Introduction
Prostate cancer (PCa) is the second leading cause of
cancer-related deaths in men in the Unites States (1). Androgen receptor (AR) signaling is
crucial not only for the normal development and physiological
functioning of the prostate gland but is also critical for
increased proliferation, survival, invasion and clonogenic ability
of the PCa cells (2,3). Therefore, the androgen-induced
protein, prostate specific antigen (PSA) is an effective biomarker
during the early stages of PCa growth (4).
Androgen deprivation therapy (ADT), achieved via the
simultaneous suppression of testicular androgen production by using
luteinizing hormone-releasing hormone (LHRH) analogues and
inhibition of AR binding with the residual androgens using AR
antagonists (anti-androgens) like casodex (bicalutamide; BIC) and
MDV3100 (enzalutamide; ENZ) remains a gold standard treatment in
PCa patients (5,6). However, despite the initial efficacy
of ADT, tumors eventually become resistant to even high doses of AR
antagonists and this hormone-refractory or castration resistant
prostate cancer (CRPC) is a significant challenge to therapy
(7–15). Indeed, these clinically approved
drugs are administered as first line anti-hormonal therapy in PCa
patients. However, the side-effects associated with chronic high
doses of these anti-androgens (16–18)
warrant the need to use adjuvant agents that can increase the
efficacy of BIC and ENZ, which will significantly reduce their
side-effects and augment their clinical effectiveness.
Various mechanisms attributable to this resistance
include ligand independent activation of AR by growth factors,
cytokines or kinases; intracrine or intratumoral androgen
production; amplification of AR gene; gain-of-function mutations
leading to AR activation by AR antagonists; overexpression of AR
coactivators and/or expression of constitutively active AR splice
variants (19–21). Thus, decreasing the incessant AR
signaling in the androgen dependent PCa cells and hampering the
transition of androgen independent PCa cells to aggressive CRPC
will be a principal approach towards increased therapeutic efficacy
of ADT. Strategies that suppress AR protein levels in PCa cells and
significantly improve the efficacy of clinically approved
anti-androgens (e.g. BIC and ENZ) will be of significance in
treating patients with CRPC tumors.
Sulforaphane (SFN), an isothiocyanate found in
cruciferous vegetables (e.g. broccoli) is a promising therapeutic
agent for metastatic PCa since it shows specific cytotoxicity
towards transformed cells without having significant adverse
effects on primary prostate epithelial cells (22–29).
Mechanistic studies have shown that SFN-induced cell death is
initiated by the release of reactive oxygen species (ROS) (30,31)
and hydrogen sulfide (32);
epigenetic modifications of Nrf-2 leading to the activation
of downstream anti-oxidative/detoxification stress pathway and by
suppression of the Akt survival pathway (33–35).
Mechanistically, SFN has been shown to decrease AR protein levels
by inhibiting the transcription of AR gene (36) and increasing the proteasomal
degradation of AR protein (37,38).
We hypothesized that combined exposure to SFN will enhance the
efficacy of anti-androgens that block AR function by competitively
inhibiting ligand (androgen) binding. Our studies demonstrate this
novel phenomenon at both the cellular and molecular levels.
Co-treatment with physiologically achievable levels of SFN
synergistically increased the anticancer efficacy of both BIC and
ENZ in both androgen dependent LNCaP cells and in its CRPC subline,
C4-2B cells.
Materials and methods
Cell culture
LNCaP, an androgen-dependent (AD) prostate cancer
cell line, was purchased from the American Type Culture Collection
(ATCC; Rockville, MD, USA). C4-2B cells, a CRPC sub-line of LNCaP,
was a kind gift from Dr Leland Chung’s laboratory (Emory
University, Atlanta, GA, USA) (39). Both cell lines were maintained in
RPMI-1640 media containing 1% penicillin/streptomycin (Cellgro,
Manassas, VA, USA) supplemented with 10% fetal bovine serum (FBS;
Atlanta Biologicals, Inc., Lawrenceville, GA, USA) in a humidified
incubator containing 5% CO2 at 37°C. To mimic steroid
hormone deprived conditions, experiments were carried out in media
supplemented with 10% charcoal-stripped FBS (CS-FBS) from Atlanta
Biologicals.
Reagents
Sulforaphane (SFN), bicalutamide (BIC) and MTT
(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) were
obtained from Sigma-Aldrich (St. Louis, MO, USA). Enzalutamide was
purchased from Apexbio Technology LLC (Houston, TX, USA).
Cycloheximide (CHX) was purchased from Cayman Chemical (Ann Arbor,
MI, USA). The synthetic androgen-agonist, R1881 was obtained from
Perkin-Elmer (Waltham, MA, USA). All drugs were dissolved in
dimethyl sulfoxide (DMSO) and final DMSO concentration used was
<0.1%. The primary antibodies including rabbit polyclonal
anti-AR (N-20) (sc-816), goat polyclonal anti-Lamin B (C-20)
(sc-6216) and mouse monoclonal anti-GAPDH (sc-47724) were purchased
from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The
horseradish peroxidase (HRP)-conjugated goat anti-rabbit (A0545)
and goat anti-mouse (A9044) secondary antibodies were purchased
from Sigma-Aldrich. The goat anti-rabbit secondary antibody tagged
with Texas Red (T-2767) was purchased from Thermo Fisher Scientific
(Rockford, IL, USA). The quantitative RT-PCR (qRT-PCR) primers were
synthesized by Integrated DNA Technologies Inc. (Coralville, IA,
USA).
Cell viability assay
MTT assays were performed to determine cell
viability post exposure to the drug(s). In brief, ~5,000 cells were
seeded in 96-well culture plates and allowed to adhere overnight.
Cells were then synchronized by overnight incubation in serum-free
medium, and then treated with desired concentrations of drug(s),
alone or in different combinations for 24–72 h. Cell viability was
determined by adding MTT solution (5 mg/ml) and incubating for 3 h
at 37°C. The formazan crystals formed were then solubilized in DMSO
and optical density was measured spectrophotometrically at 540 nm
using a μQuant plate reader (US BioTek Laboratories, Shoreline, WA,
USA). Cell survival is expressed as percent of control.
Western immunoblot
Whole cell lysates were harvested at different
time-points post-treatment(s) using RIPA lysis buffer from Santa
Cruz Biotechnology and total protein content was quantified using
the bicinchoninic acid (BCA) protein assay reagent (Thermo Fisher
Scientific). Approximately 10 μg of protein was electrophoresed in
10% SDS-PAGE gels followed by electro-transfer onto nitrocellulose
membrane. After blocking in 5% casein in TBS-T buffer
(Tris-buffered saline with 0.1% Tween-20), membranes were incubated
overnight at 4°C with the primary antibodies (1:500 dilution)
followed by incubation with the corresponding HRP-conjugated
secondary antibodies (1:2,000 dilution) for 1 h. Membranes were
developed using the SuperSignal West Femto Substrate (Thermo Fisher
Scientific). Immunoblots were scanned using ImageQuant LAS 500 (GE
Healthcare, Princeton, NJ, USA) and band intensities were
quantified using ImageJ software (NIH, Bethesda, MD, USA).
Densitometric value for AR protein was normalized to the
corresponding GAPDH level in each sample.
Nuclear and cytosolic fractions
Protein from nuclear and cytosolic fractions were
isolated using the NE-PER Nuclear and Cytoplasmic Extraction kit
(Pierce, Rockford, IL, USA) according to the manufacturer’s
instructions. Approximately 10 μg of both cytoplasmic and nuclear
protein was electrophoresed in 10% SDS-PAGE gels after
quantification and resultant immunoblots were analyzed for AR
levels as described above (cytosol to nuclei protein contents were
1:6). Densitometric values for AR protein were normalized to either
GAPDH (cytoplasmic) or Lamin B (nuclear) levels.
Immunofluorescence microscopy
Subcellular localization of AR post-treatment with
SFN in the absence or presence of R1881 (1 nM) was visualized by
immunofluorescence microscopy (IFM). Briefly, cells
(3×104) were seeded in chamber slides (EMD Millipore,
Billerica, MA, USA) and allowed to adhere overnight. After
treatment, cells were fixed in ice cold methanol followed by
permeabilization with 0.1% Triton X-100 for 1 h. After blocking in
10% goat serum, slides were incubated overnight at 4°C with the
primary antibody (1:300 dilution) followed by incubation with the
corresponding Texas Red tagged secondary antibody (1:1,000
dilution) for 1 h. The Vectashield (Vector Laboratories, Inc.,
Burlingame, CA, USA) mounting medium containing the nuclear stain
diamino-2-phenylindole (DAPI) was then added to the slides and
coverslips were mounted. Images were captured using a fluorescent
microscope from Leica Microsystems Inc. (Buffalo Grove, IL,
USA).
