Introduction
Breast cancer is one of the most common cancers
threatening women and its global incidence is still increasing.
Importantly, breast cancer is the second leading cause of
cancer-related deaths in women, which accounts for approximately
15% of all female cancer-related deaths in the United States
(1). Effective prevention and
treatment of this morbid disease remain a significant challenge.
Use of natural compounds for breast cancer prevention and treatment
is one of the major efforts in breast cancer research (2).
Genistein is a major functional component of soy
isoflavones. High intake of soy products has been associated with
low incidence of breast cancer (3). Soy-associated anticancer activities
have been attributed to soy isoflavones (4). Available evidence suggests that
genistein is a bioactive molecule with multiple functions,
including properties of phytoestrogen, tyrosine kinase and
topoisomerase inhibition (5–7). In
addition to directly binding to estrogen receptor (ER) (8), receptor tyrosine kinase (RTK)
(6) and topoisomerase (7), genistein also modulates a number of
key intracellular molecules, such as NF-κB and MAPK, to induce
growth arrest and apoptosis. Therefore, genistein functions as a
multi-targeting antitumor agent (9). Despite these advances, the mechanisms
of genistein-mediated tumor inhibition remain unclear. Hence,
identification of novel intracellular targets that mediate
genistein-induced tumor inhibition is of pivotal significance.
Cancerous inhibitor of protein phosphatase 2A
(CIP2A), encoded by the KIAA1524 gene, is a recently identified
oncoprotein (10). Overexpression
of CIP2A has been detected in tumors from various origins,
including breast, lung and colon (11,12),
which has been associated with poor prognosis and disease
progression (11,13,14).
CIP2A expression in breast cancer tissues was also significantly
related to higher tumor grades, lymph node and distant metastasis
and resistance to chemotherapeutics (15). In vitro expression of CIP2A
promotes the immortalization and malignant transformation of human
cells (12). CIP2A functions as a
protein phosphatase 2A (PP2A) inhibitor, a known tumor suppressor
(12,16). CIP2A attenuates c-Myc degradation
by inhibiting PP2A-mediated dephosphorylation of MYC at serine 62
(13). It was reported that CIP2A
and c-Myc formed a positive feed-back loop that promoted the
expression of both oncoproteins (13,17).
Collectively, CIP2A forms an ‘oncogenic nexus’ to regulate
oncogenic transformation of affected cells by virtue of its control
of PP2A and MYC stabilization in cancer cells (18,19).
Moreover, a number of recent reports indicate that CIP2A not only
plays a critical role in tumor development but also functions as a
therapeutic target of certain small molecule inhibitors and natural
compounds such as erlotinib, celastrol and bortezomib (20–22).
Downregulation of CIP2A has been associated with the antitumor
activity of these agents.
In the present report, we investigated the role of
CIP2A in genistein-treated breast cancer cells. We report here for
the first time that genistein induces downregulation of CIP2A,
which contributes to genistein-mediated growth inhibition and
apoptosis. The underlying mechanisms involve the modulation of
E2F1-mediated transcriptional regulation of CIP2A. These results
support CIP2A as a molecular target in genistein-mediated tumor
inhibition.
Materials and methods
Reagents and antibodies
Genistein, MG132, RNase A and propidium iodide (PI)
were purchased from Sigma-Aldrich. (St. Louis, MO, USA). Antibodies
against CIP2A, E2F1 and β-actin were purchased from Santa Cruz
Biotechnology (Santa Cruz, CA, USA). Antibodies against PARP,
caspase-3, cleaved caspase-3 and c-Myc were from Cell Signaling
Technology (Danvers, MA, USA).
Cell culture, transfection and
treatment
MCF-7, T47D and HEK 293T cell lines were obtained
from the American Type Culture Collection (ATCC; Rockville, MD,
USA). MCF-7-caspase-3 (MCF-7-C3) cells were a stable subline of
MCF-7 cells which were reconstituted with caspase-3. As described
in our previous report (23), this
subline was generated by transfecting MCF-7 cells with caspase-3
encoding plasmid followed by antibiotic selections.
Puromycin-resistant clones were pooled as a subline for later
studies (24). The cells were
cultured in DMEM/F12 medium supplemented with 10% fetal bovine
serum (FBS; Atlanta Biologicals, Flowery Branch, GA, USA), 100 U/ml
penicillin and 100 mg/ml streptomycin at 37°C with a 5%
CO2 atmosphere. The cells were treated with genistein or
relevant agents as specified in each experiment.
For cell transfection, cells were seeded into a
6-well plate at 2×105 cells/well 24 h prior to the
transfection. pCMV/E2F1 and the control plasmids (25) were transfected into the cells using
X-tremeGENE 9 DNA transfection reagent (Roche Disgnostics,
Indianapolis, IN, USA) according to the manufacturer’s protocol.
Twenty-four hours later, the cells were treated with genistein for
48 h before lysate collection.
Lentivirus production
Lentiviruses encoding CIP2A protein and CIP2A shRNA
were prepared from HEK 293T cells by co-transfection of pMD2.G,
psPAX2 (Addgene, Cambridge, MA, USA) and specific encoding plasmids
with X-tremeGENE 9. pLKO1-CIP2AshRNA and pLKO1/KIAA1524 were
purchased from GeneCopoeia (Rockville, MD, USA). Supernatants from
the transfected HEK 293T cells were collected at 24 and 48 h
post-transfection. Supernatants were pooled and filtered through a
0.45 μm filter. For lentiviral infection, cells were plated in
monolayer at different densities and incubated with lentivirus in
the presence of 8 μg/ml polybrene, followed by selection with 1
μg/ml puromycin. Resistant clones were pooled and expanded for
later experiments.
Cell proliferation assay
Cell proliferation was assessed with
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT) assays.
Cells were incubated in 96-well plates at 1×103
cells/well 24 h prior to treatment. The cells were then treated
with genistein at indicated concentrations for 5 days, followed by
incubation with MTT (2.5 mg/ml) for 4 h. The medium was then
removed and replaced with 50 μl/well DMSO, followed by incubation
on a shaker for 45 min. Colorimetric absorbance was read with an
ELISA reader at 540 nm. Data based on samples of 6 replicates were
statistically analyzed.
Western blot analysis
Control or treated cells were collected for lysate
preparation as previously described (26). Protein lysates were separated on
10% SDS-PAGE gels and transferred to a nitrocellulose membrane. The
membrane was blocked with 5% milk in TBS-T buffer for 2 h, followed
by incubation with primary antibodies in 5% BSA-TBS-T at 4°C
overnight. The membrane was washed with TBS-T and then incubated
with corresponding HRP-labeled secondary antibody for 1 h. Protein
signals were detected using an ECL detection kit (Thermo Fisher
Scientific, Rockford, IL, USA). The images were captured with a
FluorChem E system (Cell Biosciences, Santa Clara, CA, USA).
Flow cytometric analysis of cell
cycle
Cells were incubated in 60-mm plates
(5×105 cells/plate) overnight and then treated with
genistein at indicated concentrations for 24 h. The cells were
harvested to prepare for single cell suspension, followed by
drop-wise fixation with 70% ethanol. Fixed cells were washed with
PBS/0.1% Triton X-100 twice. The cells were then incubated with
RNase A (1 mg/ml) and propidium iodide (PI, 33 μg/ml) at 37°C for
45 min, followed analysis with a Guava easyCyte 8 flow cytometer.
The percentage of cells in cell cycle phases was analyzed with
ModFit program from Verity Software House (Topsham, ME, USA).
Quantitative real-time PCR
Total RNA was extracted from treated cells with an
RNeasy Protect Mini kit (Qiagen, Valencia, CA, USA) according to
the manufacturer’s instructions. Quality and quantity of total RNA
were determined by a NanoDrop 1000 Spectrophotometer (Thermo Fisher
Scientific, Waltham, MA USA). First-Strand cDNA Synthesis was
performed using an iScript cDNA Synthesis kit (Bio-Rad
Laboratories, Hercules, CA, USA). Primers targeting CIP2A and GAPDH
(glyceraldehydes-3-3phosphate dehydrogenase) were synthesized by
Integrated DNA Technologies (Coralville, IA, USA). The sequences
for specific primers are: GAPDH-F, 5′-TGC ACC ACC ACC TGC TTA GC-3′
and GAPDH-R, 5′-GGC ATG GAC TGT GGT CAT GAG-3′; CIP2A-F, 5′-GAA CAG
ATA AGA AAA GAG TTG AGC ATT-3′ and CIP2A-R, 5′-CGA CCT TCT AAT TGT
GCC TTT T-3′ Quantitative real-time PCR reactions were carried out
on a CFX96 Real-Time PCR Detection system (Bio-Rad Laboratories).
