Introduction
The incidence of cancer continues to increase,
largely because of an aging population and increasing rates of
cancer-causing behaviors and conditions, particularly smoking and
obesity. Colorectal cancer is the third most common cancer in males
and the second most common in females, with over 1.2 million new
cases and 608,700 deaths in 2008 (1,2). The
incidence of colorectal cancer is higher in Westernized areas such
as New Zealand, Europe and North America than in Asian countries.
However, incidence rates are rapidly increasing in several East
Asian countries such as Japan, China and Singapore (3–5). The
trend is thought to reflect a change in dietary patterns (6–8). For
this reason, interest in alternative medicine for the prevention
and treatment of colorectal cancer has increased and research on
the effects of various food extracts on colorectal cancer is in
progress (8,9).
Inducing apoptosis is one therapeutic strategy in
cancer treatment. Apoptosis is the process of programmed cell
death. In contrast to necrosis, which is a caused by acute cellular
injury, apoptosis has many biological advantages. For example, the
separation of fingers and toes in a developing human embryo occurs
because cells between the digits undergo apoptosis (10,11).
Understanding apoptosis in disease conditions is very important as
it not only offers insights into the pathogenesis of a disease but
may also give clues as to how the disease can be treated. Numerous
research studies have demonstrated the death of cancer cells
through apoptosis and compounds have been developed to take
advantage of this knowledge (7,9,12,13).
Induction of apoptosis in cancer cells is driven by
a complex interplay between several proteins. Members of the Bcl-2
family of proteins are key regulators of apoptosis. These proteins
are known to regulate mitochondrial function and control the
release of apoptosis-inducing factors such as cytochrome c
from the mitochondrial inter-membrane space (14–16).
The anti-apoptotic proteins Bcl-2 and Mcl-1 are predominantly found
in the mitochondria; they inhibit apoptosis by suppressing the
release of cytochrome c (17,18).
In contrast, the pro-apoptotic proteins Bax, Bak and PUMA mainly
induce the release of stimulators of apoptosis and bring about
mitochondrial dysfunction after translocating to the mitochondrial
outer membrane (19–21). In particular, translocation of Bax
and Bak to the mitochondrial outer membrane is required for the
release of cytochrome c during apoptosis. Bax and Bak
undergo homo- and hetero-oligomerization and bind to the
mitochondrial outer membrane. These protein complexes trigger
cytochrome c release into the cytosol by reducing
mitochondrial outer membrane permeabilization (22–25).
Cytochrome c induces apoptosis by increasing the activity of
caspases in the cytoplasm (26).
Control of this process using targeted compounds is very important
in cancer treatment.
The fruit of Torilis japonica can be used as
a substitute for She chuang zi, which is a traditional
Chinese medicine prescribed as an anti-allergenic, anti-fungal,
anti-bacterial and sedative agent. Previously, we found that a 95%
ethanol extract from Torilis japonica had beneficial effects
on metastasis through regulation of the EGFR signaling pathway in
MCF-7 breast cancer cells (27).
However, its anti-proliferative and apoptosis-inducing effects have
not yet been elucidated.
In this study, we investigated the effects of
Torilis japonica extract (TJE) extracted from the fruit of
Torilis japonica on apoptosis in HCT116 and HT-29 colon
cancer cells. TJE induced apoptosis through the generation of
intracellular reactive oxygen species (ROS) and a reduction in the
mitochondrial membrane potential via regulation of the AMPK/p38
MAPK signaling pathway. Moreover, the apoptotic effects of TJE
persisted in cells lacking p53. Taken together, our results
indicate that TJE may be a novel natural ingredient for cancer
therapy that decreases the mitochondrial membrane potential of
colorectal cancer cells, thereby inducing apoptosis.
Materials and methods
Plant material and preparation of
TJE
Dried whole fruit of Torilis japonica was
purchased from Na-num Pharmacy (Kyung-buk, Korea). Plant material
(200 g) was extracted two times with 95% ethanol at room
temperature for 3 day and was subsequently filtered. The combined
filtrate was concentrated under vacuum at 60°C, and completely
dried by freeze drying. The yield was 10% and TJE powder was
dissolved in DMSO and filtrated by 0.2 μ pore size filter for in
vitro studies.
Reagent
N-acetyl cystein (NAC),
4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT),
DCFH-DA are purchased from Sigma-Aldrich (St. Louis, MO, USA).
FITC-Annexin V apoptosis detection kit is obtained from BD
Pharmingen (San Diego, CA, USA). Mitotracker was purchased from
Molecular Probes (Eugene, OR, USA). Specific anti-bodies that
recognized p-AMPKα1, AMPKα1, p38, p-p53, caspase-3, Bcl-2, Mcl-1,
COX-4, β-actin are obtained from Cell Signaling Technology
(Beverly, MA, USA) and Bax, Bak, PUMA, p-p38 MAPK, p53, cytochrome
c are purchased from Santa Cruz Biotechnology, Inc. (Dallas,
TX, USA).
Cell culture
HCT116 and HT-29, fibroblast cells were obtained
from the American Type Culture Collection (ATCC, Rockville, MD,
USA) 3 month before experiment begun. HCT116, HT-29 cells were
grown in RPMI-1640 medium and fibroblast cells were grown in DMEM
medium containing 10% fetal bovine serum (both from HyClone,
Waltham, MA, USA) and 1% antibiotics (100 mg/l streptomycin, 100
U/ml penicillin) at 37°C in a 5% CO2 atmosphere. Cells
were suspended by Trypsin-EDTA (HyClone) and separated
1.5×105/ml at each plates, every 48 h. All the cell
lines were authenticated by each 6 month repeat analysis at Korea
Cell Line Bank (KCLB, Seoul, Korea)
Detection of intracellular ROS by
fluorescence microscope
Cells were seeded 1×105/ml in 12-well
plate with cover glasses. After treatment the indicated time and
dose at 37°C in a 5% CO2 atmosphere, the cells were
incubated with 10 μM of DCFH-DA for 30 min and fixed with 3.7%
formaldehyde for 20 min. Cells were washed with phosphate-buffered
saline (PBS) twice and fluorescence was detected by fluorescence
microscope (Carl Zeiss, Thornwood, NY, USA).
Measurement of intracellular ROS
levels
Cells were seeded 1×106/ml in 100 mm
plate and incubated for 24 h. After incubation, cells treated with
test compound for 6 h at 37°C in a 5% CO2 atmosphere.
Cells were incubated with 40 μM of DCFH-DA for 30 min and harvested
by trypsinization, collected by centrifugation, washed with PBS
twice, and resuspended in PBS. Fluorescence intensity were analyzed
by using flow cytometer (Becton-Dickinson Biosciences, Franklin
Lakes, NJ, USA).
Cell proliferation assay (MTT)
Cells were seeded at 4,000/ml each well in 96-well
plate, and incubated 24 h. After the incubation, treated with test
compound and incubate at 37°C in a 5% CO2 atmosphere.
After 24 h, cells were incubated with 20 μl MTT (5 mg/ml with PBS)
solution for 1 h. Optical densities of solution, in each well, were
determined by Microplate reader (Bio-Rad Laboratories, Inc., Tokyo,
Japan) at 595 nm.
Determination of apoptosis by Annexin
V/PI staining analysis
Cells were seeded at 1×106/ml in 100 mm
plate and incubated for 24 h. After incubation, cells treated with
test compound for 24 h at 37°C in a 5% CO2 atmosphere.
Total cells were harvested by trypsinization, collected by
centrifugation, washed with PBS, and resuspended in binding buffer.
Cells were stained with Annexin V and PI for 15 min. Fluorescence
intensity were analyzed by using flow cytometer (BD
Biosciences).