Quantitative RT-PCR
Quantitative reverse transcriptase polymerase chain
reaction (qRT-PCR) was carried out to measure the AR gene
expression. Briefly, total mRNA was isolated post treatment using
the RNeasy Plus Mini kit from Qiagen (Valencia, CA, USA) according
to manufacturer’s instructions. The complementary DNA (cDNA) was
prepared using the iScript cDNA Synthesis kit (Bio-Rad
Laboratories) according to the manufacturer’s instructions. The
following primer sequences were used: AR-forward,
5′-CAGCCTATTGCGAGAGAGCTG-3′ and AR-reverse,
5′-GAAAGGATCTTGGGCACTTGC-3′; β-actin-forward,
5′-TGAGACCTTCAACACCCCAGCCATG-3′ and β-actin-reverse,
5′-GTAGATGGGCACAGTGTGGGTG-3′. The relative AR transcript levels
were measured using iQTM SYBR Green Supermix (Bio-Rad
Laboratories) and amplification reactions were carried out using
the C1000™ thermal cycler (CFX96; Bio-Rad Laboratories). The
following amplification conditions were used: priming at 95°C for 5
min, and then 35 cycles of 95°C for 30 sec, 55°C for 30 sec and
72°C for 30 sec. Data (ΔCt values) were expressed as fold changes
in gene expression after normalization to corresponding β-actin
transcript levels.
Wound-healing assay
Wound-healing assays were carried out to measure the
migratory phenotype of PCa cells as previously described (40). Briefly, cells were seeded in 6-well
plates (1×106 cells/well) and grown until they formed a
confluent monolayer. The monolayers were scratched using a 200 μl
pipette tip, wells were washed with phosphate-buffered saline (PBS)
and images of the wound (0 time-point) were captured using a Leica
Microsystems microscope. Growth media was added back to each
culture and treatments were initiated. After appropriate drug
treatments for 24–48 h, change in wound images was captured and
cell migration (wound closures) was calculated by measuring the
distance between 4–5 random points within the wound edges in three
replicate wells.
PSA measurement
An ELISA kit (ALPCO Diagnostics, Salem, NH, USA) was
used to measure PSA levels in cell culture supernatants using the
manufacturer’s instructions. Briefly, culture media was collected
48 h post-treatment with drug(s) and samples were allowed to react
with the immobilized anti-PSA antibody on the microtiter wells.
Next, a monoclonal anti-PSA antibody conjugated with horseradish
peroxidase (HRP) was added and allowed to react with the
immobilized antigen followed by incubation with the HRP substrate.
The optical density (OD) values measured spectrophotometrically at
450 nm were converted into ng/ml of PSA by using a standard curve,
and concentrations were normalized with protein content in each
sample.
Colony forming unit assay
Cells (500 cells/dish) were seeded in 60-mm petri
dishes in 3 replicates and grown in medium supplemented with 2%
FBS. The drugs, alone or in combination, were added after 48 h and
replenished in the second week. After two weeks in culture,
colonies were fixed with 100% ethanol and stained with 0.2% crystal
violet in 20% methanol. The colony forming units (CFU) were
enumerated and grouped according to their sizes, as small, medium
and large CFUs by using the ImageJ software (NIH). Changes in CFU
number and size were compared in control and drug-exposed
cultures.
Statistical analysis
Statistical analysis was carried out using the
GraphPad Prism Software version 6 (GraphPad Software Inc., San
Diego, CA, USA). Results are expressed as the mean ± standard error
of the mean (SEM). Significant changes from controls were
determined by a two-tailed Student’s t-test and P-values of
<0.05 were considered significant. For synergy determination,
the CompuSyn software (ComboSyn, Inc., Paramus, NJ, USA) was used
and combination index (CI) was calculated based on the Chou-Talalay
method which quantitatively determines additive (CI=1), synergistic
(CI <1) or antagonistic (CI >1) effects (41).
Results
Exposure to SFN rapidly decreases AR
protein levels in both LNCaP and C4-2B cells
Dose-dependent cytotoxicity for SFN at 24–72 h of
exposure was first conducted to determine the inhibitory
concentration 50 (IC50) values in both LNCaP and C4-2B
cells (Table I). Subsequent
studies were carried out using SFN concentrations below its
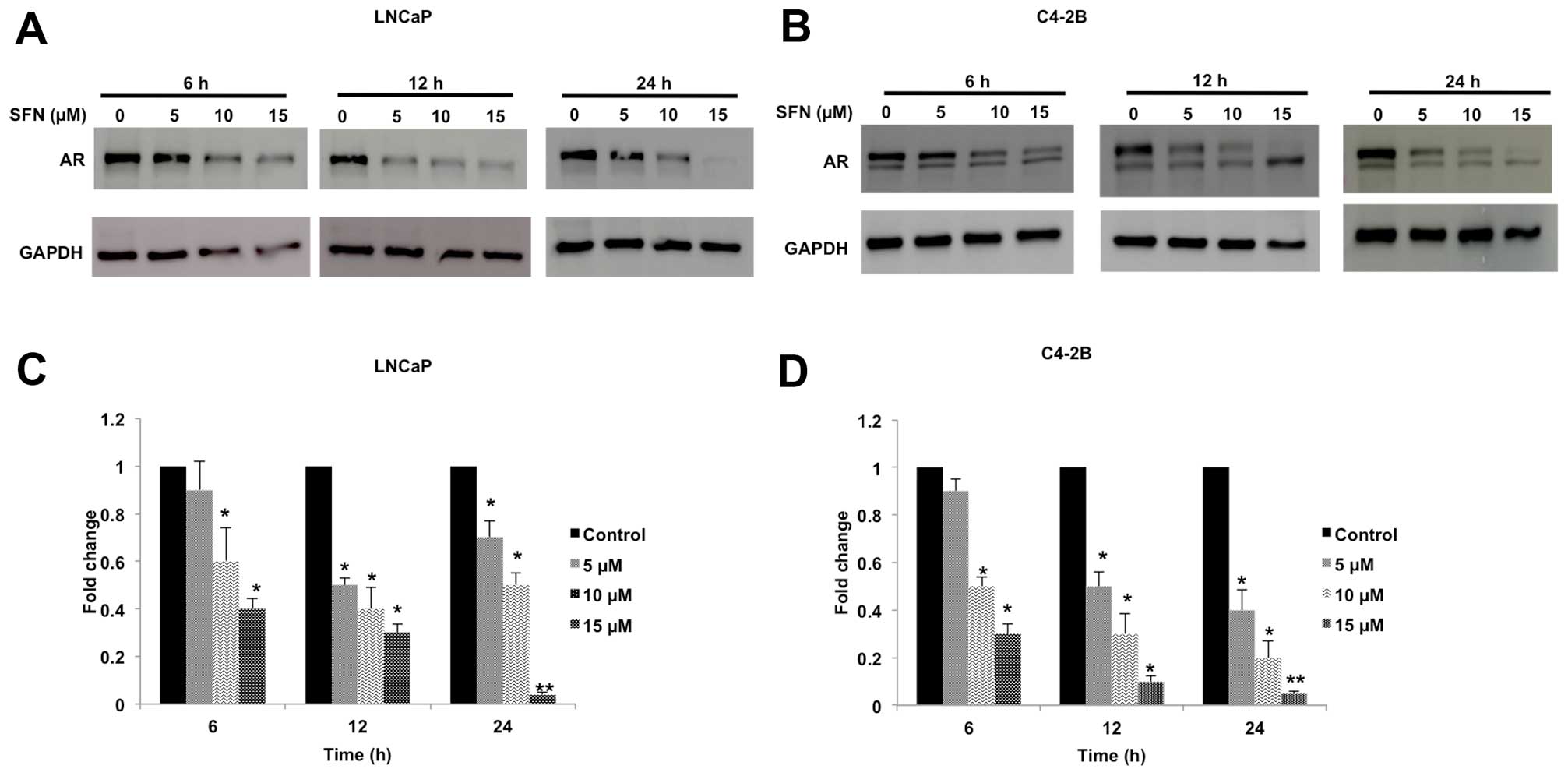
IC50 (<20 μM). Immunoblot analysis revealed that SFN
causes both time- and dose-dependent decrease of AR protein (110
kDa) in both LNCaP (Fig. 1A and C)
and C4-2B (Fig. 1B and D) cells.