The amplification started with 95°C for 10 min, followed by 95°C
for 15 sec, 95°C for 60 sec for 40 cycles. Relative mRNA levels of
CIP2A were normalized to GAPDH levels.
Apoptosis assay
The Cell Death Detection (ELISA) kit (Roche Life
Science, Indianapolis, IN, USA) was used for assessing apoptosis
according to the manufacturer’s protocol. The cell lysates were
incubated in microtiter plate modules coated with anti-histone
antibody, followed by washing. Once color developed, samples were
measured with a Synergy Mx microplate reader (BioTek Instruments,
Inc., Winooski, VT, USA) at 405 nm. All analyses were performed in
triplicate.
Statistical analysis
Calculations were performed using software from
GraphPad Prism (GraphPad) and data were expressed as means ± SEM. A
two-tailed Student’s t-test was used to compare groups. Differences
between groups were considered statistically significant at
P<0.05.
Results
Genistein-mediated downregulation of
CIP2A is associated with its growth inhibition and apoptosis
induction
To determine whether genistein induces CIP2A
regulation, we examined CIP2A protein levels in genistein-treated
MCF-7, MCF-7-caspase-3 (MCF-7-C3) and T47D breast cancer cells.
Because MCF-7 cells are deficient of caspase-3 and do not display
typical PARP cleavage and DNA fragmentation (27), we used MCF-7-C3 cells, a stable
cell line generated from pooled clones of MCF-7 cells transfected
with caspase-3, which have been well characterized in our previous
studies (23,24,28,29).
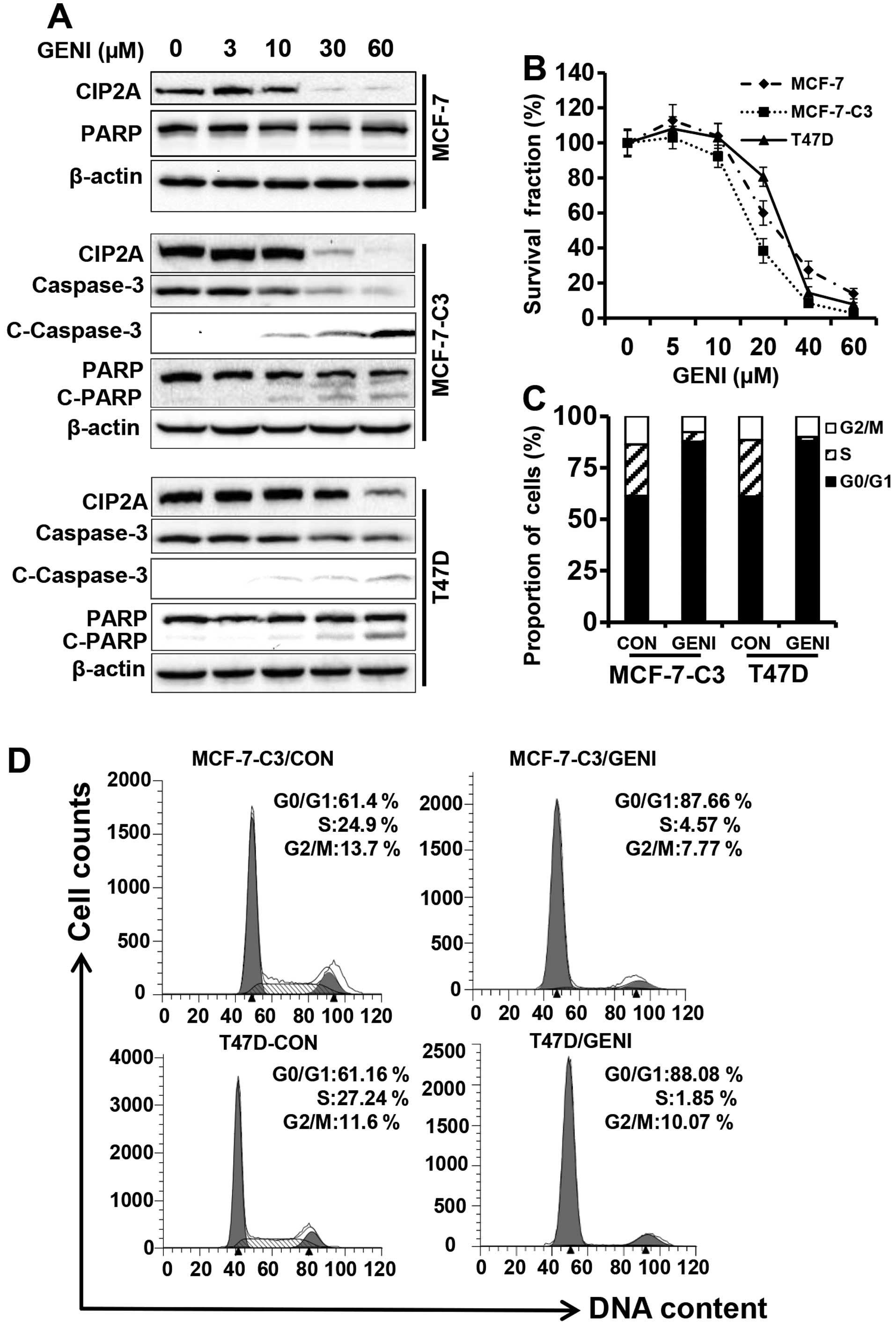
As shown in Fig. 1A, when the
cells were treated with genistein at concentrations ranging from 0
to 60 μM for 48 h, CIP2A protein levels in each cell line were
downregulated in a concentration-dependent manner, especially in
the 30 and 60 μM groups. Importantly, genistein-mediated CIP2A
downregulation was associated with growth inhibition and PARP
cleavage in MCF-7-C3 and T47D cells that express functional
caspase-3. Under similar treatment conditions, genistein also
induced inhibition of cell proliferation by MTT assay (Fig. 1B) and cell cycle arrest in G0/G1
phase by flow cytometry (Fig. 1C).
Examination of apoptotic markers indicated that genistein-induced
downregulation of CIP2A was associated with decreased total
caspase-3, and increased cleavage of caspase-3 and PARP (Fig. 1A). Taken together, these results
demonstrated that CIP2A is a cellular target of genistein. The
association between CIP2A downregulation and increased growth
inhibition and apoptosis suggests that CIP2A downregulation may
have a functional impact on genistein-induced tumor inhibition.