Measurement of mitochondria membrane
potential
Cells were seeded at 1×106/ml in 100 mm
plate and incubated for 24 h. After incubation, cells treated with
test compound for 24 h at 37°C in a 5% CO2 atmosphere.
Total cells were harvested by trypsinization, collected by
centrifugation, washed with PBS. Cell were incubated with JC-1 for
30 min 37°C in a 5% CO2 atmosphere before the flow
cytometer analysis (BD Biosciences).
Caspase-3 activity assay
We used caspase-3 activity assay kit (Abcam PLC,
Cambridge, UK). Cells were seeded at 1×106/ml in 100 mm
plate and incubated for 24 h. After incubation, cells treated with
test compound for 24 h at 37°C in a 5% CO2 atmosphere.
Total cells were harvested by trypsinization, collected by
centrifugation, washed with PBS. Cells were resuspended in lysis
buffer and mixed with 2X reaction buffer. Samples were reactivated
with DEVD-p-NA for 2 h at 37°C. Optical densities of solution, in
each well, were determined by Microplate reader (Bio-Rad
Laboratories, Inc.) at 405 nm.
Fraction of mitochondria and cytosol
proteins
We used Mitochondria/Cytosol Fraction kit (Abcam
PLC). Cells were seeded at 1×106/ml in 100 mm plate and
incubated for 24 h. After incubation, cells were treated with test
compound for 24 h at 37°C in a 5% CO2 atmosphere. Total
cells were harvested by trypsinization, collected by
centrifugation, washed with PBS, and homogenized in ice-cold
cytosol extraction buffer mix containing DTT and protease inhibitor
using a sonicator. The homogenates were centrifuged at 3,000 rpm
for 10 min at 4°C and supernatants were collection. Supernatant
were centrifuged at 13,000 rpm for 30 min at 4°C and collected
supernatant for cytosol proteins and pellets were resuspended with
ice cold mitochondria extraction buffer containing DTT and protease
inhibitor for mitochondria proteins.
Protein oligomerization
Bak and Bax oligomerization were assessed by
chemical crosslinking. Briefly, mitochondria protein fractions were
resuspended in conjugation buffer. For disulphide-bond formation,
mitochondria protein fractions were incubated with the
bis(maleimido)hexane (Thermo Fisher Scientific, Rockford, IL, USA)
for 1 h at room temperature and the samples analyzed by
non-reducing SDS-PAGE.
Immunoprecipitation (IP) assay
We used to surebead protein G magnetic beads kit
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). Cells were seeded
at 1×106/ml in 100 mm plate and incubated for 24 h.
After incubation, cells were treated with test compound for 24 h at
37°C in a 5% CO2 atmosphere. Mitochondria/cytosol
proteins were fraction and mitochondria proteins were incubated
with specific antibody bound magnetic bead. The beads were washed
using a magnet and PBS. Target proteins were elusion in 1X sample
buffer and analyzed by western blotting.
Bcl-2 activation assay
We used Muse Bcl-2 activation dual detection kit
(Merck Milipore, Darmstadt. Germany). Cells were seeded at
1×105/ml in 6-well plate and incubated for 24 h. After
incubation, cells were treated with test compound for 24 h at 37°C
in a 5% CO2 atmosphere. Total cells were harvested by
trypsinization, collected by centrifugation, washed with PBS. Cell
were fixed and permeabilized by reagent in the kit. The antibody,
which was combined with fluorescence tag, was reacted for 1 h at
room temperature with slow agitation, and it was washed once with
the assay buffer. Cells were resuspended in assay buffer and
analyzed by Muse cell analyzer.
Transient transfection with small
interfering RNA
Small interfering RNA (siRNA) was purchased by
Dharmacon (Chicago, IL, USA). For transient transfection, cells
were seeded at 5×103/ml on a 6-well plate with
antibiotics free medium. After incubation overnight, targeting
siRNA was transfected using DharmaFECT2 transfection reagent
(Dharmacon) according to manufacturer’s instructions. After
incubation for 72 h, cells were treated with TJE with NAC at
indicated doses.
Western blotting
Cells were seeded at 1×105/ml in a 6-well
plate and incubated for 24 h, and after the incubation, treated
with test compound for 6 h at 37°C in a 5% CO2
atmosphere. Cells were rinsed twice with ice-cold PBS and scraped
with lysis buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% NP-40,
0.5% sodium deoxycholate, 1 mM PMSF) and subjected to western blot
analysis. First antibody was reacted overnight at 4°C and second
antibody for 75 min at room-temperature with slow agitation.
Immunofluorescence (IF) staining
Cells were seeded 1×105/ml in a 12-well
plate with cover glasses. After treatment with the indicated time
and dose, the cells were stained with mitotracker for 30 min at
37°C in a 5% CO2 atmosphere. Cells were fixed with 3.7%
formaldehyde for 20 min and pemeabilized with 0.2% Triton X-100 for
20 min. Cells were washed with PBS twice and reacted with
cytochrome c antibody overnight at 4°C. Cells were washed
with PBS twice and reacted with secondary antibody for 1 h.
Fluorescence was detected by confocal (Olympus, Tokyo, Japan).
Xenograft model
Five-week-old male Balb/c nu/nu mice were
obtained from SLC (Tokyo, Japan) and housed in sterile filer-topped
cages. For tumor induction, HCT116 human colon cancer cells
(2.5×105 cells/0.1 ml) were subcutaneously injected into
the left flank of the mice (each group had 5 animals). One week
after the injection of cells, they were co-treated with TJE 80
mg/kg/day for 21 days. Tumor size was measured using a caliper at 2
day intervals, and the volume was calculated by the modified
formula V = 1/2 (length x width2). After the 3-week
treatment, tumor was removed and frozen in liquid nitrogen for
western blot analysis or fixed with formalin for
immunohistochemistry and H&E staining. All animal experiments
were approved by the Ethics Committee for Animal Experimentation,
Hannam University.
Immunohistochemistry
Tumor specimens from mice were fixed in 10%
formaldehyde, embedded in paraffin and sectioned into 5 μm thick
slices. Consecutive thin cryosections (5 μm) of OCT compound
(Sakura Finetek, Torrance, CA, USA) embedded tumor tissues were
fixed in acetone at 4°C for 10 min. After washing in PBS, sections
were treated with 3% H2O2 for 10 min to block
endogenous peroxidase activity, and the sections were blocked with
normal rabbit serum. Then, the sections were blocked and washed in
PBS and incubated with specific antibody overnight at 4°C. Negative
controls were incubated with the primary normal serum IgG for the
species from which the primary antibody was obtained.
TUNEL assay
Levels of apoptosis in distal colon tissue were
determined using the TdT-mediated dUTP nick-end labeling (TUNEL)
method. Tumor specimens from mice were fixed in 10% formaldehyde,
embedded in paraffin and sectioned into 5 μm thick slices. Tissue
sections were processed according to manufacturer’s instructions
for the ApopTag peroxidase in situ apoptosis detection kit
(Vector Laboratories, Burlingame, CA, USA).
Statistical analysis
Cell viability and caspase-3 activity was
statistically analyzed using unpaired t-test (SPSS, Chicago, IL,
USA). P<0.05 was considered statistically significant.
