Introduction
Colorectal cancer is a disease derived from the
epithelial cells lining the colon or rectum of the gastrointestinal
tract. Annually, >1.2 million people are diagnosed with
colorectal cancer worldwide and almost half of them die as a
consequence of disseminated disease (1). In 2011, it was the second most common
cause of cancer in women and the third most common malignancy in
men, the fourth most common cause of cancer death (2). It is more common in developed than
developing countries (3). In
addition to surgery, single-agent fluoropyrimidine in combination
with fractionated pelvic radiation remains the standard regimen for
advanced colorectal cancer treatment; however, ~30–50% of patients
with colorectal cancer will relapse and eventually die of their
disease, though many patients respond well to adequate surgery and
subsequent chemotherapy at the beginning of their treatment
(4–7). In this scenario, it is imperative to
discover novel chemotherapeutics for colorectal cancer with optimal
efficacy and safety profile.
The Aurora kinases which include Aurora A, B and C,
constitute one family of serine/threonine kinases whose activities
are essential for the mitotic progression (8,9).
Aurora A is localized to the duplicated centrosomes and the spindle
poles in mitosis. Several studies have characterized the roles of
Aurora A in many processes required for building a bipolar spindle
apparatus, including centrosome maturation and separation (10). Amplification of Aurora genes, as
well as mRNA and protein upregulation, has been frequently reported
in many human malignancies, such as prostate, breast, pancreas, and
ovarian cancers (11–13). AURKA is located on
chromosome 20q, a genomic region that is frequently amplified in
colorectal cancer, associated with adenoma-to-carcinoma
progression, and an indicator of poor prognosis (14–21).
In addition, mRNA and protein levels of Aurora A have also been
found to be upregulated in colorectal cancer, as compared with
normal tissues (22,23). Due to the key role of Aurora A in
mitotic progression and its upregulation in cancers, it has been
considered as an important molecular target for cancer therapy
(24,25).
Tanshinone I (T1) is one of the major compounds from
traditional Chinese herb Salvia miltiorrhiza Bunge (Danshen)
which has been widely used for prevention and treatment of
cardiovascular disease with minimal toxicity (26). Recently, the anticancer effect of
T1 has been discovered in prostate, breast and lung cancers
(27,28). Moreover, T1 was revealed to greatly
inhibit Aurora A expression at both gene and protein levels
(11). Since Aurora A plays a
stimulatory role in tumorigenesis and development in colorectal
cancer, it is particularly interesting to characterize the effects
of T1, an inhibitor of Aurora A kinase from plant, on the growth of
colorectal cancer cell lines. Therefore, in this study, we
evaluated the effect of T1 on colorectal cancer cells and try to
dissect the underlying mechanisms.
Materials and methods
Reagents
Tanshinone I (T1) was purchased from LKT
Laboratories (St. Paul, MN, USA). Tissue culture media DMEM with
4.5 g/l glucose, L-glutamine, and sodium pyruvate as well as 0.25%
trypsin was obtained from Mediatech, Inc. (Manassas, VA, USA).
Fetal bovine serum (FBS) was purchased from Life Technologies, Inc.
(Grand Island, NY, USA). Propidium iodide (PI) was purchased from
Sigma (St. Louis, MO, USA). Antibodies against Aurora A, cyclin D1,
CDK4, cleaved-PARP (c-PARP), cleaved-caspase-3 (c-caspase-3) were
purchased from Cell Signaling (Beverly, CA, USA). Anti-Bax,
anti-Bcl-2 and anti-survivin antibodies were from Abcam (Cambridge,
MA, USA). Mouse anti-p53 monoclonal antibody was purchased from
Novus (Littleton, CO, USA). Anti-β-actin antibody was from Santa
Cruz (Santa Cruz, CA, USA).
Cell culture
The human normal colon epithelial cell line
CCD-841CoN and colorectal adenocarcinoma cell lines, including
SW480 and HCT116, were obtained from the American Type Culture
Collection (Bethesda, MD, USA). The cells were maintained in
high-glucose DMEM supplemented with 10% FBS, L-glutamine, sodium
pyruvate and double antibiotics, in humidified CO2
incubator at 37°C.
CellTiter Blue cell viability assay
CCD-841CoN, HCT116 and SW480 cells seeded into
96-well plates were exposed to vehicle (0.1% DMSO) or different
concentrations of T1 (32, 16, 8, 4, 2, 1 and 0.5 μM) in 100 μl
media for 72 h, respectively. Cell viability was quantified via
CellTiter Blue (Promega, Madison, WI, USA) assay.
Anchorage-independent growth assay
HCT116 cells were plated in 0.35% top agarose
(SeaPlaque agarose; Lonza, Verviers, Belgium) on a surface of 0.6%
bottom agarose in 6-well cell plates (5,000 cells per well) and
treated with vehicle or T1 (1 or 4 μM) in triplicates. After 3
weeks, the number of colonies in each well was evaluated by taking
digital images through which colony numbers were counted.
PI staining for cell cycle analysis
HCT116 cells were plated and treated with 0.1% DMSO
(veh) or two doses of T1 (4 and 1 μM) for 24 h, respectively. Cells
were collected by trypsinization and washed with PBS. Cells
(1×106) were re-suspended in PBS and fixed by adding 1
ml pre-chilled ethanol dropwisely. Cells were kept at −20°C
overnight and centrifuged at 300 g for 5 min to remove supernatant.
Cells were washed twice with PBS, re-suspended in 500 μl of PBS,
stained with 50 μg/ml PI together with 50 μg/ml RNase at 37°C for
30 min in the dark and then analyzed on Gallios Flow Cytometer
(Beckman Coulter, Brea, CA, USA) to check cell cycle distribution
profile.
Apoptosis assay
HCT116 cells were plated and treated with DMSO (veh)
or two doses of T1 (1 or 4 μM) for 72 h, respectively. Briefly,
floating cells were spun down in a centrifuge tube. The attached
cells were trypsinized and combined with the floating cells, and
then centrifuged for 5 min at 300 g. Cells were washed with cold
PBS and re-suspended in 500 μl of binding buffer. Each sample was
added with 5 μl of Annexin V-FITC and 5 μl of 7-AAD reagents. The
samples were stained at room temperature for 10 min in the dark.
The samples were then analyzed using Gallios Flow Cytometer
(Beckman Coulter). Acquired data were analyzed using the FlowJo
software (Ashland, OR, USA).
