Introduction
Anaplastic thyroid cancer (ATC) is a rare orphan
disease that accounts for 1–3% of thyroid cancers. ATCs progress
rapidly and are extremely aggressive toward both adjacent organs by
invasion and distant organs by hematological dissemination. Because
of the highly malignant potential, ATC cases often become lethal
within 6 months from the initial diagnosis, despite intensive
therapeutic efforts (1–3). No standardized therapeutic strategy
has been documented to manage ATC, and experimental multimodal
therapies with surgery, chemotherapy and/or radiation therapy have
been attempted practically. Regrettably, no effective therapeutic
method for ATC has been established to date (4,5).
However, several molecular targeted therapies have
achieved successful results against ATCs (6–8).
Rosove et al (9) reported an impressive case of an ATC
patient successfully treated with a selective BRAFV600E
inhibitor, vemurafenib. Possible clinical application of this
inhibitor has been demonstrated recently in a phase 2 trial in
BRAFV600E mutation-positive ATC patients, demonstrating
an overall response rate of 29% (2/7) (10). In a study by Kim et al
(11), BRAF mutation in papillary
thyroid cancer (PTC) was found more frequently in East Asian
countries compared to the Western countries, and the proportion of
PTCs among differentiated thyroid cancers (DTCs) was higher in
Japan than Western countries. A considerable proportion of ATCs is
thought to be derived from long-lasting DTC, and BRAF mutation was
found to be maintained during the phenotypical change from DTC to
ATC (12). Although the incidence
of BRAF mutation was reported to be relatively less common in ATC
(15–24%) than that found in PTC (13–15),
a preliminary finding indicated that the rate of BRAF mutation in a
population of Japanese ATC patients was high (6 of 14 patients)
[Uchino, et al, Proceedings of the 20th Annual Meeting of
Japan Association of Endocrine Surgeons, O-11 65, 2008 (In
Japanese)]. In addition, six of seven thyroid cancer cell lines in
our series have a BRAF mutation (16). These observations suggest that the
frequency of BRAF mutation in ATC is much higher in Japan compared
to Western countries.
A previous study of our group demonstrated a
possible effect of molecular therapies targeting epidermal growth
factor receptor (EGFR), although the effect was limited to the
cells with a preserved RAS/RAF/MEK pathway (17). Our more recent study demonstrated
that part of this EGFR-targeted therapy resistance could be
overcome with an mTOR inhibitor, although we again observed that
the efficacy was limited to the cells with an altered PI3K/AKT/mTOR
pathway (18). These observations
indicated the importance of direct targeting to the RAS/RAF/MEK
pathway to manage ATC.
Another research group also demonstrated the
importance of BRAF mutation in the aggressive characteristics of
thyroid cancer and the efficacy of its inhibition on the management
of the disease (19). Several
studies described important roles of BRAF gene alteration in
genome-wide aberrant methylation (20), vascular endothelial growth factor
(VEGF) expression (21), and the
induction of epithelial-mesenchymal transformation (EMT) (22).
We conducted a preclinical investigation of the
efficacy of inhibiting the RAS/RAF/MEK pathway in a series of
authentic ATC cell lines harboring a genetic alteration in either
BRAF or NRAS (16). Our specific
aims were to determine the efficacies of BRAF/MEK inhibitors in ATC
cells and to identify possible differences in the mechanism of
blockade between the cell lines according to the differences in the
genetic alterations of the cell lines, the levels of VEGF
secretion, and/or the expression of EMT markers.
Materials and methods
Cell lines
Four human ATC cell lines, ACT-1, OCUT-2, OCUT-4 and
OCUT-6 were cultured in Dulbecco’s modified Eagle’s medium (DMEM)
supplemented with 10% fetal bovine serum (FBS), 100 IU/ml of
penicillin and 100 μg/ml of streptomycin at 37°C with 5%
CO2 in a humidified condition. The ACT-1 cell line was
kindly provided by Dr S. Ohata of Tokushima University. The other
three cell lines were established in our institute (16). The OCUT-4 cell line had a BRAF
(1799T>A; V600E) gene mutation. The OCUT-2 cell line had both
BRAF (1799T>A; V600E) and PI3KCA (3140A>G; H1047R) gene
mutations. The ACT-1 line harbored the wild-type BRAF gene and an
NRAS (181C>A; Q61K) mutation. The OCUT-6 cells had the wild-type
BRAF gene and an NRAS (182A>G; Q61R) mutation.
Inhibitors and drugs
Dabrafenib and trametinib were provided by Novartis
(Basel, Switzerland).
Cell viability after exposure to the
inhibitors
Cells (1×103) were seeded in each well of
a 96-well plastic culture plate and left overnight. They were then
treated with the intended doses of inhibitors for 72 h. After the
incubation period, MTT reagent
(3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium
bromide, Dojindo Laboratories, Kumamoto, Japan) was added to the
final concentration of 0.5 mg/ml, and the cells were incubated
again for 2 h under the same condition. The culture plate was
centrifuged at 200 g for 5 min, and the supernatant was removed.
Dimethyl sulfoxide was added for reaction, and the absorbency at
570 nm was measured with a microplate reader (Infinite F50, Tecan
Trading, Männedorf, Switzerland) and calculated using the supplied
software. The experiments were carried out three times
independently, in triplicate each time, and the average values of
the three independent experiments were calculated (17).