Results
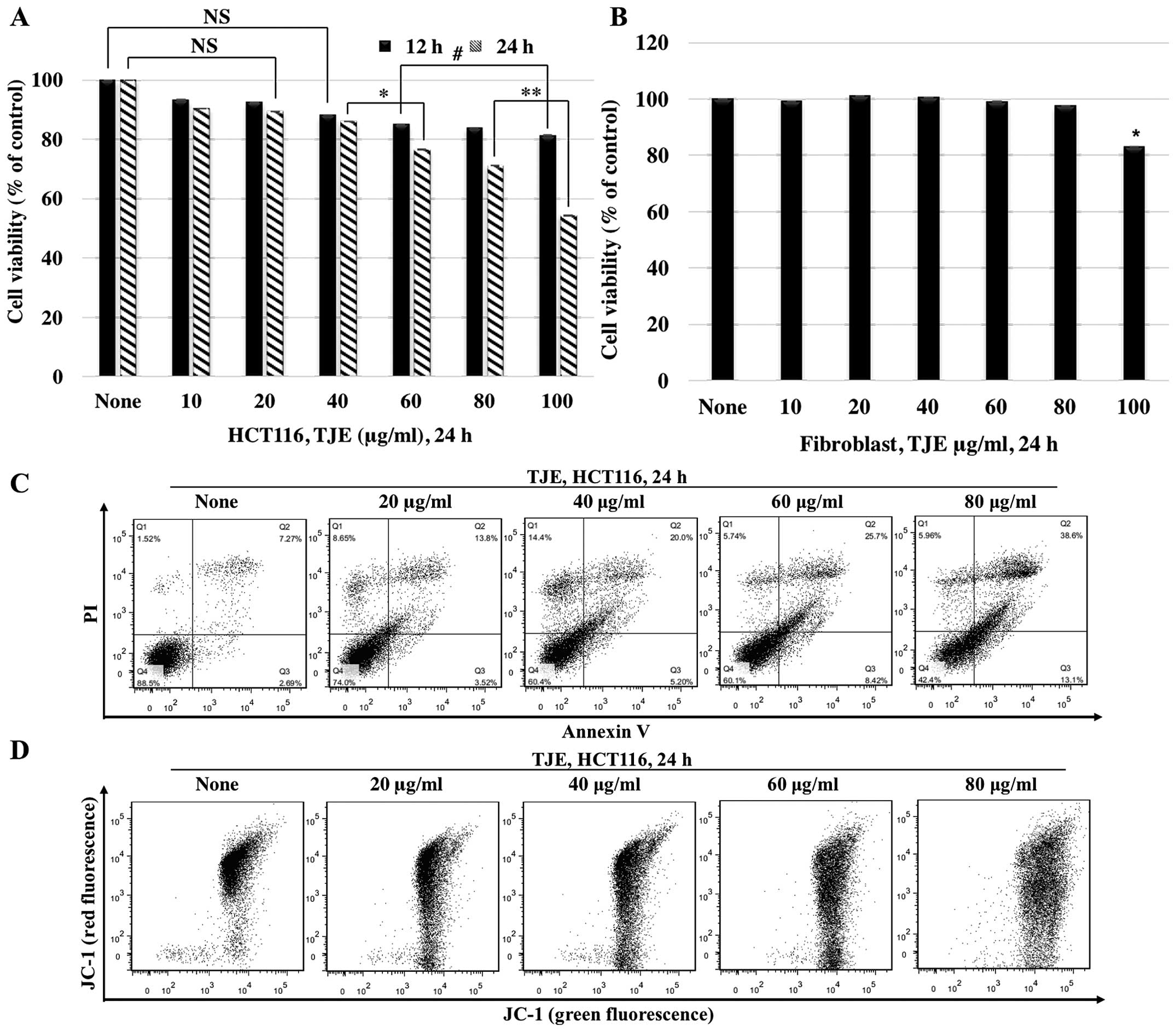
TJE suppresses cancer cell proliferation
and induces apoptosis by reducing the mitochondrial membrane
potential
We investigated the anti-proliferative and apoptotic
effects of TJE. We treated cells with TJE (10–100 μg/ml) for 12 and
24 h, and then assayed for cellular viability and apoptosis. Cells
treated with TJE showed a decrease in viability and an increase in
the number of Annexin V-positive cells in a dose-dependent manner.
In normal human fibroblasts, however, TJE had no effect on cellular
viability (Fig. 1A–C).
To understand the mechanism by which TJE induces
apoptosis, we measured the mitochondrial membrane potential after
treatment with different concentrations of TJE via JC-1 staining
(Fig. 1D). Our results showed that
TJE reduced the membrane potential dose-dependently.
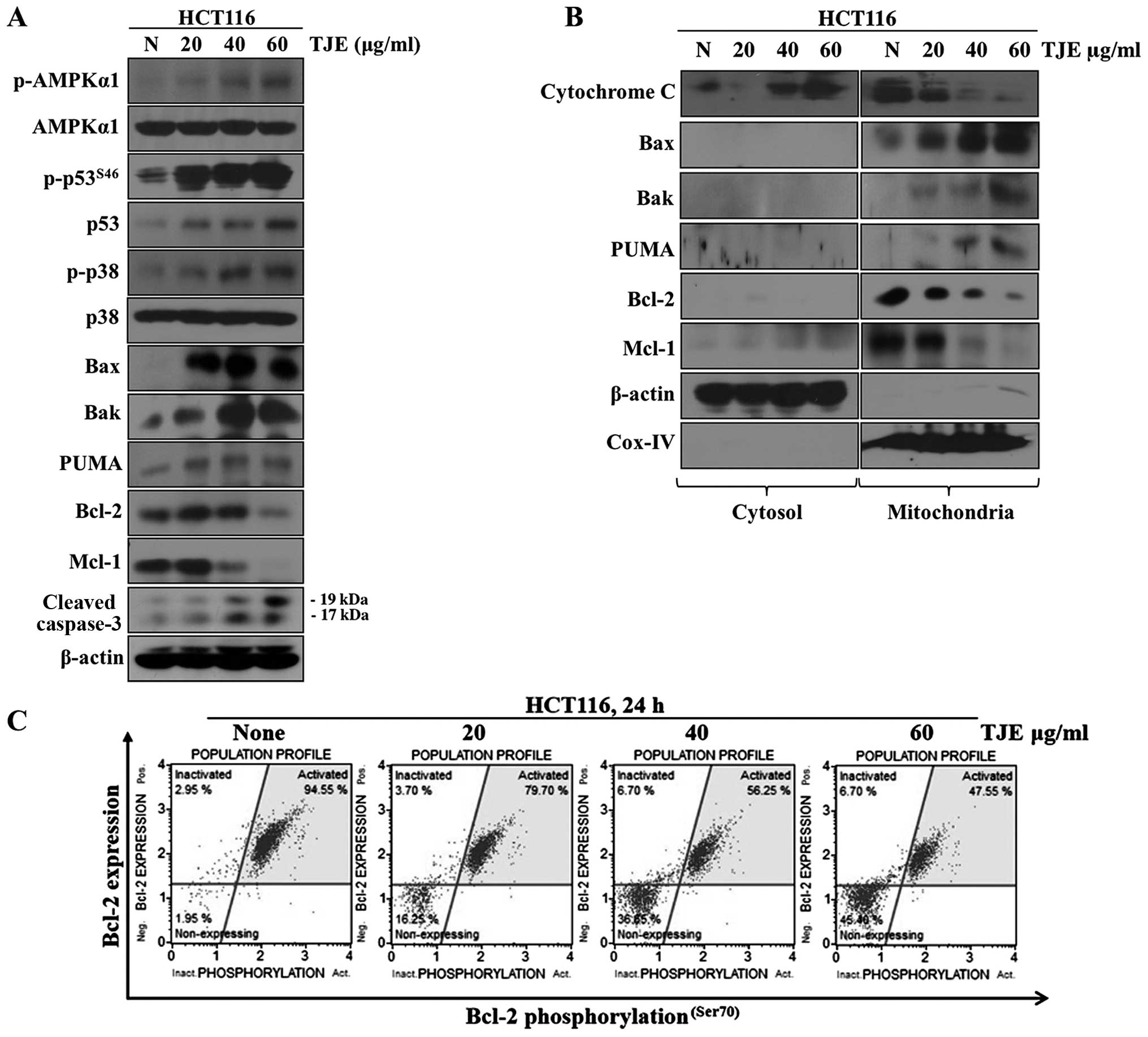
TJE regulates p53 and AMPK expression,
p38 activation and levels of pro-apoptotic proteins
We analyzed the changes in p-AMPKα1, p-p38, p53 and
p-p53 levels and the apoptosis-related proteins Bax, Bak, PUMA,
cleaved caspase-3, Mcl-1 and Bcl-2 after treatment with different
concentrations of TJE by western blotting. Our results showed that
TJE strongly activated AMPK, p38 and p53 dose-dependently (Fig. 2A). Moreover, TJE reduced the
expression of Mcl-1 and Bcl-2 and induced the expression and
mitochondrial translocation of Bax, Bak and PUMA. The latter three
proteins became localized to the outer membrane of the
mitochondria, which led to secretion of cytochrome c from
the mitochondria to the cytosol by a reduction in the mitochondrial
membrane potential (Fig. 2B and
C).
