Introduction
Lung cancer is the most frequent cancer-related
cause of death worldwide, and more than one-quarter of cancer
patients die from lung cancer annually. An estimated 1.8 million
new lung cancer cases were recorded in 2012, and these cases
accounted for approximately 13% of the total cancer cases (1). Despite the lower prevalence of
smoking in China, lung cancer rates among Chinese women (20.4 cases
per 100,000 women) are higher than those among European women. Lung
cancer treatments, including surgery, platinum doublet therapy,
radiation therapy, and targeted therapy, depend on histological
type and stage upon diagnosis; these treatment regimens account for
15% of the 5-year overall survival rate of patients in all stages
combined (2). Non-small cell lung
cancer (NSCLC) accounts for the majority (80%) of all lung cancer
cases (3). The epidermal growth
factor receptor-tyrosine kinase inhibitors (EGFR-TKIs) gefitinib
and erlotinib elicit remarkable therapeutic effects against NSCLCs,
with EGFR-activating mutations, such as exon 19 deletions and L858R
point mutations. However, cancer cells commonly develop TKI
resistance; as a result, tumor recurrence occurs (4). Therefore, acquired EGFR-TKI
resistance should be clinically resolved. Differences between
gefitinib responders and non-responders have been revealed in terms
of the frequency of secondary mutation in exon 20 of EGFR T790M.
EGFR Thr790 is an important amino acid residue accounting for the
specificity of an inhibitor in the ATP binding pocket behind the
ATP binding cleft; nevertheless, the substitution of Thr790 with
Met increases the ATP affinity and reduces the potency of any
ATP-competitive kinase inhibitor through which the T790M mutation
confers drug resistance (5). T790M
mutation has also been discovered in up to 50% of cases exhibiting
acquired resistance (6).
Therefore, strategies to overcome EGFR-TKI resistance should be
developed to prolong the survival time of lung cancer patients.
Efforts to block signaling from the second mutant
EGFR with small-molecule inhibitors has led to the conceptual
introduction of experimental, irreversible EGFR TKIs that function
primarily by covalently attaching to the cysteinyl-797 residue in
the pocket of the EGFR-kinase domain (7). Small-molecule inhibitors, such as
HKI-272, BIBW2992 (Afatinib), and PF00299804, have been mainly used
to reverse the acquired resistance to EGFR T790M mutation in
clinics and laboratories; these inhibitors have also demonstrated
modest efficacy in preclinical in vitro model studies
(8). However, in T790M
EGFR-harboring cells, the EGFR inhibition induced by currently
available second-generation EGFR-TKIs is insufficient to
physiologically prevent the emergence of cells that remain
dependent on EGFR signaling (8).
Thus, multi-targeted third-generation EGFR-TKIs have been designed
and have provided clinical advantages (9). Furthermore, novel multi-targeted
drugs that can overcome EGFR-TKI resistance should be identified
and developed to prolong the overall survival time of NSCLC
patients.
Protein phosphatase 2A (PP2A) is a tumor suppressor
enzyme that regulates cell homeostasis by counteracting most of the
kinase-driven intracellular signaling pathways (10). The aberrant expression, mutation,
and somatic alteration of the PP2A scaffold and regulatory subunits
are frequently found in human breast, lung, colon, and skin cancers
(11). Therefore, the reactivation
of PP2A on the basis of its tumor suppressor properties is a
potential therapeutic strategy to treat human cancer (12,13).
In malignant cellular growth, the cancerous inhibitor of protein
phosphatase 2A (CIP2A) protein inhibits the PP2A activity of the
oncogenic transcription factor c-Myc (14). Previous independent studies
demonstrated that aberrant CIP2A overexpression is associated with
tumor growth, apoptosis resistance, drug resistance, prognosis, and
metastasis in many human malignancies, including leukemia (15,16),
gastric (17), breast (18), tongue (19), head and neck (14), ovarian (20), and lung (21) cancers. High CIP2A expression is
also considered as a poor prognostic factor of lung cancer
(22). Thus, targeting CIP2A
protein is a potential strategy to treat cancer by reactivation of
PP2A.
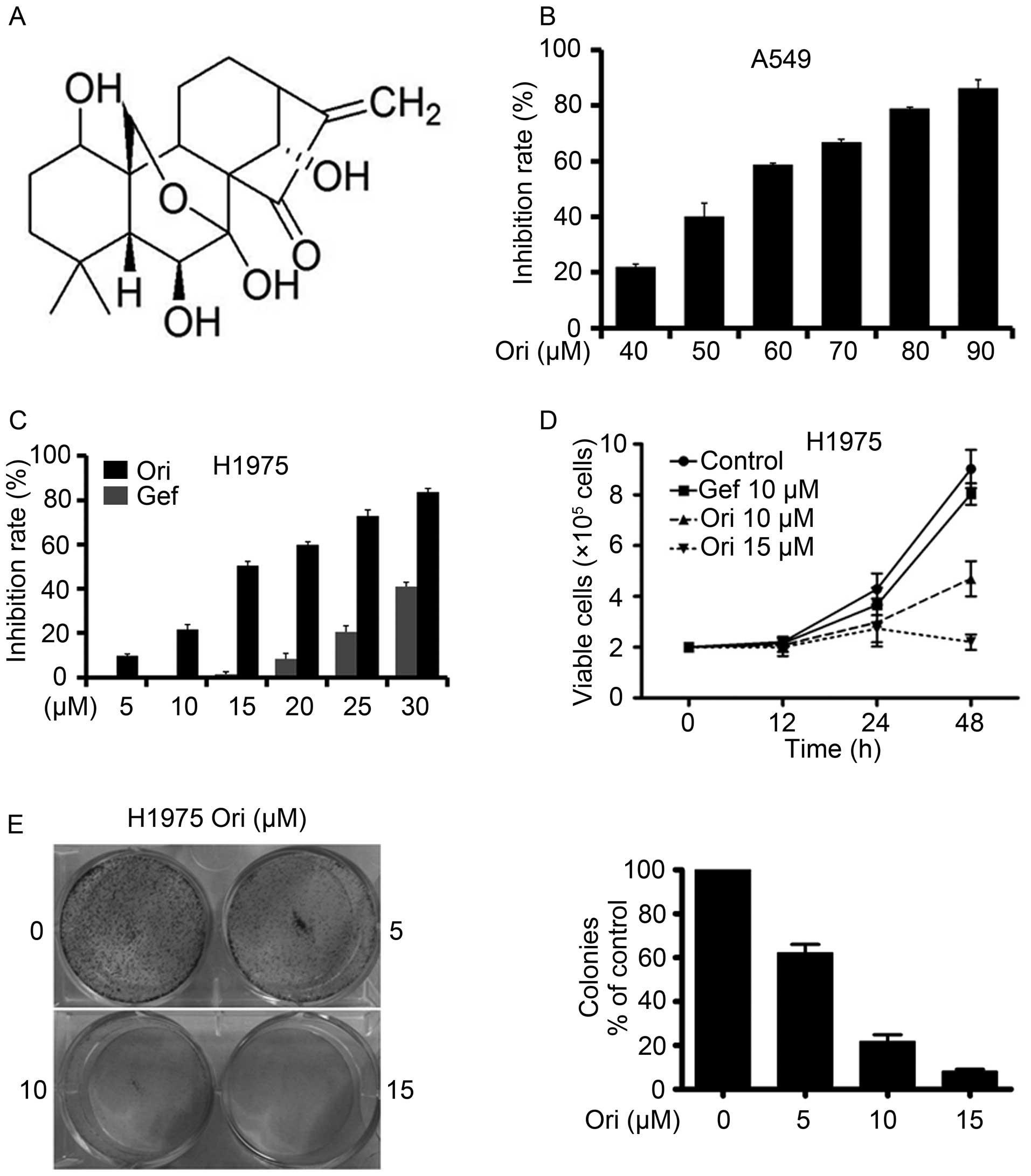
Oridonin (Ori, Fig.
1A), a bitter tetracyclin diterpenoid compound, has been
isolated from Rabdosia rubescens, Isodon japonicus
Hara, and I. trichocarpus, which are widely distributed in
China, Japan, and Korea (23). Ori
displays a potent anticancer activity against a wide range of
cancer cell types, including cells from patients with prostate and
breast cancers, NSCLC, acute leukemia, glioblastoma multiforme, and
human melanoma (23–27). This study investigated the
antitumor effects and possible mechanisms of Ori against
gefitinib-resistant EGFRL858R/T790M-driven NSCLC.