Western blotting
Cells were cultured and collected for protein
extraction. Cell lysates were prepared with RIPA buffer (Thermo
Scientific, Rockford, IL, USA) with 1% Halt Protease and
Phosphatase Inhibitor Cocktail (Thermo Scientific). Protein samples
were separated on 10 or 15% SDS-PAGE gels and transferred on PVDF
membranes. Membranes were blocked with 5% non-fat milk in
phosphate-buffered saline (PBS) containing 0.05% Tween-20 and
incubated with the prescribed primary antibody overnight at 4°C.
Anti-Aurora A antibody (1:1,000), anti-p53 antibody (1:2,000),
anti-survivin antibody (1:500) and other antibodies were
appropriately diluted in blocking buffer. After incubation with the
primary antibody, the membranes were washed 3 times with TBS-T and
were then incubated with appropriate secondary antibodies for one
hour at room temperature. Subsequently membranes were washed 4
times with TBS-T. Finally, membranes were incubated with
SuperSignal West Pico Chemiluminescent Substrate (Thermo
Scientific) and X-ray film was developed in the dark.
Real-time PCR
CCD-841CoN, HCT116 and SW480 cells were cultured and
collected for RNA extraction. Total RNA was isolated by using
Qiagen RNeasy Mini kit (Qiagen, Valencia, CA, USA) according to the
manufacturer's instructions. To synthesize cDNA, 100 ng random
primer (Invitrogen, Carlsbad, CA, USA), 1.0 μg of total RNA, 10 mM
dNTP and 200 U of reverse transcriptase (Invitrogen) per 20 μl
reaction were used. PCRs were performed in duplicates in a 25-μl
final volume by using SYBR Green master mix from Qiagen (Qiagen),
and the data were analyzed by calculating CT values. β-actin was
used as the internal control. Three independent experiments were
performed. The sequences of primers used in this study are as
follows: β-actin for RT-PCR(F): 5′-GATGA GATTGGCATGGCTTT-3′;
β-actin for RT-PCR(R): 5′-CACC TTCACCGTTCCAGTTT-3′; Aurora A for
RT-PCR(F): 5′-CAT CTTCCAGGAGGACCACT-3′; Aurora A for RT-PCR(R):
5′-CAAAGAACTCCAAGGCTCCA-3′.
Construction of plasmids, preparation and
application of lentivirus
Short hairpin RNAs targeting p53, Aurora A and
survivin were constructed using pLKO.1 vector. The DNA fragments
were synthesized (by GenScript Corporation, Nanjing, China),
annealed and sub-cloned into the vector. The sense sequence of the
sh-p53, sh-Aurora A and sh-survivin were:
5′-GTCCAGATGAAGCTCCCAGAA-3′, 5′-GAGTCTA CCTAATTCTGGAAT-3′ and
5′-CCGCATCTCTACATTCA AGAA-3′, respectively. Lentiviral particles
were produced from 293T cells using ViraPower Lentiviral expression
kit (Invitrogen). Lentivirus was applied on HCT116 or SW480 cells
for 6 h before it was changed with fresh medium. After 24 h, cells
were selected with puromycin (0.5 μg/ml) for 3 days to enrich
transducted cells. After that, cells were cultured in regular
medium and used for different purposes.
Construction of plasmids, preparation and
application of retrovirus
The DNA fragment coding full-length survivin was
amplified from cDNA of SW480 cells and sub-cloned into pBABE-Neo
vector using BamHI and EcoRI sites. The primers used
in PCR are: 5′-GCGGATCCATGGGTGCCCC GACGTTG-3′ and
5′-GCGAATTCCTAAGACATTGCTAA GGG-3′. The vector control or
survivin-coding plasmid was transfected into Phoenix cells (Phoenix
retroviral expression system). Virus was collected and applied onto
target cells according to the standard protocol. Cells were
selected with G418 (500 μg/ml) for 5 days before they were used for
different purposes.
Immuno-precipitation
To all the buffers used in the following protocol,
proteinase inhibitors were added. Treated cells were collected and
washed with cold PBS. Subsequently, cell pellete was re-suspended
in lysis buffer (150 mM NaCl, 50 mM Tris-HCl, 1 mM EDTA, 0.5% NP40,
pH 8.0) and incubated for 20 min on ice. The mixture was
centrifuged and the supernatant was transferred to new tubes.
Anti-p53 antibody (1 μg) or its isotope control was added to the
cell lysate. The mixture was incubated at 4°C for 3 h with
agitation. Subsequently, the mixture was incubated with protein
A/G-conjugated beads for one hour. After washing, the proteins were
obtained by boiling the beads in SDS sample buffer.
Statistics
The data produced in our experiments was entered
into Graphpad Prism (5.01) and analyzed by Student's t-test and
analysis of variance (ANOVA). Statistical significance was
determined according to P-values [*P<0.05;
**P<0.01; ***P<0.001 in the figures].
Results are expressed as the mean ± SD, n=3, if without specific
indication.
Results
Effects of Tanshinone I treatment on
viabilities of colorectal cancer cells
Since T1 exhibited inhibitory effects on breast and
lung cancer (27–29), we sought to treat human colorectal
cancer cell lines as well as normal colon epithelial cells with T1.