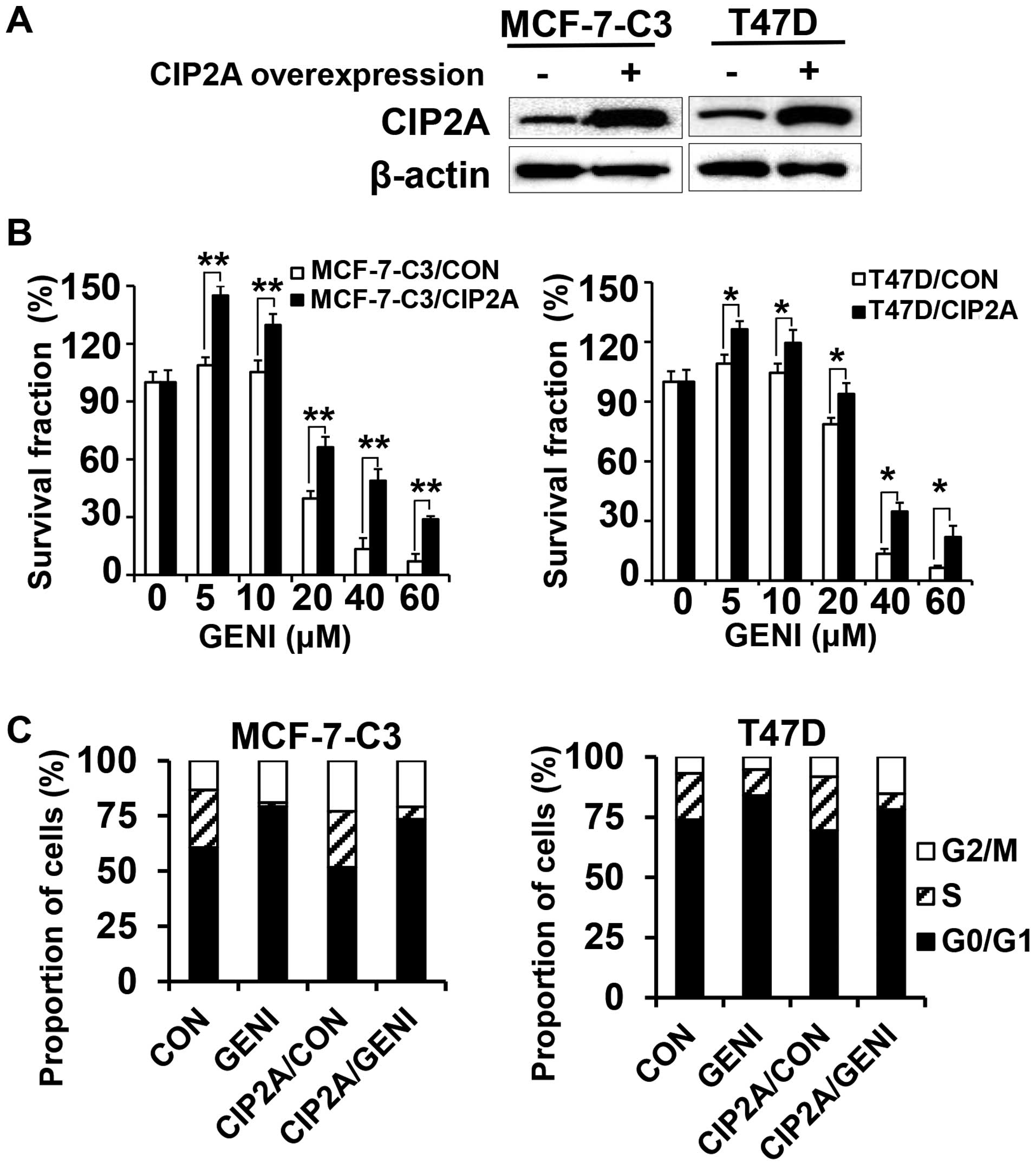
Overexpression of CIP2A attenuates
genistein-induced growth inhibition
To determine whether genistein-induced CIP2A
downregulation contributes to its growth inhibition property, we
overexpressed CIP2A in MCF-7-C3 and T47D cells and characterized
their responses to genistein. By infecting the cells with control
and CIP2A encoding lentiviruses followed by puromycin selection, we
obtained control (MCF-7-C3/CON and T47D/CON) and CIP2A
overexpressing (MCF-7-C3/CIP2A and T47D/CIP2A) stable lines
(Fig. 2A). As shown in Fig. 2B, genistein-induced growth
inhibition in MCF-7-C3/CIP2A and T47D/CIP2A cells were
significantly attenuated as compared to corresponding control
cells, indicating that CIP2A is involved in genistein-associated
anti-proliferation effect. Cell cycle analysis with flow cytometry
indicated that genistein-induced G0/G1 arrest in both
MCF-7-C3/CIP2A and T47D/CIP2A cells was decreased as compared to
the controls (Fig. 2C).
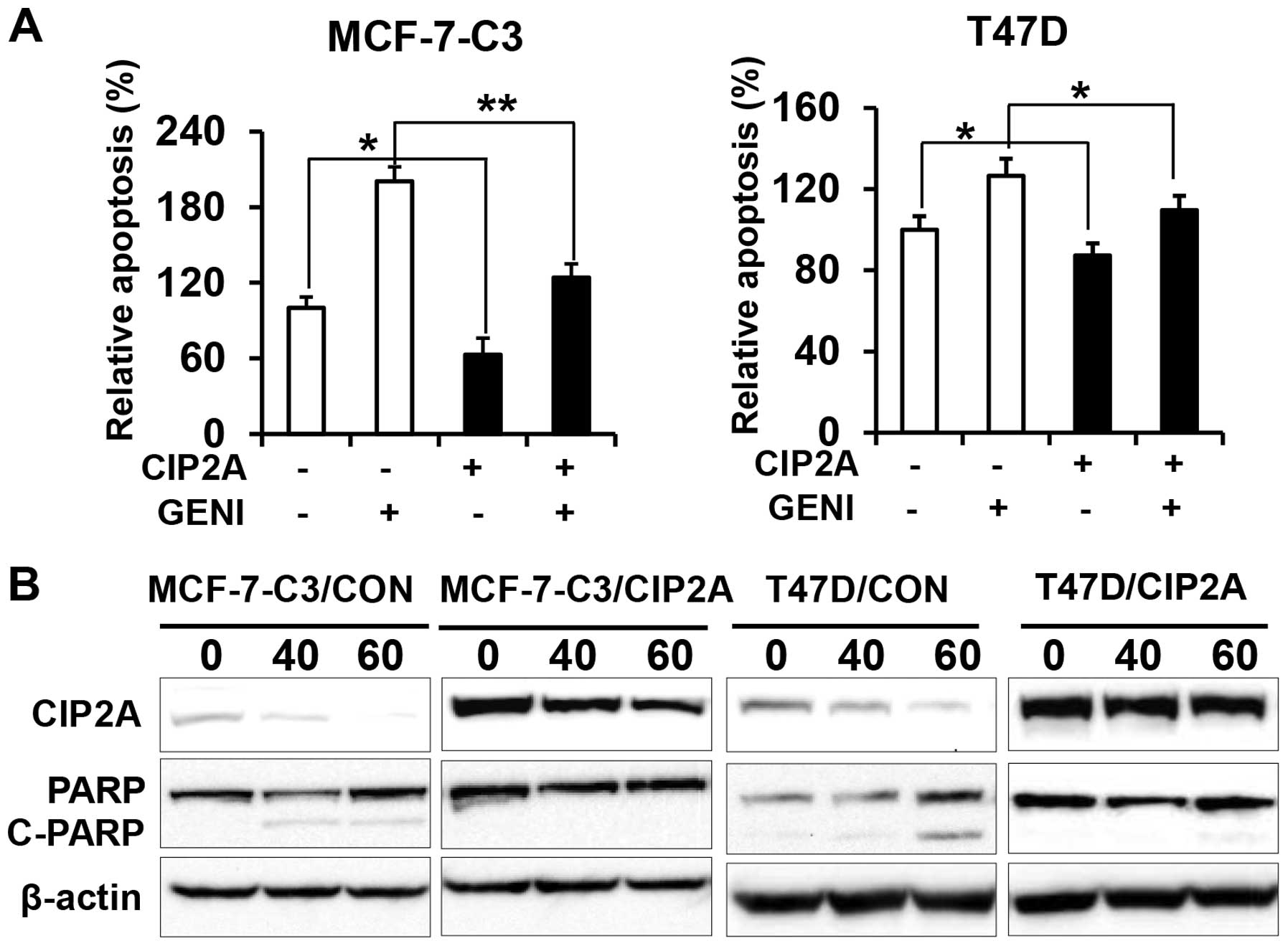