Materials and methods
Reagents
Oridonin (Ori) with a purity of up to 98% was
purchased from Shanghai Yuanye Bio-Technology Co., Ltd. Ori was
dissolved in DMSO (Sigma) at a stock solution of 100 mM and stored
at −20°C. Docetaxel (DTX) was provided by Shanghai Jinhe
Bio-Technology Co., Ltd. (Shanghai, China).
Cell culture
Human NSCLC cell lines A549 and NCI-H1975 (H1975)
were obtained from American Type Culture Collection (ATCC). A549
cells were cultured in Dulbecco modified Eagle medium (DMEM). H1975
cells were cultured in RPMI-1640 medium. DMEM and RPMI-1640 medium
were supplemented with 10% fetal bovine serum (FBS) (Hyclone), 100
U/ml penicillin, and 100 μg/ml streptomycin and cultured in a
humidified atmosphere with 5% CO2 at 37°C.
Cytotoxic assay and cell viability
Cells were seeded into 96-well plate and
pre-cultured for 24 h, then treated with Ori for 24 h. Cell
cytotoxicity was determined by MTT assay. The absorbance was
measured at 570 nm by Automated Microplated Reader (Bio-Tek,
Winooski, VT, USA), and the cell death rate was calculated as
follows: Inhibition Rate (%) = (average A570 of the
control group − average A570 of the experimental
group)/(average A570 of the control group − average
A570 of the blank group) ×100%. Cell viability was
estimated by trypan blue dye exclusion (28).
Soft-agar colony formation assay
Cells were suspended in 1 ml of RPMI-1640 containing
0.3% low-melting-point agarose (Amresco, Solon, OH, USA) and 10%
FBS, and plated on a bottom layer containing 0.6% agarose and 10%
FBS in 6-well plate in triplicate. After 2 weeks, plates were
stained with 0.2% gentian violet and the colonies were counted
under a light microscope (29).
Invasion assay
An invasion assay was carried out using a 24-well
plate (Corning). A polyvinyl-pyrrolidone-free poly-carbonate filter
(8 μm pore size) (Corning) was coated with Matrigel (BD
Biosciences). The lower chamber was filled with medium containing
20% FBS as chemoattractant. The coated filter and upper chamber
were laid over the lower chamber. Cells (1×104
cells/well) were preincubated with Ori for 30 min at room
temperature, and then cell suspension containing Ori was seeded
onto the upper chamber wells. After incubation for 20 h at 37°C,
the filter was fixed and stained with 2% ethanol containing 0.2%
crystal violet (15 min). After being dried, the stained cells were
enumerated under a light microscope at ×10 objective. For
quantification, the invaded stained cells on the other side of the
membrane were extracted with 33% acetic acid. The absorbance of the
eluted stain was determined at 570 nm.
Wound healing assay
Cells (4×105 cells/2 ml) were seeded in a
6-well plate and incubated at 37°C until 90 to 100% confluence.
After the confluent cells were scratched with a 200 μl pipette tip,
followed by washing with PBS, and then treated with Ori in a
complete medium. After 24 h of incubation, the cells were fixed and
stained with 2% ethanol containing 0.2% crystal violet powder (15
min), and randomly chosen fields were photographed under a light
microscope at ×4 objective. The number of cells migrated into the
scratched area was calculated.
Adhesion assay
Cells (5×104 cells/well) preincubated
with Ori for 30 min at 37°C were seeded in a 96-well plate coated
with matrigel for 20 min at 37°C. Unattached cells were removed by
washing with PBS. Attached cells were fixed in 4% paraformaldehyde
for 15 min, stained with 0.02% crystal violet solution for 10 min,
and randomly chosen fields were photographed under a Leica DMI 400B
microscope. Then, to quantify the number of attached cells, crystal
violet was dissolved with 70% ethanol and OD was measured by
micro-plate reader at 570 nm, reference 405 nm. The adhesion cells
were calculated as a percentage of adhesion.
Western blotting
Cell pellets were lysed in RIPA buffer containing 50
mM Tris pH 8.0, 150 mM NaCl, 0.1% SDS, 0.5% deoxycholate, 1% NP-40,
1 mM DTT, 1 mM NaF, 1 mM sodium vanadate, 1 mM PMSF (Sigma), and 1%
protease inhibitors cocktail (Merck). Protein extracts were
quantitated and loaded on 8 to 12% sodium dodecyl sulfate
polyacrylamide gel, electrophoresed, and transferred to a PVDF
membrane (Millipore). The membrane was incubated with primary
antibody, washed, and incubated with horseradish peroxidase
(HRP)-conjugated secondary antibody (Pierce). Detection was
performed by using a chemiluminescent Western detection kit (Cell
Signaling Technology). The antibodies used were anti-CIP2A,
anti-ERK2, anti-phospho-Akt (Ser473), anti-Akt (Santa Cruz
Biotechnology), anti-phospho-ERK1/2 (Thr202/Tyr204), anti-EGFR
(L858R), anti-phospho-EGFR (Y1173), anti-PP2Ac (Cell Signaling
Technology), anti-MMP-12 (Proteintech), anti-MMP-2 (Epitomics), and
anti-GAPDH (Sangon Biotech Co., Ltd., AB10016) antibodies.
Gelatin zymography
H1975 cells were seeded in 6-well plates and allowed
to grow to 80% confluency. The cells were then maintained in
serum-free medium for 12 h prior to designated treatments with Ori
for 24 h. Conditioned medium was then collected, cleared and mixed
with 5X SDS loading buffer, and subjected to electrophoresis on a
10% SDS-PAGE gel containing 0.1% gelatin. After electrophoresis,
the gels were washed in renaturing buffer (pH 7.5, 2.5% Triton
X-100) for 30 min, 4 times, and equilibrated in developing buffer
(50 mM Tris-HCl pH 7.5, 10 mM CaCl2, and 1 mM
ZnCl2) for 30 min and finally incubated in fresh
developing buffer at 37°C for 24 h to allow digestion of the
gelatin. The gelatinolytic activity of MMPs was visualized by
staining the gels with 0.5% Coomassie blue R-250 in 45% methanol,
10% acetic acid and destained with 45% methanol, 10% acetic acid
until clear bands suggestive of gelatin digestion appeared.
RNA preparation and semi-quantitative
PCR
The total RNA of the cells was isolated using the
TRIzol Reagent (Invitrogen) and the phenol-chloroform extraction
method according to the manufacturer's instruction. Total RNA (2
μg) was annealed with random primers at 65°C for 5 min. The cDNA
was synthesized using a 1st-STRAND cDNA Synthesis kit (Fermentas).
For PCR amplification, primers are as follows: GAPDH, forward
primer: 5′-TCACCAGGGCTGCTTTTA-3′, reverse primer:
5′-AAGGTCATCCCTGAGCTGAA-3′; MMP-12 forward primer:
5′-TTGTTCCTCACTGCTGTTCAC-3′; reverse primer:
5′-GTCCATCATCTGTCTCCTTTC-3′.
AP-1 luciferase reporter assay
H1975 cells were seeded in 12-well culture plates.
At a confluency of 70%, cells were transfected with the pAP-1-Luc
plasmids (Beyotime Institute of Biotechnology) using Lipofectamine
3000 (Invitrogen) according to manufacturer's instruction. After
transfection for 4 h, the cells were treated with Ori for 20 h.
Firefly luciferase activities were assayed using the Luciferase
Assay System (Promega) according to the manufacturer's
instructions.
Chromatin immunoprecipitation (ChIP)
assay
H1975 cells treated with Ori for 24 h were processed
for ChIP assay using a ChIP-IT® Express Enzymatic
Shearing kit (Active Motif). Briefly, immunoprecipitation was
performed with anti-c-Fos (component of AP-1, Cell Signaling
Technology) or rabbit IgG as a control. The AP-1 binding site of
MMP-12 promoter was detected by real-time PCR using the following
primers: 5′-GCTAATTGATCCATTGT-3′ (forward) and
5′-TCTAGCCTAAGTTCC-3′ (reverse).