Western blotting
Cells were incubated in 10 ml of DMEM containing
1,000 nM dabrafenib or 500 nM trametinib for 1 h. The cells were
then rinsed with phosphate-buffered saline (PBS) and lysed with
Pro-Prep (iNtRON Biotechnology, Kyungki-Do, Korea). After the
protein concentration of each sample was adjusted, the lysates were
electrophoretically separated using 4–12% Tris/Gly gels (Novex,
Carlsbad, CA, USA) and transferred to a polyvinylidene difluoride
membrane (Trans-Blot Turbo Transfer Pack, Bio-Rad, Hercules, CA,
USA). Membranes were blocked with skim milk and incubated either
with anti-human p44/42 MAPK antibody (#4695S; Cell Signaling
Technology, Beverly, MA, USA), anti-human phospho-p44/42 MAPK
antibody (T202/Y204) (#9101S; Cell Signaling Technology),
anti-human MEK1/2 antibody (#8727S; Cell Signaling Technology),
anti-human phospho-MEK1/2 (S217/221) (#9154S; Cell Signaling
Technology) and anti-human β-actin antibody (#4963; Cell Signaling
Technology) using SNAP i.d. (Merck, Darmstadt, Germany). The bands
were detected using an enhanced chemiluminescence system
(ImageQuant LAS 4000mini, General Electric, Fairfield, CA,
USA).
Cell cycle analysis by flow
cytometry
The cells treated with 100 nM of dabrafenib or 5 nM
of trametinib for 24 h were collected after brief trypsinization,
washed with PBS and fixed with 70% cold ethanol. The samples were
then treated with ribonuclease (R6513: Sigma-Aldrich, St. Louis,
MO, USA), stained with 10 mg/ml propidium iodine and analyzed by a
cell sorter (FACScan, Becton-Dickinson, Mountain View, CA, USA).
The cell cycle distributions were quantified using CellQuest
software (17).
Measurement of VEGF secretion
Approximately 1×105 cells were seeded on
a 10-mm plastic culture plate in 5 ml of culture medium, and
treated with either or both 100 nM of dabrafenib or 5 nM of
trametinib for 24 h. The conditioned medium was then sampled, and
the concentrations of VEGF were measured by an enzyme-linked