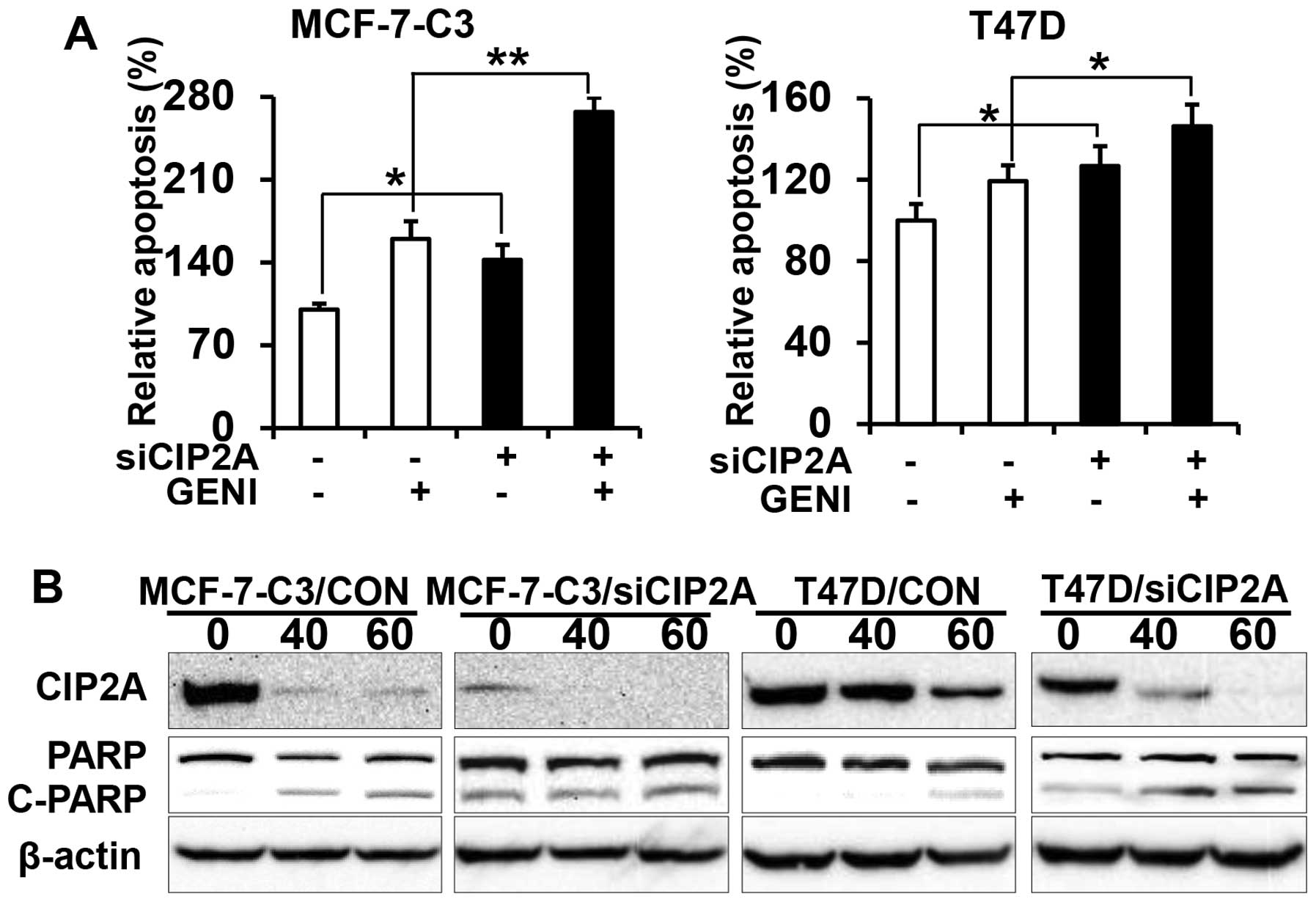
Overexpression of CIP2A renders breast
cancer cells resistant to genistein-induced apoptosis
To determine the role of CIP2A in genistein-induced
apoptosis, we examined apoptotic responses in the two pairs of cell
lines in the presence or absence of genistein. Data from apoptosis
ELISA assays showed that genistein-induced apoptosis in
MCF-7-C3/CIP2A and T47D/CIP2A cells was significantly decreased as
compared to MCF-7-C3/CON and T47D/CON cells (Fig. 3A). Consistently, we found that PARP
cleavage in genistein-treated MCF-7-C3/CIP2A and T47D/CIP2A cells
was also decreased as compared to the control (Fig. 3B). The data indicate that CIP2A
downregulation contributes to genistein-induced apoptosis.
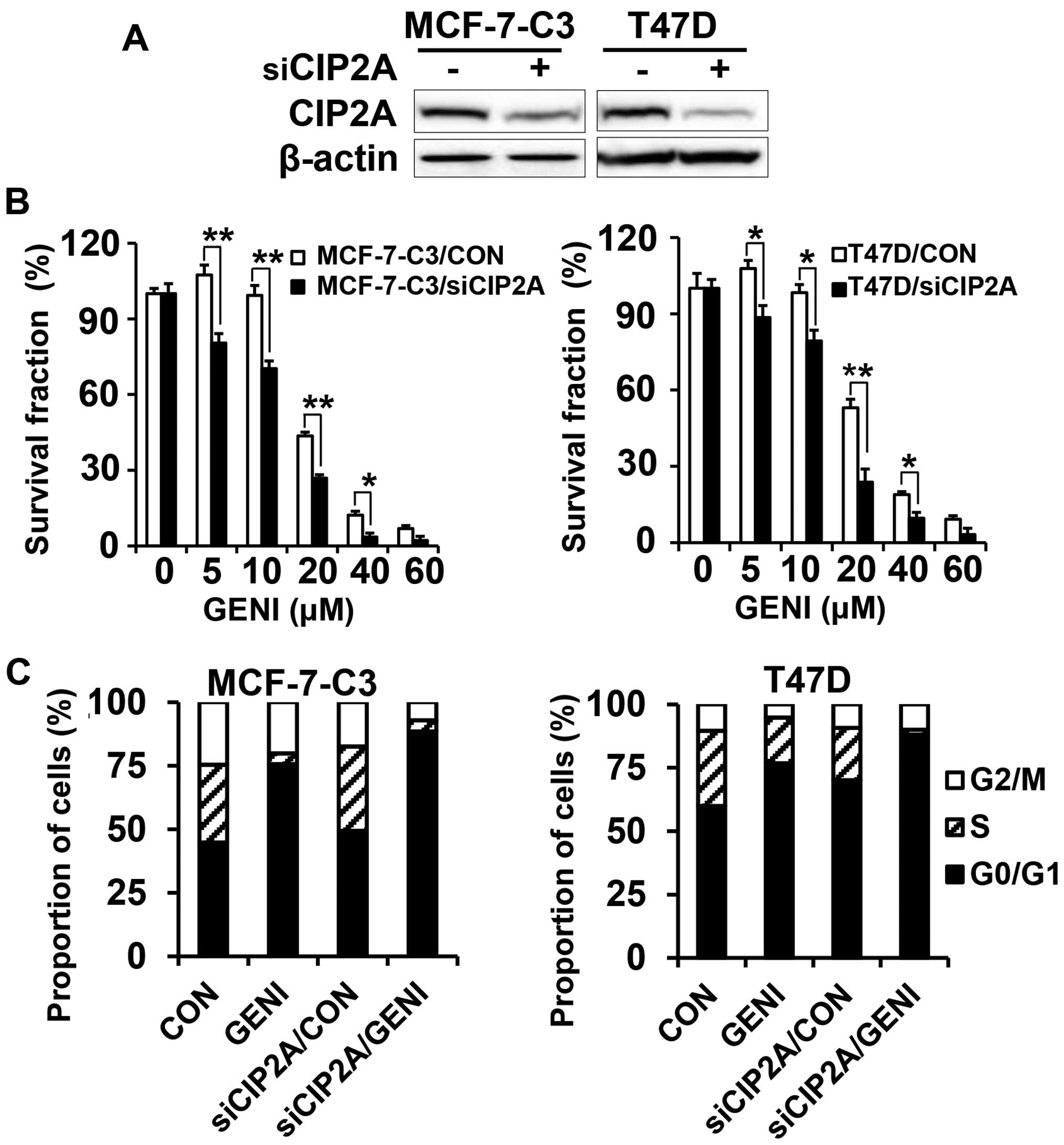
CIP2A knockdown enhances
genistein-induced growth inhibition
To confirm the functional role of CIP2A in
genistein-induced growth inhibition and apoptosis, we also
established stable MCF-7-C3 and T47D sublines with CIP2A knockdown.