PP2A activity assay
PP2A immunoprecipitation phosphatase assay kit
(Upstate Biotechnology, Temecula, CA, USA) was used to measure
phosphate release as an index of phosphatase activity according to
the manufacturer's instructions. Briefly, 100 μg of protein
isolated from cells was incubated with 4 μg anti-PP2A monoclonal
antibody overnight. 40 μl of protein A agarose beads were added and
the mixture was incubated at 4°C for 2 h. Subsequently, the beads
were collected and washed three times with 700 μl of ice-cold TBS
and one time with 500 μl of Ser/Thr Assay buffer. The beads were
further incubated with 750 mM phosphopeptide in assay buffer for 10
min at 30°C with constant agitation. Malachite Green Phosphate
Detection Solution (100 μl) was added and the absorbance at 650 nm
was measured on a microplate reader (30).
Drug combination assay
Drug combination is widely used in cancer treatment
to achieve synergistic therapeutic effect and overcome drug
resistance in clinic. To estimate the effect of Ori and DTX
combination, the combination index (CI) was calculated by the
Chou-Talalay equation (29). H1975
cells were seeded in 96-well plates. Drugs were added alone or
together at indicated concentration. The inhibition effect was
measured by MTT assay as mentioned above. The formula of CI = (D)
Ori/(Dx) Ori + (D)DTX/(Dx)DTX ((D)Ori and (D)DTX: the doses of
compounds Ori and DTX, respectively, necessary to produce the same
effect in combination. Dx: the dose of one compound alone required
producing an effect). With this formula and assistance of CalcuSyn
software (version 2.1), the combined effects of the two compounds
can be assessed as follows: CI<1 indicates synergism; CI=1
indicates additive effect; and CI>1 indicates antagonism.
Human NSCLC xenograft experiments
Equal number of female and male nude immunodeficient
mice (nu/nu), 6–8 weeks old, were purchased from Hunan SJA
Laboratory Animal Co., Ltd., and maintained and monitored in a
specific pathogen-free environment. All animal studies were
conducted according to protocols approved by the Hubei University
of Medicine Animal Care and Use Committee, complying with the rules
of Regulations for the Administration of Affairs Concerning
Ex-Perimental Animals (Approved by the State Council of China). The
mice were injected subcutaneously with NSCLC H1975 cells
(2.5×106) suspended in 100 μl RPMI-1640 medium into the
right flank of each mouse. Treatments were started when the tumors
reached a palpable size. Mice were randomly divided into three
groups and treated with Ori (30 mg/kg, n=8), Gefitinib (30 mg/kg,
n=7) or vehicle control (n=7) for 3 weeks. Caliper measurements of
the longest perpendicular tumor diameters were performed twice a
week to estimate the tumor volume, using the following formula:
4π/3 × (width/2)2 × (length/2), representing the
3-dimensional volume of an ellipse. Animals were sacrificed when
tumors reached 1.5 cm or if the mice appeared moribund to prevent
unnecessary morbidity to the mice. At the time of sacrifice, tumors
were excised for immunohistochemistry.
Immunohistochemistry of tissues
For malin-fixed, paraffin-embedded tissues from mice
were selected for immunohistochemical examination by using an
indirect immunoperoxidase method. The antibodies used for
immunohistochemical staining were pEGFR, CIP2A, and MMP-12.
Statistical analysis
All experiments were repeated at least three times
and the data are presented as the mean ± SD unless noted otherwise.
Differences between data groups were evaluated for significance
using Student t-test of unpaired data or one way analysis of
variance and Bonferroni post-test. P-values <0.05 indicate
statistical significance.
Results
Ori inhibits A549 and H1975 NSCLC
cells
A549 and H1975 cells were seeded in 96-well plates
for 24 h and then treated with different Ori concentrations
(Fig. 1B and C). After 24 h, cell
viability was evaluated through an MTT assay in accordance with the
manufacturer's protocol. The absorbance at 570 nm was determined by
using an automated microplate reader. Ori exerted moderate
cytotoxicity against the A549 and H1975 cells with IC50
of 55.91 and 15.53 μM, respectively (Table I). Trypan blue exclusion assay
revealed that Ori rapidly reduced the number of viable H1975 cells
(Fig. 1D) in a dose- and
time-dependent manner. We also investigated the effect of Ori on
cell colony formation and found that Ori significantly inhibited
the clonogenic ability of H1975 (Fig.
1E). These results suggested that Ori inhibited the
anchorage-dependent (cell proliferation) and anchorage-independent
(colony formation) growth of H1975 cells.
 | Table IThe IC50 of Ori on lung
cancer cell lines. |
Table I
The IC50 of Ori on lung
cancer cell lines.
| Cell lines | A549 | NCI-H1975 |
|---|
| IC50
(μM) | 55.91±5.43 | 15.53±3.57 |
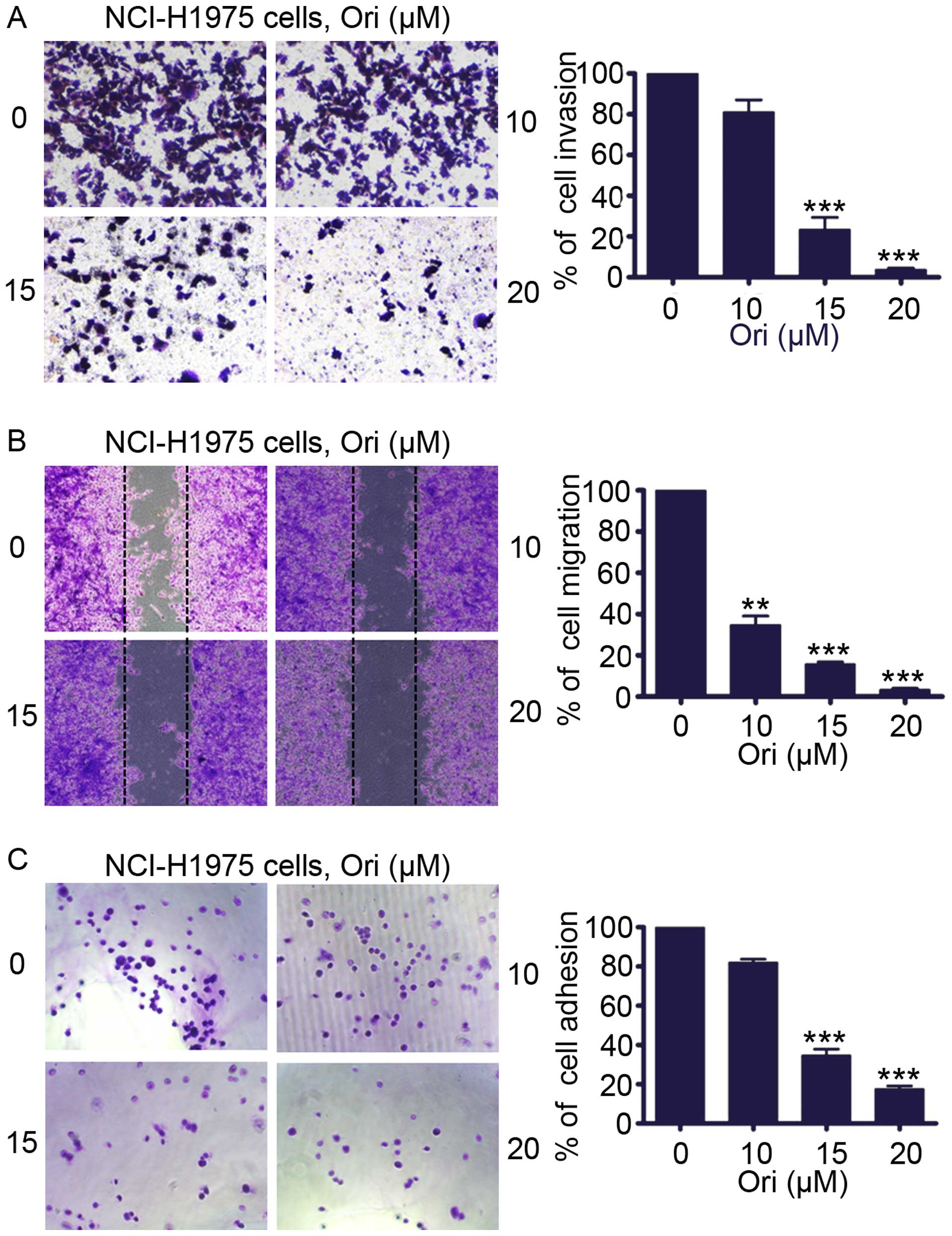
Ori inhibits invasion, migration, and
adhesion of H1975 cells
We determined whether Ori inhibited the invasive
behavior of H1975 cells. An invasion assay was performed using
Matrigel-coated 24-well microchemotaxis chambers in the presence of
Ori. Fig. 2A shows that Ori (0–20
μM) markedly suppressed H1975 cell invasion. To explore the effect
of Ori on migration, we treated the H1975 cells with Ori (0–20 μM)
and determined cell migration after 24 h. Fig. 2B shows that Ori significantly and
dose-dependently reduced H1975 cell migration. We evaluated the
effect of Ori on cell adhesion. Fig.