immunosorbent assay (ELISA; Mitsubishi, Tokyo, Japan). Culture
medium without cells was used to measure the baseline
concentrations (16).
Reverse transcription-polymerase chain
reaction (RT-PCR)
The cells were treated with 1,000 nM of dabrafenib
or 500 nM of trametinib for 1 h. After incubation, total cellular
RNA was isolated using an RNeasy Mini kit (Qiagen, Hilden, Germany)
and was reverse transcribed into cDNA with the use of ReverTra Ace
qPCR RT Master Mix (Toyobo, Osaka, Japan) according to the
manufacturer’s instructions. Reverse transcription-polymerase chain
reaction (RT-PCR) was performed using a StepOnePlus™ Real-Time PCR
system (Applied Biosystems, Foster City, CA, USA), with
TaqMan® Gene Expression Assays (Thermo Fisher
Scientific, Waltham, MA, USA) for GAPDH (Hs02758991), SNAI1
(Hs00195591), SNAI2 (Hs00950344) and TWIST1 (Hs01675818). The
threshold cycle (CT) values were used to calculate the relative
expression ratios between control and treated cells. We performed
the relative quantification of gene expression by the
2−ΔΔCT method (23).
Results
Cell viability after exposure to the
inhibitors
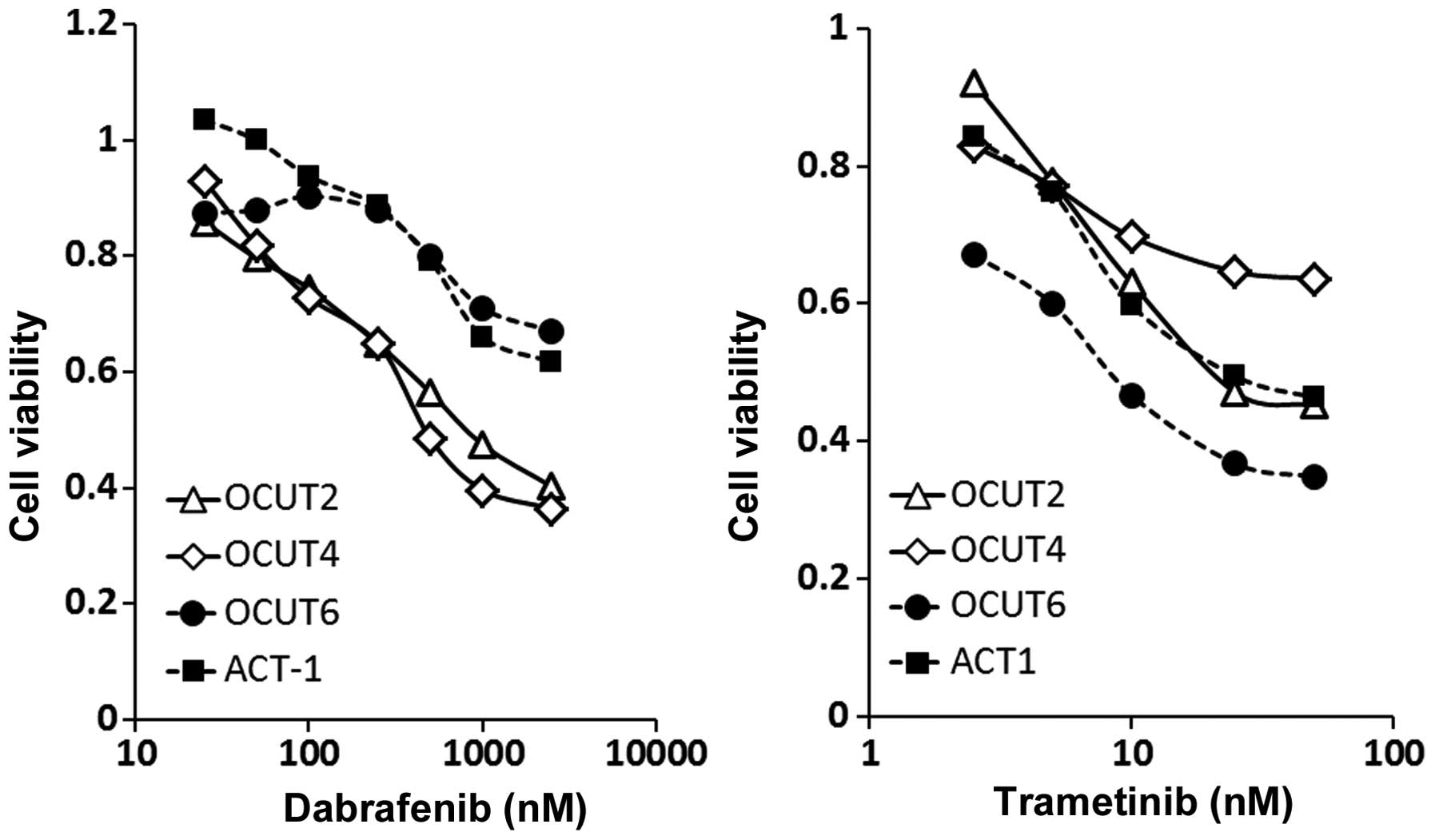
The dabrafenib treatment resulted in dose-dependent
inhibitions of cell viability. The cellular viability was
significantly more strongly inhibited in the OCUT-2 and OCUT-4
cells, which harbor a BRAF V600E mutation, compared to the ACT-1
and OCUT-6 cells, which have the wild-type BRAF gene (Fig. 1, left). The efficacy of trametinib
was found in all four cell lines, with no relationship to the gene
mutation status. The OCUT-6 line (the NRAS mutant) showed the
weakest sensitivity to dabrafenib and the highest sensitivity to
trametinib among all of the cell lines. The OCUT-4 line (the BRAF
mutant) showed the weakest sensitivity to trametinib (Fig. 1, right). Significant impairment of
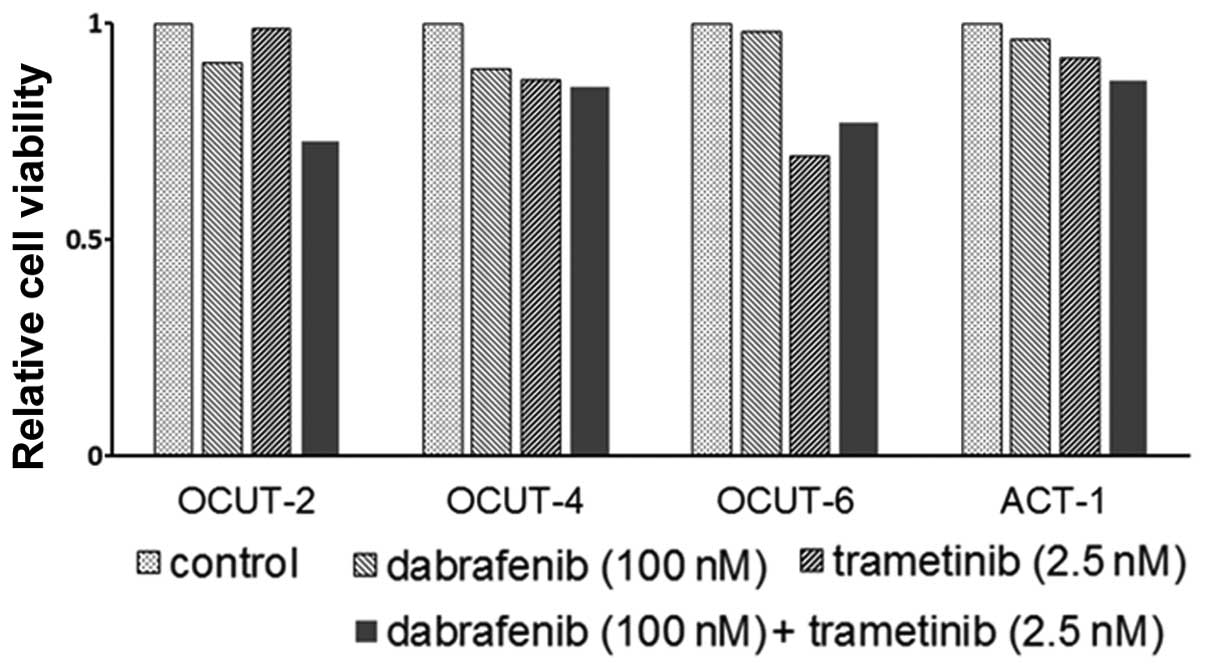
the cellular viability by trametinib in addition to that by
dabrafenib was observed in all cell lines tested (Fig. 2).
