Introduction
Cancer is a major cause of human death in modern
industrialized countries. Non-small cell lung cancer (NSCLC), in
particular, is associated with air pollution and smoking, and
presents a low 5-year survival rate due to difficulties of
diagnosis and limited therapeutic options (1). NSCLC therapy typically involves
surgery, radiotherapy, and/or drug treatment. However, drugs and
radiation therapy frequently result in therapeutic resistance, the
primary obstacle to effective treatment for most cancers (2). Development of combinations of
therapeutic drugs forms an essential element of strategies to
improve patient survival. The rationale for combined drug therapy
is based on the concept that combinations of therapeutic modalities
with different mechanisms of action are more effective at
eradicating cancer than a single agent, while minimizing toxicity
and requiring lower drug doses (3).
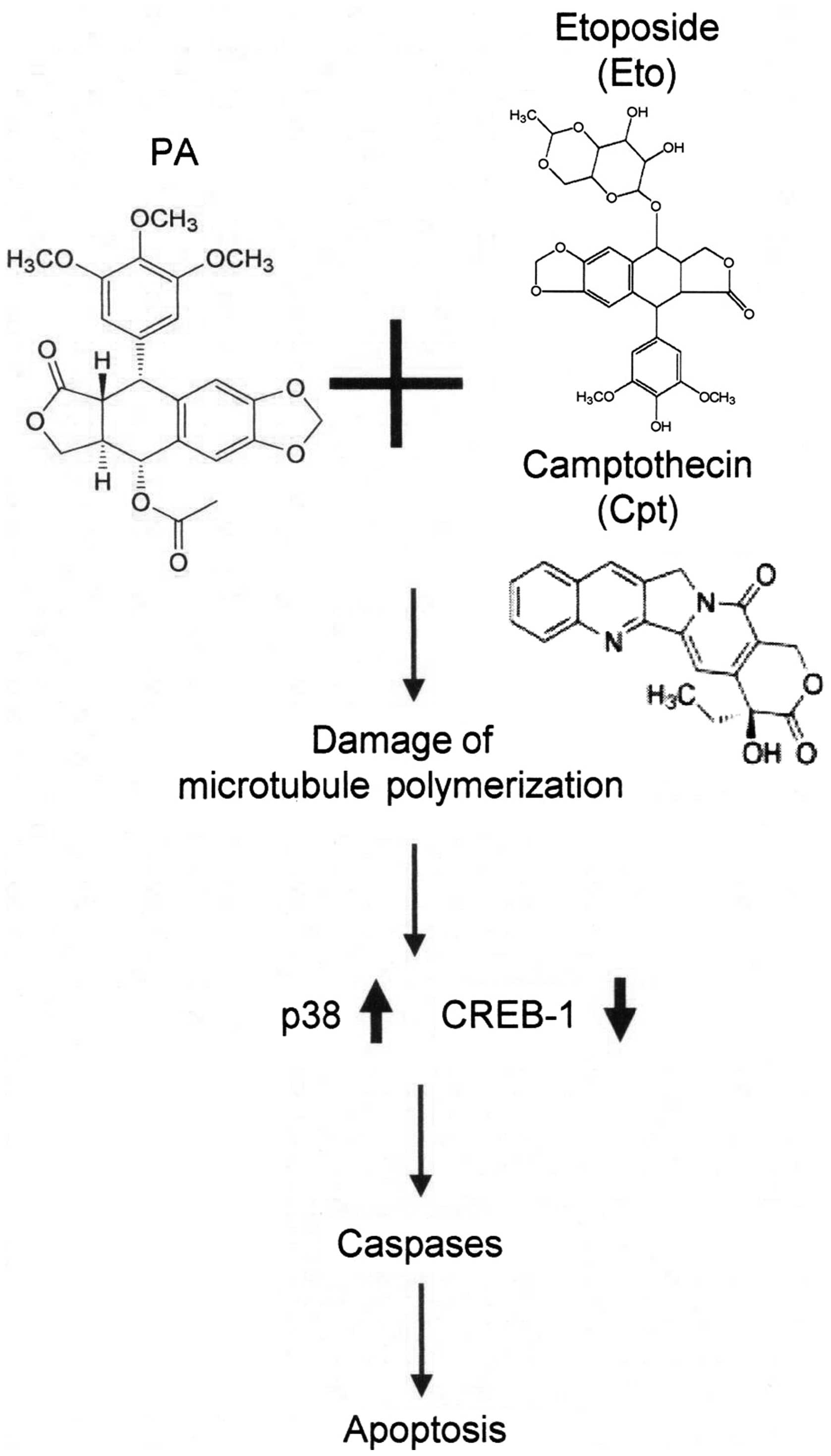
PA is a derivative of podophyllotoxin, previously
isolated from the natural product library as an anticancer drug
candidate and radiosensitizer (4).
Podophyllotoxin, one of the lignans isolated from podophyllin, is a
type of resin from Podophyllum. These lignans are secondary
metabolites consisting of two phenylpropane units produced via the
shikimic acid pathway. Podophyllotoxin exhibits the aryl-tetralin
structure of cyclolignans, a carbocycle between the two
phenylpropane units comprising two single carbon-carbon bonds
through the side-chains, one of which is located between the β-β′
positions. The lignan is historically reported to exert antiviral
and immunosuppressive effects as well as activity against venereal
warts (5). Additionally,
podophyllotoxin mediates antitumor activity via reversible binding
of tubulin and disruption of tubulin polymerization. This destroys
the dynamic equilibrium of microtubules and triggers disruption of
mitotic-spindle microtubule formation, inducing cell cycle arrest.
Several investigators have synthesized various derivatives, with
the aim of improving the antitumor effects of podophyllotoxin.
Studies to date led to the development of three representative
semi-synthetic epipodophyllotoxin derivatives, etoposide (Eto),
teniposide and etopophos (6).
Interestingly, the main anticancer target of Eto among these drugs
is not tubulin polymerization but topoisomerase (TOP) II in the DNA
TOP family, since etoposide does not prevent microtubule formation
due to the presence of a bulky glucoside moiety in its chemical
structure. DNA Tops are ubiquitous enzymes controlling the
topological state of DNA during replication that exist in two
forms, type I and II. Type I breaks a single strand of
double-stranded DNA, while type II cleaves both strands, resulting
in inhibition of religation of the nucleic acid-drug-enzyme complex
(5,6). Camptothecin (Cpt), an inhibitor of
TOP I initially isolated from the bark of Camptotheca
acuminate in 1966 is a cytotoxic quinoline alkaloid (7,8) that
possesses a planar pentacyclic ring structure. The A-D rings are
required for maintaining activity and the ‘E-ring lactone interacts
with the binding site in TOP I. Therefore, hydrolysis or removal of
lactone disrupts Cpt activity (9).
Induction of single- and double-stranded DNA breaks by TOP
inhibitors leads to effective cell death or apoptosis in rapidly
growing cell populations, such as cancer cells. Accordingly, TOP
inhibitors and derivatives have been widely employed to treat a
range of cancers, either as sole agents or combinations (10).
In this study, we examined whether PA promotes cell
death in combination with TOP inhibitors and assessed its potential
as a chemosensitizer to enhance cancer treatment efficiency. Our
results collectively demonstrated a synergistic effect between PA
and TOP inhibitors leading to enhanced apoptosis in NSCLC cell
lines, which was attributable to activation of p38 and caspases,
and inactivation of CREB-1.