2C illustrates that Ori also inhibited the adhesion of H1975
cells onto the Matrigel in a concentration-dependent manner
compared with the untreated control cells. These results suggested
that Ori exerted an anti-invasive behavior toward lung cancer at
moderate concentrations.
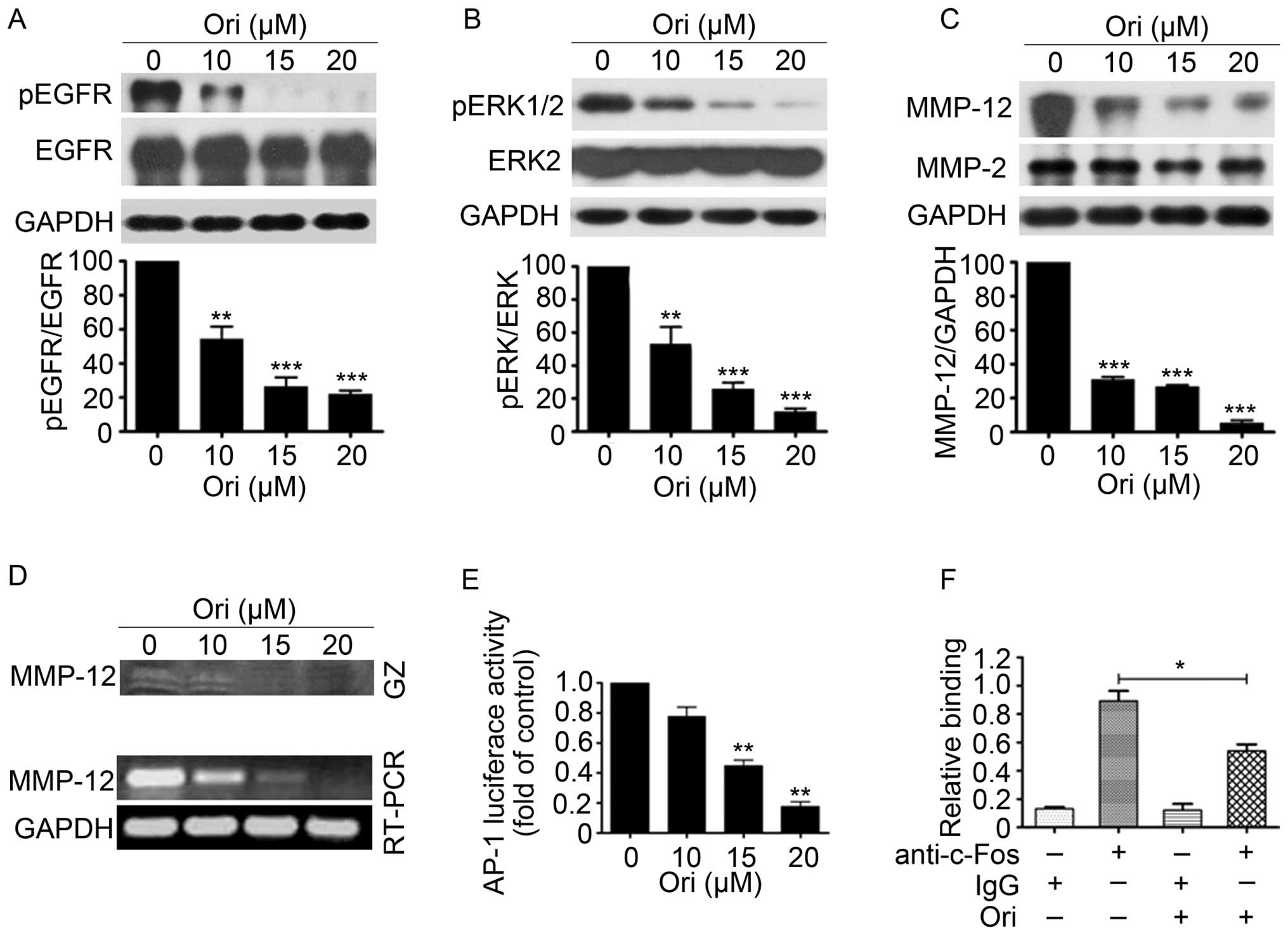
Ori suppresses EGFR and ERK1/2 signaling
in H1975 cells
EGFR and extracellular regulated protein kinases
(ERK) play important roles in the proliferation and invasion of
NSCLC cells (31). Ori can
dose-dependently reduce the phosphorylation levels of EGFR and ERK
without causing evident changes in the total EGFR and total ERK
expression levels in H1975 cells (Fig.
3A and B). Considering that MMP-12, which can be regulated by
ERK1/2 pathway, plays an important role in the migration and
invasion of cancer cells (32), we
examined the expression and gelatinolytic activity of MMP-12 in
H1975 cells exposed to different Ori concentrations (Fig. 3C and D). The results showed that
the gelatinolytic activity and expression levels of MMP-12 were
significantly reduced in the Ori-treated H1975 cells in a
dose-dependent manner compared with the control groups. AP-1, which
is regulated by MAPKs, such as ERK, JNK, and P38 kinase, is a
critical transcription factor that activates MMP-12 transcription
(33). We also examined the effect
of Ori on the transcriptional activity of AP-1 through luciferase
reporter assay and found that 15 and 20 μM of Ori significantly
inhibited AP-1 transcription activity (Fig. 3E). Chromatin immunoprecipitation
(ChIP) assays also revealed that 15 μM of Ori inhibited the DNA
binding of c-Fos (component of AP-1) to MMP-12 promoter (Fig. 3F). These results indicated that Ori
can significantly suppress the invasive behavior (invasion,
migration, and adhesion) of H1975 cells via the
EGFR/ERK/AP-1/MMP-12 signaling pathway.
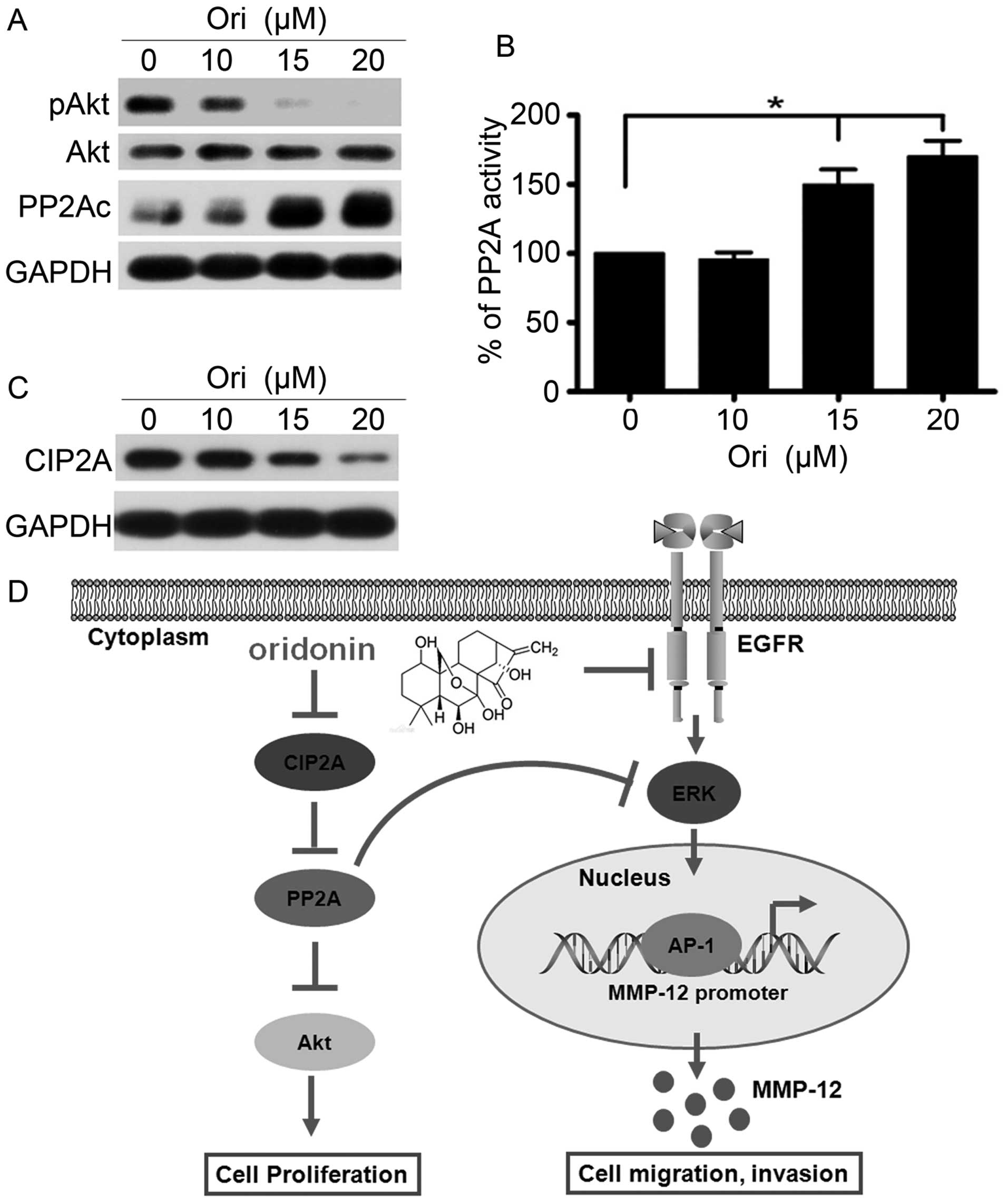
Ori inhibits CIP2A and regulates the
PP2A/Akt pathway
The Akt pathway controls tumor cell growth,
survival, progression, apoptosis, invasion, and metastasis, and Akt
is a critical locus of cancer multidrug resistance and fragility
(34). Ori selectively targets the
Akt signaling pathway and inhibits apoptosis of cervical carcinoma
HeLa cell line (35). Therefore,
we investigated the effect of Ori on Akt phosphorylation in H1975
cells. Our results showed that the phosphorylation level of Akt was
dose-dependently downregulated, and the total Akt level did not
evidently change in the Ori-treated H1975 cells compared with the
control group (Fig. 4A).