As shown in Fig. 4A, CIP2A
expression was efficiently knocked down in MCF-7-C3 and T47D cells
transfected with CIP2A shRNA encoding lentiviruses. With these
paired cell lines, we found that CIP2A knockdown significantly
enhanced genistein-induced growth inhibition, as measured with MTT
assays (Fig. 4B). Cell cycle
analysis showed that CIP2A knockdown alone was able to induce
increased G0/G1 arrest. Genistein-induced G0/G1 arrest was further
enhanced in genistein-treated MCF-7-C3/siCIP2A and T47D/siCIP2A
cells (Fig. 4C). These results
demonstrated that CIP2A plays a role in genistein-induced cell
cycle regulation and growth inhibition.
CIP2A knockdown enhances
genistein-induced apoptosis
Next, we examined the effect of CIP2A knockdown on
genistein-induced apoptosis. The apoptosis ELISA assay showed that
genistein induced apoptosis in MCF-7-C3/siCIP2A cells and
T47D/siCIP2A cells significantly, as compared to the control
(Fig. 5A). Examination of PARP
cleavage indicated that genistein induced a significant increase in
PARP cleavage in MCF-7-C3/siCIP2A and T47D/siCIP2A cells (Fig. 5B). The data confirm CIP2A’s role in
genistein-induced apoptosis.
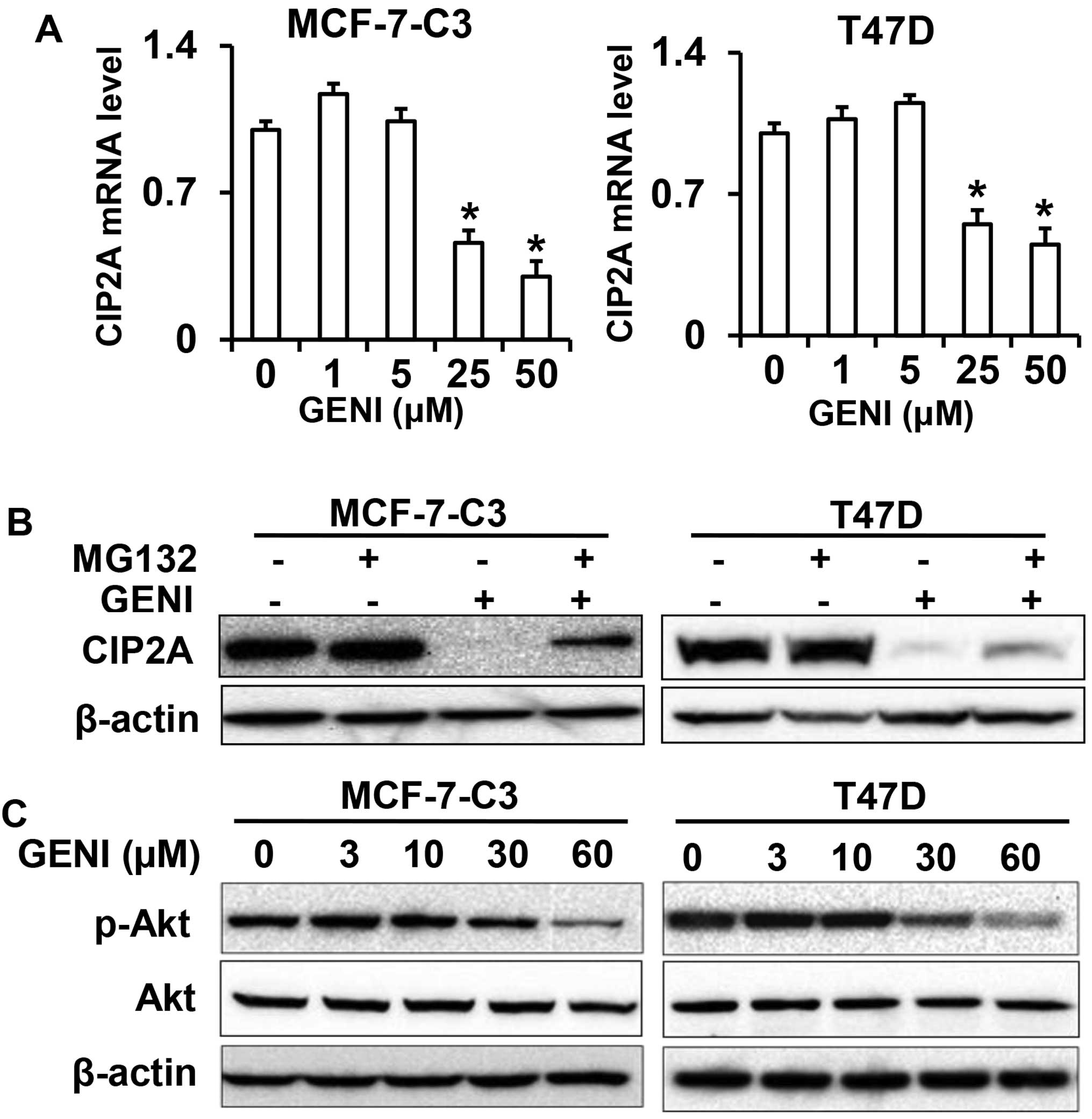
Genistein regulates CIP2A at both mRNA
and protein levels
To understand the mechanism of genistein-mediated
downregulation of CIP2A, we first examined CIP2A mRNA levels in
genistein-treated MCF-7-C3 and T47D cells. As shown in Fig. 6A, CIP2A mRNA levels in the cells
treated with 25 or 50 μM genistein were significantly decreased.
Notably, CIP2A mRNA levels in the cells treated with genistein at
lower concentrations were slightly increased. This dose-effect
pattern was consistent with the protein level changes observed
above. These results indicate that genistein-mediated inhibition of
CIP2A transcription is involved in its downregulation of CIP2A.
As it was reported that CIP2A could be regulated by
proteasomal degradation in celastrol treated cancer cells (21), we then examined whether modulation
of proteasomal degradation contributes to genistein-mediated
downregulation of CIP2A. We found that treating the cells with
proteasomal inhibitor MG132 significantly reversed
genistein-induced downregulation of CIP2A, suggesting that
genistein also regulates CIP2A at the protein level (Fig. 6B). As CIP2A protein stability could
be regulated by Akt activation (21), we examined phospho-Akt levels in
genistein-treated cells (Fig. 6C).
Inhibition of phospho-Akt in the treated cells suggests a link
between these changes and CIP2A protein degradation.