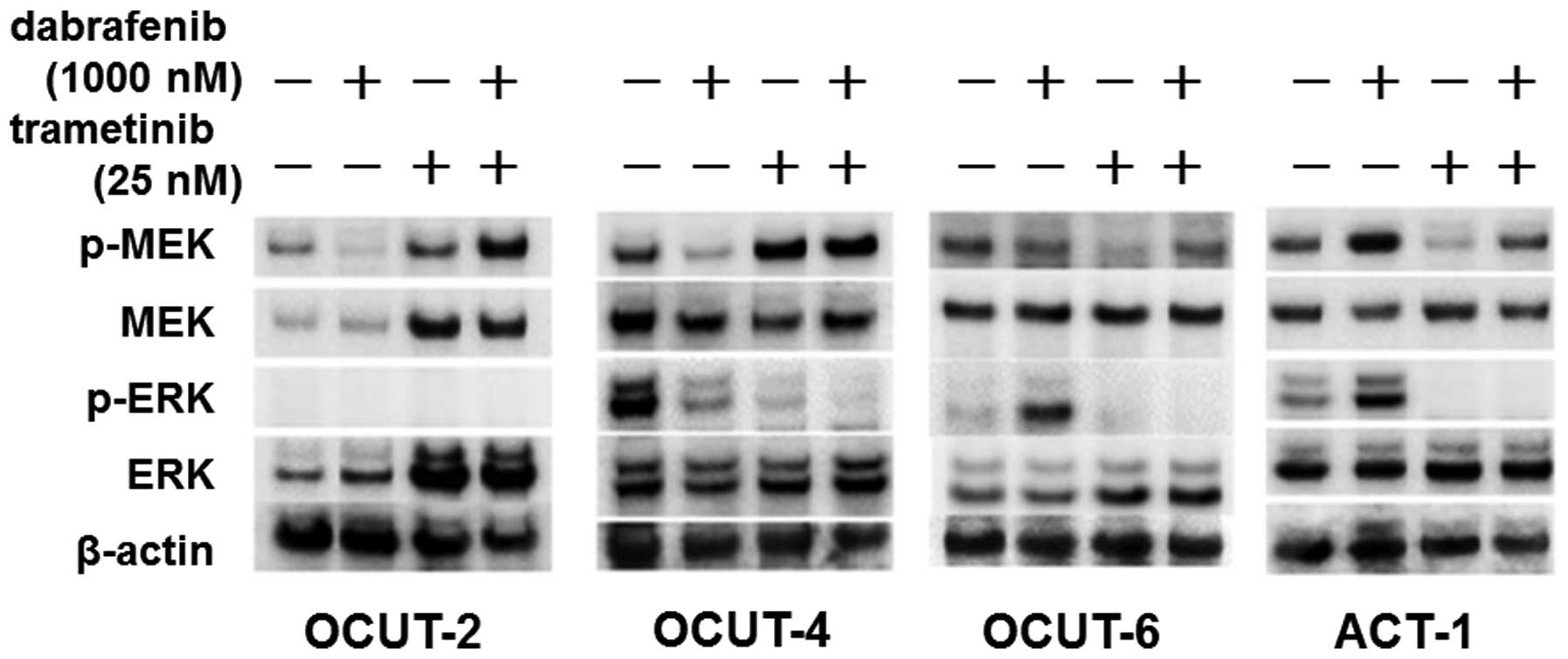
Alteration of the phosphorylation status
of ERK and MEK after exposure to inhibitors
There was a clear downregulation in the
phosphorylation of ERK and MEK after exposure to dabrafenib in the
BRAF mutant cell lines, OCUT-2 and -4. The phosphorylation of ERK
was also significantly decreased after trametinib exposure in these
cell lines, but the phosphorylation of MEK was increased at the
same time. The combination treatment with dabrafenib and trametinib
resulted in the additional shut-down of ERK phosphorylation. In
contrast, an upregulation of the phosphorylation of ERK was
observed in the two NRAS mutant cell lines ACT-1 and OCUT-6 after
exposure to dabrafenib. Trametinib clearly inhibited the
phosphorylation of MEK in these NRAS mutant cells, and significant
shutdown of ERK phosphorylation was observed after the dual
blockade with dabrafenib and trametinib (Fig. 3).
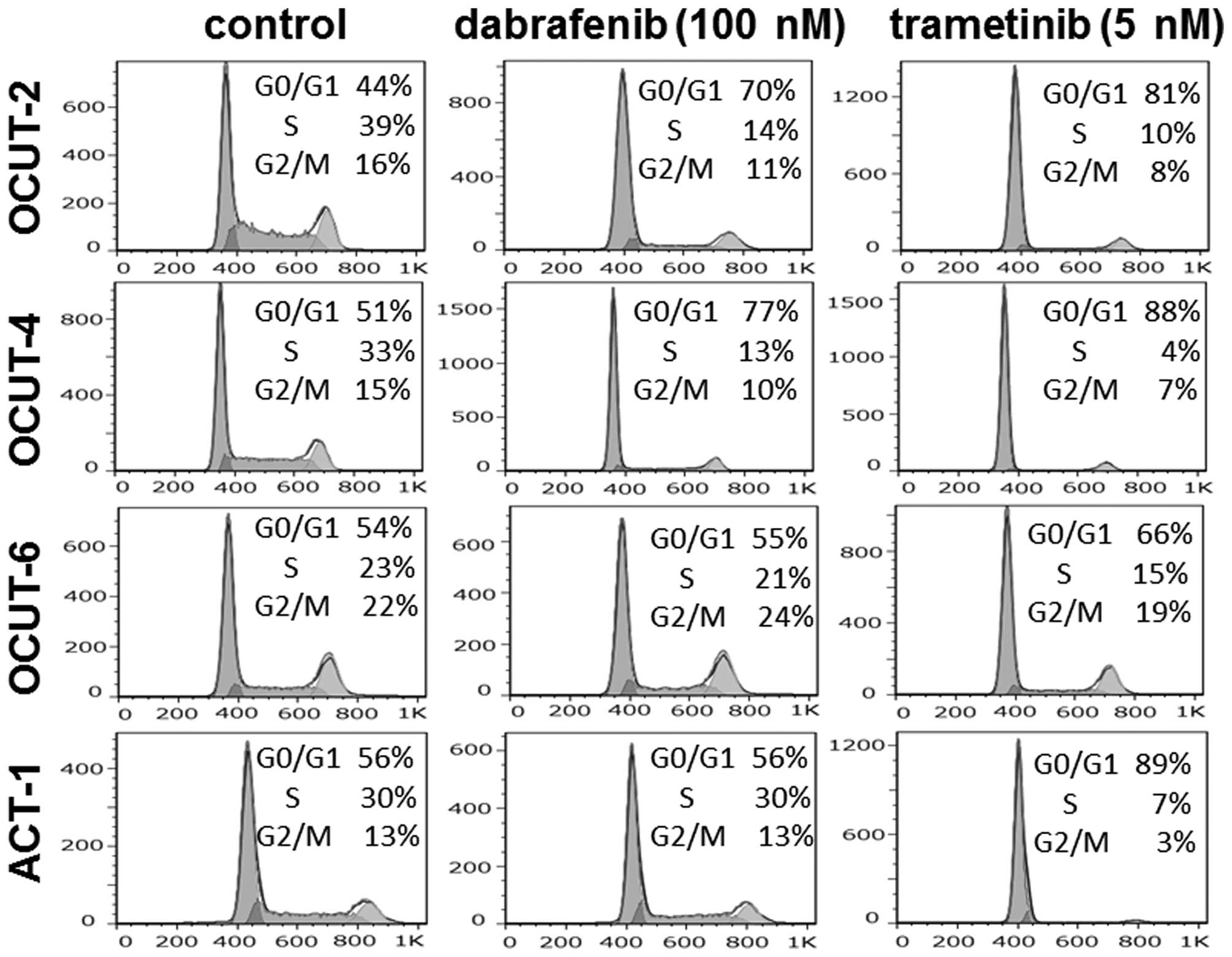
The effects of the inhibitors on cell
cycle progression
Significant increases in the proportion of cells in