Furthermore, we determined the changes in the expression of PP2A,
which is an upstream phosphatase of Akt. The results suggested that
Ori could upregulate the PP2Ac (catalytic subunit) expression
level. To elucidate the mechanism of Ori-induced PP2Ac
upregulation, we examined the PP2A activity and found that this
activity was upregulated in the Ori-treated H1975 cells compared
with that in the control group, which is possibly one of the
reasons leading to the Ori-mediated downregulation of Akt
phosphorylation (Fig. 4B).
Moreover, CIP2A is an endogenous inhibitory protein of PP2A, which
demonstrates cancer-promoting profile in NSCLC (21). We subsequently detected the CIP2A
expression and found that Ori significantly downregulated the
expression of the CIP2A protein (Fig.
4C). Thus, Ori elicits an inhibitory effect on H1975 cells by
regulating the CIP2A/PP2A/Akt signal cascade.
On the basis of these results, we concluded that Ori
inhibited gefitinib-resistant NSCLC growth and cell invasion by
inhibiting the EGFR/ERK/AP-1/MMP-12 signaling pathway and the
CIP2A/PP2A/Akt signal cascade (Fig.
4D).
Ori and docetaxel (DTX) synergistically
inhibit H1975 cells
One or more drugs are commonly used in clinics to
improve the therapeutic efficacy and reduce the side effects on
patients and non-target tissues. Furthermore, an optimal combined
treatment may reduce or postpone drug resistance.
Microtubular-targeted chemotherapy agents, such as DTX, are among
the most widely prescribed first- and second-line chemotherapeutic
options for patients suffering from common malignancies, including
lung cancer (36). Unfortunately,
these malignancies usually develop primary resistance to DTX; thus,
drug resistance poses an important clinical problem. We examined
whether the combination of Ori and DTX synergistically inhibits the
NSCLC treatment. We further chose the low-toxicity (2.5 and 5 μM)
doses of Ori and DTX (4 and 8 nM) to detect (Fig. 5A). The combination of Ori and DTX
elicited a greater effect on the H1975 cells than the sole
treatment of DTX or Ori (P<0.05). We also analyzed the
combination index (CI) by using a formula assisted by CalcuSyn
software (version 2.1) and found that the CIs were <1 (Fig. 5B, Table II). This finding indicated that
Ori and DTX synergistically affected the H1975 cells. Western blot
analysis further confirmed this synergistic effect and revealed
that the combination of Ori and DTX reduced the expression levels
of pEGFR, pERK, and MMP-12 (Fig.
5C). This combination also influenced the CIP2A/PP2A/Akt signal
cascade (Fig. 5D).
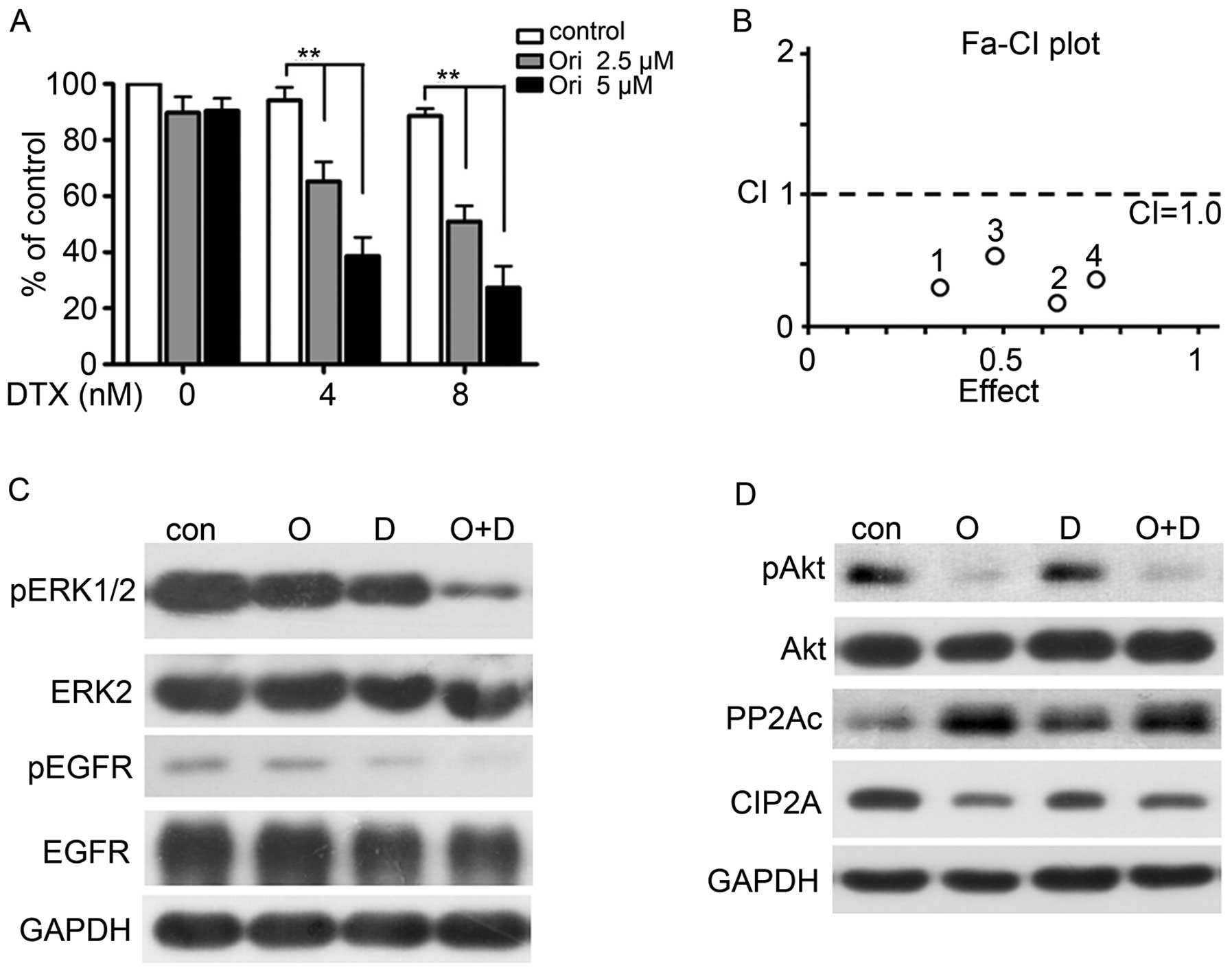 | Figure 5Ori and docetaxel (DTX)
synergistically inhibit H1975 cells. (A–B) H1975 cells were treated
for 24 h with DTX (0, 4, 8 nM) in the presence of Ori at 0, 2.5, 5
μM. MTT assay was used to test the proliferation of cells (A), and
the combined effects were evaluated by the Chou-Talay method and
Calcusyn software (B). *P<0.05,
**P<0.001. (C–D) H1975 cells were cultured with
control media (Con), Ori (O, 5 μM), DTX (D, 8 nM), or Ori (5 μM)
plus DTX (8 nM) (O+D) for 24 h. Cells were then lysed and subjected
to western blotting using indicated antibodies. |
| Table IIOri and DTX combination index (CI)
values. |
Table II
Ori and DTX combination index (CI)
values.