This suppressive effect was more pronounced in the latter cell
line. Decrease in AR protein was apparent within 6 h post-exposure
and even with the lowest concentration of SFN used (5 μM) and the
AR band was virtually undetectable following 24 h exposure to 15 μM
SFN. It is worth noting that we consistently detected a lower
molecular weight AR band (~100 kDa) in the C4-2B cells, but not in
LNCaP cells. Notably, SFN did not suppress the level of this
putative AR variant.
 | Table IIC50 values for drugs at
24–72 h (MTT assay). |
Table I
IC50 values for drugs at
24–72 h (MTT assay).
| LNCaP
(IC50 ± SEM) |
|---|
|
|---|
| Drugs | SFN (μM) | BIC (μM) | ENZ (μM) |
|---|
| Time (h) |
| 24 | 23.8±2.33 | 140±5.66 | 62±2.71 |
| 48 | 22±2.28 | 105±4.62 | 57±3.44 |
| 72 | 17.2±1.3 | 80±3.67 | 49±2.13 |
|
| C4-2B
(IC50 ± SEM) |
|
| Drugs | SFN (μM) | BIC (μM) | ENZ (μM) |
|
| Time (h) |
| 24 | 26±3.12 | 151±6.32 | 65±3.08 |
| 48 | 24±2.81 | 107±5.89 | 61±4.23 |
| 72 | 19±1.6 | 87±4.12 | 53±5.21 |
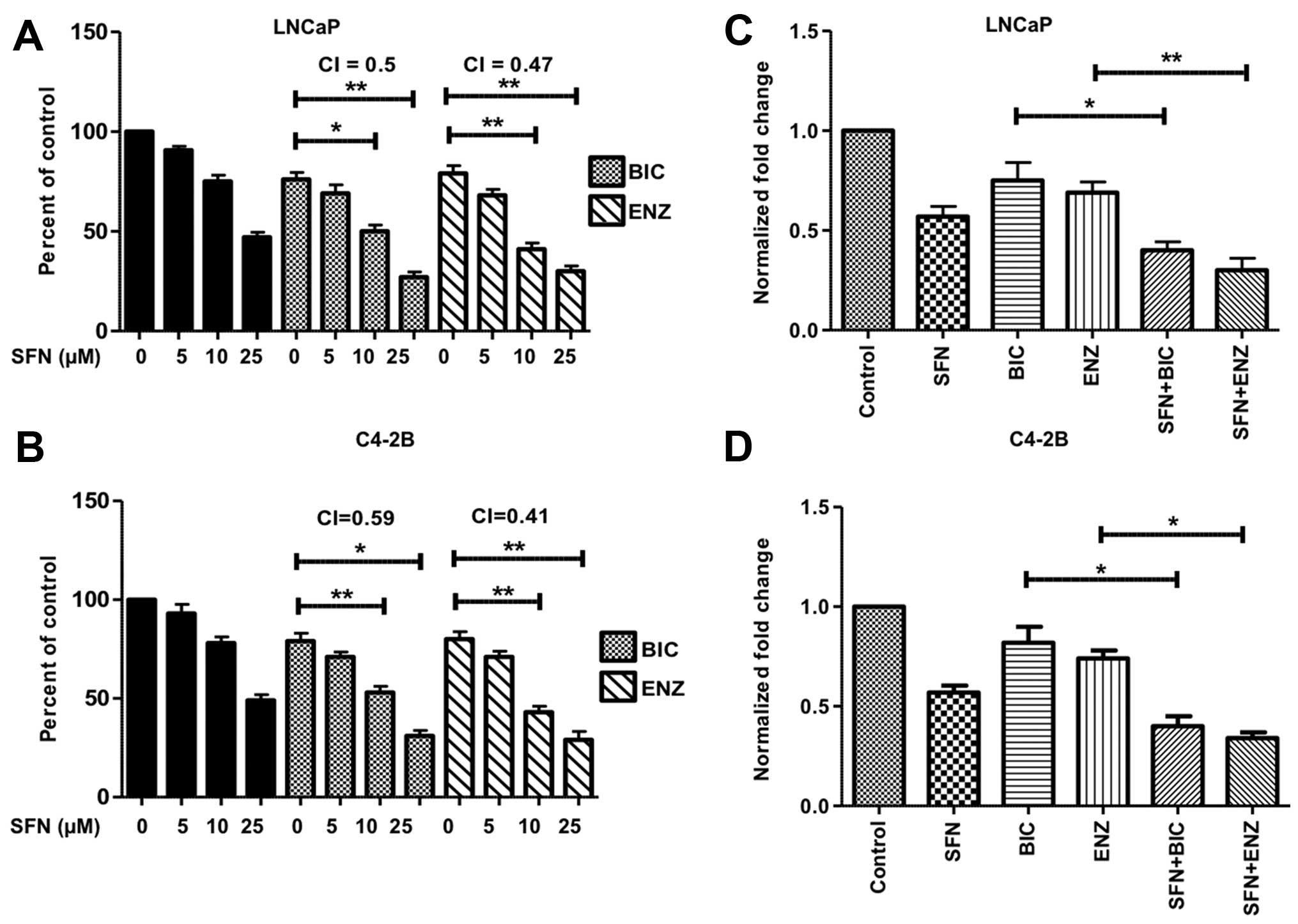
Co-exposure to SFN increases the
suppressive effects of anti-androgens on proliferation and PSA
expression
As expected, exposure of both LNCaP and C4-2B cells
to the anti-androgens for 24–72 h showed lower IC50
values for the more potent agent, ENZ (Table I). To assess possible synergistic
effects in combination with SFN, subsequent studies utilized the
sub-IC50 values of BIC (50 μM) and ENZ (20 μM). The
highest level of synergy was observed after 48 h of treatment and
the data are presented in the bar graphs (Fig. 2). More than 50% decrease in
viability was observed in both LNCaP (Fig. 2A) and C4-2B (Fig. 2B) cells when SFN (10–25 μM) was
used in combination with BIC or ENZ. The combination index (CI)
values suggested synergistic increase in cytotoxicity in both LNCaP
and C4-2B cells. Similarly, SFN co-exposure significantly enhanced
the efficacy of BIC or ENZ in reducing the secretory levels of PSA
in both LNCaP (Fig. 2C) and C4-2B
(Fig. 2D) cells.