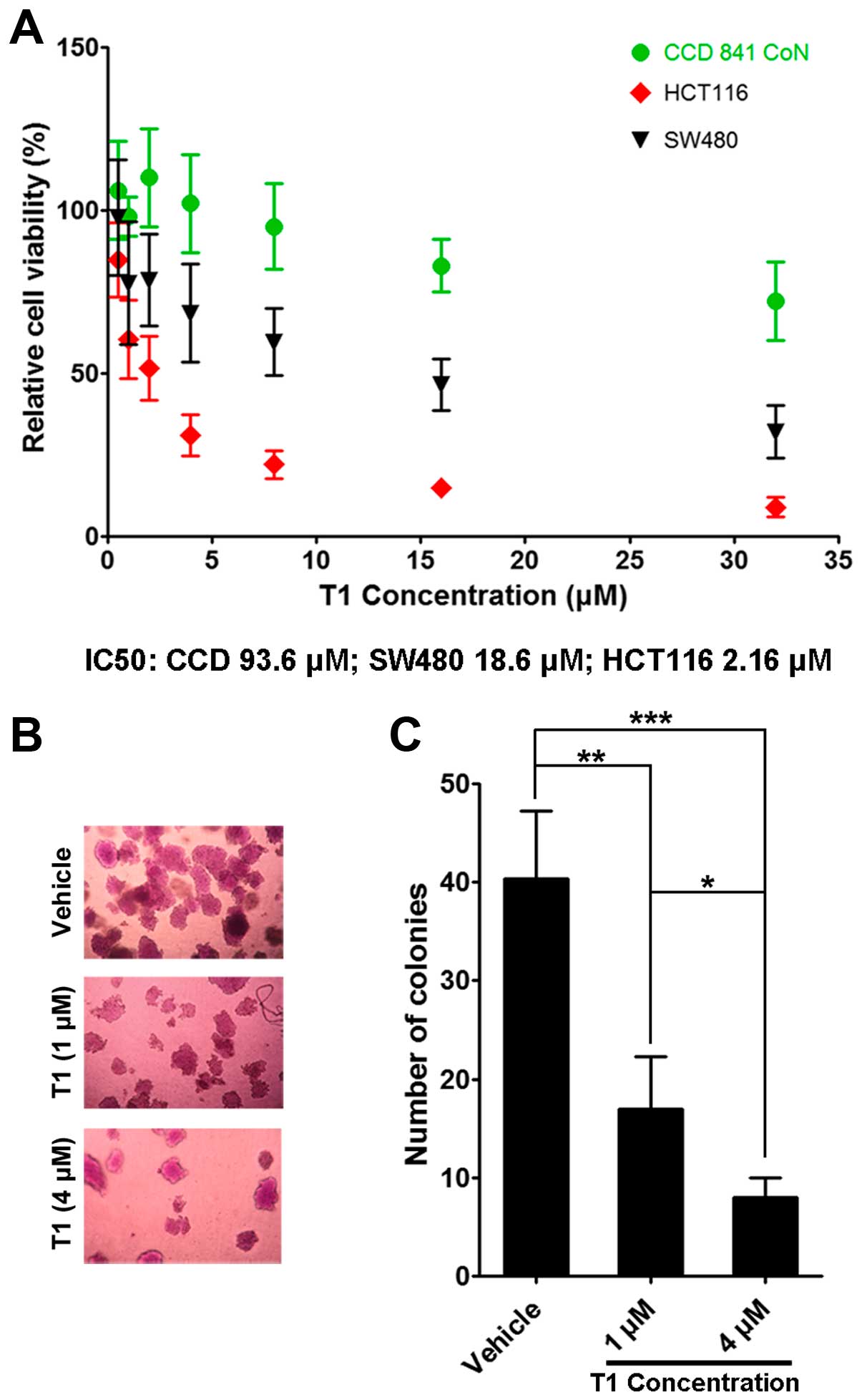
After 72 h of T1 treatment at various concentrations, HCT116
colorectal cell with wild-type p53 protein displayed decreased
viability in a dose-dependent manner. In contrast, this effect was
partially ameliorated in SW480 cells and was largely abolished in
CCD841 CoN cells (Fig. 1A).
Consistently, we found that anchorage-independent growth of HCT116
cells in soft agarose was significantly inhibited following T1
treatment in a dose-dependent manner (Fig. 1B and C).
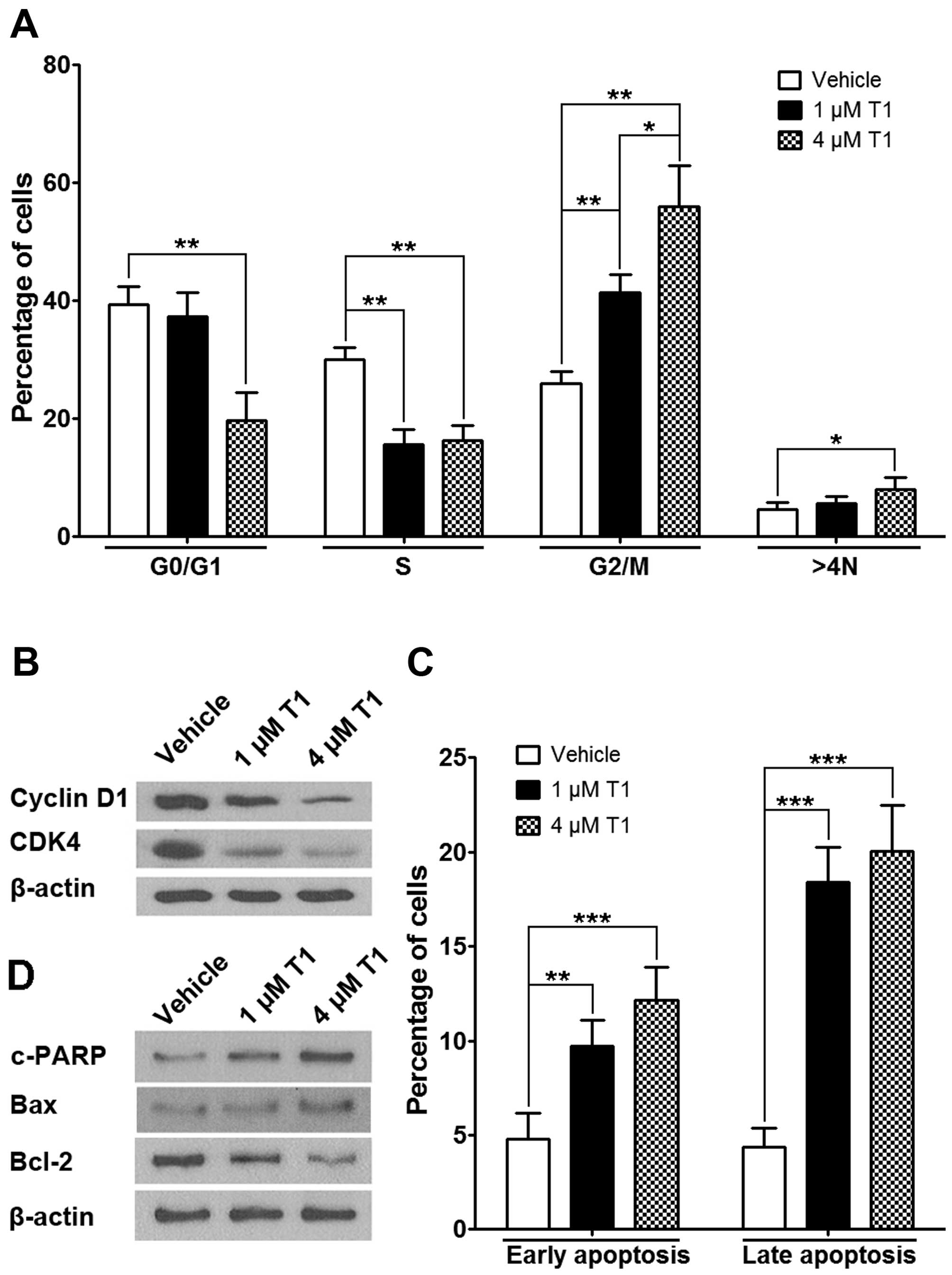
To analyze how HCT116 cancer cells were affected by
T1, we conducted flow cytometric analysis to detect cell cycle
profiles following T1 exposure. Results showed that when treated
for 24 h, T1 significantly increased the proportion of G2/M and 4N
cells and decreased the proportion of S and G0/G1 cells (Fig. 2A), which is consistent with the
down-regulation of CDK4 and cyclin D1 protein levels, indicating a
cell cycle arrest (Fig. 2B).