| Ori (μM) | DTX (nM) | Effect | CI (Ori+DTX) |
|---|
| 2.5 | 4 | 0.32 | 0.32 |
| 2.5 | 8 | 0.62 | 0.22 |
| 5 | 4 | 0.49 | 0.52 |
| 5 | 8 | 0.73 | 0.39 |
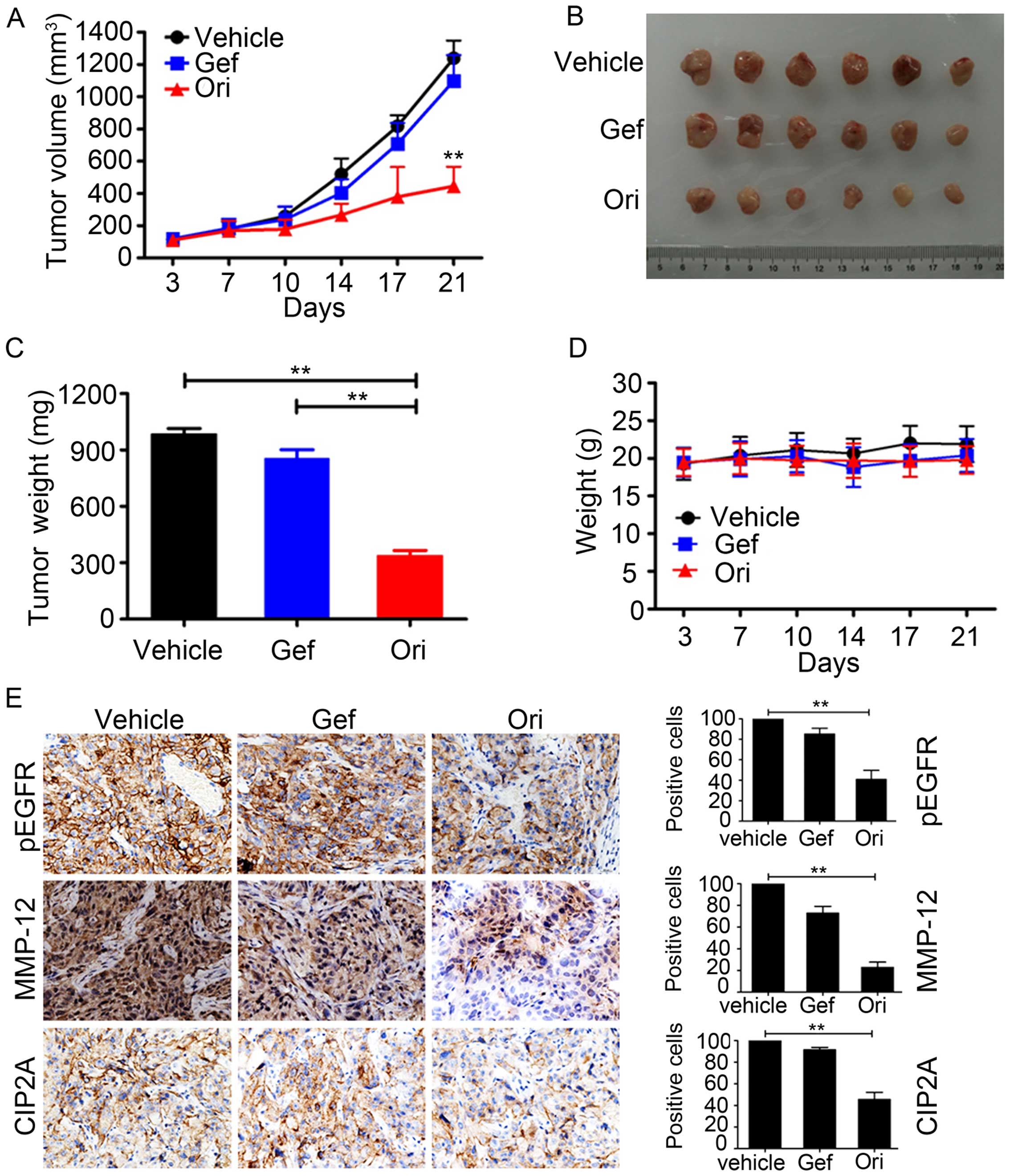
Ori inhibits tumor growth in murine
models
To determine the antitumor effect of Ori on NSCLC
in vivo, we subcutaneously inoculated 2.5×106
H1975 cells in 100 μl of RPMI-1640 medium in the right flank of
nude mice to generate xenografted murine models. Once the tumors
grew to a measurable size, each group was administered with the
vehicle control, gefitinib (30 mg/kg), and Ori (30 mg/kg) 5 times
per week for 21 days. The tumor-bearing mice were humanely
sacrificed when their tumors reached 1.5 cm in diameter or when
paralysis or major compromise in their quality of life occurred. We
found that Ori efficiently repressed tumor growth compared with the
vehicle control or gefitinib (P<0.01; Fig. 6A and B). Ori treatment also
significantly reduced the tumor weight of the mice (Fig. 6C). In addition, Ori treatment did
not significantly reduce the body weight of the mice. This finding
suggested that Ori did not cause evident side effects (Fig. 6D). All of the mice were euthanized,
and the tumor specimens were examined through immunohistochemistry;
the results showed that the expression levels of p-EGFR, MMP-12,
and CIP2A were downregulated in the Ori-treated groups (Fig. 6E). Therefore, Ori could be a
potential therapeutic agent against gefitinib-resistant NSCLC.
Discussion
Ori elicits an anti-proliferative effect on lung
cancer cell lines in vitro and in vivo (26,37).
However, the antitumor effects of Ori on gefitinib-resistant lung
cancer cells remain poorly understood. This study is the first to
demonstrate that Ori inhibited the metastasis of
gefitinib-resistant lung cancer cells in vitro by
downregulating the EGFR/ERK-mediated MMP-12 expression, by
reactivating PP2A, and by downregulating CIP2A. These data strongly
suggested the possible beneficial effects of Ori on patients
suffering from gefitinib-resistant lung cancer.
Over 90% of deaths caused by solid tumors are
attributed to tumor metastasis (38). Thus, therapeutic strategies to
suppress or prevent cancer metastasis will greatly improve the
survival of cancer patients. Our data suggested that Ori remarkably
inhibited the invasive (Fig. 2A)
and migratory (Fig. 2B) abilities
of H1975 cells. In addition, the adhesion of cancer cells onto
extracellular matrix (ECM) and their interactions are crucial steps
during metastasis and invasion. Our result showed that Ori
treatment remarkably inhibited cell adhesion onto matrigel
(Fig. 2C).
Cell invasion and migration involve various growth
factors, which bind to their receptors on the cell surface and then
activate downstream signaling pathways; as a result, the
cytoskeleton is reorganized and the cellular motility machinery is
stimulated (39). Aberrations in
EGFR expression and downstream signaling pathways contribute to the
progression, invasion, and maintenance of the malignant phenotype
of many human cancers (40). We
found that the phosphorylation level of EGFR remarkably decreased
as the Ori concentrations increased. This finding suggested that
Ori affected the H1975 cells possibly by suppressing the EGFR
phosphorylation and the EGFR-mediated signaling pathway (Fig. 3A). We subsequently explored the
signal transduction pathways that potentially mediate EGF signal
transduction. ERK has been implicated in the control of diverse
biological processes, such as cell proliferation, differentiation,
and apoptosis; ERK also plays complementary function on cell
survival. ERK is rapidly activated in cells stimulated by various
mitogens or motogens, including EGF (41). This study found that Ori could
inhibit ERK1/2 phosphorylation (Fig.