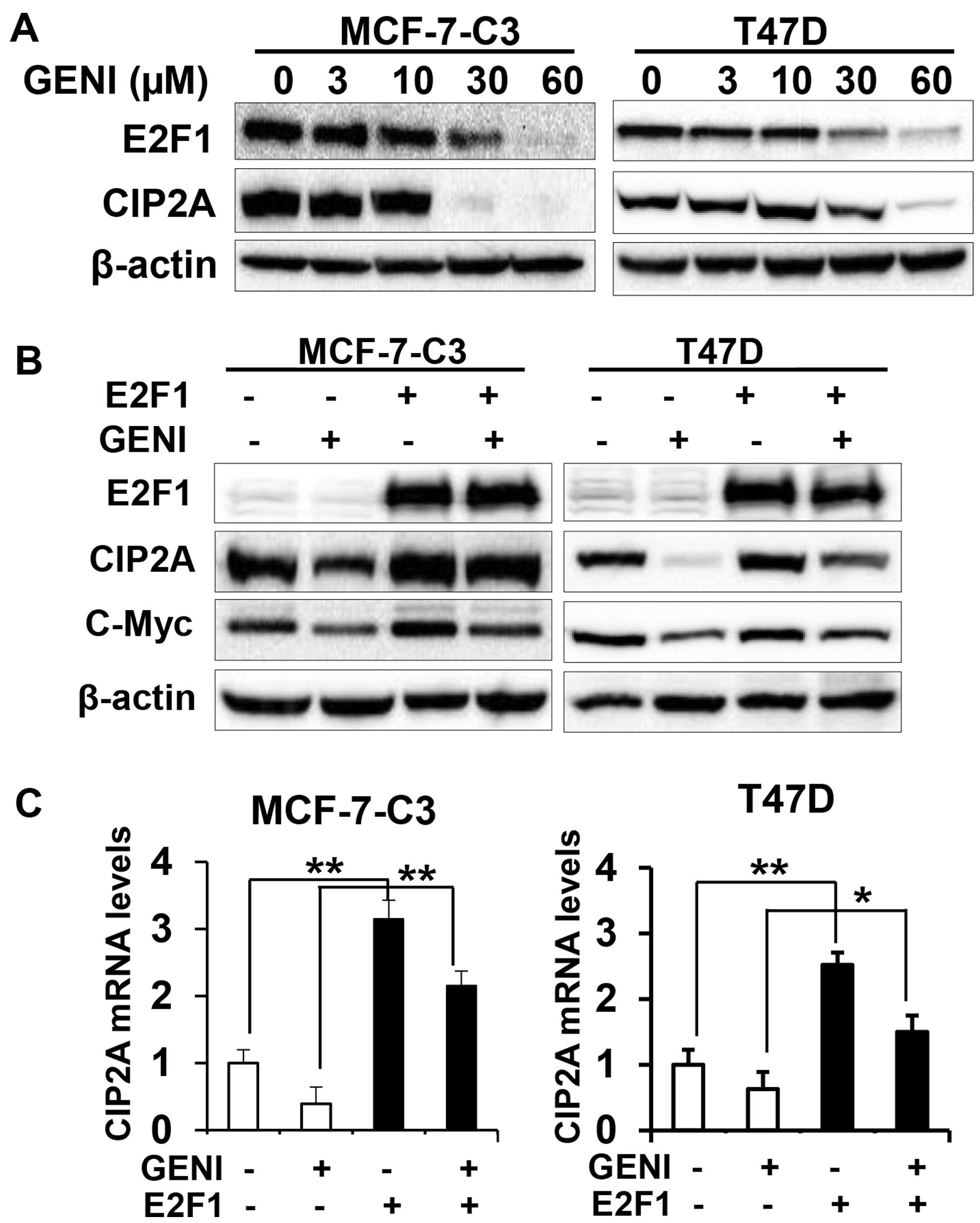
Genistein-induced downregulation of CIP2A
involves E2F1-mediated transcriptional regulation
To further investigate the underlying mechanism of
genistein-induced downregulation of CIP2A, we focused on
genistein-induced transcriptional downregulation. It was reported
that E2F1 is a major transcription factor that regulates CIP2A
transcription (30,31). We therefore examined the role of
E2F1 in genistein-mediated transcriptional regulation of CIP2A. As
shown in Fig. 7A,
genistein-induced concurrent decrease of CIP2A and E2F1 protein
levels in both MCF-7-C3 and T47D cells, suggesting a correlation
between the two molecules. To define the specific role of E2F1, we
examined the effect of E2F1 overexpression on genistein-modulated
CIP2A mRNA and protein levels. We found that E2F1 overexpression
attenuated genistein-induced downregulation of CIP2A protein
(Fig. 7B). Examination of mRNA
levels in the cells with different E2F1 status and genistein
treatments indicated that E2F1 overexpression induced a dramatic
increase in CIP2A mRNA, whereas genistein-induced downregulation of
CIP2A mRNA was abolished in both cell lines with E2F1
overexpression (Fig. 7C). The
results suggest that E2F1 plays a critical role in
genistein-mediated downregulation of CIP2A mRNA, which contribute
to the overall downregulation of CIP2A protein.
Discussion
In the present study, we demonstrated that genistein
induced downregulation of CIP2A in MCF-7-C3 and T47D breast cancer
cells, which was associated with its growth inhibition and
apoptosis induction. We further demonstrated that overexpression of
CIP2A attenuated genistein-induced growth inhibition and apoptosis,
and conversely, CIP2A knockdown sensitized genistein-induced
cellular effects. Our results indicate that genistein specifically
regulates CIP2A in breast cancer cells and the consequent CIP2A
downregulation may play a critical role in its antitumor effects.
This study identifies CIP2A as a novel cellular target of
genistein. Although previous studies have established ER, EGFR and
topoisomerase as the major targets of genistein-mediated inhibition
in breast cancer cells (5–7), the intracellular mediators of
genistein-associated cellular activities remain obscure. Although
CIP2A might not be a direct binding target of genistein, it is a
critical intracellular target/mediator of genistein. This study is
an advancement in the understanding of genistein-mediated growth
inhibition and apoptosis.
The functional impact of genistein-mediated CIP2A
downregulation is supported by its correlation with
genistein-induced growth inhibition and apoptosis. We showed that
CIP2A overexpression reduced genistein-induced overall growth
inhibition, cell cycle arrest in G0/G1 phase and apoptosis in each
cell line. In contrast, CIP2A knockdown enhanced genistein-induced
cell cycle arrest and apoptosis. These data demonstrated the
specific role of CIP2A modulation in genistein-associated tumor
inhibition. Previously, it was reported that genistein could induce
G2/M (32,33) or G0/G1 arrest (34–37).
Under the conditions in the present study, CIP2A modulation was a
critical factor affecting genistein-induced G0/G1 arrest. As an
oncogene, CIP2A overexpression or knockdown alone could increase
and reduce S phase cells (19),
respectively, as it was observed in T47D cells in this study
(Figs. 2D and 4D). Although CIP2A overexpression or
knockdown modified genistein-induced cell cycle progression, the
differences between non-genistein treated control (CON) and
CIP2A/control (CIP2A/CON) cells were not evident (Figs. 2C and 4C). This could be because these cells
were stable lines derived from pooled resistant clones, the cells
might have developed adaptations to the new environment under
non-stress conditions. Nevertheless, the effect of CIP2A modulation
in genistein-treated control and the overexpressing/knockdown cells
was evident.
Our findings on genistein-mediated downregulation of
CIP2A open new avenues to understand genistein-mediated antitumor
effects. Increasing evidence has indicated that CIP2A is a pivotal
molecule involved in cancer development and progression (12,13,38).
It acts as an endogenous inhibitor of PP2A and has a broad impact
on cellular proliferation and survival (12,39–41).
Deregulation of CIP2A leads to the upregulation/activation of Myc
and Akt (42,43). Indeed, our recent data indicate the
existence of a positive CIP2A-Akt feedback loop (Zhao et al,
unpublished data). Whether genistein directly acts on Akt kinase
activity is an intriguing question to be addressed in future
studies. Given the critical role of CIP2A in the regulation of the
PP2A network, genistein-induced downregulation of CIP2A would
inhibit these pathways and contribute to its overall inhibition of
cancer cells. This is supported by our data showing that CIP2A
overexpression or knockdown resulted in significant changes in
genistein-induced cell cycle progression, growth inhibition and
apoptosis. Moreover, our results also support CIP2A as a
therapeutic target as it was reported in the studies on other
anticancer agents (44,45).
Our results showed that genistein induced
downregulation of CIP2A mRNA levels, whereas proteasomal inhibitor
MG132 treatment also attenuated genistein-induced downregulation of
CIP2A protein, indicating that genistein regulates CIP2A expression
at both transcriptional and post-translational levels. Previously,
induction of CIP2A degradation through proteasomal pathways in
response to celastrol has been reported (21). This could be triggered by direct
activation of E3 ligase such as CHIP or through inactivation of Akt
(21). Based on our data that
genistein inhibited Akt phosphorylation (Fig. 6C), it is possible that
genistein-induced inhibition of Akt activation at higher
concentrations may contribute to this process. Notably, it was
reported that genistein may function as a proteasomal inhibitor to
induce p27 and Bax in SV-40 transformed cells (46), suggesting that genistein-induced
proteasomal regulation is complicated and multi-level,
multi-faceted mechanisms may exist. Whether genistein has direct
effect on E3 ligase activities and whether genistein directly binds
and inhibits Akt will be investigated in future studies.