TJE modulates signaling pathways and
mitochondrial membrane potential through generation of
intracellular ROS
We examined whether TJE promotes the generation of
ROS in HCT116 colon cancer cells. We measured intracellular ROS
levels following treatment of cells with TJE (20–80 μg/ml) for 6 h.
As shown in Fig. 3A (left panel),
TJE increased ROS levels at the indicated concentrations. These
effects were completely blocked by co-treatment with NAC, a ROS
scavenger (right panel).
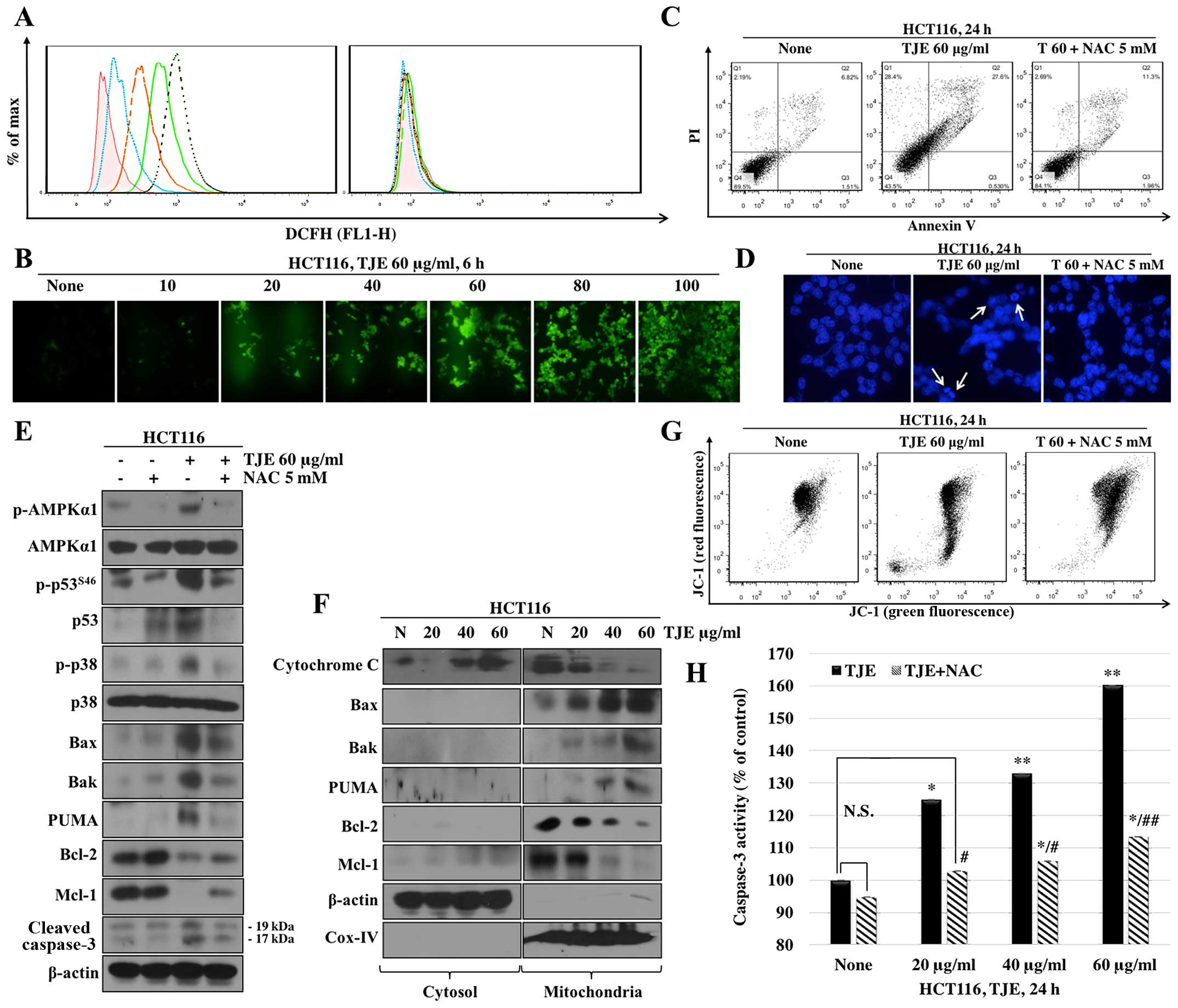 | Figure 3TJE modulates signaling pathways and
mitochondrial membrane potential through generation of
intracellular ROS. (A) Cells were treated with the indicated
concentrations of TJE for 6 h (left panel), pretreated with 5 mM
NAC for 30 min, and then exposed to quercetin (right panel). After
6 h, cells were treated with 40 μM DCFH-DA for 30 min, and
fluorescence intensity was measured by flow a cytometer. Red line,
control; cyan line, TJE 20 μg/ml; orange line, TJE 40 μg/ml; green
line, TJE 60 μg/ml; dark green line, TJE 80 μg/ml. (B) In addition,
cells were treated with the indicated concentrations and times of
TJE and fluorescence was detected using a fluorescence microscope.
Cells were treated with TJE after pretreatment 5 mM NAC for 30 min.