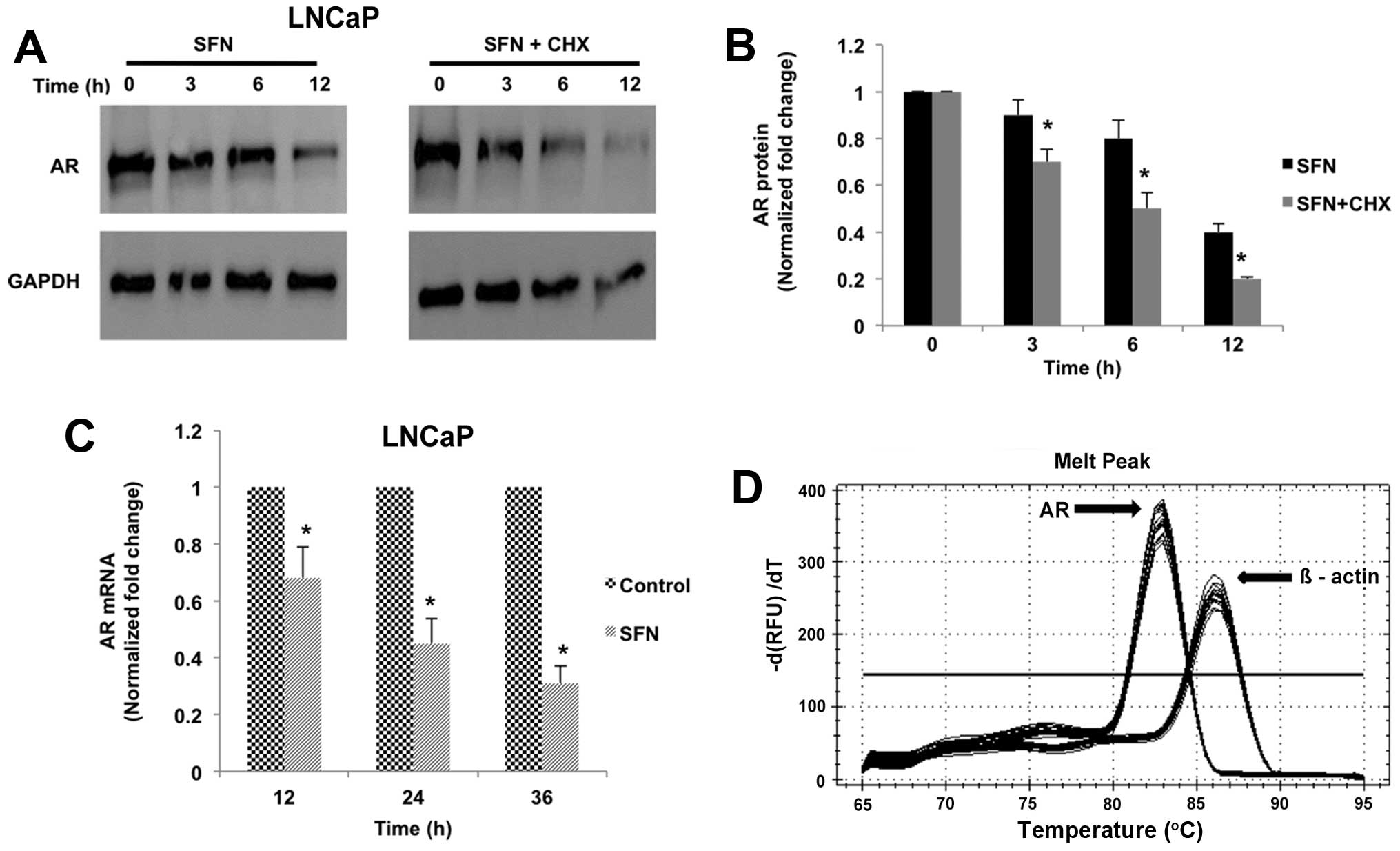
SFN-mediated AR suppression is regulated
at both post-translational and transcriptional levels
Immunoblot analysis in LNCaP cells showed that SFN
reduced AR protein levels in a time-dependent manner (3, 6 and 12
h); however, pre-treatment with the protein-synthesis inhibitor,
CHX did not significantly alter this reduction (Fig. 3A and B). This suggested that SFN
regulates AR at the post-translational level, possibly via
promoting AR protein degradation. Notably, qRT-PCR analysis showed
that continued SFN treatment (12, 24 and 36 h) also suppressed AR
gene expression in LNCaP cells (Fig.
3C and D). Taken together, these results suggest that the
potent AR suppressive effects of SFN may be manifested via its
actions on both pre- and post-translational mechanisms of AR
regulation.
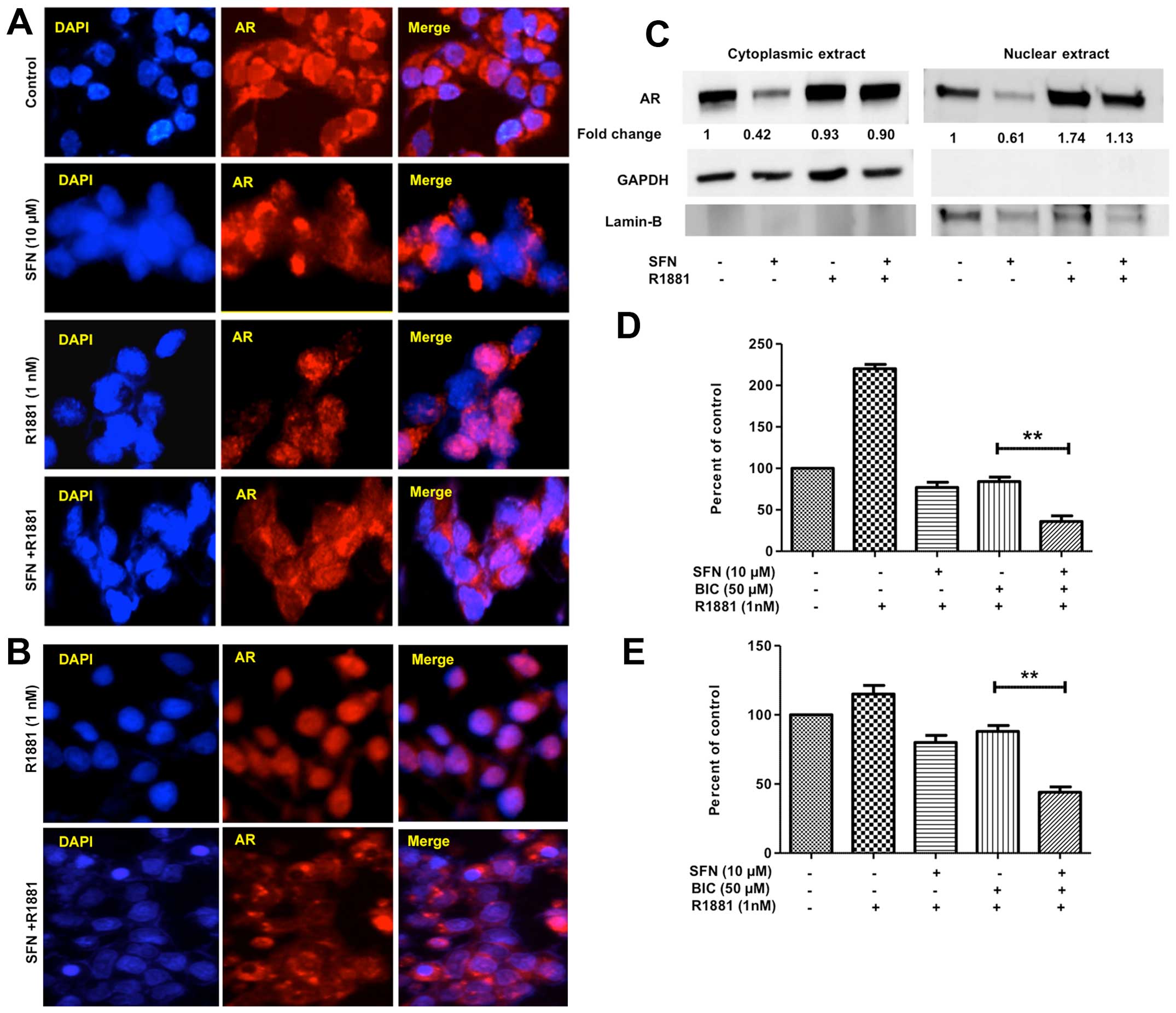
SFN suppresses R1881-induced nuclear AR
translocation and cell growth
Since AR is a transcription factor and localizes to
the nucleus following ligand (androgen) binding (2), we monitored the effect of SFN on both
basal AR levels and androgen-induced nuclear AR translocation
(Fig. 4A–C). We also monitored the
effect of SFN on androgen-induced cell proliferation in PCa cells
(Fig. 4D and E). Consistent with
SFN-mediated reduction in total AR protein levels (Fig. 1), immunofluorescence studies
revealed that 6 h of pre-exposure to SFN (10 μM) decreased both
basal as well as R1881-induced AR levels in both cytoplasm and
nuclei of LNCaP and C4-2B cells (Fig.
4A and B). These observations were confirmed by immunoblotting
studies to document AR levels in cytoplasmic and nuclear fractions
of LNCaP cells (Fig. 4C). In LNCaP
cells (Fig. 4D) long-term exposure
to R1881 increased cell growth by >2-fold, and to a lesser
extent in the C4-2B cells (Fig.
4E). This growth stimulatory effect of R1881 was partially
inhibited by treatment with SFN or BIC alone, and was significantly
abrogated with combined exposure to SFN and BIC, as evident in both
LNCaP and C4-2B cells (Fig. 4D and
E).