Further study revealed that T1 significantly induced both early and
late apoptotic events in HCT116 cells (Fig. 2C), which is consistent with the
subsequent observation showing T1 induces c-PARP and Bax (both
pro-apoptotic) protein expression as well as Bcl-2 (anti-apoptotic)
downregulation in a dose-dependent manner (Fig. 2D).
The role of Aurora A and p53 in
Tanshinone I mediated HCT116 cell apoptosis
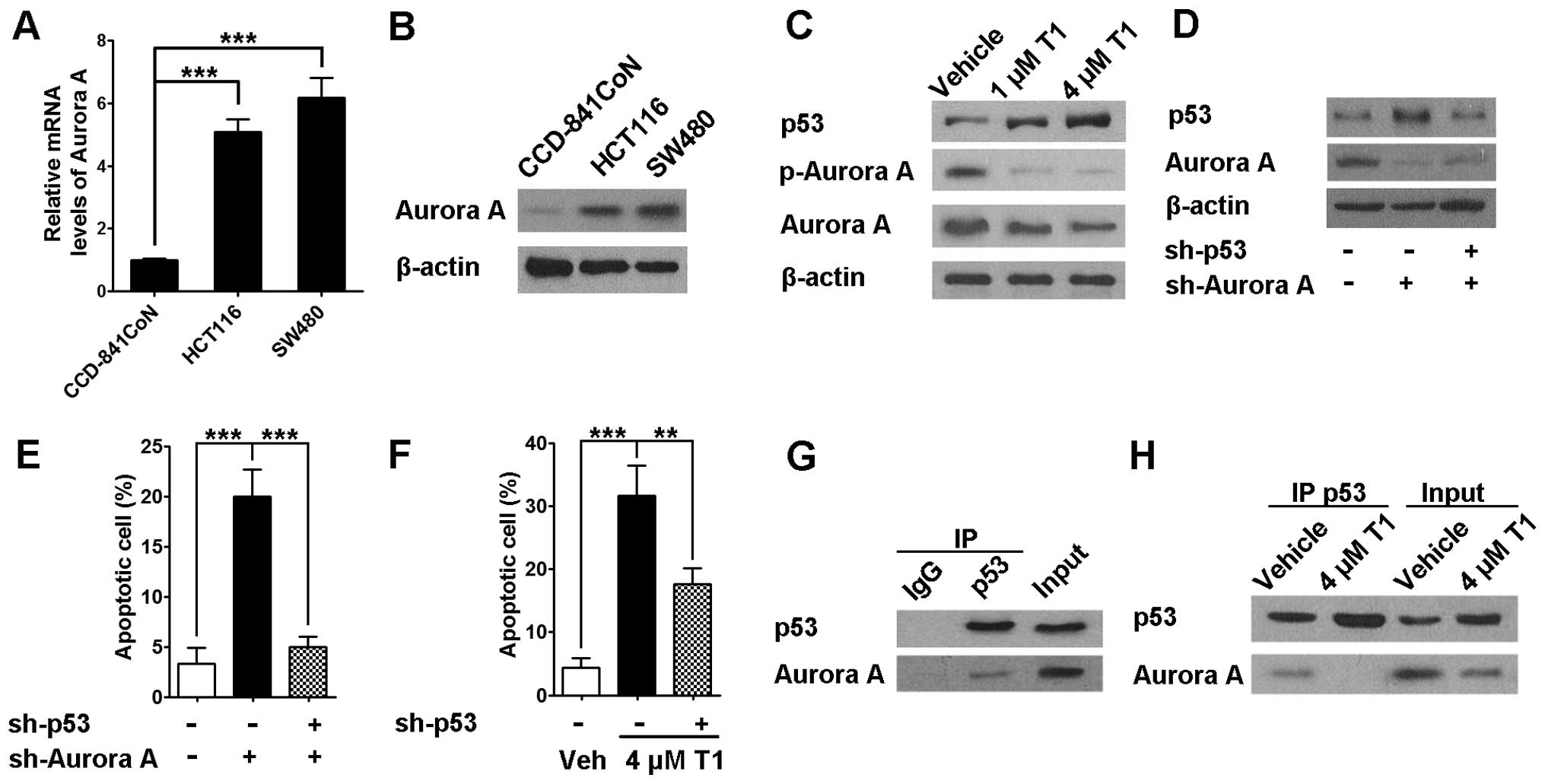
As the next step, we sought to identify the
underlying mechanism of T1 on CRC growth and cell cycle
progression. As described above, existing data show the potential
roles of Aurora A on numerous malignancies other than CRC. In our
experiments, we found that compared to CCD-841CoN colon epithelial
cells, both HCT116 and SW480 cells exhibited significantly higher
Aurora A mRNA and protein levels (Fig.
3A and B). In addition, data also revealed that T1
significantly downregulated Aurora A and phosphorylated-Aurora A
protein expression in a dose-dependent manner, which seems to be
strongly associated with significant p53 protein upregulation
(Fig. 3C). Consistently,
engineered Aurora A knock-down in HCT116 cells resulted in a
similar p53 upregulation and a strong induction of apoptosis
(Fig. 3D and E). Interestingly,
when p53 was knocked down in HCT116 cells, Aurora A knock-down
mediated apoptosis was almost completely abolished, indicating p53
is required and pivotal in this Aurora A mediated action (Fig. 3E). However, our data also suggested
that p53 knock-down only partially abolished T1 mediated HCT116
cell apoptosis, indicating signaling machineries other than Aurora
A-p53 pathway may exist for T1 induced apoptosis (Fig. 3F). It was reported by Katayama
et al that Aurora A interacts with and phosphorylates p53,
leading to its proteolysis (30).