Materials and methods
Cell culture and chemical reagents
NCI-H1299 and A549 human NSCLC cell lines were
purchased from American Type Culture Collection (Rockville, MD,
USA). SB203580, U0126, JNK inhibitor II, Eto, Cpt, z-VAD-fmk and
CBP-CREB interaction inhibitor (CREB inhibitor) were obtained from
EMD Millipore Corp. (Billerica, MA, USA). PA was acquired from
MicroSource Discovery Systems, Inc. (Gaylordsville, CT, USA).
MTT assay
NCI-H1299 and A549 cells (4×103
cells/well in 96-well plates) were seeded and treated with
different concentrations of PA and Eto/Cpt. After 72 h, 50 μl of
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
solution (2 mg/ml) was added to each well, and plates incubated at
37°C for 2 h. Formazan crystals formed by living cells were
dissolved in 200 μl/well dimethyl sulf-oxide (DMSO), and the
absorbance of individual wells read at 545 nm using a Multiskan EX
ELISA reader (Thermo Fisher Scientific, Waltham, MA, USA). The 50%
inhibitory concentration (IC50) of PA on HCI-H1299 and
A549 cells was calculated from a concentration-response analysis
performed using Softmax Pro software (Molecular Devices, Sunnyvale,
CA, USA).
Drug combination analysis
CompuSyn software (Ver. 1.0) and its manual were
downloaded from http://www.combosyn.com to calculate the synergistic
effects of combinations of PA and Eto/Cpt. Cell survival percentage
data were acquired from the MTT assay and inserted into CompuSyn
software. The effects of the PA and Eto/Cpt combinations were
analyzed, and the combination index (CI) for each treatment
calculated as described in the product manual.
Microtubule assembly assay
The microtubule assembly assay was performed as
described previously by our group (11). Briefly, NCI-H1299 and A549 cells
were seeded (1×106 cells per 100-mm culture dish) and
treated with 7.5 or 15 nM PA and 0.5 μM Eto/10 nM Cpt,
respectively, for 24 h. Cells were lysed and samples collected via
centrifugation. The supernatant fractions contained soluble αβ
tubulin dimers while the pellets contained polymerized
microtubules. Each fraction was subjected to immunoblot
analysis.
Immunocytochemical staining
NCI-H1299 and A549 cells (1×104) were
seeded in chamber slides and treated with 7.5 or 15 nM PA and 0.5
μM Eto/10 nM Cpt, respectively, for 24 h. Treated cells were fixed
with 1% paraformaldehyde and subsequently stained with an
anti-α-tubulin antibody and DAPI. Images of stained cells were
acquired with a 710 confocal microscope (Carl Zeiss, Germany).
Propidium iodide uptake assay
Cells were seeded at a density of 1×105
cells and incubated with or without 7.5 or 15 nM PA and 0.5 μM
Eto/10 nM Cpt. After 72 h, cells were harvested via trypsinization,
washed twice with cold PBS, and resuspended in 300 μl of 5 μg/ml
propidium iodide (PI, Sigma-Aldrich, St. Louis, MO, USA). The
apoptotic fraction was evaluated using a FACSort flow cytometer
(Becton-Dickinson, Franklin Lakes, NJ, USA).
Immunoblot analysis
Immunoblots were performed as described previously
(12). Membranes were probed with
antibodies against caspase-3, -8, and -9, phospho-p38, and p38
(Cell Signaling Technology, Inc., Beverly, MA, USA). An
anti-β-actin antibody (Sigma-Aldrich) was used a control for equal
loading. Relative band densities of targets, determined
densitometrically and normalized to that of β-actin or p38, were
analyzed using ImageJ software (NIH, USA).
ELISA detection assay of p38 enzyme
activity
To detect the phosphorylation level of p38, the p38
MAPK alpha (pT180/pY182) + total p38 MAPK alpha ELISA kit was
purchased from Abcam® (Cambridge, UK). NCI-H1299 and
A549 cells were seeded onto a 100-mm plate (1×106) and
treated with control, Eto, Cpt, PA only or combinations of PA/Eto
or PA/Cpt for 24 h. After discarding media, cells were rinsed with
PBS and dissolved with 1X cell lysate buffer containing protease
and phosphatase inhibitors provided in the kit. ELISA was performed
as described by the manufacturer. Supernatant fractions of each
sample were collected via centrifugation, followed by sequential
incubation with the relevant primary/secondary antibodies. Samples
were treated with TMB One-Step substrate reagent for 30 min and
Stop solution for detection of OD values at 450 nm in a Multiskan
EX ELISA reader (Thermo Fisher Scientific). Each OD value was
calculated as a phosphor-p38/p38 activity ratio and graphically
illustrated.
Electrophoretic mobility shift (EMSA)
assay
NCI-H1299 and A549 cells were harvested and nuclei
isolated as described previously (13). The EMSA system was purchased from
Promega (Madison, WI, USA), and all procedures performed as
described by the manufacturer.
Statistical analysis
Data were analyzed using GraphPad Prism software
(GraphPad Software, La Jolla, CA, USA), and the significance of
differences between experimental groups determined using the
Student's t-test. Data were considered significant at p-values
<0.05. Individual p-values in figures are denoted by asterisks
(*p<0.05,
**p<0.01,***p<0.001). The numbers above
each point or bar in the graphs represent the means of three
independent experiments, and error bars indicate standard
deviations (SD).
Results
The combination of PA and TOP inhibitors
has a synergistic effect
PA was initially screened from a natural product
library as an anticancer drug candidate (Fig. 1A). The IC50 values of PA
in NCI-H1299 and A549 cells were determined as 7.6 and 16.1 nM
after 72 h of treatment, respectively. Effects of combined
treatment with PA and TOP inhibitors (Eto and Cpt) on NSCLC cells
were assessed with CompuSyn software (Fig. 1B and C). NCI-H1299 and A549 cell
lines were treated with 7.5, 15, 30 and 60 nM PA and 0.5, 1, 2 and
4 μM or 0.25, 0.5, 1 and 2 μM Eto, respectively, and the
synergistic effects of PA/Eto examined. Treatment of NCI-H1299 and
A549 cells with 7.5, 15, 30 and 60 nM PA and 10, 20, 40 and 80 μM
or 5, 10, 20 and 40 nM Cpt was performed to detect synergistic
effects. Specifically, 7.5 nM PA + 0.5 μM Eto and 15 nM PA + 1 μM
Eto in NCI-H1299 cells and 7.5 nM PA + 0.25 μM Eto and 15 nM PA +
0.5 μM Eto in A549 cells were determined as combinations displaying
effective synergistic activity (Fig.
1B). All combinations of PA+Cpt, except 30 nM PA + 40 nM Cpt in
NCI-H1299 cells and 60 nM PA + 40 nM Cpt in A549 cells, exerted
synergistic effects (Fig. 1C). Our
results clearly indicate that PA and TOP inhibitors exert
synergistic effects in vitro. Accordingly, the intracellular
machineries involved in synergism were subsequently examined.