SFN enhances the ability of
anti-androgens to suppress PCa cell migration
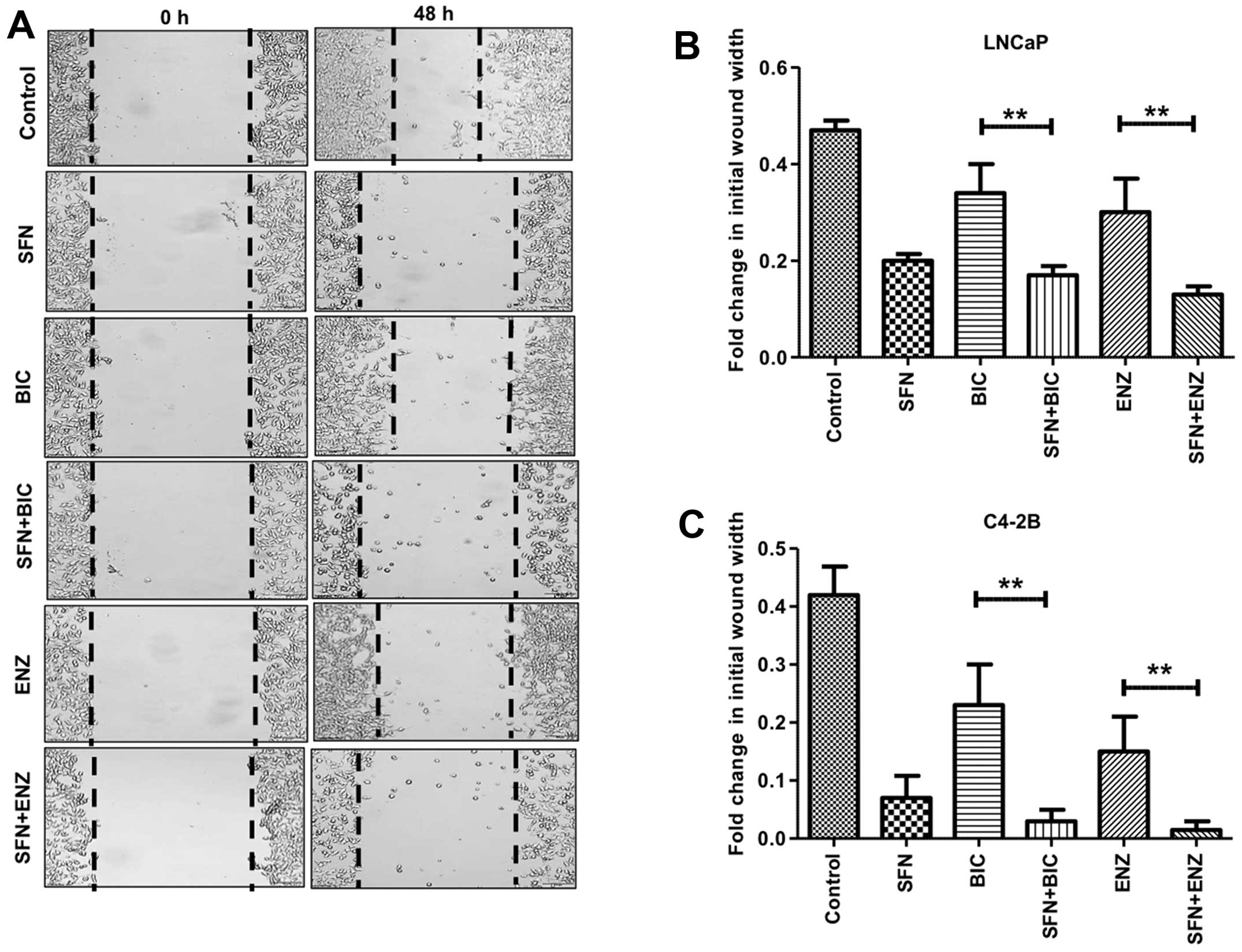
We carried out wound-healing assays to monitor the
effect of SFN, alone and/or in combination with anti-androgens
(BIC/ENZ) on cell migration behavior (Fig. 5). In control cells, the wound-width
decreased by ~50% after 48 h in culture, indicating robust cell
migration under normal conditions. Exposure to BIC or ENZ alone
showed little effect in decreasing cell migration in LNCaP cells
(Fig. 5B). However, exposure to
even SFN alone enabled <50% wound closure and combined exposure
to SFN+BIC or SFN+ENZ showed as little as 10–15% wound closure.
Interestingly, this suppressive effect of SFN on migratory behavior
of PCa was more pronounced in the C4-2B cells (Fig. 5A and C). Although, BIC or ENZ alone
showed only slight decrease in migratory behavior, exposure to SFN
alone decreased migration to <10% and combined treatment with
SFN and anti-androgens completely suppressed cell migration. In
fact, as compared to the initial time-point of wounding, very
little wound-healing was evident at 48 h in the combined drug
treatment groups. These experiments demonstrated the efficacy of
SFN in suppressing the migratory phenotype of antiandrogen treated
PCa cells.
SFN enhances the ability of
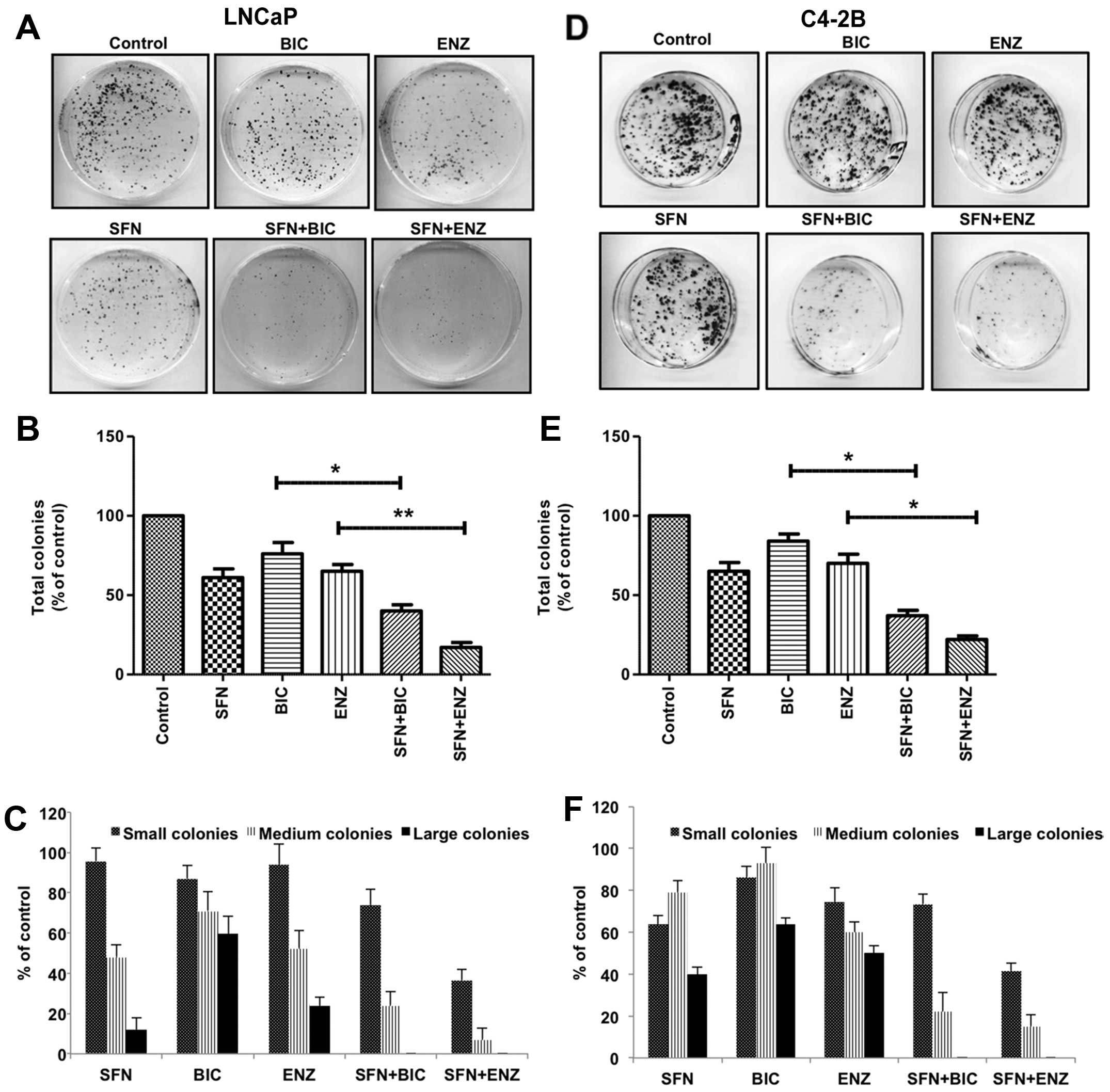
anti-androgens to suppress clonogenic ability of PCa cells
To examine the long-term (14 days) effects of
drug(s), IC50 values for SFN, BIC and ENZ on
colony-forming ability for LNCaP and C4-2B cells was determined
(Table II); the doses that caused
<50% decrease in CFUs were used for combination studies. SFN
exposure caused ~40–45% decrease in the total number of CFUs in PCa
cells, which were higher than that observed with BIC or ENZ alone.