(C) Cells were stained with Annexin V/PI and fluorescence intensity
was measured by a flow cytometer. (D) Cells were treated with 10 μM
Hoechst 33342 for 30 min, and fluorescence was detected using a
fluorescence microscope. Arrow indicate apoptotic bodies. (E) The
expression of apoptosis related-proteins and the activation of
AMPKα1, p38, p53 were analyzed by western blotting. (F) Cell were
stained with JC-1 and fluorescence intensity was measured by a flow
cytometer. (G) Fraction of mitochondria/cytosol proteins were
analyzed by western blotting. (H) Caspase-3 activities were
measured by caspase-3 activity assay. *P<0.05 and
**P<0.01 compared to control; #P<0.05
and ##P<0.01 compared to TJE treated only groups. NS,
not significant (each experiment n=3). |
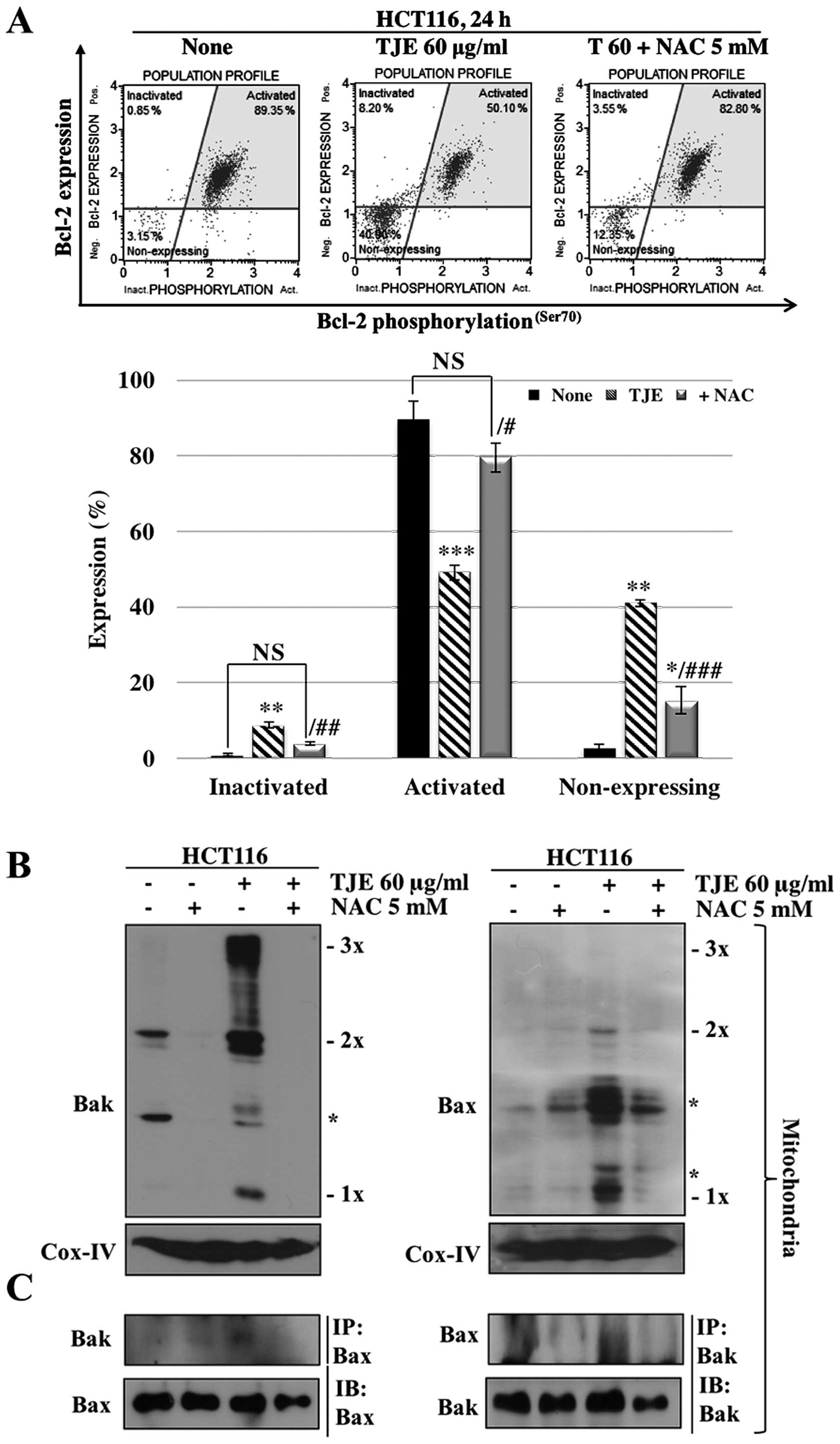
To make sure that the increase in intracellular ROS
levels was related to TJE-regulated signaling proteins and the
induction of apoptosis, we co-treated cells with NAC and then
analyzed protein levels and the concentration of Annexin V-positive
cells. The cells co-treated with NAC and TJE did not undergo
increases in AMPK, p38 or p53 phosphorylation and showed decreased
expression, oligomerization, and binding of apoptosis-related
proteins and increases in the expression of anti-apoptotic proteins
in comparison to cells treated with TJE alone (Figs. 3E and 4). In addition, cells co-treated with TJE
and NAC did not undergo cell death and did not exhibit a reduction
in the mitochondrial membrane potential (Fig. 3F). By contrast, TJE-treated cells
displayed increased levels of apoptotic cell death through changes
in the mitochondrial membrane potential which led to secretion of
cytochrome c from the mitochondria to the cytosol (Fig. 3G). Generally, cytochrome c
in the cytosol initiates caspase cleavage and activates effector
caspases such as caspase-3, which in turn induce apoptosis. We
demonstrated that TJE induced caspase-3 activation
dose-dependently. Co-treatment with NAC abolished this effect
(Fig. 3H).
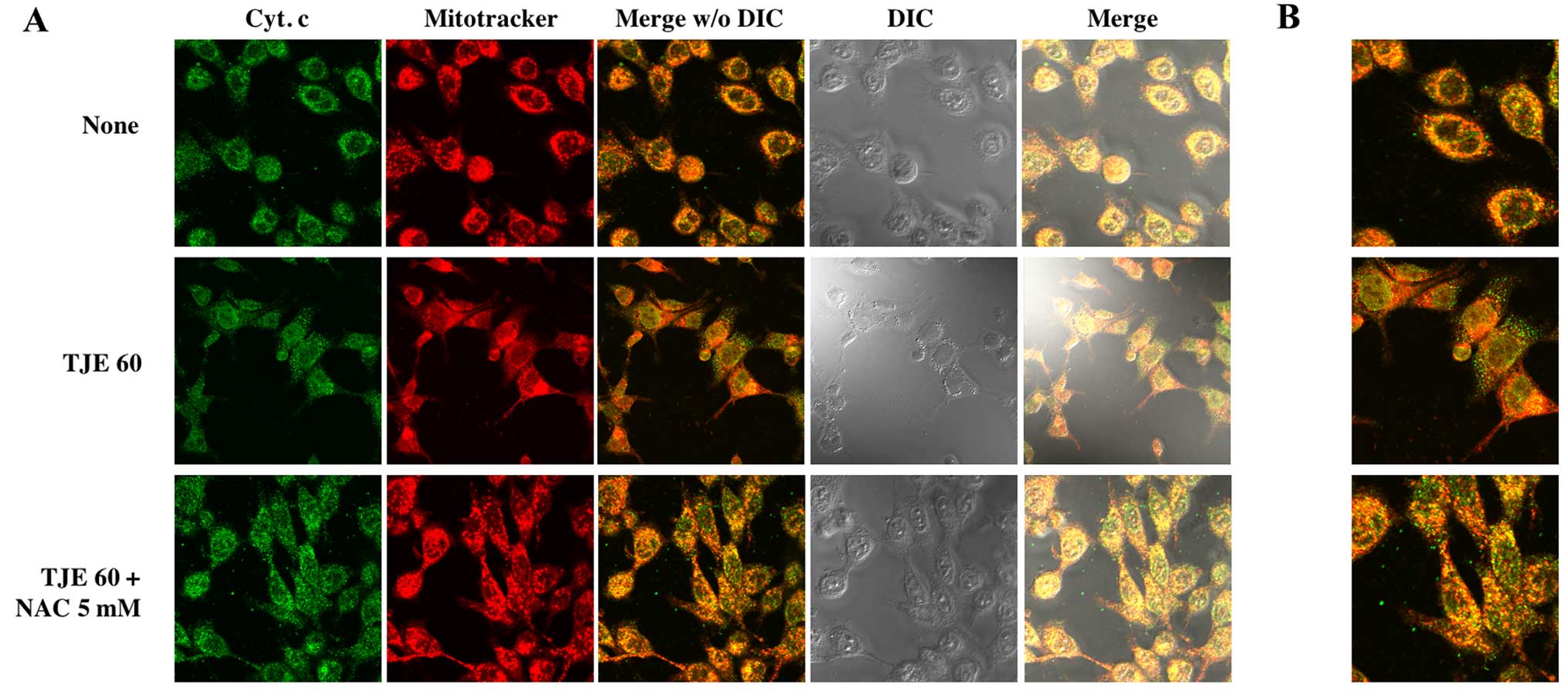
TJE regulates cytochrome c translocation
to the cytoplasm
To show that TJE treatment induced cytochrome
c release from the mitochondria to the cytosol, we
co-treated cells with TJE and NAC for 24 h and stained cells in
order to visualize the mitochondria and cytochrome c. In
TJE-treated cells, cytochrome c translocated from the
mitochondria to the cytosol; in contrast, in control cells and
those co-treated with both TJE and NAC, cytochrome c
remained in the mitochondria (Fig.