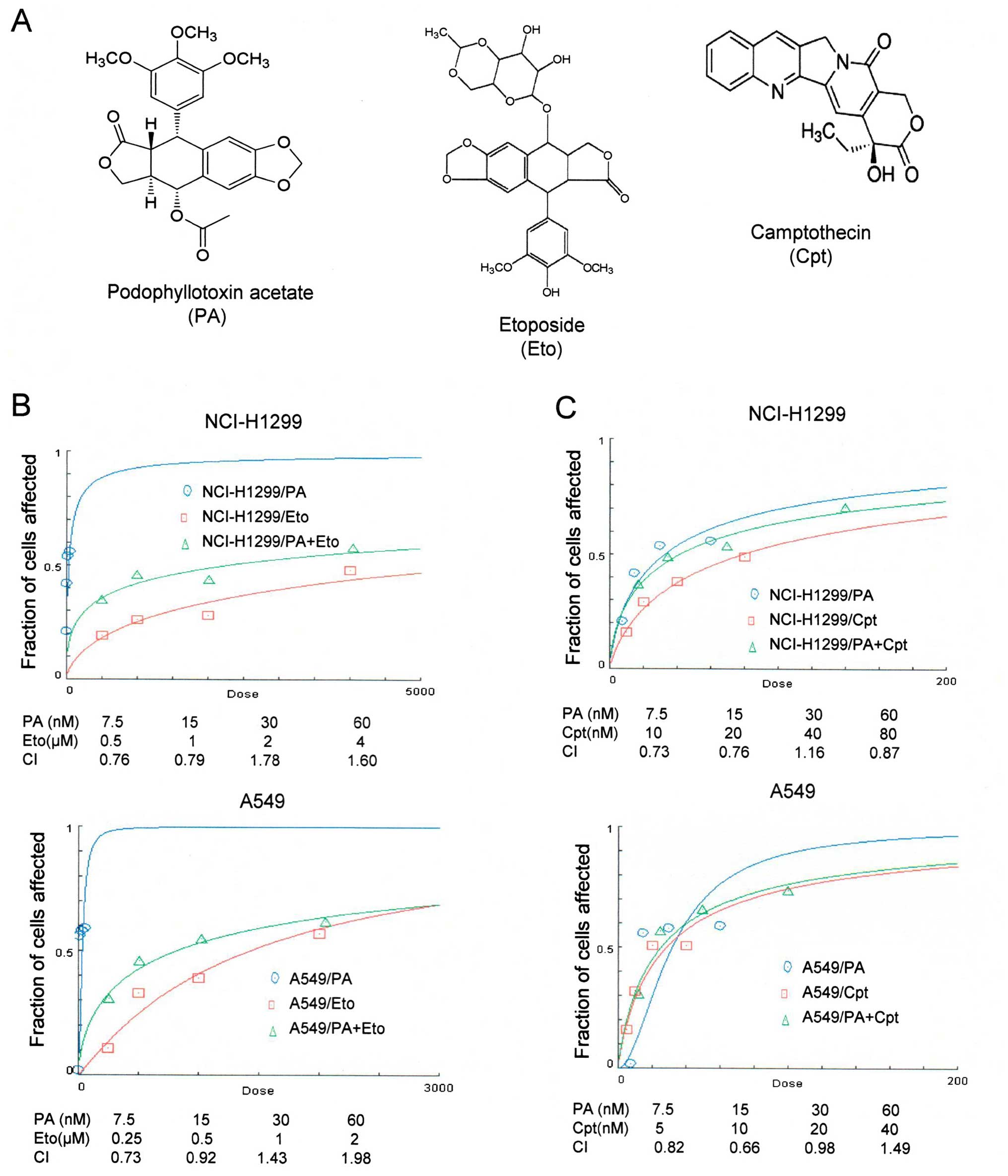 | Figure 1Combined treatment with PA and TOP
inhibitors (Eto and Cpt) synergistically induces apoptosis of
NCI-H1299 and A549 cells. (A) Chemical structures of PA (http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?sid=93087),
Eto (ref. 5) and Cpt (ref.
10). (B and C) Calculation of the
combination index (CI) of PA and Eto/Cpt. The MTT assay was
performed to determine the fraction of cells affected on the
y-axis. NCI-H1299 cells were co-treated with 0, 7.5, 15, 30 or 60
nM PA and 0, 0.5, 1, 2 or 4 μM Eto or 0, 10, 20, 40 or 80 nM Cpt.
A549 cells treated with 0, 7.5, 15, 30 or 60 nM PA and 0, 0.25,
0.5, 1 or 2 μM Eto or 0, 5, 10, 20 or 40 nM Cpt. |
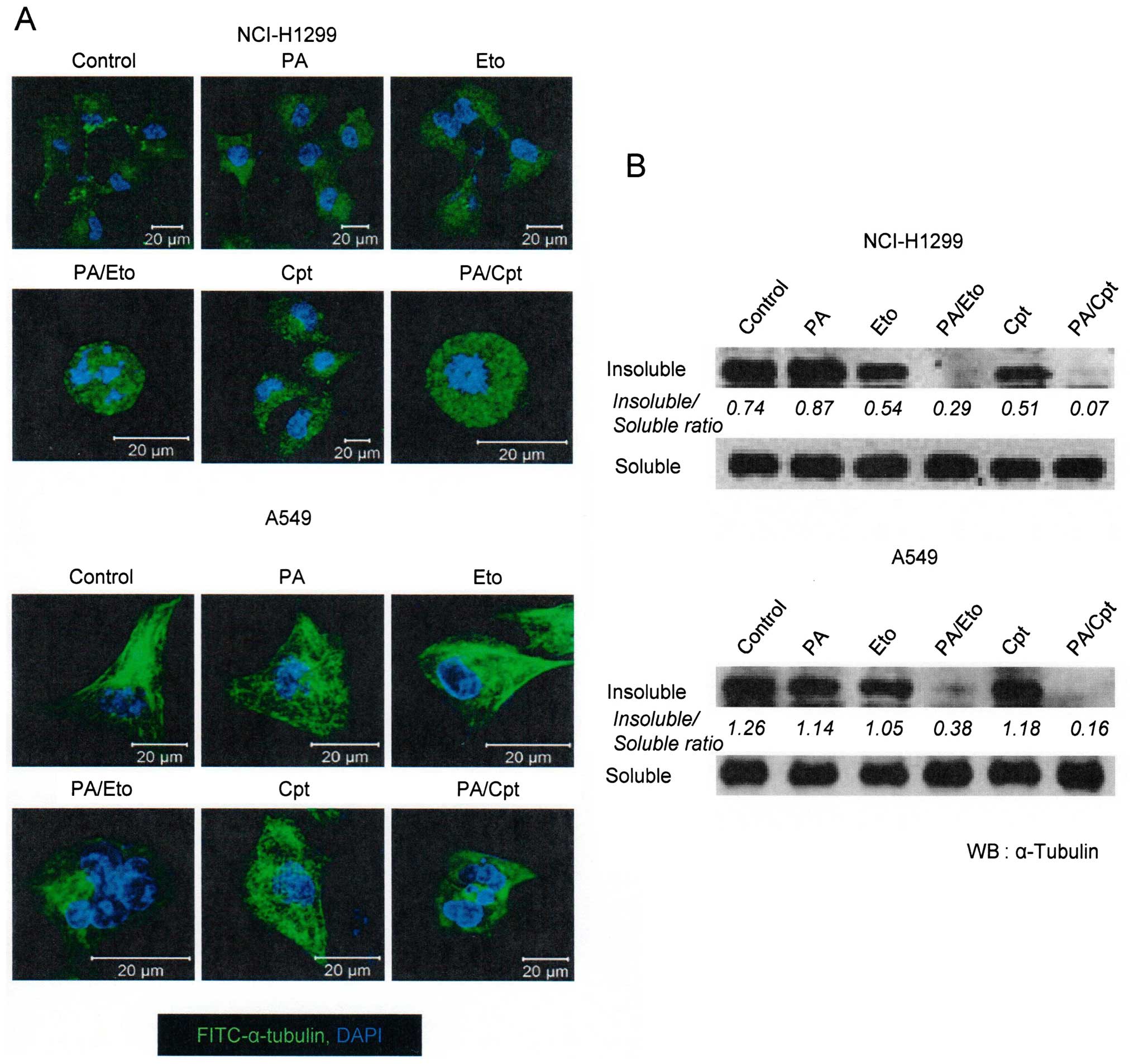
PA and TOP inhibitor combinations enhance
microtubule polymerization and apoptotic cell death
In view of the ability of PA to disrupt tubulin
polymerization (11), we examined
whether the combination of PA and TOP inhibitors exerts similar
effects. Microtubule assembly assays and immunocytochemical
staining using an anti-α-tubulin antibody revealed that combination
treatment led to decreased microtubule polymerization and
disruption of microtubule organization in NCI-H1299 and A549 cells
in a synergistic manner (Fig. 2).