Both SFN+BIC and SFN+ENZ combinations showed significantly striking
effect in suppressing the total number of CFUs in both LNCaP
(Fig. 6A and B) and C4-2B
(Fig. 6D and E) cells, as compared
to anti-androgens alone. Notably, the inhibitory effect of SFN
(either alone or in combination with BIC/ENZ) was more pronounced
for large size CFUs as compared to small and medium size CFUs in
both LNCaP (Fig. 6C) and C4-2B
cells (Fig. 6F). These results
suggest that co-exposure to SFN can augment the efficacy of
anti-androgens to suppress the clonogenic ability of aggressive PCa
cells.
| Table IIIC50 values for drugs at
14 days (CFU assay). |
Table II
IC50 values for drugs at
14 days (CFU assay).
| LNCaP | C4-2B |
|---|
|
|
|
|---|
| Drugs | IC50 ±
SEM | IC50 ±
SEM |
|---|
| SFN (μM) | 0.3±0.01 | 0.38±0.02 |
| BIC (μM) | 2±0.12 | 2.4±0.2 |
| ENZ (μM) | 0.55±0.029 | 0.6±0.035 |
Discussion
Following anti-androgen therapy, the more aggressive
PCa cells select for the resistant CRPC phenotype. A continuous AR
signaling is considered to be a key factor in this inevitable
transition (2,3,5–7). A
better ablation of AR signaling will enhance the initial anticancer
efficacy of clinically approved anti-androgens and thus, delay the
outgrowth of CRPC. Although a number of studies have previously
shown the anti-cancer effects of SFN in PCa cells (26,27,30–33),
this study is the first to demonstrate the utility of SFN in
combination with anti-androgens like BIC and ENZ. In the present
in vitro investigations, we show that a rapid, substantial
and synergistic sensitization of both androgen-dependent (LNCaP) as
well as androgen-independent (C4-2B) PCa cells can be accomplished
when SFN is used in combination with anti-androgens. This
sensitizing ability of SFN functions via the suppression of AR
levels, and decreasing effects of androgen signaling on growth,
migration and clonogenic ability.
Previous studies have shown that SFN can suppress AR
protein levels in different PCa cell lines (36–38).
This study further supported a concentration- and time-dependent
effect of sub-toxic SFN doses on AR in both LNCaP and C4-2B cells
(Fig. 1). LNCaP is an
androgen-dependent cell line expressing both AR and PSA
mRNA/protein which was first isolated from a human metastatic
prostate adenocarcinoma in the lymph node (42,43).
C4-2B is AR/PSA expressing bone metastatic CRPC subline derived
from LNCaP (39,43). Although these cells can grow in
androgen depleted conditions, they need AR both for cell growth and
for PSA expression (44). They
have also been shown to grow persistently both in intact as well as
castrated mice (43). Thus, C4-2B
cells are androgen-independent, but AR dependent similar to LNCaP
cells. AR has been reported to remain fully functional in hormone
refractory PCa. Ligand-independent activation of AR by
cytokines/growth factors, intratumoral/intracrine androgen
production and overexpression of AR are mainly responsible for the
survival of androgen-independent cells even in the castrate
concentrations of androgens (19–21,44).
Thus, SFN acts in a similar fashion to degrade AR through the most
recognized proteasomal pathway in both androgen-dependent LNCaP and
androgen-independent C4-2B cells. Interestingly, we observed that
the downregulation of AR protein is more pronounced in the C4-2B
cells, as compared to LNCaP cells. We established that
sub-IC50 doses of SFN can rapidly suppress AR in both
cell lines and these concentrations of SFN significantly decreased
the IC50 of both anti-androgens (Fig. 2A and B).
A number of earlier studies have also shown
multimodal actions of SFN in suppressing AR levels in PCa cells
(36–38). AR protein is protected from
proteasomal degradation via the multi-chaperone complex of heat
shock proteins (HSPs) (45).
Indeed, HSP-90 inhibitors are known to target AR for proteasomal
degradation (46). Previous
studies have shown that SFN inhibits HDAC-6. This leads to the
hyperacetylation and inactivation of HSP-90 that is responsible for
suppressing AR levels (38,47).
Androgen binding to AR also dissociates AR from HSP-90 and enables
its nuclear translocation (45,46).
ADT increases oxidative stress and proteasomal
inhibition, which increases AR stability (48). Antioxidants such as SFN can
suppress this effect by increasing Nrf-2 (27,33,35).
Previous studies from our laboratory have shown that Nrf-2
downregulates AR transactivation (34). Our current observations further
support the conclusions that SFN co-exposure with BIC and ENZ in
both androgen-dependent as well as androgen-independent cells leads
to: i) significant increase in the anti-proliferative effects (MTT
assay); ii) decrease in the secreted levels of AR-regulated gene
product PSA (ELISA); iii) decrease in the colony number and size
(CFU assay); iv) decrease in the migration ability (wound healing
assay); and (v) decrease in the stimulatory growth effects of
synthetic AR agonist (R1881) compared to anti-androgens (BIC and
ENZ) alone.
The PSA gene is a well-established target of AR
signaling, and secreted levels of PSA is often used as a biomarker
of PCa growth (4). Anti-androgens,
both BIC and ENZ, are known to suppress serum PSA levels (49) but their efficacy in combination
with SFN has not been tested before. In the present study, we
reported a significant suppression in the secretory levels of PSA
in the conditioned media of both LNCaP and C4-2B cells after
combined exposure to SFN and anti-androgens (Fig. 2C and D). Thus, our findings
corroborate that SFN potentiates the efficiency of clinically
approved anti-androgens by decreasing AR protein levels and the
transactivation function of AR.
We observed that the rapid effects of SFN in
suppressing AR is primarily regulated at the post-translational
level. Previous studies have shown that SFN enhances AR protein
degradation (37,38) and AR gene expression (36). In this study, pre-treatment with
the protein synthesis inhibitor, CHX could not rescue the decrease
in AR protein by SFN, suggesting regulation at the post-translation
level (Fig. 3A and B).
Investigations on the effect of SFN on AR mRNA levels suggested
that long-term exposure to SFN can result in the downregulation of
AR gene expression as well (Fig.
3C). This may be highly beneficial towards the potent
therapeutic effects of SFN towards long-term abrogation of all
AR-mediated effects on proliferation, invasion and clonogenic
ability of aggressive PCa cells. Past studies have indeed
demonstrated that AR signaling can regulate its own gene expression
(50). Therefore, this dual effect
would potentiate the sensitizing ability of SFN when combined with
anti-androgens.
The potent AR level suppression by SFN will be of
significant advantage in the targeting of CRPC tumors, especially
since low levels of androgen have been reported within tumor
microenvironments even after castration. This has been attributed
to the intratumoral production of androgens by PCa cells via its
de novo biosynthesis from cholesterol and other precursors
(8). Residual androgens are
sufficient to activate AR signaling, stimulate PCa growth, and play
a crucial role in the progression towards CRPC tumors (51,52).
Thus, the continuous nuclear localization of AR and transcription
of AR regulated genes remains a significant problem despite
systemic ADT. We observed that SFN exposure could inhibit the
nuclear translocation of AR even in the presence of the synthetic
AR agonist, R1881 (Fig. 4A and B).
This AR sequestration in the cytosol by SFN was corroborated by
both immunofluorescence microscopy and immunoblot analysis of
nuclear/cytoplasmic fractions (Fig.
4C). Numerous studies have shown that the continuous AR
signaling activates mitogenic pathways in PCa cells (2,3).