the G0/G1 phase were observed after exposure to dabrafenib in both
the OCUT-2 and -4 lines. The G0/G1 arrest was not observed in the
RAS mutant ACT-1 or OCUT-6 cells after dabrafenib treatment.
Trametinib induced G0/G1 arrest in all four cell lines. Sub-G1
population cells were scarcely observed (0.7–4.4%) after treatment
(Fig. 4).
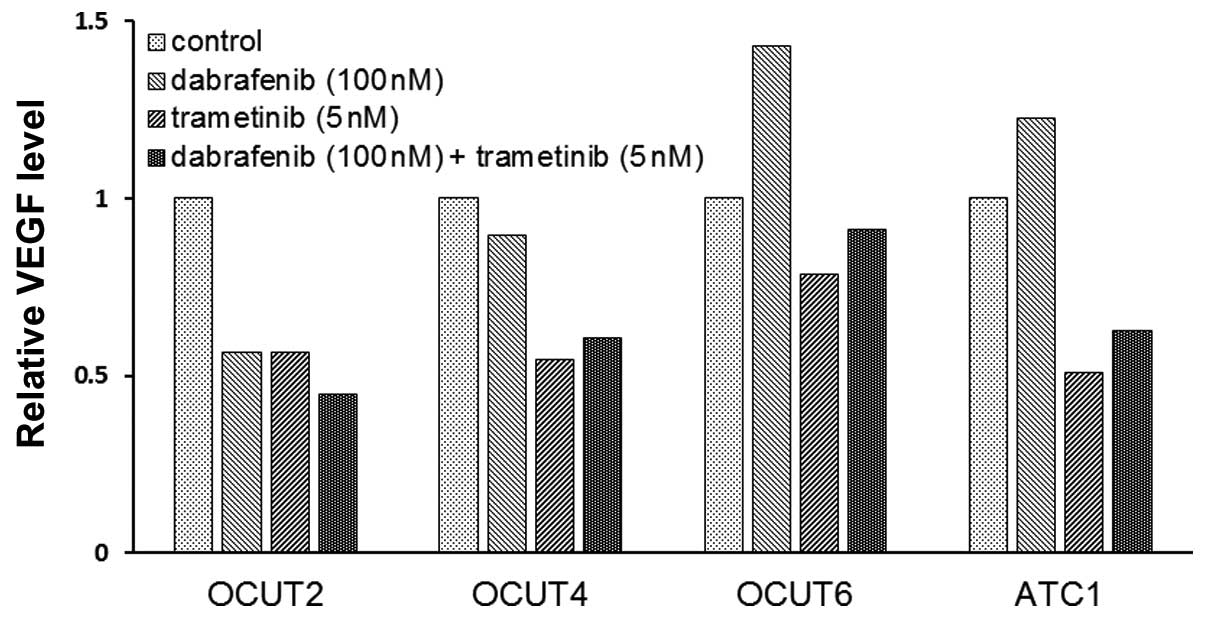
The effects of the inhibitors on VEGF
secretion of the cell lines
The VEGF concentration in the conditioned medium
varied among the cell lines. The OCUT-2 cells demonstrated the
highest concentration at 13,500 pg/ml and the OCUT-4 cells showed
the lowest concentration at 384 pg/ml in the stable condition. The
concentration of VEGF decreased after dabrafenib treatment in the
OCUT-2 and -4 cells, whereas it increased after dabrafenib
treatment in the two cell lines with wild-type BRAF, i.e., the
OCUT-6 and ACT-1 cells. A decrease in the VEGF concentration in the
conditioned medium was observed in all four cell lines after
trametinib treatment (Fig. 5).