Consistent with this observation, we found that Aurora A interacted
with p53 in regularly cultured HCT116 cells as shown in Fig. 3G. In addition, when the cells were
treated with T1, the interaction between Aurora A and p53 was
disrupted along with decrease of Aurora A expression (Fig. 3H).
In SW480 cells, survivin-inhibition is
pivotal for Tanshinone I action
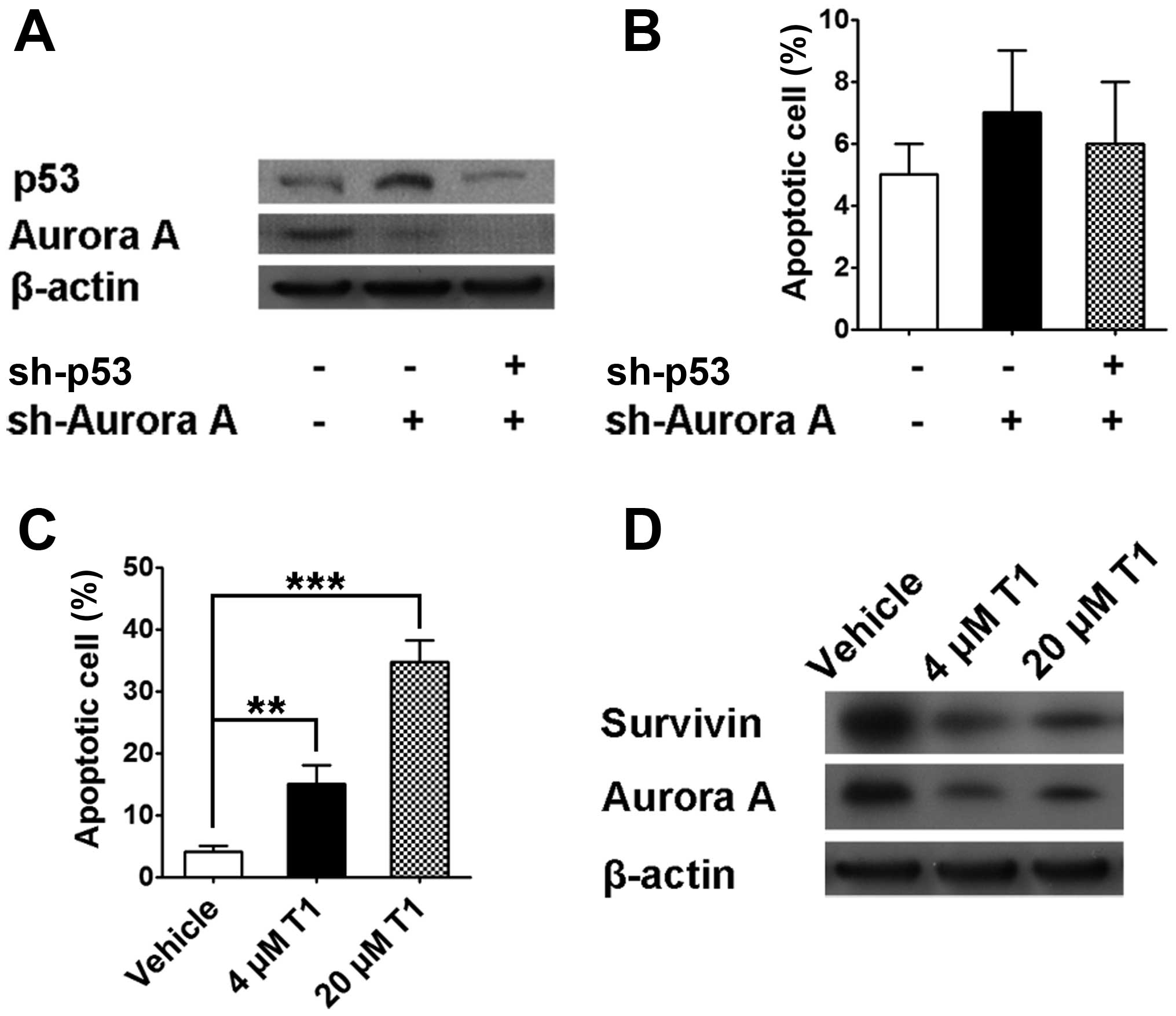
In SW480 cell line with p53 point mutation, Aurora A
knock-down resulted in p53 protein upregulation, which is similar
to data shown in Fig. 3. In
contrast, we were not able to observe a similar Aurora A knock-down
mediated apoptosis in SW480 cells and engineered p53 knock-down
neither enhanced nor decreased cell apoptosis, indicating Aurora
A-p53 axis does not play a pivotal role if mutant p53 protein is
not able to localize to the nucleus (Fig. 4A and B). Further studies showed
that higher concentration T1 did induce SW480 cell apoptosis (4 and
20 μm) as well as Aurora A downregulation (Fig. 4C). Interestingly, T1 also mediated
survivin downregulation, which potentially provides a distinct
mechanism for T1 action other than p53-mediated pathway (Fig. 4D).