Therefore, its effective blockade should enable suppression of
androgen-stimulated PCa growth. Our investigations showed that SFN
co-exposure significantly augmented the anti-proliferative effects
of BIC, in both LNCaP (Fig. 4D)
and C4-2B cells (Fig. 4E) which
were stimulated with R1881. These findings underscore the
importance of using SFN as an adjunct agent to suppress the effects
of intratumoral androgens in CRPC tumors.
Androgen signaling has also been shown to increase
the migratory behavior of PCa cells (2–4). The
characteristics of migration include higher invasive ability of
cells, elevated apoptotic resistance and increased epithelial
mesenchymal transition (EMT) phenotype, crucial determinants of
tumor metastasis (40,53). Interestingly, despite their potent
anti-proliferative effects, anti-androgens have not shown
significant effects towards suppressing PCa cell migration
(49). The present study clearly
shows that SFN co-treatment can impair the migratory ability of
both LNCaP and C4-2B cells, and this effect is potentiated in the
presence of anti-androgens (Fig.
5). In tumor xenograft models, SFN has been reported to inhibit
PCa progression and pulmonary metastasis in vivo (54). Our in vitro finding further
suggests the advantage of using SFN in combination with
anti-androgens to decrease the metastatic behavior of PCa.
The acute cytotoxic effects observed with this drug
combination were further corroborated by chronic exposure in the
CFU assays (Fig. 6). These in
vitro clonogenic assays mimic the seeding and proliferation of
tumor initiating cells (cancer stem cells) and the number of
colonies formed post-drug exposure is proportional to the number of
viable progenitors (55). The
suppressive effect of SFN on tumor initiating cells was reported
earlier (56). This study further
demonstrated that SFN can potentiate the chronic long-term efficacy
of anti-androgens in suppressing the clonogenic potential of both
LNCaP and C4-2B cells. Although we did not investigate the
efficiency of this combination in vivo, the long-term
synergistic effects apparent in our CFU assays, especially with
much lower concentrations of each of the agents, clearly implicate
the potential of combination therapy in suppressing the seeding and
outgrowth of metastatic tumors. Our findings suggest that adjunct
therapy with SFN will be highly beneficial in increasing the
effectiveness of ADT, both at the initiation of therapy in
androgen-dependent PCa cells and during the later stages when PCa
cells are selecting for the androgen-independent phenotype. Chronic
exposure to this safe phytochemical may impede the progression to
CRPC.
Acknowledgements
The present study was supported by funds from the
Louisiana Cancer Research Consortium (LCRC) and the Department of
Defense (DoD, #PC0810811) to D.M. and from the Laboratory Training
funds to S.C.S.
References
|
1
|
Yap TA, Zivi A, Omlin A and de Bono JS:
The changing therapeutic landscape of castration-resistant prostate
cancer. Nat Rev Clin Oncol. 8:597–610. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hodgson MC, Bowden WA and Agoulnik IU:
Androgen receptor footprint on the way to prostate cancer
progression. World J Urol. 30:279–285. 2012. View Article : Google Scholar :
|
|
3
|
Chang KH, Ercole CE and Sharifi N:
Androgen metabolism in prostate cancer: From molecular mechanisms
to clinical consequences. Br J Cancer. 111:1249–1254. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ryan CJ, Smith A, Lal P, Satagopan J,
Reuter V, Scardino P, Gerald W and Scher HI: Persistent
prostate-specific antigen expression after neoadjuvant androgen
depletion: An early predictor of relapse or incomplete androgen
suppression. Urology. 68:834–839. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Harris WP, Mostaghel EA, Nelson PS and
Montgomery B: Androgen deprivation therapy: Progress in
understanding mechanisms of resistance and optimizing androgen
depletion. Nat Clin Pract Urol. 6:76–85. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Godbole AM and Njar VC: New insights into
the androgen-targeted therapies and epigenetic therapies in
prostate cancer. Prostate Cancer. 2011:9187072011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim W and Ryan CJ: Androgen receptor
directed therapies in castration-resistant metastatic prostate
cancer. Curr Treat Options Oncol. 13:189–200. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu C, Lou W, Zhu Y, Yang JC, Nadiminty N,
Gaikwad NW, Evans CP and Gao AC: Intracrine androgens and AKR1C3
activation confer resistance to enzalutamide in prostate cancer.
Cancer Res. 75:1413–1422. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schrader AJ, Schrader MG and Cronauer MV:
Words of wisdom. Re: Androgen receptor splice variants mediate
enzalutamide resistance in castration-resistant prostate cancer
cell lines. Eur Urol. 64:169–170. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Arora VK, Schenkein E, Murali R, Subudhi
SK, Wongvipat J, Balbas MD, Shah N, Cai L, Efstathiou E, Logothetis
C, et al: Glucocorticoid receptor confers resistance to
anti-androgens by bypassing androgen receptor blockade. Cell.
155:1309–1322. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Joseph JD, Lu N, Qian J, Sensintaffar J,
Shao G, Brigham D, Moon M, Maneval EC, Chen I, Darimont B, et al: A
clinically relevant androgen receptor mutation confers resistance
to second-generation anti-androgens enzalutamide and ARN-509.
Cancer Discov. 3:1020–1029. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Korpal M, Korn JM, Gao X, Rakiec DP, Ruddy
DA, Doshi S, Yuan J, Kovats SG, Kim S, Cooke VG, et al: An F876L
mutation in androgen receptor confers genetic and phenotypic
resistance to MDV3100 (enzalutamide). Cancer Discov. 3:1030–1043.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kawata H, Ishikura N, Watanabe M,
Nishimoto A, Tsunenari T and Aoki Y: Prolonged treatment with
bicalutamide induces androgen receptor overexpression and androgen
hypersensitivity. Prostate. 70:745–754. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Colabufo NA, Pagliarulo V, Berardi F,
Contino M, Inglese C, Niso M, Ancona P, Albo G, Pagliarulo A and
Perrone R: Bicalutamide failure in prostate cancer treatment:
Involvement of Multi Drug Resistance proteins. Eur J Pharmacol.
601:38–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bohl CE, Gao W, Miller DD, Bell CE and
Dalton JT: Structural basis for antagonism and resistance of
bicalutamide in prostate cancer. Proc Natl Acad Sci USA.
102:6201–6206. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Saad F, Adachi JD, Brown JP, Canning LA,
Gelmon KA, Josse RG and Pritchard KI: Cancer treatment-induced bone
loss in breast and prostate cancer. J Clin Oncol. 26:5465–5476.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Salturk Z, Çakır O, Kumral TL, Yıldırım G,
Ötünçtemur A, Aydoğdu Ï and Uyar Y: Subjective and objective
effects of androgen ablation therapy on voice. J Voice. 29:490–493.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
McCarty MF, Hejazi J and Rastmanesh R:
Beyond androgen deprivation: Ancillary integrative strategies for
targeting the androgen receptor addiction of prostate cancer.
Integr Cancer Ther. 13:386–395. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lamont KR and Tindall DJ: Minireview:
Alternative activation pathways for the androgen receptor in
prostate cancer. Mol Endocrinol. 25:897–907. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Brooke GN and Bevan CL: The role of
androgen receptor mutations in prostate cancer progression. Curr
Genomics. 10:18–25. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Armstrong CM and Gao AC: Drug resistance
in castration resistant prostate cancer: Resistance mechanisms and
emerging treatment strategies. Am J Clin Exp Urol. 3:64–76.
2015.PubMed/NCBI
|
|
22
|
Elbarbry F and Elrody N: Potential health
benefits of sulforaphane: A review of the experimental, clinical
and epidemiological evidences and underlying mechanisms. J Med
Plants Res. 5:473–484. 2011.
|
|
23
|
Zhang Y and Tang L: Discovery and
development of sulforaphane as a cancer chemopreventive
phytochemical. Acta Pharmacol Sin. 28:1343–1354. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Clarke JD, Dashwood RH and Ho E:
Multi-targeted prevention of cancer by sulforaphane. Cancer Lett.
269:291–304. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cheung KL and Kong AN: Molecular targets
of dietary phenethyl isothiocyanate and sulforaphane for cancer
chemoprevention. AAPS J. 12:87–97. 2010. View Article : Google Scholar :
|
|
26
|
Traka MH, Melchini A and Mithen RF:
Sulforaphane and prostate cancer interception. Drug Discov Today.
