
Rhotekin RBD agarose beads was performed to pull the active RhoA.
The results revealed an increase in the activation of RhoA
suggesting that LeTx could be acting on RhoA to induce the changes
observed in cell migration and invasion. The pull-down results also
explain the changes in cellular morphology observed though
time-lapse imaging whereby cells seemed to exhibit longer cellular
extensions. This is due to the high RhoA activity causing
stabilization of focal adhesions at the leading edge and the tail
preventing forward protrusion and tail retraction. High RhoA
activity also explains the remarkable presence of stress fibers
observed upon staining for actin.
Our findings come in parallel with a decrease in
MAPK activity upon toxin treatment, as compared to control and
treatment using MAPK inhibitor U0126. Moreover, LeTx seems to have
no effect on the activation of phosphatidylinositol 3-kinase
(PI3K). This is in accordance with previously published works
depicting the PI3K LeTx-resistance and the critical role of the
PI3K/Akt/glycogen synthase kinase-3β signaling pathway in the
protection and the recovery from LeTx-induced MEK cleavage in
macrophages (36). Given its
effect on RhoA, collectively this suggests that LeTx is not
exerting its effect on RhoA through PI3K.
U0126 is a specific inhibitor of the MEK1/2 pathway
whereas LeTx has a broader effect. Both agents led to a complete
inhibition of Rac which is one of the main effectors of cell
migration (9,18,37).
This would suggest that LeTx leads to the inhibition of cell
migration and invasion, at least in part, through its inhibition of
MEK1/2 and Rac downstream. However, as shown the MEK1/2 had no
effect on RhoA and did not mimic the elongated phenotype seen in
the LeTx-treated cells. This indicates that, in addition to its
effect on MEK1/2-Rac, LeTx also leads to the overactivation of RhoA
through the inhibition of another MAPK pathway, leading to the
inhibition of cell migration and invasion.
Our data show that LeTx is a potent in vitro
targeted toxin that could inhibit astrocytoma cell motility and
invasion. This study only paves the way for prospectively thorough
studies and opens new horizons to novel therapeutic approaches.
Consequently, further studies should be conducted to investigate
the underlying mechanism behind the effect of LeTx on astrocytoma
cell motility and the specific MAPK pathway downstream from LeTx.
Deciphering signaling pathways impacted by LeTx are of great
importance. It will also be significant to further dissect the
direct relationship between the MAPK pathways and Rho GTPases in
these cells.
Acknowledgements
The ROI_Tracker software was supplied by David
Entenberg and John Condeelis as supported by CA100324 and GM064346.
The present study was supported by the Natural Science Department
at the Lebanese American University.
References
|
1
|
Deorah S, Lynch CF, Sibenaller ZA and
Ryken TC: Trends in brain cancer incidence and survival in the
United States: Surveillance, Epidemiology, and End Results Program,
1973 to 2001. Neurosurg Focus. 20:E12006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
von Deimling A, von Ammon K, Schoenfeld D,
Wiestler OD, Seizinger BR and Louis DN: Subsets of glioblastoma
multiforme defined by molecular genetic analysis. Brain Pathol.
3:19–26. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
DeAngelis LM: Brain tumors. N Engl J Med.
344:114–123. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Moon SY and Zheng Y: Rho GTPase-activating
proteins in cell regulation. Trends Cell Biol. 13:13–22. 2003.
View Article : Google Scholar
|
|
5
|
Yamazaki D, Kurisu S and Takenawa T:
Regulation of cancer cell motility through actin reorganization.
Cancer Sci. 96:379–386. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
El-Sibai M, Pertz O, Pang H, Yip SC,
Lorenz M, Symons M, Condeelis JS, Hahn KM and Backer JM:
RhoA/ROCK-mediated switching between Cdc42- and Rac1-dependent
protrusion in MTLn3 carcinoma cells. Exp Cell Res. 314:1540–1552.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ananthakrishnan R and Ehrlicher A: The
forces behind cell movement. Int J Biol Sci. 3:303–317. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vega FM, Fruhwirth G, Ng T and Ridley AJ:
RhoA and RhoC have distinct roles in migration and invasion by
acting through different targets. J Cell Biol. 193:655–665. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
El-Sibai M and Backer JM: Phospholipase C
γ negatively regulates Rac/Cdc42 activation in antigen-stimulated
mast cells. Eur J Immunol. 37:261–270. 2007. View Article : Google Scholar
|
|
10
|
Moorman JP, Luu D, Wickham J, Bobak DA and
Hahn CS: A balance of signaling by Rho family small GTPases RhoA,
Rac1 and Cdc42 coordinates cytoskeletal morphology but not cell
survival. Oncogene. 18:47–57. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ridley AJ: RhoA, RhoB and RhoC have
different roles in cancer cell migration. J Microsc. 251:242–249.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Buchsbaum RJ: Rho activation at a glance.
J Cell Sci. 120:1149–1152. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
O'Connor K and Chen M: Dynamic functions
of RhoA in tumor cell migration and invasion. Small GTPases.
4:141–147. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ridley AJ: Life at the leading edge. Cell.
145:1012–1022. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ellenbroek SI and Collard JG: Rho GTPases:
Functions and association with cancer. Clin Exp Metastasis.