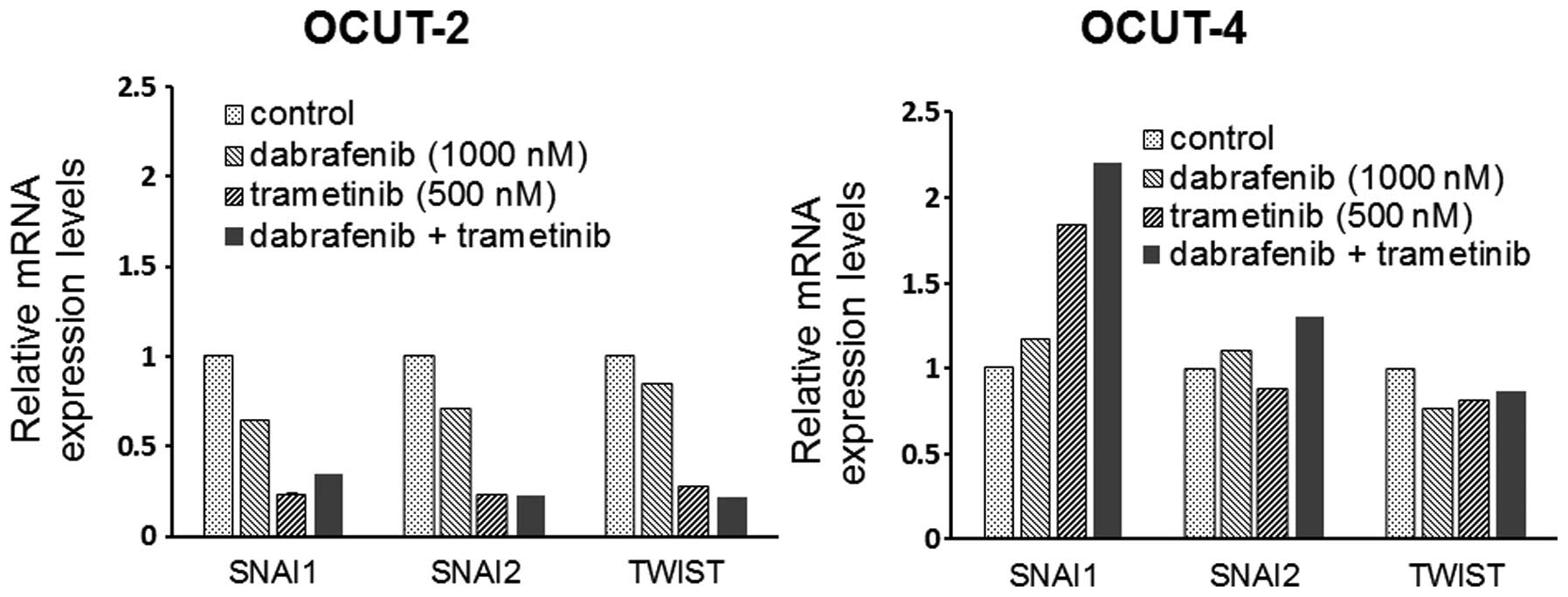
The effects of the inhibitors on the
expression of EMT markers
A significant decrease in the mRNA expression of the
EMT markers snail (SNAI1), slug (SNAI2) and twist (TWIST) was
observed in the OCUT-2 cells after exposure to dabrafenib and
trametinib, alone and in combination. Increased expression of SNAI1
mRNA was seen in the OCUT-4 cell line after exposure to either and
both inhibitors. The changes of SNAI2 and TWIST expressions after
treatment were not significant in the OCUT-4 line (Fig. 6).
Discussion
Dabrafenib is a reversible and potent
ATP-competitive inhibitor that selectively inhibits the
BRAFV600E kinase (24).
In the OCUT-2 and -4 cell lines, which harbor a
BRAFV600E mutation, dabrafenib clearly inhibited
cellular growth by demonstrating G0/G1 arrest. The strongest
inhibitory effect was in the OCUT-4 cells, in which the marked
activation of a downstream pathway from BRAF gene was observed in
the stable culturing condition. The phosphorylations of MEK and ERK
were strongly downregulated by exposure to dabrafenib in OCUT-4
cells, causing a significant G0/G1 arrest. In the OCUT-2 line, the
mutation in PI3KCA gene in addition to BRAFV600E
mutation (16) and signaling
through the PI3K/AKT/mTOR pathway can also be expected to
contribute to aberrant cell proliferation to some extent.
Nevertheless, the dabrafenib treatment resulted in a degree of
growth inhibition by G0/G1 arrest in the OCUT-2 cells that was
similar to that observed in the OCUT-4 cells. This observation
suggested that the activated MAPK/ERK pathway, and not the
PI3K/AKT/mTOR pathway, was the main driver for aggressive cell
proliferation in OCUT-2 cell line. The results indicate that the
inhibition of BRAFV600E by dabrafenib might be effective
against cancer cells harboring active alterations in both the
MAPK/ERK and PI3K/AKT/mTOR pathways.
We observed an upregulation of phosphorylated ERK
after dabrafenib exposure in ACT-1 and OCUT-6 cells, which have an
NRAS mutation. The mechanism of the upregulation of p-ERK in RAS
mutant cells after treatment with a selective BRAFV600E
kinase inhibitor has been investigated. Dimeric complexes with
wild-type BRAF, CRAF or kinase-dead BRAF is able to generate
excessive downstream signaling under stimulation by mutant RAS
enzyme (25). This mechanism
resulted in paradoxical phosphorylation in ERK after BRAF
inhibition, but did not contribute to the cell cycle progression or
cell growth in the present study. However, the VEGF secretion was
clearly stimulated in the NRAS mutant cells after dabrafenib
treatment in our study. VEGF is well known as a strong inducer of
cancer neo-vasculature that contributes to the arrangement of the
cancer microenvironment for aggressive growth. Our present findings
indicated one of the potential mechanisms of tumor growth in
dabrafenib-resistant NRAS mutant cancer cells.