5).
TJE induces apoptosis via the AMPK/p38
MAPK signaling pathway in a p53-independent manner
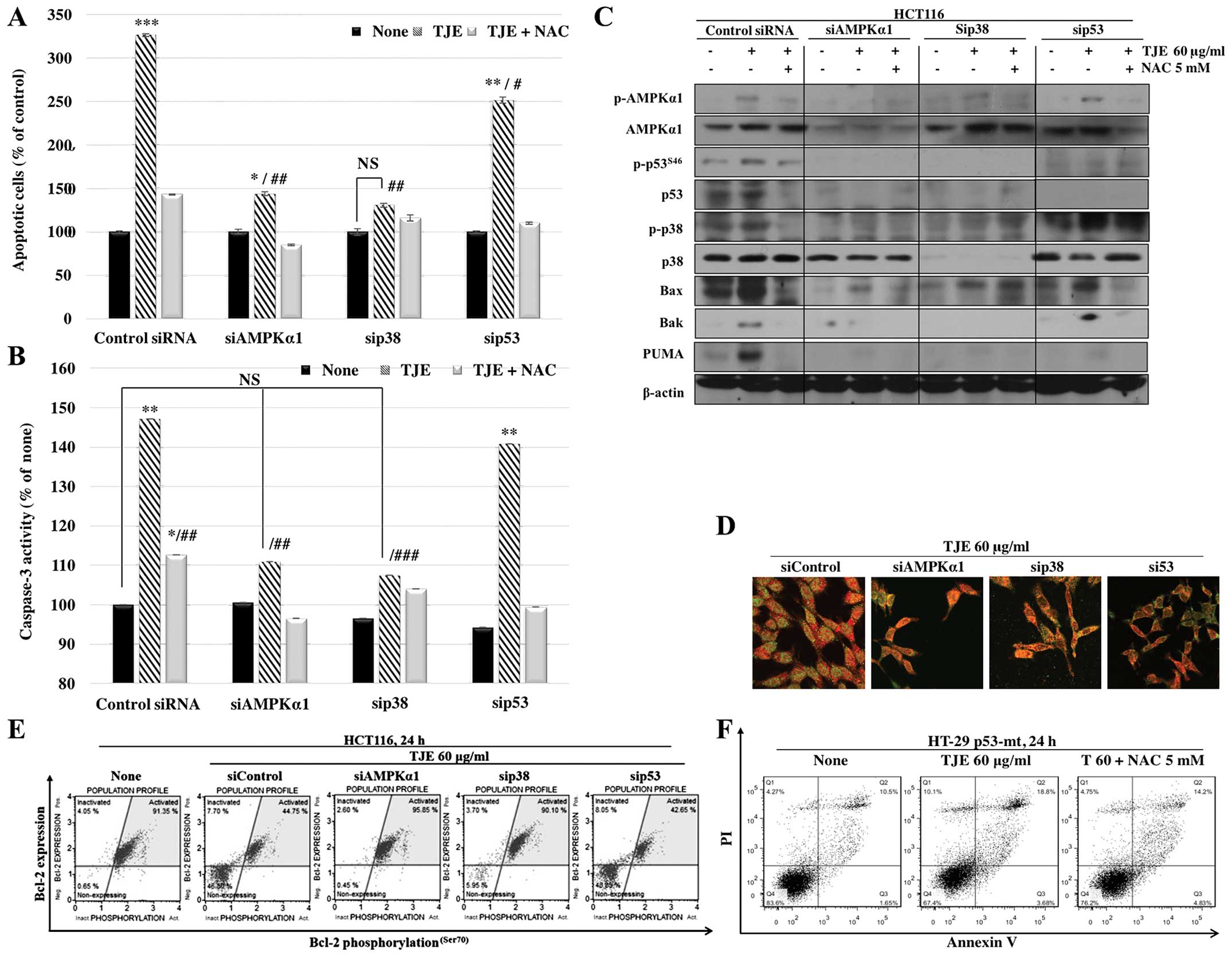
In order to identify the intracellular signaling
pathway that mediates the effects of TJE, we transfected cells with
siRNAs against AMPK, p38 and p53. The cells in which AMPK and p38
were knocked down did not undergo apoptosis following TJE
treatment, but the cells treated with p53 siRNA still showed an
increase in apoptotic cell death (Fig.
6A). In addition, TJE did not affect caspase-3 activity or
cytochrome c translocation in the cells treated with AMPK or
p38 siRNA, but did affect those activities in the p53 siRNA-treated
group (Fig. 6B).
To confirm that apoptotic cell death following TJE
treatment is p53-independent, we analyzed apoptosis-related protein
expression and translocation to the mitochondria in HCT116 cells
transfected with siRNAs against apoptosis-related proteins. Our
results showed that TJE did not induce pro-apoptotic protein
expression or downregulation of anti-apoptotic proteins in AMPK
siRNA- or p38 siRNA-transfected cells, but the effects of TJE were
still seen in p53 siRNA-transfected cells, except those treated
with PUMA; PUMA cannot act independently of p53 (Fig. 6C and D). Moreover, TJE induced
apoptotic cell death in HT-29 colon cancer cells, which have a
mutation in the p53 gene (Fig.
6F).