3B). Cancer invasion and migration are a multistep process
involving many types of cell-cell interactions.
A critical step in tumor invasion and migration is
the basement membrane degradation, which is catalyzed by
proteolytic enzymes, such as matrix metalloproteinases (MMPs) and
tissue inhibitor of metalloproteinases (TIMPs). MMPs comprise a
multigene family, which includes more than 22 members. MMP-1,
MMP-2, MMP-9, MMP-10, MMP-13, and MMP-14 are expressed in human
lung cancer cells (42). MMP-2 and
MMP-9 are the most studied MMPs in lung cancer; they are
upregulated in locally invasive tumors and their expression is
correlated with lung cancer invasiveness (43,44).
MMP-12 expression is also induced in human lung cancer during
malignant transformation. However, limited information is available
regarding the potential contribution of MMP-12 to NSCLC invasion.
In addition, MMP-12 is regulated by the ERK pathway (32). Our study demonstrated that Ori
treatment inhibited the gelatinolytic activity and expression of
MMP-12 at protein and mRNA levels in H1975 cells compared with
those in the untreated control cells (Fig. 3C and D). Therefore, MMP-12 is
possibly an Ori-responsive mediator that likely degrades ECM; as a
result, subsequent cancer migration and invasion may occur. With
the activation of EGFR, the AP-1 signaling pathway in cancer cells
is also activated (45). The
activation of AP-1, which is found downstream of the ERK pathway,
is involved in many pathological processes, such as inflammation,
cancer cell adhesion, invasion, metastasis, and angiogenesis
(40). Moreover, the MMP-12
promoter region contains a cis-regulatory element of two AP-1
binding sites. Thus, we investigated the effects of Ori on the AP-1
transcription activity and DNA binding to MMP-12 promoter; we found
that Ori could inhibit the transcription activity (Fig. 3E) and DNA binding of AP-1 to MMP-12
promoter (Fig. 3F). Therefore, Ori
is a novel anti-metastatic agent that may treat gefitinib-resistant
NSCLC by suppressing the EGFR/ERK/MMP-12 pathway.
The Akt signaling pathway is one of the most
critical cancer-promoting pathways and is frequently activated in
many types of human cancers, including NSCLC. We then examined the
p-Akt level in H1975 cells after Ori was administered; the results
showed that the p-Akt expression was reduced in a dose-dependent
manner (Fig. 4A). To clarify its
mechanism, we evaluated the upstream Akt regulatory factors,
including PP2A and EGFR. PP2A is a tumor suppressor protein that
can exhibit protein phosphatase activity and can block the PI3K/Akt
pathway; this protein also dephosphorylates and inactivates MEK1
and ERK family kinases (30). Our
study also demonstrated that Ori treatment upregulated the PP2A
activity in H1975 cells (Fig. 4B),
and the PP2A upregulation inactivated the Akt and ERK pathways. As
a result, the proliferation, migration, and invasion of in H975
cells were suppressed and the apoptosis of these cells were
induced. To determine whether CIP2A, a critical upstream molecule
of PP2A, is the target of Ori, we examined the CIP2A expression and
found that the CIP2A expression was downregulated (Fig. 4C). CIP2A is also overexpressed and
is related to poor clinical outcome in lung cancer. Several
compounds targeting the CIP2A protein have shown potential activity
in lung cancer in vivo and in vitro (21,22).
Our results indicated that the CIP2A/PP2A/Akt pathway may serve as
an alternative mechanism underlying the effects of Ori.
Ori elicits enhanced cytotoxicity against H1975
cells when it is combined with the conventional agent DTX, a drug
currently used in clinical trials to treat solid malignancies
(Fig. 5). In a xenograft murine
model of H1975, Ori significantly inhibited tumor growth, but the
body weight of the mice remained unaffected (Fig. 6A–D). Our results also showed that
Ori inhibited pEGFR, MMP-12, and CIP2A in vivo (Fig. 6E). Our results indicated that Ori
directly affected not only the EGFR pathway but also the CIP2A/PP2A
pathway. As a result, the growth of H1975 cells was suppressed.
Therefore, Ori may be considered as a new antitumor agent to
prevent and treat gefitinib-resistant lung cancer.
Acknowledgements
This work was supported by grants from the National
Natural Sciences Foundation of China (81400157); grants from the
Foundation for Innovative Research Team of Hubei University of
Medicine (2014CXX05); the Natural Science Foundation of Hubei
Provincial Department of Education (Q20152106); the Key Discipline
Project of Hubei Province; the Faculty Development Grant
2014QDJZR08, 2015QDJZR16 from Hubei University of Medicine.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhang B, Jiao J, Liu Y, Guo LX, Zhou B, Li
GQ, Yao ZJ and Zhou GB: Gefitinib analogue V1801 induces apoptosis
of T790M EGFR-harboring lung cancer cells by up-regulation of the
BH-3 only protein Noxa. PLoS One. 7:e487482012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kwon T, Rho JK, Lee JC, Park YH, Shin HJ,
Cho S, Kang YK, Kim BY, Yoon DY and Yu DY: An important role for
peroxiredoxin II in survival of A549 lung cancer cells resistant to
gefitinib. Exp Mol Med. 47:e1652015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yun CH, Mengwasser KE, Toms AV, Woo MS,
Greulich H, Wong KK, Meyerson M and Eck MJ: The T790M mutation in
EGFR kinase causes drug resistance by increasing the affinity for
ATP. Proc Natl Acad Sci USA. 105:2070–2075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Engelman JA and Jänne PA: Mechanisms of
acquired resistance to epidermal growth factor receptor tyrosine
kinase inhibitors in non-small cell lung cancer. Clin Cancer Res.
14:2895–2899. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kwak EL, Sordella R, Bell DW,
Godin-Heymann N, Okimoto RA, Brannigan BW, Harris PL, Driscoll DR,
Fidias P, Lynch TJ, et al: Irreversible inhibitors of the EGF
receptor may circumvent acquired resistance to gefitinib. Proc Natl
Acad Sci USA. 102:7665–7670. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kim Y, Ko J, Cui Z, Abolhoda A, Ahn JS, Ou
SH, Ahn MJ and Park K: The EGFR T790M mutation in acquired
resistance to an irreversible second-generation EGFR inhibitor. Mol
Cancer Ther. 11:784–791. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Finlay MR, Anderton M, Ashton S, Ballard
P, Bethel PA, Box MR, Bradbury RH, Brown SJ, Butterworth S,
Campbell A, et al: Discovery of a potent and selective EGFR
inhibitor (AZD9291) of both sensitizing and T790M resistance
mutations that spares the wild type form of the receptor. J Med
Chem. 57:8249–8267. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Perrotti D and Neviani P: Protein
phosphatase 2A: A target for anticancer therapy. Lancet Oncol.
14:e229–e238. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Seshacharyulu P, Pandey P, Datta K and
Batra SK: Phosphatase: PP2A structural importance, regulation and
its aberrant expression in cancer. Cancer Lett. 335:9–18. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schönthal AH: Role of serine/threonine
protein phosphatase 2A in cancer. Cancer Lett. 170:1–13. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lv P, Wang Y, Ma J, Wang Z, Li JL, Hong
CS, Zhuang Z and Zeng YX: Inhibition of protein phosphatase 2A with
a small molecule LB100 radiosensitizes nasopharyngeal carcinoma
xenografts by inducing mitotic catastrophe and blocking DNA damage
repair. Oncotarget. 5:7512–7524. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Junttila MR, Puustinen P, Niemelä M, Ahola
R, Arnold H, Böttzauw T, Ala-aho R, Nielsen C, Ivaska J, Taya Y, et
al: CIP2A inhibits PP2A in human malignancies. Cell. 130:51–62.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lucas CM, Harris RJ, Giannoudis A, Copland
M, Slupsky JR and Clark RE: Cancerous inhibitor of PP2A (CIP2A) at
diagnosis of chronic myeloid leukemia is a critical determinant of
disease progression. Blood. 117:6660–6668. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang J, Li W, Li L, Yu X, Jia J and Chen
C: CIP2A is over-expressed in acute myeloid leukaemia and
associated with HL60 cells proliferation and differentiation. Int J
Lab Hematol. 33:290–298. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li W, Ge Z, Liu C, Liu Z, Björkholm M, Jia
J and Xu D: CIP2A is overexpressed in gastric cancer and its
depletion leads to impaired clonogenicity, senescence, or
differentiation of tumor cells. Clin Cancer Res. 14:3722–3728.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tseng LM, Liu CY, Chang KC, Chu PY, Shiau
CW and Chen KF: CIP2A is a target of bortezomib in human triple
negative breast cancer cells. Breast Cancer Res. 14:R682012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Böckelman C, Hagström J, Mäkinen LK,
Keski-Säntti H, Häyry V, Lundin J, Atula T, Ristimäki A and Haglund
C: High CIP2A immunoreactivity is an independent prognostic
indicator in early-stage tongue cancer. Br J Cancer. 104:1890–1895.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Böckelman C, Lassus H, Hemmes A, Leminen
A, Westermarck J, Haglund C, Bützow R and Ristimäki A: Prognostic
role of CIP2A expression in serous ovarian cancer. Br J Cancer.