Immunocytochemical staining disclosed rounded morphology of
NCI-H1299 cells and fragmented nuclei of A549 cells (Fig. 2A, upper panel). Based on results
showing that the drug combinations enhance the microtubule damage
effects of PA, studies on other cell death machinery were
performed. In subsequent experiments, apoptotic cell death
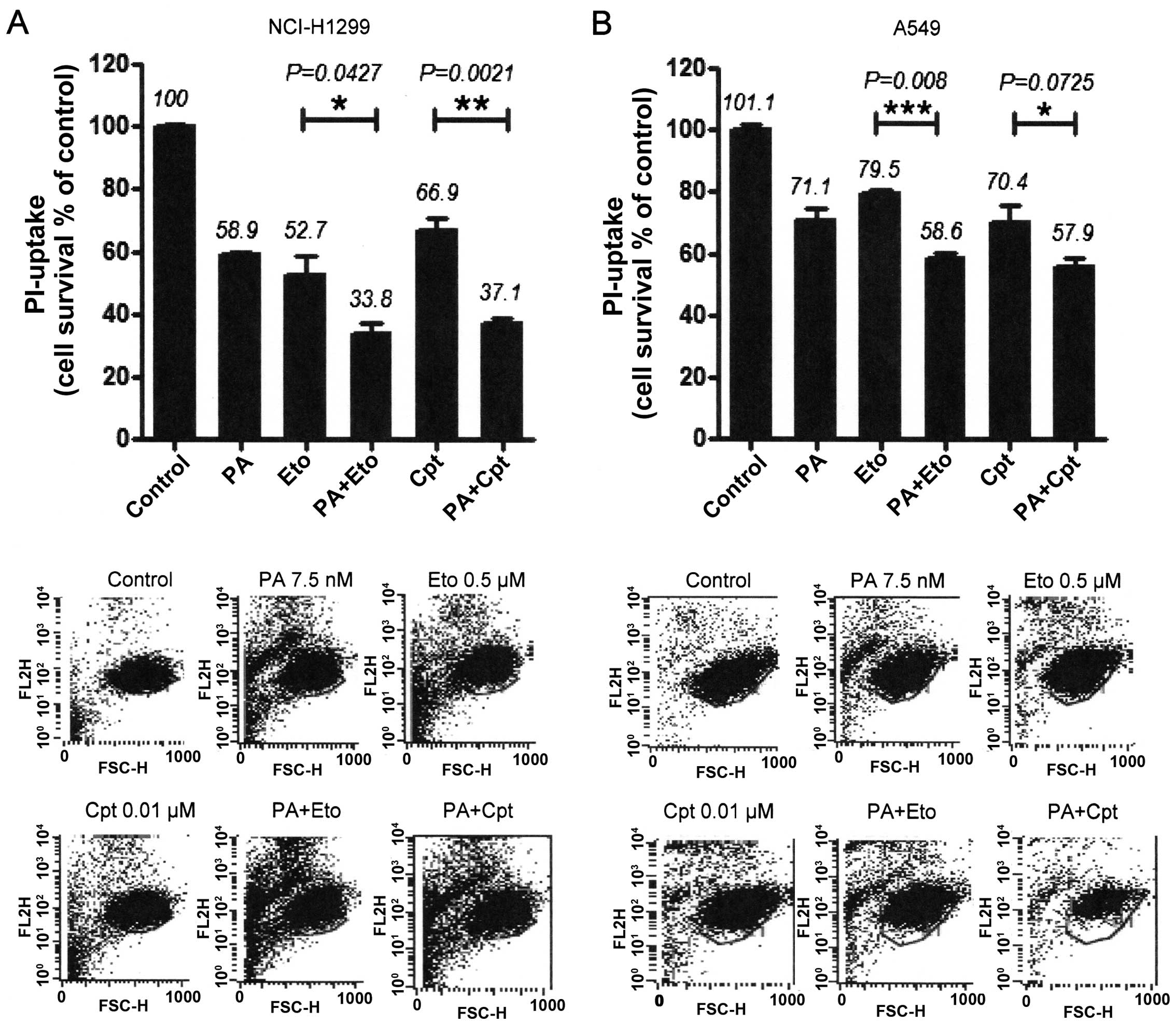
induction by the combinations was assessed with PI uptake (11). In NCI-H1299 cells, a combination of
PA and Eto (7.5 nM PA + 0.5 μM Eto) enhanced apoptosis by ~25 and
21%, compared with PA (7.5 nM) only and Eto (0.5 μM) only
treatment, while a combination of PA and Cpt (7.5 nM PA + 10 nM
Cpt) increased cell death by ~21 and 30%, respectively (Fig. 4A). In A549 cells, PA and Eto (15 nM
PA + 0.5 μM Eto) synergistically enhanced apoptosis by ~14 and 21%,
compared with PA (15 nM) only and Eto (0.5 μM) only, while PA and
Cpt (15 nM PA + 10 nM Cpt) increased cell death by ~15 and 24%,
respectively, relative to treatment with the individual drugs
(Fig. 4B). Treatment with the
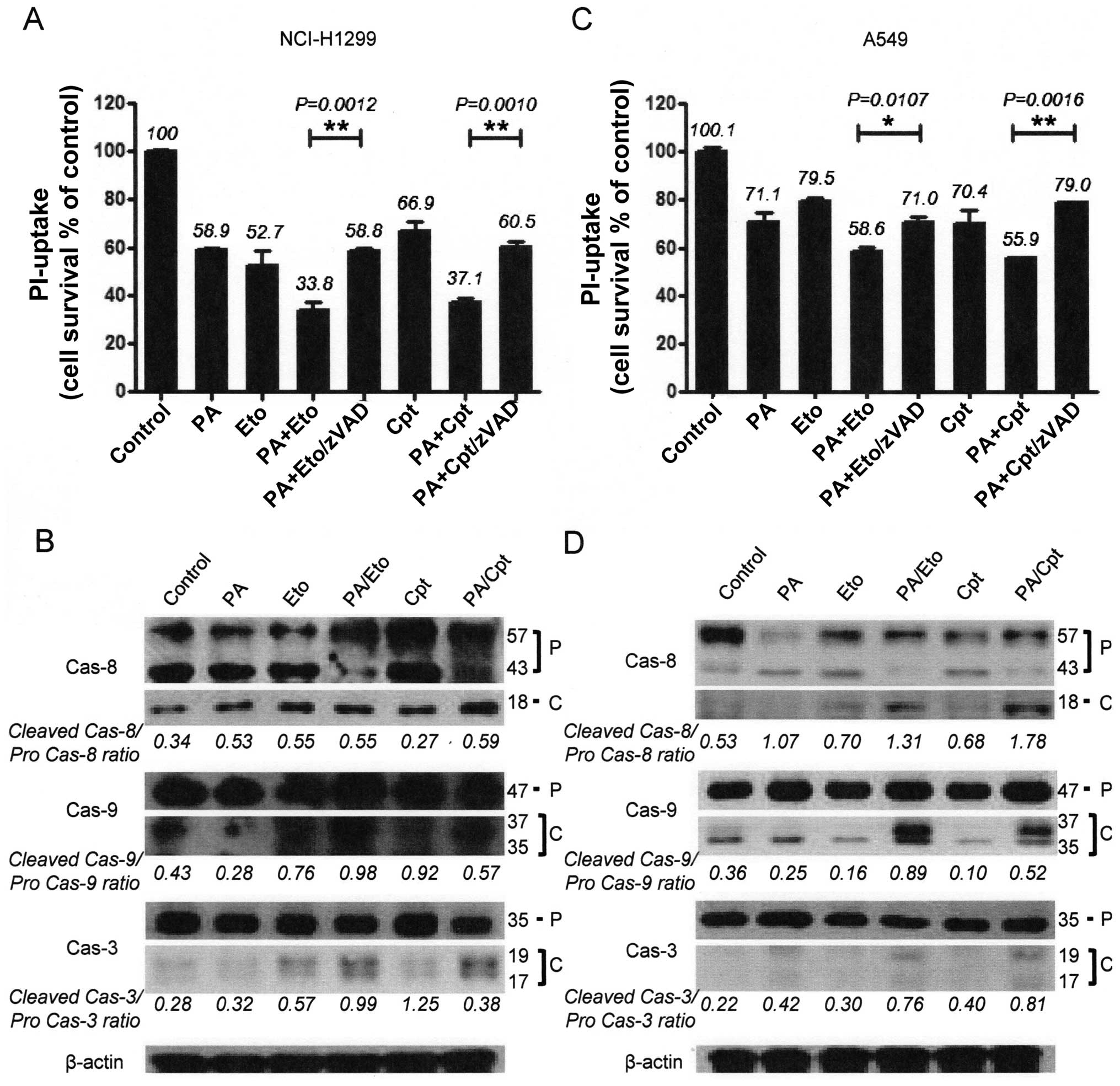
pan-caspase inhibitor, z-VAD-fmk, blocked the enhancement of cell