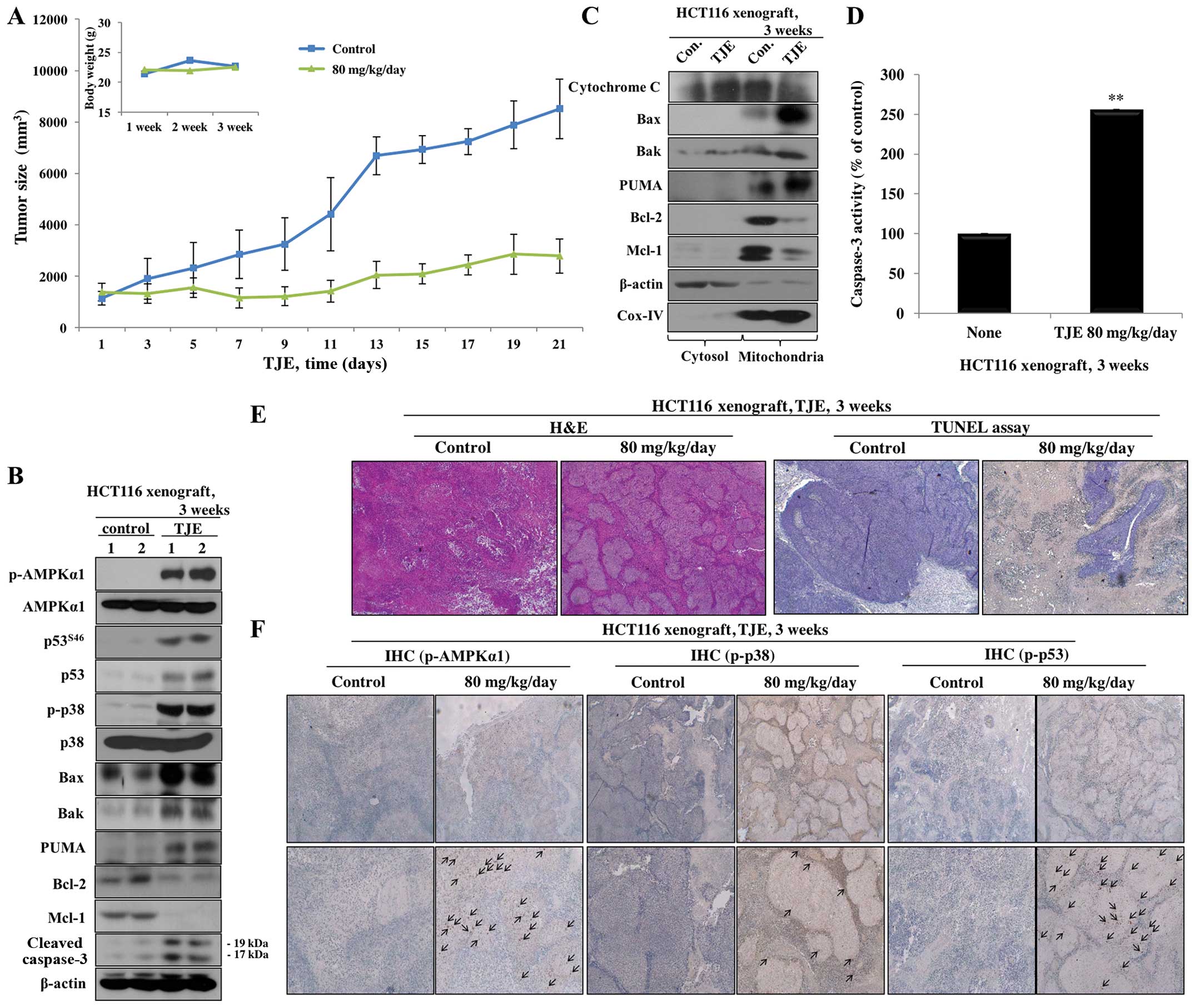
TJE induces cell death through regulation
of intracellular signaling pathways in an HCT116 xenograft
model
To analyze the consequences of TJE treatment in an
HCT116 xenograft model of tumor growth, we performed histological
analysis from control- and TJE (80 mg/kg/day)-treated tumor tissue
stained with H&E using the TUNEL assay. The amount of tumor
tissue in TJE-treated samples was considerably less than that of
control samples (Fig. 7A). The
number of TUNEL-positive cells was significantly increased and
cancer tissue was degraded in TJE-treated samples (Fig. 7E). In addition, in a similar in
vitro experiment, the AMPK/p38 MAPK and p53 signaling pathways
were activated, and apoptosis-related proteins showed an increase
in expression and translocation to the cytosol (Fig. 7B–D and F).
Discussion
The incidence of colorectal cancer has increased
because of changes in dietary patterns and lifestyles (1–5). For
this reason, there has been an increased interest in the effect of
compounds extracted from natural sources on the prevention and
treatment of colorectal cancer. Such compounds induce apoptosis and
arrest metastasis through the regulation of intracellular signaling
in cancer cells (27–33). Many researchers have identified
food extracts that have been used in cancer chemotherapy
experiments.
The fruit of Torilis japonica is used as a
substitute for She chuang zi, which is a traditional Chinese
medicine. Previously, we found that ethanol extracts from the fruit
of Torilis japonica arrested abnormal metastasis through
regulation of the EGFR signaling pathway in MCF-7 breast cancer
cells (27). In this study, we
focused on the effects of TJE on the induction of apoptosis through
alterations in the mitochondrial membrane potential in colorectal
cancer cells.
In this study, we confirmed that TJE negatively
regulated cancer cell viability. Then, we showed that TJE reduced
cell viability dose-dependently. Also, it induced apoptotic cell
death by altering the mitochondrial membrane potential and
regulating apoptosis related-proteins including Bax, Bak and PUMA.
Induction of intracellular ROS in cancer cells by treatment with
natural compounds is an appealing option in the development of
anticancer agents. Increasing ROS by treatment with natural
products not only controls the cell cycle but also induces
apoptotic cell death by regulating intracellular signaling
molecules such as the Bcl-2 family and caspases (12,13,28,32–35).
Thus, we hypothesized that TJE treatment effects cancer cells
through an increase in intracellular ROS. Indeed, our results
showed that treatment of cancer cells with TJE induces
intracellular ROS and affects apoptosis via a reduction in the
mitochondrial outer membrane potential through the regulation of
protein signaling.
Previous study demonstrated that AMPK activation by
ROS induces apoptosis via the ASK1/p38 MAPK pathway in MCF-7 breast
cancer cells (35). In the present
study, we found that activation of p38 MAPK by phosphorylation of
AMPK regulates the mitochondrial membrane potential and induces
apoptosis in a p53-independent manner (28). p53 is known to be involved in
cancer cell death and arrest of abnormal cellular proliferation
(36–39). However, ~50% of cancer cells have
p53 mutations (40). Thus, a
p53-independent method of inducing apoptosis would be extremely
valuable in cancer treatment. Our results showed that TJE regulates
the AMPK/p38 MAPK signaling pathway and induces apoptosis via
regulation of the mitochondrial outer membrane potential and
translocation of pro-apoptotic proteins to the mitochondria. When
we silenced AMPK and p38 using specific siRNAs, TJE did not affect
apoptotic cell death or pro-apoptotic protein expression.
Interestingly, in cells transfected with p53 siRNA, apoptosis was
induced and apoptosis-related proteins were affected, as in cells
treated with only TJE. Moreover, in cells in which AMPK and p38
MAPK were silenced, p53 was not activated. Previous studies found
that p38 MAPK activation may be regulated by p53 expression and
activation via downstream signaling pathways (41,42).
Also, the AMPK/p38 MAPK pathway controls apoptosis-related protein
expression and induces apoptotic cell death without p53 activation
(28). From our results and those
of previous studies, we conclude that TJE induces apoptosis and
downregulates the mitochondrial outer membrane potential via the
AMPK/p38 MAPK signaling pathway in a p53-indepent manner.
As in our in vitro studies, TJE induced
apoptosis in a mouse xenograft model. The rate of tumor growth was
reduced in the TJE-injected group as compared with the control
group. Moreover, the TJE-treated group exhibited an increase in
pro-apoptosis protein expression and translocation to the
mitochondria. Thus, our in vivo studies confirm that TJE
regulates the AMPK/p38 MAPK signaling pathway.
In conclusion, we demonstrated that TJE, a natural
compound, has potential as an anticancer agent and may provide a
substitute for chemotherapeutic drugs.
Acknowledgements
This study was supported by the 2016 Hannam
University Research Fund.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jemal A, Center MM, DeSantis C and Ward
EM: Global patterns of cancer incidence and mortality rates and
trends. Cancer Epidemiol Biomarkers Prev. 19:1893–1907. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Center MM, Jemal A and Ward E:
International trends in colorectal cancer incidence rates. Cancer
Epidemiol Biomarkers Prev. 18:1688–1694. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Parkin DM, Whelan SL, Ferlay J and Storm
H: Cancer Incidence in Five Continents. I to VIII. Cancer Base 7.
IARC Press; Lyon: 2005
|
|
5
|
Ito Y, Ioka A, Tanaka M, Nakayama T and
Tsukuma H: Trends in cancer incidence and mortality in Osaka,
Japan: Evaluation of cancer control activities. Cancer Sci.
100:2390–2395. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Giovannucci E: Modifiable risk factors for
colon cancer. Gastroenterol Clin North Am. 31:925–943. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kushi LH, Byers T, Doyle C, Bandera EV,
McCullough M, McTiernan A, Gansler T, Andrews KS and Thun MJ;
American Cancer Society 2006 Nutrition and Physical Activity
Guidelines Advisory Committee: American Cancer Society Guidelines
on Nutrition and Physical Activity for cancer prevention. Reducing
the risk of cancer with healthy food choices and physical activity.
CA Cancer J Clin. 56:254–281; quiz 313–314. 2006. View Article : Google Scholar
|
|
8
|
Colditz GA, Sellers TA and Trapido E:
Epidemiology - identifying the causes and preventability of cancer?