105:989–995. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu Z, Ma L, Wen ZS, Hu Z, Wu FQ, Li W,
Liu J and Zhou GB: Cancerous inhibitor of PP2A is targeted by
natural compound celastrol for degradation in non-small-cell lung
cancer. Carcinogenesis. 35:905–914. 2014. View Article : Google Scholar
|
|
22
|
Ma L, Wen ZS, Liu Z, Hu Z, Ma J, Chen XQ,
Liu YQ, Pu JX, Xiao WL, Sun HD, et al: Overexpression and small
molecule-triggered downregulation of CIP2A in lung cancer. PLoS
One. 6:e201592011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhou GB, Kang H, Wang L, Gao L, Liu P, Xie
J, Zhang FX, Weng XQ, Shen ZX, Chen J, et al: Oridonin, a
diterpenoid extracted from medicinal herbs, targets AML1-ETO fusion
protein and shows potent antitumor activity with low adverse
effects on t(8;21) leukemia in vitro and in vivo. Blood.
109:3441–3450. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li Y, Wang Y, Wang S, Gao Y, Zhang X and
Lu C: Oridonin phosphate-induced autophagy effectively enhances
cell apoptosis of human breast cancer cells. Med Oncol. 32:3652015.
View Article : Google Scholar
|
|
25
|
Zhao Z and Chen Y: Oridonin, a promising
antitumor natural product in the chemotherapy of hematological
malignancies. Curr Pharm Biotechnol. 15:1083–1092. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang YY, Lv YF, Lu L and Cai L: Oridonin
inhibits mTOR signaling and the growth of lung cancer tumors.
Anticancer Drugs. 25:1192–1200. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xu B, Shen W, Liu X, Zhang T, Ren J, Fan Y
and Xu J: Oridonin inhibits BxPC-3 cell growth through cell
apoptosis. Acta Biochim Biophys Sin (Shanghai). 47:164–173. 2015.
View Article : Google Scholar
|
|
28
|
Liu Y, Cao W, Zhang B, Liu YQ, Wang ZY, Wu
YP, Yu XJ, Zhang XD, Ming PH, Zhou GB, et al: The natural compound
magnolol inhibits invasion and exhibits potential in human breast
cancer therapy. Sci Rep. 3:30982013.PubMed/NCBI
|
|
29
|
Cao W, Liu Y, Zhang R, Zhang B, Wang T,
Zhu X, Mei L, Chen H, Zhang H, Ming P, et al: Homoharringtonine
induces apoptosis and inhibits STAT3 via IL-6/JAK1/STAT3 signal
pathway in Gefitinib-resistant lung cancer cells. Sci Rep.
5:84772015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu H, Gu Y, Wang H, Yin J, Zheng G, Zhang
Z, Lu M, Wang C and He Z: Overexpression of PP2A inhibitor SET
oncoprotein is associated with tumor progression and poor prognosis
in human non-small cell lung cancer. Oncotarget. 6:14913–14925.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gao J, Liu X, Yang F, Liu T, Yan Q and
Yang X: By inhibiting Ras/Raf/ERK and MMP-9, knockdown of EpCAM
inhibits breast cancer cell growth and metastasis. Oncotarget.
6:27187–27198. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yang XS, Liu SA, Liu JW and Yan Q:
Fucosyltransferase IV enhances expression of MMP-12 stimulated by
EGF via the ERK1/2, p38 and NF-κB pathways in A431 cells. Asian
Pacific journal of cancer prevention Asian Pac J Cancer Prev.
13:1657–1662. 2012. View Article : Google Scholar
|
|
33
|
Hofmann HS, Hansen G, Richter G, Taege C,
Simm A, Silber RE and Burdach S: Matrix metalloproteinase-12
expression correlates with local recurrence and metastatic disease
in non-small cell lung cancer patients. Clin Cancer Res.
11:1086–1092. 2005.PubMed/NCBI
|
|
34
|
Radisavljevic Z: AKT as locus of cancer
multidrug resistance and fragility. J Cell Physiol. 228:671–674.
2013. View Article : Google Scholar
|
|
35
|
Hu HZ, Yang YB, Xu XD, Shen HW, Shu YM,
Ren Z, Li XM, Shen HM and Zeng HT: Oridonin induces apoptosis via
PI3K/Akt pathway in cervical carcinoma HeLa cell line. Acta
Pharmacol Sin. 28:1819–1826. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Brodie SA, Li G, Harvey D, Khuri FR,
Vertino PM and Brandes JC: Small molecule inhibition of the
CHFR-PARP1 interaction as novel approach to overcome intrinsic
taxane resistance in cancer. Oncotarget. 6:30773–30786.
2015.PubMed/NCBI
|
|
37
|
Liu Y, Liu JH, Chai K, Tashiro S, Onodera
S and Ikejima T: Inhibition of c-Met promoted apoptosis, autophagy
and loss of the mitochondrial transmembrane potential in
oridonin-induced A549 lung cancer cells. J Pharm Pharmacol.
65:1622–1642. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Pritchard VL, Dimond L, Harrison JS,
Velázquez CC S, Zieba JT, Burton RS and Edmands S: Interpopulation
hybridization results in widespread viability selection across the
genome in Tigriopus californicus. BMC Genet. 12:542011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang S, Yu S, Shi W, Ge L, Yu X, Fan J and
Zhang J: Curcumin inhibits the migration and invasion of mouse
hepatoma Hca-F cells through down-regulating caveolin-1 expression
and epidermal growth factor receptor signaling. IUBMB Life.
63:775–782. 2011. View Article : Google Scholar
|
|
40
|
Hsieh CY, Tsai PC, Tseng CH, Chen YL,
Chang LS and Lin SR: Inhibition of EGF/EGFR activation with
naphtho[1,2-b]furan-4,5-dione blocks migration and invasion of
MDA-MB-231 cells. Toxicol In Vitro. 27:1–10. 2013. View Article : Google Scholar
|
|
41
|
Hu Y, Chen F, Liu F, Liu X, Huang N, Cai
X, Sun Y, Li A and Luo R: Overexpression of TIP30 inhibits the
growth and invasion of glioma cells. Mol Med Rep. 13:605–612.
2016.PubMed/NCBI
|
|
42
|
Tsai JR, Liu PL, Chen YH, Chou SH, Cheng
YJ, Hwang JJ and Chong IW: Curcumin Inhibits non-small cell lung
cancer cells metastasis through the adiponectin/NF-κB/MMPs
signaling pathway. PLoS One. 10:e01444622015. View Article : Google Scholar
|
|
43
|
Hwang J, Kim Y, Kang HB, Jaroszewski L,
Deacon AM, Lee H, Choi WC, Kim KJ, Kim CH, Kang BS, et al: Crystal
structure of the human N-Myc downstream-regulated gene 2 protein
provides insight into its role as a tumor suppressor. J Biol Chem.
286:12450–12460. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cai J, Li R, Xu X, Zhang L, Wu S, Yang T,
Fang L, Wu J, Zhu X, Li M, et al: URGCP promotes non-small cell
lung cancer invasiveness by activating the NF-κB-MMP-9 pathway.
Oncotarget. 6:36489–36504. 2015.PubMed/NCBI
|
|
45
|
Sen T and Chatterjee A:
Epigallocatechin-3-gallate (EGCG) downregulates EGF-induced MMP-9
in breast cancer cells: Involvement of integrin receptor α5β1 in
the process. Eur J Nutr. 50:465–478. 2011. View Article : Google Scholar
|