death induced by the drug combinations in both cell lines (Fig. 4A and C). We confirmed that the
combination treatments promote activation of apoptosis via
immunoblot assays for caspase-3, -8, or -9 (Fig. 4B and D). These results imply that
PA acts as a chemosensitizer that enhances the cytotoxicity of TOP
inhibitors by promoting apoptosis.
Modulation of MAPKs is involved in the
chemosensitizing effect of PA
To elucidate the mechanisms underlying
chemo-sensitization by PA, we initially examined the activities of
the mitogen-activated protein kinase (MAPK) family, including p38,
ERK and c-Jun N-terminal kinase (JNK), following treatment with the
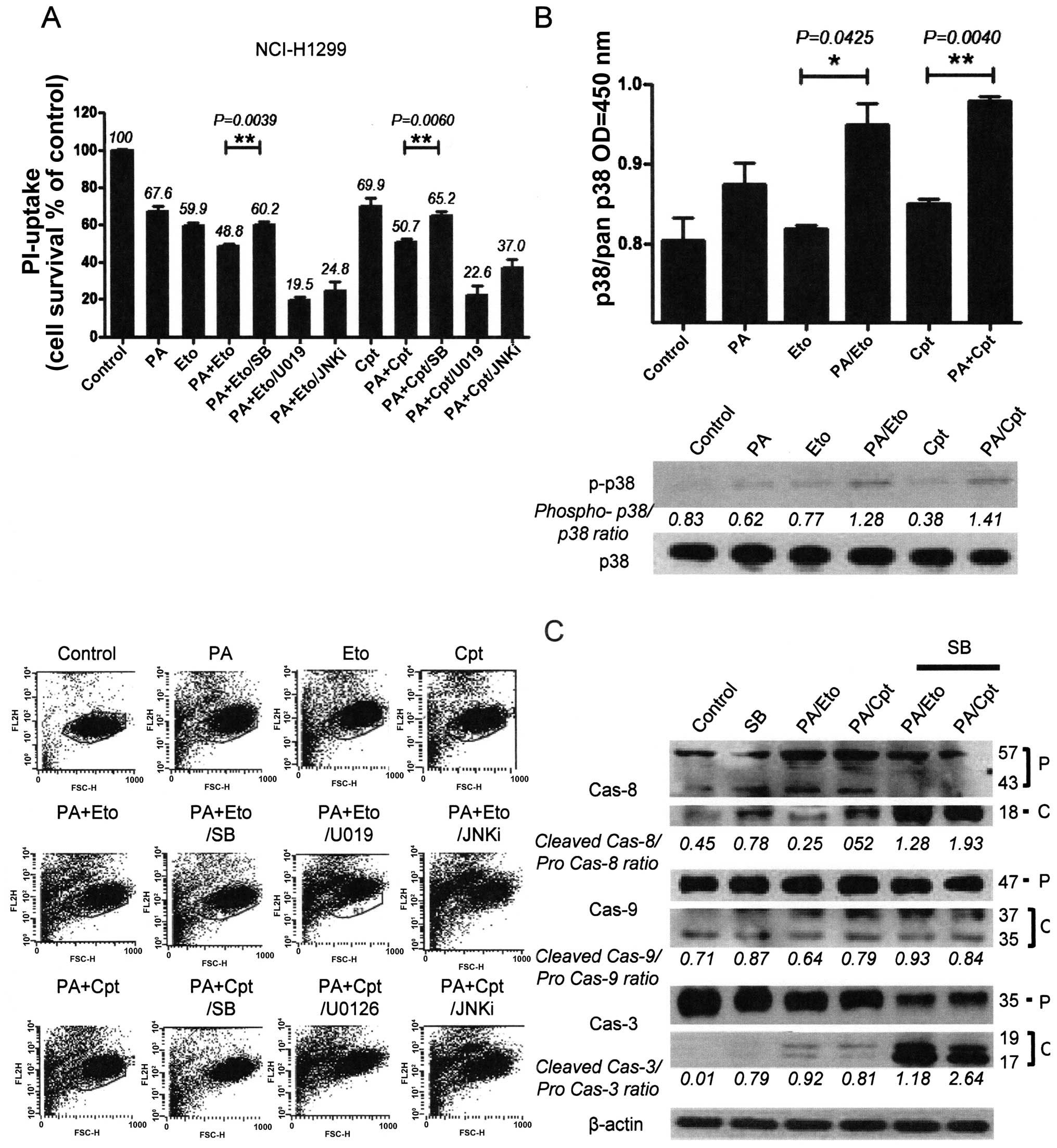
drug combinations. Pre-treatment of combined treatment cells with
chemical inhibitors (SB203580, U0126, JNK inhibitor II) of each
kinase revealed that only p38 inhibition by SB203580 blocked cell
death, indicating that p38 kinase is specifically involved in the
chemosensitization effect of PA. In contrast, pre-treatment with
U0126 and JNK inhibitor II enhanced cell death induced by the
combination (Fig. 5A and D).