The treatment with trametinib, a reversible
allosteric inhibitor of MEK1 and MEK2 activation and kinase
activity, resulted in universal growth suppression in all four cell
lines independent of the mutational status of BRAF or NRAS. A weak
growth-inhibitory effect was observed in the BRAF mutant OCUT-4
cells. After trametinib exposure, inhibition of the phosphorylation
of ERK was clear, and cell cycle arrest was obviously identified.
Nevertheless, the phosphorylation of MEK was strongly induced in
OCUT-4 cells by trametinib treatment, suggesting that resistance to
trametinib could be caused by a mechanism other than one downstream
of the MAPK/ERK pathway. This hypothesis was also suggested by the
result of our dual blockade by dabrafenib and trametinib. There was
no additional effect of either inhibitor in combination with the
other in the OCUT-4 cell line, suggesting a limited effect of
inhibiting the MAPK/MEK pathway. In addition, the OCUT-4 cells
showed a different EMT marker expression profile after exposure to
the inhibitors. Only this cell line showed an upregulation in the
expression of the mRNA of SNAI1.
The expression of EMT markers is thought to have a
role in the acquisition of resistance to a cytotoxic drug (26). Our present findings indicated that
the phenotypical change through the EMT also contributed to the
mechanism of resistance to these inhibitors. Several mechanisms
have been confirmed to trigger resistance to BRAF inhibition
(27–30). Additional investigations are needed
to clarify the involvement of the EMT in the effect of BRAF
inhibition.
Dabrafenib and trametinib, as monotherapy or in
combination, were approved for the treatment of melanomas by the US
Food and Drug Administration. Dabrafenib as a treatment for
advanced thyroid cancer resulted in durable responses in
BRAF-mutant DTC patients (31). A
recent report suggested the re-differentiation of iodine-refractory
thyroid cancer after dabrafenib treatment (32). These observations clearly indicate
the usefulness of BRAF/MEK inhibitors for the management of
advanced and inoperable thyroid cancer. The results of the present
study suggest the importance of selecting ATC patients in accord
with the mutation status of BRAF and RAS when applying inhibitors
(33).
Our present findings demonstrated the efficacy of a
mutation-selective BRAF inhibitor and a MEK inhibitor in human ATC
cell lines. Our observations indicated the existence of a unique
driver gene for the aggressive proliferation of ATC cancer cells,
and we observed that a cellular growth inhibitory effect can be
expected when appropriate inhibitor(s) are selected.
Acknowledgements
This study was supported in part by Grants-in-Aid
for Scientific Research (JSPS KAKENHI, #25461992). N. Onoda
received honoraria from Bayer and Eisai, research grant from Bayer.
The English of this manuscript has been edited and proofread by Ms.
Mary Stewart of KN International Inc.
References
|
1
|
Sugitani I, Miyauchi A, Sugino K, Okamoto
T, Yoshida A and Suzuki S: Prognostic factors and treatment
outcomes for anaplastic thyroid carcinoma: ATC Research Consortium
of Japan cohort study of 677 patients. World J Surg. 36:1247–1254.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kebebew E, Greenspan FS, Clark OH, Woeber
KA and McMillan A: Anaplastic thyroid carcinoma. Treatment outcome
and prognostic factors. Cancer. 103:1330–1335. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Haymart MR, Banerjee M, Yin H, Worden F
and Griggs JJ: Marginal treatment benefit in anaplastic thyroid
cancer. Cancer. 119:3133–3139. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Smallridge RC, Ain KB, Asa SL, Bible KC,
Brierley JD, Burman KD, Kebebew E, Lee NY, Nikiforov YE, Rosenthal
MS, et al; American Thyroid Association Anaplastic Thyroid Cancer
Guidelines Taskforce. American Thyroid Association guidelines for
management of patients with anaplastic thyroid cancer. Thyroid.
22:1104–1139. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sugino K and Onoda N: Part VII. Anaplastic
carcinoma. Treatment of Thyroid cancer. Japanese Clinical
Guidelines. Takami H, Ito Y, Noguchi H, Yoshida A and Okamoto T:
Springer; Japan, Tokyo: pp. 203–230. 2012
|
|
6
|
Haugen BR and Sherman SI: Evolving
approaches to patients with advanced differentiated thyroid cancer.
Endocr Rev. 34:439–455. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Onoda N, Ito Y, Ito K, Sugitani I,
Takahashi S, Yamaguchi I, Kawakami Y and Tsukada K: Phase II
clinical trial of sorafenib in Japanese patients with anaplastic
thyroid carcinoma and locally advanced or metastatic medullary
thyroid carcinoma. Thyroid. 25:A1202015.
|
|
8
|
Takahashi S, Tahara M, Kiyota N, Yamazaki
T, Chayahara N, Nakano K, Inagaki R, Toda K, Enokida T, Minami H,
et al: Phase II study of lenvatinib, a multi-targeted tyrosine
kinase inhibitor, in patients with all histologic subtypes of
advanced thyroid cancer. Ann Oncol. 25(Suppl_4): iv340–iv356.
2014.
|
|
9
|
Rosove MH, Peddi PF and Glaspy JA: BRAF
V600E inhibition in anaplastic thyroid cancer. N Engl J Med.
368:684–685. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hyman DM, Puzanov I, Subbiah V, Faris JE,
Chau I, Blay JY, Wolf J, Raje NS, Diamond EL, Hollebecque A, et al:
Vemurafenib in multiple nonmelanoma cancers with BRAF V600
mutations. N Engl J Med. 373:726–736. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim TH, Park YJ, Lim JA, Ahn HY, Lee EK,
Lee YJ, Kim KW, Hahn SK, Youn YK, Kim KH, et al: The association of
the BRAF(V600E) mutation with prognostic factors and poor clinical
outcome in papillary thyroid cancer: A meta-analysis. Cancer.