19:1488–1492. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Keum YS, Khor TO, Lin W, Shen G, Kwon KH,
Barve A, Li W and Kong AN: Pharmacokinetics and pharmacodynamics of
broccoli sprouts on the suppression of prostate cancer in
transgenic adenocarcinoma of mouse prostate (TRAMP) mice:
Implication of induction of Nrf2, HO-1 and apoptosis and the
suppression of Akt-dependent kinase pathway. Pharm Res.
26:2324–2331. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shapiro TA, Fahey JW, Dinkova-Kostova AT,
Holtzclaw WD, Stephenson KK, Wade KL, Ye L and Talalay P: Safety,
tolerance, and metabolism of broccoli sprout glucosinolates and
isothiocyanates: a clinical phase I study. Nutr Cancer. 55:53–62.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Petri N, Tannergren C, Holst B, Mellon FA,
Bao Y, Plumb GW, Bacon J, O’Leary KA, Kroon PA, Knutson L, et al:
Absorption/metabolism of sulforaphane and quercetin, and regulation
of phase II enzymes, in human jejunum in vivo. Drug Metab Dispos.
31:805–813. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Singh SV, Srivastava SK, Choi S, Lew KL,
Antosiewicz J, Xiao D, Zeng Y, Watkins SC, Johnson CS, Trump DL, et
al: Sulforaphane-induced cell death in human prostate cancer cells
is initiated by reactive oxygen species. J Biol Chem.
280:19911–19924. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xiao D, Powolny AA, Antosiewicz J, Hahm
ER, Bommareddy A, Zeng Y, Desai D, Amin S, Herman-Antosiewicz A and
Singh SV: Cellular responses to cancer chemopreventive agent
D,L-sulforaphane in human prostate cancer cells are initiated by
mitochondrial reactive oxygen species. Pharm Res. 26:1729–1738.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pei Y, Wu B, Cao Q, Wu L and Yang G:
Hydrogen sulfide mediates the anti-survival effect of sulforaphane
on human prostate cancer cells. Toxicol Appl Pharmacol.
257:420–428. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang C, Su ZY, Khor TO, Shu L and Kong
AN: Sulforaphane enhances Nrf2 expression in prostate cancer TRAMP
C1 cells through epigenetic regulation. Biochem Pharmacol.
85:1398–1404. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schultz MA, Hagan SS, Datta A, Zhang Y,
Freeman ML, Sikka SC, Abdel-Mageed AB and Mondal D: Nrf1 and Nrf2
transcription factors regulate androgen receptor transactivation in
prostate cancer cells. PLoS One. 9:e872042014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kensler TW, Egner PA, Agyeman AS,
Visvanathan K, Groopman JD, Chen JG, Chen TY, Fahey JW and Talalay
P: Keap1-nrf2 signaling: A target for cancer prevention by
sulforaphane. Top Curr Chem. 329:163–177. 2013. View Article : Google Scholar :
|
|
36
|
Kim SH and Singh SV: D,L-Sulforaphane
causes transcriptional repression of androgen receptor in human
prostate cancer cells. Mol Cancer Ther. 8:1946–1954. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wiczk A, Hofman D, Konopa G and
Herman-Antosiewicz A: Sulforaphane, a cruciferous vegetable-derived
isothiocyanate, inhibits protein synthesis in human prostate cancer
cells. Biochim Biophys Acta. 1823:1295–1305. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gibbs A, Schwartzman J, Deng V and Alumkal
J: Sulforaphane destabilizes the androgen receptor in prostate
cancer cells by inactivating histone deacetylase 6. Proc Natl Acad
Sci USA. 106:16663–16668. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wu HC, Hsieh JT, Gleave ME, Brown NM,
Pathak S and Chung LW: Derivation of androgen-independent human
LNCaP prostatic cancer cell sublines: Role of bone stromal cells.
Int J Cancer. 57:406–412. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Uygur B and Wu WS: SLUG promotes prostate
cancer cell migration and invasion via CXCR4/CXCL12 axis. Mol
Cancer. 10:1392011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chou TC: Drug combination studies and
their synergy quantification using the Chou-Talalay method. Cancer
Res. 70:440–446. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Horoszewicz JS, Leong SS, Chu TM, Wajsman
ZL, Friedman M, Papsidero L, Kim U, Chai LS, Kakati S, Arya SK, et
al: The LNCaP cell line - a new model for studies on human
prostatic carcinoma. Prog Clin Biol Res. 37:115–132. 1980.
|
|
43
|
Cunningham D and You Z: In vitro and in
vivo model systems used in prostate cancer research. J Biol
Methods. 2:pii. e172015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Agoulnik IU, Vaid A, Bingman WE, Erdeme H,
Frolov A, Smith CL, Ayala G, Ittmann MM and Weigel NL: Role of
SRC-1 in the promotion of prostate cancer cell growth and tumor
progression. Cancer Res. 65:7959–7967. 2005.PubMed/NCBI
|
|
45
|
He S, Zhang C, Shafi AA, Sequeira M,
Acquaviva J, Friedland JC, Sang J, Smith DL, Weigel NL, Wada Y, et
al: Potent activity of the Hsp90 inhibitor ganetespib in prostate
cancer cells irrespective of androgen receptor status or variant
receptor expression. Int J Oncol. 42:35–43. 2013.
|
|
46
|
Ai J, Wang Y, Dar JA, Liu J, Liu L, Nelson
JB and Wang Z: HDAC6 regulates androgen receptor hypersensitivity
and nuclear localization via modulating Hsp90 acetylation in
castration-resistant prostate cancer. Mol Endocrinol. 23:1963–1972.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Myzak MC, Tong P, Dashwood WM, Dashwood RH
and Ho E: Sulforaphane retards the growth of human PC-3 xenografts
and inhibits HDAC activity in human subjects. Exp Biol Med
(Maywood). 232:227–234. 2007.
|
|
48
|
Shiota M, Yokomizo A and Naito S:
Oxidative stress and androgen receptor signaling in the development
and progression of castration-resistant prostate cancer. Free Radic
Biol Med. 51:1320–1328. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Demir A, Cecen K, Karadag MA, Kocaaslan R
and Turkeri L: The course of metastatic prostate cancer under
treatment. Springerplus. 3:7252014. View Article : Google Scholar
|
|
50
|
Lee JG, Zheng R, McCafferty-Cepero JM,
Burnstein KL, Nanus DM and Shen R: Endothelin-1 enhances the
expression of the androgen receptor via activation of the c-myc
pathway in prostate cancer cells. Mol Carcinog. 48:141–149. 2009.
View Article : Google Scholar
|
|
51
|
Mostaghel EA and Nelson PS: Intracrine
androgen metabolism in prostate cancer progression: Mechanisms of
castration resistance and therapeutic implications. Best Pract Res
Clin Endocrinol Metab. 22:243–258. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Mostaghel EA, Page ST, Lin DW, Fazli L,
Coleman IM, True LD, Knudsen B, Hess DL, Nelson CC, Matsumoto AM,
et al: Intraprostatic androgens and androgen-regulated gene
expression persist after testosterone suppression: Therapeutic
implications for castration-resistant prostate cancer. Cancer Res.
67:5033–5041. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Huo C, Kao YH and Chuu CP: Androgen
receptor inhibits epithelial-mesenchymal transition, migration, and
invasion of PC-3 prostate cancer cells. Cancer Lett. 369:103–111.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Singh SV, Warin R, Xiao D, Powolny AA,
Stan SD, Arlotti JA, Zeng Y, Hahm ER, Marynowski SW, Bommareddy A,
et al: Sulforaphane inhibits prostate carcinogenesis and pulmonary
metastasis in TRAMP mice in association with increased cytotoxicity
of natural killer cells. Cancer Res. 69:2117–2125. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Aapro MS, Eliason JF, Krauer F and Alberto
P: Colony formation in vitro as a prognostic indicator for primary
breast cancer. J Clin Oncol. 5:890–896. 1987.PubMed/NCBI
|
|
56
|
Kallifatidis G1, Rausch V, Baumann B, Apel
A, Beckermann BM, Groth A, Mattern J, Li Z, Kolb A, Moldenhauer G,
et al: Sulforaphane targets pancreatic tumour-initiating cells by
NF-κB-induced antiapoptotic signaling. Gut. 58:949–963. 2009.
View Article : Google Scholar
|