Nat Rev Cancer. 6:75–83. 2006. View
Article : Google Scholar
|
|
9
|
Pan SY, Zhou SF, Gao SH, Yu ZL, Zhang SF,
Tang MK, Sun JN, Ma DL, Han YF, Fong WF, et al: New perspectives on
how to discover drugs from Herbal medicines: CAM’s outstanding
contribution to modern therapeutics. Evid Based Complement Alternat
Med. 2013:6273752013. View Article : Google Scholar
|
|
10
|
Prindull G: Apoptosis in the embryo and
tumorigenesis. Eur J Cancer. 31A:116–123. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Meier P, Finch A and Evan G: Apoptosis in
development. Nature. 407:796–801. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee YK, Park SY, Kim YM, Kim DC, Lee WS,
Surh YJ and Park OJ: Suppression of mTOR via Akt-dependent and
-independent mechanisms in selenium-treated colon cancer cells:
Involvement of AMPKalpha1. Carcinogenesis. 31:1092–1099. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chien SY, Wu YC, Chung JG, Yang JS, Lu HF,
Tsou MF, Wood WG, Kuo SJ and Chen DR: Quercetin-induced apoptosis
acts through mitochondrial- and caspase-3-dependent pathways in
human breast cancer MDA-MB-231 cells. Hum Exp Toxicol. 28:493–503.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Adams JM and Cory S: Life-or-death
decisions by the Bcl-2 protein family. Trends Biochem Sci.
26:61–66. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Harris MH and Thompson CB: The role of the
Bcl-2 family in the regulation of outer mitochondrial membrane
permeability. Cell Death Differ. 7:1182–1191. 2000. View Article : Google Scholar
|
|
16
|
Martinou JC and Green DR: Breaking the
mitochondrial barrier. Nat Rev Mol Cell Biol. 2:63–67. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jia L, Macey MG, Yin Y, Newland AC and
Kelsey SM: Subcellular distribution and redistribution of Bcl-2
family proteins in human leukemia cells undergoing apoptosis.
Blood. 93:2353–2359. 1999.PubMed/NCBI
|
|
18
|
Griffin DE and Hardwick JM: Regulators of
apoptosis on the road to persistent alphavirus infection. Annu Rev
Microbiol. 51:565–592. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hsu YT, Wolter KG and Youle RJ:
Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during
apoptosis. Proc Natl Acad Sci USA. 94:3668–3672. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Luo X, Budihardjo I, Zou H, Slaughter C
and Wang X: Bid, a Bcl2 interacting protein, mediates cytochrome c
release from mitochondria in response to activation of cell surface
death receptors. Cell. 94:481–490. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li H, Zhu H, Xu CJ and Yuan J: Cleavage of
BID by caspase 8 mediates the mitochondrial damage in the Fas
pathway of apoptosis. Cell. 94:491–501. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mikhailov V, Mikhailova M, Pulkrabek DJ,
Dong Z, Venkatachalam MA and Saikumar P: Bcl-2 prevents Bax
oligomerization in the mitochondrial outer membrane. J Biol Chem.
276:18361–18374. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Antonsson B, Montessuit S, Lauper S, Eskes
R and Martinou JC: Bax oligomerization is required for
channel-forming activity in liposomes and to trigger cytochrome c
release from mitochondria. Biochem J. 345:271–278. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Antonsson B, Montessuit S, Sanchez B and
Martinou JC: Bax is present as a high molecular weight
oligomer/complex in the mitochondrial membrane of apoptotic cells.
J Biol Chem. 276:11615–11623. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sundararajan R and White E: E1B 19K blocks
Bax oligomerization and tumor necrosis factor alpha-mediated
apoptosis. J Virol. 75:7506–7516. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Degterev A, Boyce M and Yuan J: A decade
of caspases. Oncogene. 22:8543–8567. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kim GT, Lee SH and Kim YM: Torilis
japonica extract, a new potential EMT suppressor agent by
regulation of EGFR signaling pathways. Int J Oncol. 45:1673–1679.
2014.PubMed/NCBI
|
|
28
|
Kim GT, Lee SH, Kim JI and Kim YM:
Quercetin regulates the sestrin 2-AMPK-p38 MAPK signaling pathway
and induces apoptosis by increasing the generation of intracellular
ROS in a p53-independent manner. Int J Mol Med. 33:863–869.
2014.PubMed/NCBI
|
|
29
|
Lee YK, Park SY, Kim YM, Lee WS and Park
OJ: AMP kinase/cyclooxygenase-2 pathway regulates proliferation and
apoptosis of cancer cells treated with quercetin. Exp Mol Med.
41:201–207. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gibellini L, Pinti M, Nasi M, Montagna JP,
De Biasi S, Roat E, Bertoncelli L, Cooper EL and Cossarizza A:
Quercetin and cancer chemoprevention. Evid Based Complement
Alternat Med. 2011:5913562011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Weidner C, Rousseau M, Micikas RJ, Fischer
C, Plauth A, Wowro SJ, Siems K, Hetterling G, Kliem M, Schroeder
FC, et al: Amorfrutin C induced apoptosis and inhibits
proliferation in colon cancer cells through targeting mitochondria.
J Nat Prod. 79:2–12. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang X, Chen M, Zou P, Kanchana K, Weng
Q, Chen W, Zhong P, Ji J, Zhou H, He L, et al: Curcumin analog WZ35
induced cell death via ROS-dependent ER stress and G2/M cell cycle
arrest in human prostate cancer cells. BMC Cancer. 15:8662015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
He G, He G, Zhou R, Pi Z, Zhu T, Jiang L
and Xie Y: Enhancement of cisplatin-induced colon cancer cells
apoptosis by shikonin, a natural inducer of ROS in vitro and in
vivo. Biochem Biophys Res Commun. 469:1075–1082. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tanigawa S, Fujii M and Hou DX:
Stabilization of p53 is involved in quercetin-induced cell cycle
arrest and apoptosis in HepG2 cells. Biosci Biotechnol Biochem.
72:797–804. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lee YK, Hwang JT, Kwon DY, Surh YJ and
Park OJ: Induction of apoptosis by quercetin is mediated through
AMPKalpha1/ASK1/p38 pathway. Cancer Lett. 292:228–236. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bouchet BP, Caron de Fromentel C, Puisieux
A and Galmarini CM: p53 as a target for anticancer drug
development. Crit Rev Oncol Hematol. 58:190–207. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Moll UM, Marchenko N and Zhang XK: p53 and
Nur77/TR3 - transcription factors that directly target mitochondria
for cell death induction. Oncogene. 25:4725–4743. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gorgoulis VG, Vassiliou LV, Karakaidos P,
Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA Jr,
Kastrinakis NG, Levy B, et al: Activation of the DNA damage
checkpoint and genomic instability in human precancerous lesions.
Nature. 434:907–913. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Oren M: Decision making by p53: Life,
death and cancer. Cell Death Differ. 10:431–442. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Olivier M, Hollstein M and Hainaut P: TP53
mutations in human cancers: Origins, consequences, and clinical
use. Cold Spring Harb Perspect Biol. 2:a0010082010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cuadrado A, Lafarga V, Cheung PC, Dolado
I, Llanos S, Cohen P and Nebreda AR: A new p38 MAP kinase-regulated
transcriptional coactivator that stimulates p53-dependent
apoptosis. EMBO J. 26:2115–2126. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sanchez-Prieto R, Rojas JM, Taya Y and
Gutkind JS: A role for the p38 mitogen-acitvated protein kinase
pathway in the transcriptional activation of p53 on genotoxic
stress by chemotherapeutic agents. Cancer Res. 60:2464–2472.
2000.PubMed/NCBI
|