Increased activity and phosphorylation of p38 were also detected
with activity (upper panel) and immunoblot assays (lower panel) in
the combination treatment groups (Fig.
5B and E). Moreover, inhibition of p38 blocked caspase
activation by the drug combinations in both NCI-H1299 and A549
cells, as shown in Fig. 5C and F.
Our findings suggest that the chemosensitization effect of PA on
TOP inhibitors is derived from enhancement of apoptosis via p38
activation.
Modulation of CREB-1 is involved in the
chemosensitizing effect of PA
To further elucidate the mechanism underlying the
chemosensitization activity of PA, we examined several
transcriptional factors, in view of the finding that
transcriptional factors, such as NF-κB, are activated downstream of
various stress signals (14).
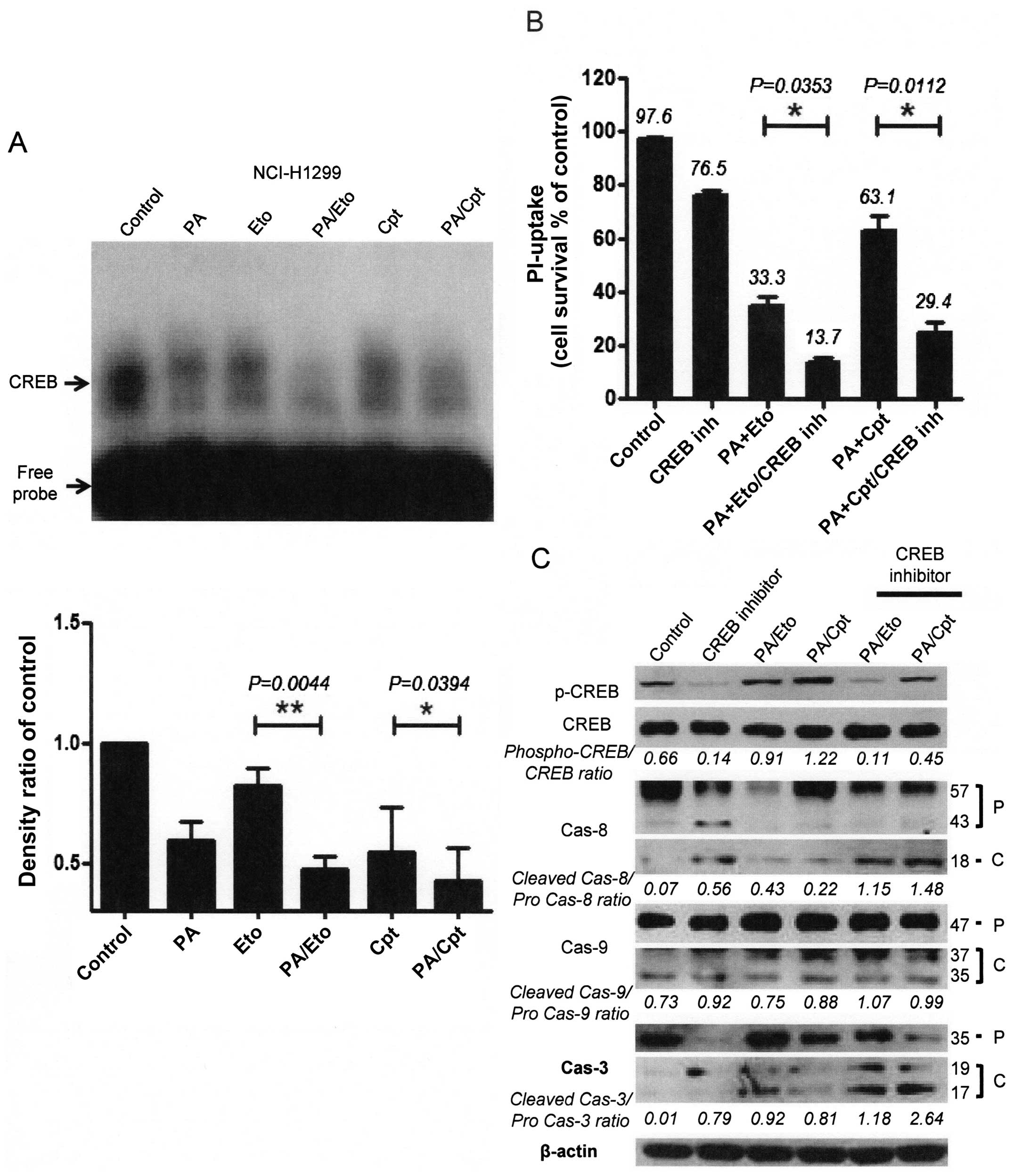
CREB-1 was constitutively activated in both NCI-H1299 and A549
cells, which was attenuated upon treatment with PA, Eto, or Cpt
only. Interestingly, combinations of PA/TOP inhibitors exerted
synergistic suppressive effects on activation of CREB-1 (Fig. 6A and D). Treatment with CREB
inhibitors enhanced cell death (Fig.
6B and E) and apoptotic pathway activation in cells treated
with the PA/TOP inhibitor combinations (Fig. 6C and F). Based on these findings,
we suggest that activation of CREB-1 is an essential step for NSCLC
cell survival and PA exerts its chemosensitizing effect through
inhibition of CREB-1.
Discussion
Improving chemotherapy and minimizing side-effects
may entail modulation of different facets of cancer-specific
processes involving chemoresistance. The mechanisms of
chemoresistance in cancer are classified into intrinsic (cells are
resistant before treatment) or acquired (resistance develops during
treatment). Significant mechanisms of chemoresistance include
overexpression of drug efflux pumps, increased activity of DNA
repair mechanisms, altered drug target enzymes, and overexpression
of enzymes involved in drug detoxification and elimination
(15). Since almost all anticancer
reagents induce elimination of cancer cells via apoptosis, the
chemoresistance mechanism is focused on modulation of the apoptosis
signal and molecules, for example, mutation or deletion of tumor
suppressor genes, such as p53, and overexpression of Bcl-2 or IAP
family proteins (16,17). Therefore, development of new
anticancer drugs or therapeutic strategies that induce activation
of apoptosis may present a key approach to overcoming
chemoresistance (18).
Data from this study demonstrated chemosensitization
activity of PA in combination with TOP inhibitors against NSCLC
cell lines, leading to enhanced apoptotic cell death and
attenuation of transcription factors involved in cell survival.