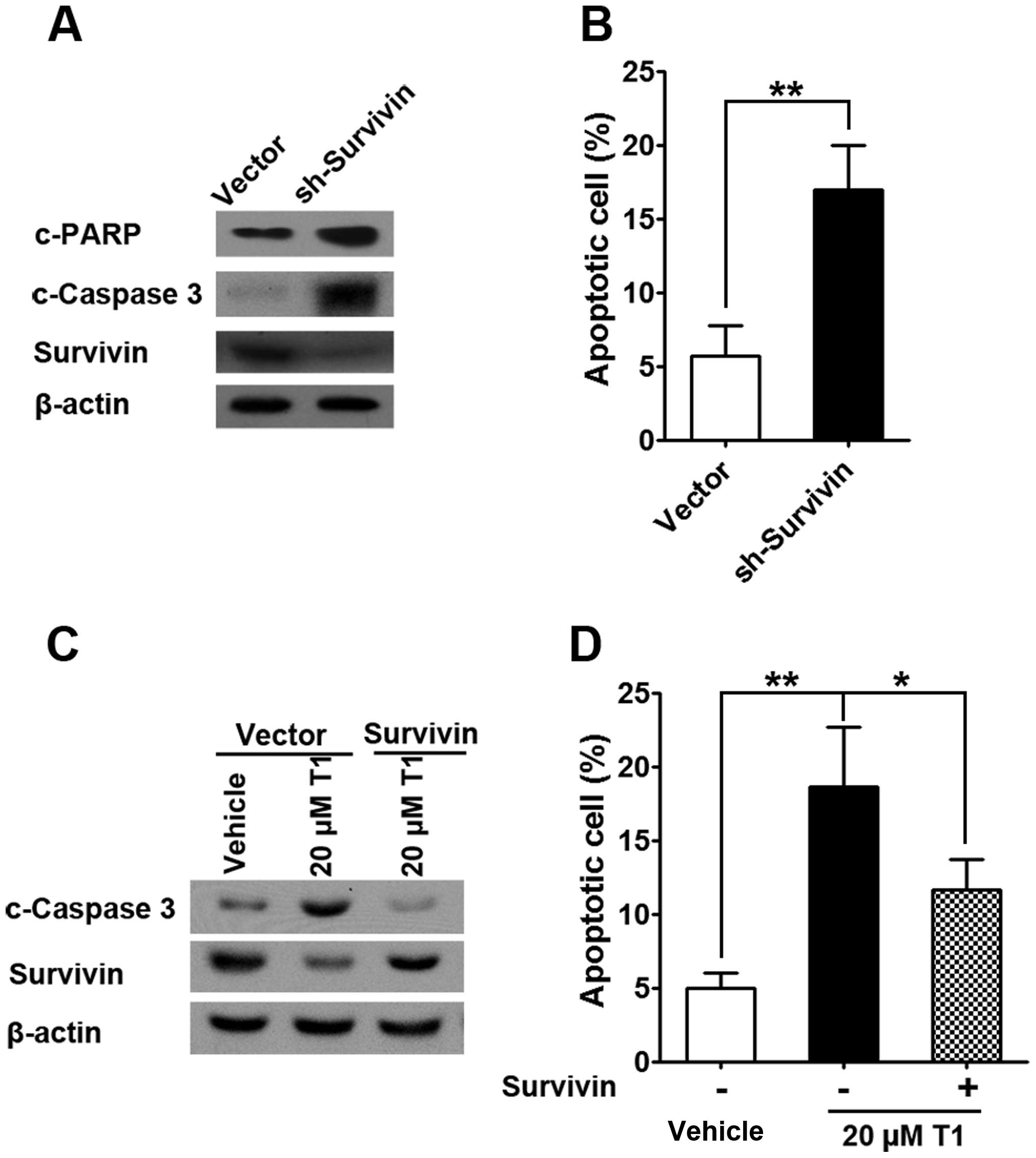
To confirm the role of survivin in T1 action, we
successfully engineered survivin knock-down in SW480 cells and
found significant upregulation of apoptotic proteins such as c-PARP
and c-caspase-3 (Fig. 5A), which
is consistent with enhanced apoptosis in survivin knock-down cells
(Fig. 5B). On the other hand, when
SW480 cells were treated with T1, c-caspase-3 protein expression as
well as SW480 apoptosis was significantly upregulated and this
effect was reversed by survivin overexpression, which highlights
survivin as a governor for T1 mediated SW480 cell apoptosis.
Discussion
Salvia miltiorrhiza Bunge (Danshen) is a
traditional Chinese herb that has been widely adopted in the
traditional Chinese medical therapy. With 20 phenolic acids, and 30
diterpene compounds, T1 is a relatively abundant component. In
addition to the function in cardiovascular systems, tanshinones
have been recently shown to possess activities against human
cancers. In this study, we examined the effects of T1 on human
colorectal cells in vitro. Results showed that T1 markedly
inhibited CRC cell growth and induced apoptosis, particularly in
CRC cells with functional p53 protein. Interestingly, T1 did not
exert as much inhibitory effect on normal colon epithelial cells or
CRC cells with mutant p53, indicating relative selectivity toward
CRC cells with full presence of functional p53. In cells with
wild-type p53 protein, Aurora A is critical for T1 action and p53
is indispensable for Aurora A-mediated signaling. In contrast, in
CRC cells with mutant p53 protein (not able to localize to the
nucleus), Aurora A knock-down, although it increased p53 protein
level, failed to induce cell apoptosis. Instead, it seems that
downregulation of survivin, an anti-apoptotic protein (31), induced apoptosis of SW480 cells
following T1 treatment. These observations were further
substantiated by the pivotal role of survivin in T1 mediated
apoptosis shown in Fig. 5.
Aurora A is a member of the Aurora kinase family
(Aurora A, B and C) (32–34). Aurora A protein overexpression and
gene amplification have been frequently observed in CRC and other
cancers. Bischoff et al were the first to report
over-expression of Aurora A mRNA in >50% of CRC tumors (15). It was later reported that
overexpression of Aurora A protein was found in 19% of CRC samples
of various disease stages by immunohistochemistry (IHC) (35). Another group identified Aurora A
overexpression by IHC in 48.5% of early-stage CRC samples (23). Aurora A has been recently
considered as one of the most promising molecular targets for CRC
therapy (24).
In light of previous notions indicating that p53 may
serve as a downstream effector for Aurora A, we hypothesized and
successfully confirmed that it is also true in CRC scenario in
terms of T1 action. Although we should admit that significant
distinctions other than p53 protein expression exist between SW480,
HCT116 and CCD-841CoN cells and p53 may not be the only explanation
for the differential pattern of Tanshinone action observed, our
argument was still strongly substantiated with a series of
subsequent data from p53 knockdown cells.
P53 has been extensively implicated in regulation of
cell cycle arrest, senescence and apoptosis (36). In malignant cells, higher Aurora-A
level is usually associated with minimal expression of p53
(37). There are several signaling
machineries which mediate Aurora-A induced p53 inactivation. First,
Aurora-A phosphorylates p53 at Serine residue 315 which triggers
MDM2-mediated p53 degradation (30). Alternatively, Aurora-A can also
phosphorylate p53 at Serine residue 215, which disrupts its DNA
binding and transactivation of downstream molecules such as p21 and
PTEN (38). In addition, Nair
et al showed that engineered Aurora-A silencing with siRNA
promotes p53 phosphorylation at Serine residue 15, which in turn
enhances G1/S cell cycle arrest (39). Furthermore, Aurora-A phosphorylates
a p53 family member-p73 at Serine residue 235 and ruins
physiological DNA damage-repair response (25). Taken together, it seems that
Aurora-A exerts potent inhibitory effects on p53 action via
phosphorylation on numerous important regulatory sites, which in
turn disrupt p53-mediated innate DNA damage repair response and
subsequent apoptotic events.