24:657–672. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vega FM and Ridley AJ: Rho GTPases in
cancer cell biology. FEBS Lett. 582:2093–2101. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Boettner B and Van Aelst L: The role of
Rho GTPases in disease development. Gene. 286:155–174. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Khalil BD, Hanna S, Saykali BA, El-Sitt S,
Nasrallah A, Marston D, El-Sabban M, Hahn KM, Symons M and El-Sibai
M: The regulation of RhoA at focal adhesions by StarD13 is
important for astrocytoma cell motility. Exp Cell Res. 321:109–122.
2014. View Article : Google Scholar
|
|
19
|
Lauffenburger DA and Horwitz AF: Cell
migration: A physically integrated molecular process. Cell.
84:359–369. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bermudez O, Pagès G and Gimond C: The
dual-specificity MAP kinase phosphatases: Critical roles in
development and cancer. Am J Physiol Cell Physiol. 299:C189–C202.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vial E, Sahai E and Marshall CJ: ERK-MAPK
signaling coordinately regulates activity of Rac1 and RhoA for
tumor cell motility. Cancer Cell. 4:67–79. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sebolt-Leopold JS, Dudley DT, Herrera R,
Van Becelaere K, Wiland A, Gowan RC, Tecle H, Barrett SD, Bridges
A, Przybranowski S, et al: Blockade of the MAP kinase pathway
suppresses growth of colon tumors in vivo. Nat Med. 5:810–816.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Krueger JS, Keshamouni VG, Atanaskova N
and Reddy KB: Temporal and quantitative regulation of
mitogen-activated protein kinase (MAPK) modulates cell motility and
invasion. Oncogene. 20:4209–4218. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zohrabian VM, Forzani B, Chau Z, Murali R
and Jhanwar-Uniyal M: Rho/ROCK and MAPK signaling pathways are
involved in glioblastoma cell migration and proliferation.
Anticancer Res. 29:119–123. 2009.PubMed/NCBI
|
|
25
|
Bradley KA, Mogridge J, Mourez M, Collier
RJ and Young JA: Identification of the cellular receptor for
anthrax toxin. Nature. 414:225–229. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Scobie HM, Rainey GJ, Bradley KA and Young
JA: Human capillary morphogenesis protein 2 functions as an anthrax
toxin receptor. Proc Natl Acad Sci USA. 100:5170–5174. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Abi-Habib RJ, Urieto JO, Liu S, Leppla SH,
Duesbery NS and Frankel AE: BRAF status and mitogen-activated
protein/extracellular signal-regulated kinase kinase 1/2 activity
indicate sensitivity of melanoma cells to anthrax lethal toxin. Mol
Cancer Ther. 4:1303–1310. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Abrami L, Liu S, Cosson P, Leppla SH and
van der Goot FG: Anthrax toxin triggers endocytosis of its receptor
via a lipid raft-mediated clathrin-dependent process. J Cell Biol.
160:321–328. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Melnyk RA and Collier RJ: A loop network
within the anthrax toxin pore positions the phenylalanine clamp in
an active conformation. Proc Natl Acad Sci USA. 103:9802–9807.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Huang D, Ding Y, Luo WM, Bender S, Qian
CN, Kort E, Zhang ZF, VandenBeldt K, Duesbery NS, Resau JH, et al:
Inhibition of MAPK kinase signaling pathways suppressed renal cell
carcinoma growth and angiogenesis in vivo. Cancer Res. 68:81–88.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
During RL, Li W, Hao B, Koenig JM,
Stephens DS, Quinn CP and Southwick FS: Anthrax lethal toxin
paralyzes neutrophil actin-based motility. J Infect Dis.
192:837–845. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dudley DT, Pang L, Decker SJ, Bridges AJ
and Saltiel AR: A synthetic inhibitor of the mitogen-activated
protein kinase cascade. Proc Natl Acad Sci USA. 92:7686–7689. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Duesbery NS, Webb CP, Leppla SH, Gordon
VM, Klimpel KR, Copeland TD, Ahn NG, Oskarsson MK, Fukasawa K,
Paull KD, et al: Proteolytic inactivation of MAP-kinase-kinase by
anthrax lethal factor. Science. 280:734–737. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Abi-Habib RJ, Singh R, Leppla SH, Greene
JJ, Ding Y, Berghuis B, Duesbery NS and Frankel AE: Systemic
anthrax lethal toxin therapy produces regressions of subcutaneous
human melanoma tumors in athymic nude mice. Clin Cancer Res.
12:7437–7443. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chopra AP, Boone SA, Liang X and Duesbery
NS: Anthrax lethal factor proteolysis and inactivation of MAPK
kinase. J Biol Chem. 278:9402–9406. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ha SD, Ng D, Pelech SL and Kim SO:
Critical role of the phosphatidylinositol 3-kinase/Akt/glycogen
synthase kinase-3 signaling pathway in recovery from anthrax lethal
toxin-induced cell cycle arrest and MEK cleavage in macrophages. J
Biol Chem. 282:36230–36239. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hanna S and El-Sibai M: Signaling networks
of Rho GTPases in cell motility. Cell Signal. 25:1955–1961. 2013.
View Article : Google Scholar : PubMed/NCBI
|