We explored the mechanisms of genistein-mediated
inhibition of CIP2A transcription by focusing on E2F1-mediated
regulation of CIP2A. E2F1 is a critical regulator of cell cycle
progression (47), although it may
induce apoptosis under certain cellular contexts (48). It was reported that E2F1 is a major
transcription factor that promotes CIP2A expression (49), and CIP2A has a positive feedback
effect on E2F1 by stabilizing serine 364 phosphorylation of E2F1
(49). We showed that
genistein-induced concurrent downregulation of E2F1 and CIP2A at
higher concentrations. Importantly, overexpression of E2F1
attenuated genistein-induced downregulation of CIP2A protein and
mRNA levels as well. These data indicate that modulation of
E2F1-mediated regulation of CIP2A transcription and E2F1-CIP2A
feedback loop is involved in genistein-induced downregulation of
CIP2A. While the immediate mediators between genistein treatment
and E2F1 downregulation require further investigation, the
regulation may be determined by the functional interaction among
E2F1, c-Myc and CIP2A, a mutual interacting multi-factorial
network. Recently, it was proposed that CIP2A promotes
carcinogenesis via an ‘oncogenic nexus’, which involves its
intersection with PP2A, Akt, Myc and other factors (18,19).
Hence, our data suggest that genistein-mediated tumor inhibition
involves the suppression of the ‘oncogenic nexus’ of CIP2A.
The present study provided proof of concept for
CIP2A downregulation in genistein-induced inhibition of breast
cancer cells. However, how genistein regulates CIP2A degradation
and transcription appears to be complicated. More work is needed to
understand genistein-mediated regulation of proteasomal pathways in
CIP2A degradation. Genistein-induced inhibition of CIP2A
transcription may also involve the regulation of other mechanisms.
For example, we observed that genistein may induce CIP2A mRNA
increases at lower concentrations. This might be mediated by its
estrogenic activity, because it was reported that activation of ER
also promotes CIP2A expression (50). Although genistein-induced
inhibition of CIP2A transcription at higher concentrations may also
be related to genistein-associated anti-estrogenic activity at
higher concentrations, genistein-induced downregulation of E2F1 and
the consequent CIP2A mRNA decrease could be independent of estrogen
signaling, as it was also observed in ER negative MDA-MB-231 breast
cancer cells (data not shown). Further analysis of the relative
contributions of individual mechanisms in genistein-mediated
downregulation of CIP2A will be followed in future studies.
In conclusion, our results identified CIP2A as an
intracellular target of genistein, which has a functional impact on
genistein-induced growth inhibition and apoptosis induction.
Genistein-mediated downregulation of CIP2A involves both
transcriptional and proteasomal regulation. While multiple factors
might contribute to genistein-mediated transcriptional
downregulation of CIP2A, we demonstrated that modulation of
E2F1-mediated CIP2A transcription plays a critical role in
genistein-induced cellular responses. While being consistent with
previous reports (31), these data
also advance our understanding of genistein-associated antitumor
activities and support its implication in breast cancer prevention
and treatment. The study also supports the previous implication of
CIP2A as a therapeutic target in the development of novel
anticancer agents (51).
Acknowledgements
The present study was supported in part by a grant
from NIEHS (R21ES025337) and a pilot project of a U54 grant from
NCI (5U54CA156735). X.Y. is also supported by a grant from American
Cancer Society (RSG-08-138-01-CNE) and a pilot project grant of the
NIH U54 AA019765 grant. We thank Dr Erin Witalison for assistance
in manuscript editing.
References
|
1
|
Ferlay J1, Soerjomataram I, Dikshit R,
Eser S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–386. 2015.
View Article : Google Scholar
|
|
2
|
Bonofiglio D, Giordano C, De Amicis F,
Lanzino M and Ando S: Natural products as promising antitumoral
agents in breast cancer: Mechanisms of action and molecular
targets. Mini Rev Med Chem. 15:12015. View Article : Google Scholar
|
|
3
|
Nagata C, Mizoue T, Tanaka K, Tsuji I,
Tamakoshi A, Matsuo K, Wakai K, Inoue M, Tsugane S, Sasazuki S, et
al: Research Group for the Development and Evaluation of Cancer
Prevention Strategies in Japan: Soy intake and breast cancer risk:
An evaluation based on a systematic review of epidemiologic
evidence among the Japanese population. Jpn J Clin Oncol.
44:282–295. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lamartiniere CA: Protection against breast
cancer with genistein: a component of soy. Am J Clin Nutr.
71:1705S–1707S; discussion 1708S–1709S. 2000.PubMed/NCBI
|
|
5
|
Shao ZM, Shen ZZ, Fontana JA and Barsky
SH: Genistein’s ‘ER-dependent and independent’ actions are mediated
through ER pathways in ER-positive breast carcinoma cell lines.
Anticancer Res. 20:2409–2416. 2000.PubMed/NCBI
|
|
6
|
Akiyama T, Ishida J, Nakagawa S, Ogawara
H, Watanabe S, Itoh N, Shibuya M and Fukami Y: Genistein, a
specific inhibitor of tyrosine-specific protein kinases. J Biol
Chem. 262:5592–5595. 1987.PubMed/NCBI
|
|
7
|
Corbett AH, Hong D and Osheroff N:
Exploiting mechanistic differences between drug classes to define
functional drug interaction domains on topoisomerase II. Evidence
that several diverse DNA cleavage-enhancing agents share a common
site of action on the enzyme. J Biol Chem. 268:14394–14398.
1993.PubMed/NCBI
|
|
8
|
Wang TT, Sathyamoorthy N and Phang JM:
Molecular effects of genistein on estrogen receptor mediated
pathways. Carcinogenesis. 17:271–275. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sarkar FH and Li Y: Mechanisms of cancer
chemoprevention by soy isoflavone genistein. Cancer Metastasis Rev.
21:265–280. 2002. View Article : Google Scholar
|
|
10
|
Khanna A, Pimanda JE and Westermarck J:
Cancerous inhibitor of protein phosphatase 2A, an emerging human
oncoprotein and a potential cancer therapy target. Cancer Res.
73:6548–6553. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Côme C1, Laine A, Chanrion M, Edgren H,
Mattila E, Liu X, Jonkers J, Ivaska J, Isola J, Darbon JM, et al:
CIP2A is associated with human breast cancer aggressivity. Clin
Cancer Res. 15:5092–5100. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Junttila MR, Puustinen P, Niemelä M, Ahola
R, Arnold H, Böttzauw T, Ala-aho R, Nielsen C, Ivaska J, Taya Y, et
al: CIP2A inhibits PP2A in human malignancies. Cell. 130:51–62.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Khanna A, Böckelman C, Hemmes A, Junttila
MR, Wiksten JP, Lundin M, Junnila S, Murphy DJ, Evan GI, Haglund C,
et al: MYC-dependent regulation and prognostic role of CIP2A in
gastric cancer. J Natl Cancer Inst. 101:793–805. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Khanna A and Pimanda JE: Clinical
significance of cancerous inhibitor of protein phosphatase 2A in
human cancers. Int J Cancer. 138:525–532. 2016. View Article : Google Scholar
|
|
15
|
Yu G, Liu G, Dong J and Jin Y: Clinical
implications of CIP2A protein expression in breast cancer. Med
Oncol. 30:5242013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mumby M: PP2A: Unveiling a reluctant tumor
suppressor. Cell. 130:21–24. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mathiasen DP, Egebjerg C, Andersen SH,
Rafn B, Puustinen P, Khanna A, Daugaard M, Valo E, Tuomela S,
Bøttzauw T, et al: Identification of a c-Jun N-terminal
kinase-2-dependent signal amplification cascade that regulates
c-Myc levels in ras transformation. Oncogene. 31:390–401. 2012.