On the other hand, in case p53 does not function
properly such as in SW480 cells, which also commonly occurred in
many types of malignancies including CRC (40), our data suggested that
survivin-involved pathway is responsible for T1 action.
Survivin is a member of the so-called inhibitor of
apoptosis (IAP) family and exerts its regulatory effects via
inhibiting caspase activation, which is required for apoptosis.
Data showed that disruption of survivin induction results in
increased apoptosis and decreased tumor growth. Survivin is
abundantly expressed in a wide spectrum of human malignancies, but
is completely absent in well differentiated cells (41). Mechanistically, survivin
co-localizes with the mitotic spindle via binding to tubulin and
exerts regulatory effects during mitosis. Emerging evidence
suggested survivin could be regulated by p53 protein as well as Wnt
pathway and β-catenin (42),
indicating p53 and survivin may co-exist in the same signaling
machinery. However, our data argues that survivin-involved pathway
may work independently from p53-mediated pathway. In this scenario,
either p53 activation or survivin inhibition may subsequently
result in a similar regulatory effect.
In conclusion, our study supported that T1, a novel
natural compound, exerts significant inhibitory effect on CRC cell
growth. The underlying mechanism involves either Aurora A-p53 axis
or survivin-involving mechanism depending on intrinsic
characteristics of tumor cells. Although more human efficacy and
safety data are needed before clinical application of T1 on CRC
patients, our preliminary data seem to support the notion that a
pre-screening for full presence of functional p53 protein in CRC
tissue may be required to ensure an optimal response.
Future studies will be directed to: i)
identification of the potential roles of Aurora A-p53 axis and
survivin-mediated pathway in various subtypes of human CRC
specimen, which highlights the significance of individualized CRC
therapy and ii) screening novel herbal compounds which exert
optimal inhibitory effect on CRC cell via survivin signaling.
Acknowledgements
This study was supported by intramural funding of
the First Affiliated Hospital of Nanjing Medical Univeristy.
References
|
1
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar
|
|
2
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Merika E, Saif MW, Katz A, Syrigos K and
Morse M: Review. Colon cancer vaccines: An update. In Vivo.
24:607–628. 2010.PubMed/NCBI
|
|
4
|
Lee YT: Local and regional recurrence of
carcinoma of the colon and rectum: I. Tumour-host factors and
adjuvant therapy. Surg Oncol. 4:283–293. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Declan Fleming RY: Colorectal cancer
screening and follow-up. Surg Oncol. 7:125–137. 1998. View Article : Google Scholar
|
|
6
|
Figueredo A, Rumble RB, Maroun J, Earle
CC, Cummings B, McLeod R, Zuraw L and Zwaal C; Gastrointestinal
Cancer Disease Site Group of Cancer Care Ontario's Program in
Evidence-based Care. Follow-up of patients with curatively resected
colorectal cancer: A practice guideline. BMC Cancer. 3:262003.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chambers WM and Mortensen NJ:
Postoperative leakage and abscess formation after colorectal
surgery. Best Pract Res Clin Gastroenterol. 18:865–880. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Carmena M and Earnshaw WC: The cellular
geography of aurora kinases. Nat Rev Mol Cell Biol. 4:842–854.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nigg EA: Mitotic kinases as regulators of
cell division and its checkpoints. Nat Rev Mol Cell Biol. 2:21–32.
2001. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hirota T, Kunitoku N, Sasayama T, Marumoto
T, Zhang D, Nitta M, Hatakeyama K and Saya H: Aurora-A and an
interacting activator, the LIM protein Ajuba, are required for
mitotic commitment in human cells. Cell. 114:585–598. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li D, Zhu J, Firozi PF, Abbruzzese JL,
Evans DB, Cleary K, Friess H and Sen S: Overexpression of oncogenic
STK15/BTAK/Aurora A kinase in human pancreatic cancer. Clin Cancer
Res. 9:991–997. 2003.PubMed/NCBI
|
|
12
|
Matarasso N, Bar-Shira A, Rozovski U,
Rosner S and Orr-Urtreger A: Functional analysis of the Aurora
Kinase A Ile31 allelic variant in human prostate. Neoplasia.
9:707–715. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yamamoto S, Yamamoto-Ibusuki M, Yamamoto
Y, Fujiwara S and Iwase H: A comprehensive analysis of Aurora A;
transcript levels are the most reliable in association with
proliferation and prognosis in breast cancer. BMC Cancer.
13:2172013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Aust DE, Muders M, Köhler A, Schmidt M,
Diebold J, Müller C, Löhrs U, Waldman FM and Baretton GB:
Prognostic relevance of 20q13 gains in sporadic colorectal cancers:
A FISH analysis. Scand J Gastroenterol. 39:766–772. 2004.
View Article : Google Scholar
|
|
15
|
Bischoff JR, Anderson L, Zhu Y, Mossie K,
Ng L, Souza B, Schryver B, Flanagan P, Clairvoyant F, Ginther C, et
al: A homologue of Drosophila aurora kinase is oncogenic and
amplified in human colorectal cancers. EMBO J. 17:3052–3065. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Carvalho B, Postma C, Mongera S, Hopmans
E, Diskin S, van de Wiel MA, van Criekinge W, Thas O, Matthäi A,
Cuesta MA, et al: Multiple putative oncogenes at the chromosome 20q
amplicon contribute to colorectal adenoma to carcinoma progression.
Gut. 58:79–89. 2009. View Article : Google Scholar
|
|
17
|
Goos JA, Coupe VM, Diosdado B, Delis-Van
Diemen PM, Karga C, Beliën JA, Carvalho B, van den Tol MP, Verheul
HM, Geldof AA, et al; DeCoDe PET group. Aurora kinase A (AURKA)
expression in colorectal cancer liver metastasis is associated with
poor prognosis. Br J Cancer. 109:2445–2452. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hermsen M, Postma C, Baak J, Weiss M,
Rapallo A, Sciutto A, Roemen G, Arends JW, Williams R, Giaretti W,
et al: Colorectal adenoma to carcinoma progression follows multiple
pathways of chromosomal instability. Gastroenterology.