Topoisomerase I inhibitors, such as topotecan and irinotecan, are
usually components of combination chemotherapy against NSCLC
(19–21). Eto and Cpt have been identified as
powerful and dynamic cytostatic anticancer reagents under both
experimental and clinical conditions. These chemicals target a
group of microtubules, coincident with our present results
(Fig. 3), and act as apoptosis
inducers operating via distinct mechanisms. Therefore, treatment
with combinations of these agents should theoretically enhance the
final therapeutic outcomes (19).
The activities of Eto and Cpt as apoptosis inducers were confirmed
in our experiments (Fig. 4),
whereby two (intrinsic and extrinsic) major apoptotic pathways were
activated. The intrinsic pathway is induced by external stress and
leads to changes in mitochondrial permeability and activation of
caspase-9. The extrinsic pathway mainly begins with death
receptor/ligand binding and proceeds through caspase-8 activation.
Caspases-8 and -9 in extrinsic and intrinsic pathways are starting
‘initiator’ caspases, and both pathways activate caspase-3, a
common ‘executioner’ caspase (22). Inhibition of caspase activation
prevented apoptosis in both cell types and blocked synergistic cell
death (Fig. 4A and C). We further
showed that increased cell death induced by the combination of PA
and TOP inhibitors is accompanied by enhanced phosphorylation of
p38, which has been shown to evoke cell death secondary to DNA
damage and membrane oxidative damage (23,24).
Previous reports have shown that disruption of tubulin dynamics
leads to activation of p38, inducing apoptosis (11). Coincident with these findings,
combination treatment with PA and TOP inhibitors promoted
phosphorylation and consequent activation of p38 resulting in
increased apoptosis (Fig. 5).
Suppression of p38 with the specific inhibitor, SB203580,
attenuated caspase activation and apoptosis increase induced by the
combination of PA and TOP inhibitors. In contrast, prevention of
ERK and JNK activity with U0126 and JNK inhibitor II, respectively,
did not block enhancement of apoptosis in either NCI-H1299 or A549
cell line. Based on these findings, we propose that p38 lies
downstream of tubulin damage and is responsible for caspase
activation and induction of apoptosis. Additionally, the
radiosensitizer effect of PA was shown to be related to increased
reactive oxygen species (ROS) in our previous study (11), but the chemosensitizer effect of PA
with TOP inhibitors was not established (data not shown). Other
studies have also shown that p38 is a stress response molecule
involved in apoptotic cell death induced by numerous
chemotherapeutic agents and natural products, including
cyclophosphamide (CTX) commonly used for breast cancer,
anthocyanins for human colon cancer cells and oxaliplatin for human
colorectal cancer cells (25–27).
The increased chemosensitivity to CDDP induced by Met inactivation
is attributable to p38 MAPK activation (28). Interestingly, the combination of PA
and TOP inhibitors reduced phosphorylation of CREB-1, a 43-kDa
basic/leucine zipper (bZIP) transcription factor. CREB-1 is found
in most tissues and shown to be overexpressed in several cancer
tissues, including leukemic blast cells of acute myeloid leukemia
patients and lung cancer, compared to normal tissues (29–32).
CREB can form both homo and heterodimeric complexes. CREB binds to
the octanucleotide cAMP response element (CRE), TGANNTCA, as a
homodimer and to other members of the CREB/ATF transcriptional
factor superfamily as a heterodimer (33,34).
CREB becomes transcriptionally activated after phosphorylation at
serine 133, which induces activity by promoting interactions with
the 256-kDa coactivator CREB-binding protein (CBP) (35). Following its activation, the
transcription factor is reported to perform various physiological
functions, including integration of various stimuli, such as IR, in
a p53-dependent manner (34–36),
and promote pro-survival signaling important in cancer development
and progression (37,38). We detected phosphorylated and
activated CREB-1 in both NCI-H1299 and A549 cells (Fig. 6). Sole treatment with PA or TOP
inhibitor attenuated endogenous activation of CREB-1. Combined
treatments with PA/TOP inhibitors further enhanced suppression of
CREB-1 phosphorylation and activation. Our results imply that
CREB-1 facilitates cell survival in both cell types, and thus
potentially presents a major target of the PA/TOP inhibitor
combination.
We did not determine whether PA protects normal
tissue from chemotherapy-induced side-effects or whether modulation
of p38 and CREB-1 mediates whole-cell death in response to PA and
TOP inhibitor combinations in this study. Nonetheless, our current
results suggest a novel role for PA as a chemosensitizer that
enhances apoptotic death of NCI-H1299 and A549 NSCLC cells through
promotion of tubulin degradation, activation of the p38/caspase
pathway and suppression of CREB-1 activation (Fig. 7). Our findings provide new insights
supporting the utility of podophyllotoxin derivatives as
chemosensitizers and induction of apoptosis via p38/CREB-1/caspase
activation and suppression of CREB-1 as a useful strategy for the
development of new therapeutic agents.
Acknowledgements
This study was supported by the Nuclear Research and
Development Program of the National Research Foundation of Korea
(NRF) grant funded by the Korean government (MEST)
(2012M2A2A7010422) and the Basic Science Research Program through
the NRF (NRF-2014R1A1A2054985).
References
|
1
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mitsudomi T, Suda K and Yatabe Y: Surgery
for NSCLC in the era of personalized medicine. Nat Rev Clin Oncol.
10:235–244. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ismael GF, Rosa DD, Mano MS and Awada A:
Novel cytotoxic drugs: Old challenges, new solutions. Cancer Treat
Rev. 34:81–91. 2008. View Article : Google Scholar
|
|
4
|
Choi JY, Cho HJ, Hwang SG, Kim WJ, Kim JI,
Um HD and Park JK: Podophyllotoxin acetate enhances γ-ionizing
radiation-induced apoptotic cell death by stimulating the
ROS/p38/caspase pathway. Biomed Pharmacother. 70:111–118. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gordaliza M, García PA, del Corral JM,
Castro MA and Gómez-Zurita MA: Podophyllotoxin: Distribution,
sources, applications and new cytotoxic derivatives. Toxicon.
44:441–459. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Guerram M, Jiang Z-Z and Zhang L-Y:
Podophyllotoxin, a medicinal agent of plant origin: Past, present
and future. Chin J Nat Med. 10:161–169. 2012. View Article : Google Scholar
|
|
7
|
Wall ME, Wani MC, Cooke CE, Palmer KH,
McPhail AT and Sim GA: Plant antitumor agents. I. The isolation and
structure of camptothecin, a novel alkaloidal leukemia and tumor
nhibitor from Camptotheca acuminata. J Am Chem Soc. 88:3888–3890.
1966. View Article : Google Scholar
|
|
8
|
Hsiang Y-H, Hertzberg R, Hecht S and Liu
LF: Camptothecin induces protein-linked DNA breaks via mammalian
DNA topoisomerase I. J Biol Chem. 260:14873–14878. 1985.PubMed/NCBI
|
|
9
|
Gabr A, Kuin A, Aalders M, El-Gawly H and
Smets LA: Cellular pharmacokinetics and cytotoxicity of
camptothecin and topotecan at normal and acidic pH. Cancer Res.