View Article : Google Scholar
|
|
18
|
De P, Carlson J, Leyland-Jones B and Dey
N: Oncogenic nexus of cancerous inhibitor of protein phosphatase 2A
(CIP2A): An oncoprotein with many hands. Oncotarget. 5:4581–4602.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
De P, Carlson JH, Leyland-Jones B and Dey
N: Role of ‘oncogenic nexus’ of CIP2A in breast oncogenesis: How
does it work? Am J Cancer Res. 5:2872–2891. 2015.
|
|
20
|
Chao TT, Wang CY, Lai CC, Chen YL, Tsai
YT, Chen PT, Lin HI, Huang YC, Shiau CW, Yu CJ, et al: TD-19, an
erlotinib derivative, induces epidermal growth factor receptor
wild-type nonsmall-cell lung cancer apoptosis through
CIP2A-mediated pathway. J Pharmacol Exp Ther. 351:352–358. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu Z, Ma L, Wen ZS, Hu Z, Wu FQ, Li W,
Liu J and Zhou GB: Cancerous inhibitor of PP2A is targeted by
natural compound celastrol for degradation in non-small-cell lung
cancer. Carcinogenesis. 35:905–914. 2014. View Article : Google Scholar
|
|
22
|
Ding Y, Wang Y, Ju S, Wu X, Zhu W, Shi F
and Mao L: Role of CIP2A in the antitumor effect of bortezomib in
colon cancer. Mol Med Rep. 10:387–392. 2014.PubMed/NCBI
|
|
23
|
Yang XH, Edgerton S and Thor AD:
Reconstitution of caspase-3 sensitizes MCF-7 breast cancer cells to
radiation therapy. Int J Oncol. 26:1675–1680. 2005.PubMed/NCBI
|
|
24
|
Yang XH, Sladek TL, Liu X, Butler BR,
Froelich CJ and Thor AD: Reconstitution of caspase 3 sensitizes
MCF-7 breast cancer cells to doxorubicin- and etoposide-induced
apoptosis. Cancer Res. 61:348–354. 2001.PubMed/NCBI
|
|
25
|
Yang XH and Sladek TL: Overexpression of
the E2F-1 transcription factor gene mediates cell transformation.
Gene Expr. 4:195–204. 1995.PubMed/NCBI
|
|
26
|
Yang X, Yang S, McKimmey C, Liu B,
Edgerton SM, Bales W, Archer LT and Thor AD: Genistein induces
enhanced growth promotion in ER-positive/erbB-2-overexpressing
breast cancers by ER-erbB-2 cross talk and p27/kip1 downregulation.
Carcinogenesis. 31:695–702. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jänicke RU, Sprengart ML, Wati MR and
Porter AG: Caspase-3 is required for DNA fragmentation and
morphological changes associated with apoptosis. J Biol Chem.
273:9357–9360. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Talanian RV, Yang X, Turbov J, Seth P,
Ghayur T, Casiano CA, Orth K and Froelich CJ: Granule-mediated
killing: Pathways for granzyme B-initiated apoptosis. J Exp Med.
186:1323–1331. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang S, Thor AD, Edgerton S and Yang X:
Caspase-3 mediated feedback activation of apical caspases in
doxorubicin and TNF-alpha induced apoptosis. Apoptosis.
11:1987–1997. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang W, Chen H, Chen Y, Liu J, Wang X, Yu
X, Chen JJ and Zhao W: Cancerous inhibitor of protein phosphatase
2A contributes to human papillomavirus oncoprotein E7-induced cell
proliferation via E2F1. Oncotarget. 6:5253–5262. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Laine A, Sihto H, Come C, Rosenfeldt MT,
Zwolinska A, Niemelä M, Khanna A, Chan EK, Kähäri VM,
Kellokumpu-Lehtinen PL, et al: Senescence sensitivity of breast
cancer cells is defined by positive feedback loop between CIP2A and
E2F1. Cancer Discov. 3:182–197. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Constantinou AI, Kamath N and Murley JS:
Genistein inactivates bcl-2, delays the G2/M phase of the cell
cycle, and induces apoptosis of human breast adenocarcinoma MCF-7
cells. Eur J Cancer. 34:1927–1934. 1998. View Article : Google Scholar
|
|
33
|
Oki T, Sowa Y, Hirose T, Takagaki N,
Horinaka M, Nakanishi R, Yasuda C, Yoshida T, Kanazawa M, Satomi Y,
et al: Genistein induces Gadd45 gene and G2/M cell cycle arrest in
the DU145 human prostate cancer cell line. FEBS Lett. 577:55–59.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yu JY, Lee JJ, Lim Y, Kim TJ, Jin YR,
Sheen YY and Yun YP: Genistein inhibits rat aortic smooth muscle
cell proliferation through the induction of p27kip1. J
Pharmacol Sci. 107:90–98. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kuzumaki T, Kobayashi T and Ishikawa K:
Genistein induces p21Cip1/WAF1 expression and blocks the
G1 to S phase transition in mouse fibroblast and melanoma cells.
Biochem Biophys Res Commun. 251:291–295. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shen JC, Klein RD, Wei Q, Guan Y, Contois
JH, Wang TT, Chang S and Hursting SD: Low-dose genistein induces
cyclin-dependent kinase inhibitors and G1 cell-cycle
arrest in human prostate cancer cells. Mol Carcinog. 29:92–102.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yang S, Zhou Q and Yang X: Caspase-3
status is a determinant of the differential responses to genistein
between MDA-MB-231 and MCF-7 breast cancer cells. Biochim Biophys
Acta. 1773:903–911. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Guo Z, Liu D and Su Z: CIP2A mediates
prostate cancer progression via the c-MYC signaling pathway. Tumour
Biol. 36:3583–3589. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kim JS, Kim EJ, Oh JS, Park IC and Hwang
SG: CIP2A modulates cell-cycle progression in human cancer cells by
regulating the stability and activity of Plk1. Cancer Res.
73:6667–6678. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ventelä S, Côme C, Mäkelä JA, Hobbs RM,
Mannermaa L, Kallajoki M, Chan EK, Pandolfi PP, Toppari J and
Westermarck J: CIP2A promotes proliferation of spermatogonial
progenitor cells and spermatogenesis in mice. PLoS One.
7:e332092012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang J, Huang T, Sun J, Yu Y, Liu Z, Li W,
Jia J and Chen C: CIP2A is overexpressed and involved in the
pathogenesis of chronic myelocytic leukemia by interacting with
breakpoint cluster region-Abelson leukemia virus. Med Oncol.
31:1122014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lei N, Peng B and Zhang JY: CIP2A
regulates cell proliferation via the AKT signaling pathway in human
lung cancer. Oncol Rep. 32:1689–1694. 2014.PubMed/NCBI
|
|
43
|
Niemelä M, Kauko O, Sihto H, Mpindi JP,
Nicorici D, Pernilä P, Kallioniemi OP, Joensuu H, Hautaniemi S and
Westermarck J: CIP2A signature reveals the MYC dependency of
CIP2A-regulated phenotypes and its clinical association with breast
cancer subtypes. Oncogene. 31:4266–4278. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Khanna A, Rane JK, Kivinummi KK, Urbanucci
A, Helenius MA, Tolonen TT, Saramäki OR, Latonen L, Manni V,
Pimanda JE, et al: CIP2A is a candidate therapeutic target in
clinically challenging prostate cancer cell populations.
Oncotarget. 6:19661–19670. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gonzalez-Alonso P, Cristobal I, Manso R,
Madoz-Gurpide J, Garcia-Foncillas J and Rojo F: PP2A inhibition as
a novel therapeutic target in castration-resistant prostate cancer.
Tumour Biol. 36:5753–5755. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kazi A, Daniel KG, Smith DM, Kumar NB and
Dou QP: Inhibition of the proteasome activity, a novel mechanism
associated with the tumor cell apoptosis-inducing ability of
genistein. Biochem Pharmacol. 66:965–976. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mundle SD and Saberwal G: Evolving
intricacies and implications of E2F1 regulation. FASEB J.
17:569–574. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Moon NS, Frolov MV, Kwon EJ, Di Stefano L,
Dimova DK, Morris EJ, Taylor-Harding B, White K and Dyson NJ:
Drosophila E2F1 has context-specific pro- and antiapoptotic
properties during development. Dev Cell. 9:463–475. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Laine A and Westermarck J: Molecular
pathways: harnessing E2F1 regulation for prosenescence therapy in
p53-defective cancer cells. Clin Cancer Res. 20:3644–3650. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Choi YA, Koo JS, Park JS, Park MY, Jeong
AL, Oh KS and Yang Y: Estradiol enhances CIP2A expression by the
activation of p70 S6 kinase. Endocr Relat Cancer. 21:189–202. 2014.
View Article : Google Scholar
|
|
51
|
Xue Y, Wu G, Wang X, Zou X, Zhang G, Xiao
R, Yuan Y, Long D, Yang J, Wu Y, et al: CIP2A is a predictor of
survival and a novel therapeutic target in bladder urothelial cell
carcinoma. Med Oncol. 30:4062013. View Article : Google Scholar : PubMed/NCBI
|