123:1109–1119. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nakao K, Mehta KR, Fridlyand J, Moore DH,
Jain AN, Lafuente A, Wiencke JW, Terdiman JP and Waldman FM:
High-resolution analysis of DNA copy number alterations in
colorectal cancer by array-based comparative genomic hybridization.
Carcinogenesis. 25:1345–1357. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Postma C, Terwischa S, Hermsen MA, van der
Sijp JR and Meijer GA: Gain of chromosome 20q is an indicator of
poor prognosis in colorectal cancer. Cell Oncol. 29:73–75.
2007.PubMed/NCBI
|
|
21
|
Sillars-Hardebol AH, Carvalho B, de Wit M,
Postma C, Delis-van Diemen PM, Mongera S, Ylstra B, van de Wiel MA,
Meijer GA and Fijneman RJ: Identification of key genes for
carcinogenic pathways associated with colorectal
adenoma-to-carcinoma progression. Tumour Biol. 31:89–96. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gerlach U, Kayser G, Walch A, Hopt U,
Schulte-Mönting J, Werner M and Lassmann S: Centrosome-,
chromosomal-passenger- and cell-cycle-associated mRNAs are
differentially regulated in the development of sporadic colorectal
cancer. J Pathol. 208:462–472. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lam AK, Ong K and Ho YH: Aurora kinase
expression in colorectal adenocarcinoma: Correlations with
clinicopathological features, p16 expression, and telomerase
activity. Hum Pathol. 39:599–604. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dar AA, Goff LW, Majid S, Berlin J and
El-Rifai W: Aurora kinase inhibitors - rising stars in cancer
therapeutics? Mol Cancer Ther. 9:268–278. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Görgün G, Calabrese E, Hideshima T, Ecsedy
J, Perrone G, Mani M, Ikeda H, Bianchi G, Hu Y, Cirstea D, et al: A
novel Aurora-A kinase inhibitor MLN8237 induces cytotoxicity and
cell-cycle arrest in multiple myeloma. Blood. 115:5202–5213. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cheng TO: Cardiovascular effects of
Danshen. Int J Cardiol. 121:9–22. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gong Y, Li Y, Abdolmaleky HM, Li L and
Zhou JR: Tanshinones inhibit the growth of breast cancer cells
through epigenetic modification of Aurora A expression and
function. PLoS One. 7:e336562012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nizamutdinova IT, Lee GW, Lee JS, Cho MK,
Son KH, Jeon SJ, Kang SS, Kim YS, Lee JH, Seo HG, et al: Tanshinone
I suppresses growth and invasion of human breast cancer cells,
MDA-MB-231, through regulation of adhesion molecules.
Carcinogenesis. 29:1885–1892. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lee CY, Sher HF, Chen HW, Liu CC, Chen CH,
Lin CS, Yang PC, Tsay HS and Chen JJ: Anticancer effects of
tanshinone I in human non-small cell lung cancer. Mol Cancer Ther.
7:3527–3538. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Katayama H, Sasai K, Kawai H, Yuan ZM,
Bondaruk J, Suzuki F, Fujii S, Arlinghaus RB, Czerniak BA and Sen
S: Phosphorylation by aurora kinase A induces Mdm2-mediated
destabilization and inhibition of p53. Nat Genet. 36:55–62. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tamm I, Wang Y, Sausville E, Scudiero DA,
Vigna N, Oltersdorf T and Reed JC: IAP-family protein survivin
inhibits caspase activity and apoptosis induced by Fas (CD95), Bax,
caspases, and anticancer drugs. Cancer Res. 58:5315–5320. 1998.
|
|
32
|
Giet R and Prigent C: Aurora/Ipl1p-related
kinases, a new oncogenic family of mitotic serine-threonine
kinases. J Cell Sci. 112:3591–3601. 1999.PubMed/NCBI
|
|
33
|
Kaestner P, Stolz A and Bastians H:
Determinants for the efficiency of anticancer drugs targeting
either Aurora-A or Aurora-B kinases in human colon carcinoma cells.
Mol Cancer Ther. 8:2046–2056. 2009. View Article : Google Scholar
|
|
34
|
Vankayalapati H, Bearss DJ, Saldanha JW,
Muñoz RM, Rojanala S, Von Hoff DD and Mahadevan D: Targeting
aurora2 kinase in oncogenesis: A structural bioinformatics approach
to target validation and rational drug design. Mol Cancer Ther.
2:283–294. 2003.PubMed/NCBI
|
|
35
|
Baba Y, Nosho K, Shima K, Irahara N, Kure
S, Toyoda S, Kirkner GJ, Goel A, Fuchs CS and Ogino S: Aurora-A
expression is independently associated with chromosomal instability
in colorectal cancer. Neoplasia. 11:418–425. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang Y, Sun H, Wang Z, Liu M, Qi Z, Meng
J, Sun J and Yang G: Aurora-A: A potential DNA repair modulator.
Tumour Biol. 35:2831–2836. 2014. View Article : Google Scholar
|
|
37
|
Hu W, Kavanagh JJ, Deaver M, Johnston DA,
Freedman RS, Verschraegen CF and Sen S: Frequent overexpression of
STK15/Aurora-A/BTAK and chromosomal instability in tumorigenic cell
cultures derived from human ovarian cancer. Oncol Res. 15:49–57.
2005.PubMed/NCBI
|
|
38
|
Liu Q, Kaneko S, Yang L, Feldman RI,
Nicosia SV, Chen J and Cheng JQ: Aurora-A abrogation of p53 DNA
binding and trans-activation activity by phosphorylation of serine
215. J Biol Chem. 279:52175–52182. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nair JS, Ho AL and Schwartz GK: The
induction of polyploidy or apoptosis by the Aurora A kinase
inhibitor MK8745 is p53-dependent. Cell Cycle. 11:807–817. 2012.
View Article : Google Scholar :
|
|
40
|
Liu MC and Gelmann EP: P53 gene mutations:
Case study of a clinical marker for solid tumors. Semin Oncol.
29:246–257. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sah NK, Khan Z, Khan GJ and Bisen PS:
Structural, functional and therapeutic biology of survivin. Cancer
Lett. 244:164–171. 2006. View Article : Google Scholar
|
|
42
|
Olie RA, Simões-Wüst AP, Baumann B, Leech
SH, Fabbro D, Stahel RA and Zangemeister-Wittke U: A novel
antisense oligo-nucleotide targeting survivin expression induces
apoptosis and sensitizes lung cancer cells to chemotherapy. Cancer
Res. 60:2805–2809. 2000.PubMed/NCBI
|