57:4811–4816. 1997.PubMed/NCBI
|
|
10
|
Venditto VJ and Simanek EE: Cancer
therapies utilizing the camptothecins: A review of the in vivo
literature. Mol Pharm. 7:307–349. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Choi JY, Hong WG, Cho JH, Kim EM, Kim J,
Jung CH, Hwang SG, Um HD and Park JK: Podophyllotoxin acetate
triggers anticancer effects against non-small cell lung cancer
cells by promoting cell death via cell cycle arrest, ER stress and
autophagy. Int J Oncol. 47:1257–1265. 2015.PubMed/NCBI
|
|
12
|
Park JK, Jung HY, Park SH, Kang SY, Yi MR,
Um HD and Hong SH: Combination of PTEN and gamma-ionizing radiation
enhances cell death and G(2)/M arrest through regulation of AKT
activity and p21 induction in non-small-cell lung cancer cells. Int
J Radiat Oncol Biol Phys. 70:1552–1560. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Park JK, Park SH, So K, Bae IH, Yoo YD and
Um HD: ICAM-3 enhances the migratory and invasive potential of
human non-small cell lung cancer cells by inducing MMP-2 and MMP-9
via Akt and CREB. Int J Oncol. 36:181–192. 2010.
|
|
14
|
Piva R, Belardo G and Santoro MG:
NF-kappaB: A stress-regulated switch for cell survival. Antioxid
Redox Signal. 8:478–486. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shabbits JA, Hu Y and Mayer LD: Tumor
chemosensitization strategies based on apoptosis manipulations. Mol
Cancer Ther. 2:805–813. 2003.PubMed/NCBI
|
|
16
|
Reed JC: Regulation of apoptosis by bcl-2
family proteins and its role in cancer and chemoresistance. Curr
Opin Oncol. 7:541–546. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
LaCasse EC, Baird S, Korneluk RG and
MacKenzie AE: The inhibitors of apoptosis (IAPs) and their emerging
role in cancer. Oncogene. 17:3247–3259. 1998. View Article : Google Scholar
|
|
18
|
Turrini E, Ferruzzi L and Fimognari C:
Natural compounds to overcome cancer chemoresistance: Toxicological
and clinical issues. Expert Opin Drug Metab Toxicol. 10:1677–1690.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rudolf E and Cervinka M: Topoisomerases
and tubulin inhibitors: A promising combination for cancer
treatment. Curr Med Chem Anticancer Agents. 3:421–429. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Houghton PJ, Cheshire PJ, Hallman JD II,
Lutz L, Friedman HS, Danks MK and Houghton JA: Efficacy of
topoisomerase I inhibitors, topotecan and irinotecan, administered
at low dose levels in protracted schedules to mice bearing
xenografts of human tumors. Cancer Chemother Pharmacol. 36:393–403.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bunn PA Jr and Kelly K: New
chemotherapeutic agents prolong survival and improve quality of
life in non-small cell lung cancer: A review of the literature and
future directions. Clin Cancer Res. 4:1087–1100. 1998.PubMed/NCBI
|
|
22
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lawenda BD, Kelly KM, Ladas EJ, Sagar SM,
Vickers A and Blumberg JB: Should supplemental antioxidant
administration be avoided during chemotherapy and radiation
therapy? J Natl Cancer Inst. 100:773–783. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Van Laethem A, Nys K, Van Kelst S,
Claerhout S, Ichijo H, Vandenheede JR, Garmyn M and Agostinis P:
Apoptosis signal regulating kinase-1 connects reactive oxygen
species to p38 MAPK-induced mitochondrial apoptosis in
UVB-irradiated human keratinocytes. Free Radic Biol Med.
41:1361–1371. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pang H, Cai L, Yang Y, Chen X, Sui G and
Zhao C: Knockdown of osteopontin chemosensitizes MDA-MB-231 cells
to cyclophosphamide by enhancing apoptosis through activating p38
MAPK pathway. Cancer Biother Radiopharm. 26:165–173. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shin DY, Lee WS, Lu JN, Kang MH, Ryu CH,
Kim GY, Kang HS, Shin SC and Choi YH: Induction of apoptosis in
human colon cancer HCT-116 cells by anthocyanins through
suppression of Akt and activation of p38-MAPK. Int J Oncol.
35:1499–1504. 2009.PubMed/NCBI
|
|
27
|
Chiu SJ, Chao JI, Lee YJ and Hsu TS:
Regulation of gamma-H2AX and securin contribute to apoptosis by
oxaliplatin via a p38 mitogen-activated protein kinase-dependent
pathway in human colorectal cancer cells. Toxicol Lett. 179:63–70.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lou X, Zhou Q, Yin Y, Zhou C and Shen Y:
Inhibition of the met receptor tyrosine kinase signaling enhances
the chemosensitivity of glioma cell lines to CDDP through
activation of p38 MAPK pathway. Mol Cancer Ther. 8:1126–1136. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Crans HN and Sakamoto KM: Transcription
factors and translocations in lymphoid and myeloid leukemia.
Leukemia. 15:313–331. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pigazzi M, Ricotti E, Germano G, Faggian
D, Aricò M and Basso G: cAMP response element binding protein
(CREB) over-expression CREB has been described as critical for
leukemia progression. Haematologica. 92:1435–1437. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shankar DB, Cheng JC, Kinjo K, Federman N,
Moore TB, Gill A, Rao NP, Landaw EM and Sakamoto KM: The role of
CREB as a proto-oncogene in hematopoiesis and in acute myeloid
leukemia. Cancer Cell. 7:351–362. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Seo H-S, Liu DD, Bekele BN, Kim MK,
Pisters K, Lippman SM, Wistuba II and Koo JS: Cyclic AMP response
element-binding protein overexpression: A feature associated with
negative prognosis in never smokers with non-small cell lung
cancer. Cancer Res. 68:6065–6073. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shaywitz AJ and Greenberg ME: CREB: A
stimulus-induced transcription factor activated by a diverse array
of extracellular signals. Annu Rev Biochem. 68:821–861. 1999.
View Article : Google Scholar
|
|
34
|
Mayr B and Montminy M: Transcriptional
regulation by the phosphorylation-dependent factor CREB. Nat Rev
Mol Cell Biol. 2:599–609. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mayr BM, Canettieri G and Montminy MR:
Distinct effects of cAMP and mitogenic signals on CREB-binding
protein recruitment impart specificity to target gene activation
via CREB. Proc Natl Acad Sci USA. 98:10936–10941. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Amorino GP, Hamilton VM, Valerie K, Dent
P, Lammering G and Schmidt-Ullrich RK: Epidermal growth factor
receptor dependence of radiation-induced transcription factor
activation in human breast carcinoma cells. Mol Biol Cell.
13:2233–2244. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Matsumoto K, Yamamoto T, Kurachi H, Nishio
Y, Takeda T, Homma H, Morishige K, Miyake A and Murata Y: Human
chorionic gonadotropin-alpha gene is transcriptionally activated by
epidermal growth factor through cAMP response element in
trophoblast cells. J Biol Chem. 273:7800–7806. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Swarthout JT, Tyson DR, Jefcoat SC Jr and
Partridge NC: Induction of transcriptional activity of the cyclic
adenosine monophosphate response element binding protein by
parathyroid hormone and epidermal growth factor in osteoblastic
cells. J Bone Miner Res. 17:1401–1407. 2002. View Article : Google Scholar : PubMed/NCBI
|