118:1764–1773. 2012. View Article : Google Scholar
|
|
12
|
Smallridge RC and Copland JA: Anaplastic
thyroid carcinoma: Pathogenesis and emerging therapies. Clin Oncol
(R Coll Radiol). 22:486–497. 2010. View Article : Google Scholar
|
|
13
|
Xing M: BRAF mutation in thyroid cancer.
Endocr Relat Cancer. 12:245–262. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang HM, Huang YW, Huang JS, Wang CH, Kok
VC, Hung CM, Chen HM and Tzen CY: Anaplastic carcinoma of the
thyroid arising more often from follicular carcinoma than papillary
carcinoma. Ann Surg Oncol. 14:3011–3018. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shi X, Liu R, Qu S, Zhu G, Bishop J, Liu
X, Sun H, Shan Z, Wang E, Luo Y, et al: Association of TERT
promoter mutation 1,295,228 C>T with BRAF V600E mutation, older
patient age, and distant metastasis in anaplastic thyroid cancer. J
Clin Endocrinol Metab. 100:E632–E637. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Onoda N, Nakamura M, Aomatsu N, Noda S,
Kashiwagi S and Hirakawa K: Establishment, characterization and
comparison of seven authentic anaplastic thyroid cancer cell lines
retaining clinical features of the original tumors. World J Surg.
38:688–695. 2014. View Article : Google Scholar
|
|
17
|
Nobuhara Y, Onoda N, Yamashita Y, Yamasaki
M, Ogisawa K, Takashima T, Ishikawa T and Hirakawa K: Efficacy of
epidermal growth factor receptor-targeted molecular therapy in
anaplastic thyroid cancer cell lines. Br J Cancer. 92:1110–1116.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Onoda N, Nakamura M, Aomatsu N, Noda S,
Kashiwagi S, Kurata K, Uchino S and Hirakawa K: Significant
cytostatic effect of everolimus on a gefitinib-resistant anaplastic
thyroid cancer cell line harboring PI3KCA gene mutation. Mol Clin
Oncol. 3:522–526. 2015.PubMed/NCBI
|
|
19
|
Salvatore G, De Falco V, Salerno P, Nappi
TC, Pepe S, Troncone G, Carlomagno F, Melillo RM, Wilhelm SM and
Santoro M: BRAF is a therapeutic target in aggressive thyroid
carcinoma. Clin Cancer Res. 12:1623–1629. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hou P, Liu D and Xing M: Genome-wide
alterations in gene methylation by the BRAF V600E mutation in
papillary thyroid cancer cells. Endocr Relat Cancer. 18:687–697.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jo YS, Li S, Song JH, Kwon KH, Lee JC, Rha
SY, Lee HJ, Sul JY, Kweon GR, Ro HK, et al: Influence of the BRAF
V600E mutation on expression of vascular endothelial growth factor
in papillary thyroid cancer. J Clin Endocrinol Metab. 91:3667–3670.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rusinek D, Szpak-Ulczok S and Jarzab B:
Gene expression profile of human thyroid cancer in relation to its
mutational status. J Mol Endocrinol. 47:R91–R103. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
24
|
Menzies AM and Long GV: Dabrafenib and
trametinib, alone and in combination for BRAF-mutant metastatic
melanoma. Clin Cancer Res. 20:2035–2043. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cichowski K and Jänne PA: Drug discovery:
Inhibitors that activate. Nature. 464:358–359. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fischer KR, Durrans A, Lee S, Sheng J, Li
F, Wong ST, Choi H, El Rayes T, Ryu S, Troeger J, et al:
Epithelial-to-mesenchymal transition is not required for lung
metastasis but contributes to chemoresistance. Nature. 527:472–476.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Carlino MS, Todd JR, Gowrishankar K,
Mijatov B, Pupo GM, Fung C, Snoyman S, Hersey P, Long GV, Kefford
RF, et al: Differential activity of MEK and ERK inhibitors in BRAF
inhibitor resistant melanoma. Mol Oncol. 8:544–554. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Johannessen CM, Boehm JS, Kim SY, Thomas
SR, Wardwell L, Johnson LA, Emery CM, Stransky N, Cogdill AP,
Barretina J, et al: COT drives resistance to RAF inhibition through
MAP kinase pathway reactivation. Nature. 468:968–972. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nazarian R, Shi H, Wang Q, Kong X, Koya
RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, et al: Melanomas
acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS
upregulation. Nature. 468:973–977. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wagle N, Emery C, Berger MF, Davis MJ,
Sawyer A, Pochanard P, Kehoe SM, Johannessen CM, Macconaill LE,
Hahn WC, et al: Dissecting therapeutic resistance to RAF inhibition
in melanoma by tumor genomic profiling. J Clin Oncol. 29:3085–3096.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Falchook GS, Millward M, Hong D, Naing A,
Piha-Paul S, Waguespack SG, Cabanillas ME, Sherman SI, Ma B, Curtis
M, et al: BRAF inhibitor dabrafenib in patients with metastatic
BRAF-mutant thyroid cancer. Thyroid. 25:71–77. 2015. View Article : Google Scholar :
|
|
32
|
Rothenberg SM, McFadden DG, Palmer EL,
Daniels GH and Wirth LJ: Redifferentiation of iodine-refractory
BRAF V600E-mutant metastatic papillary thyroid cancer with
dabrafenib. Clin Cancer Res. 21:1028–1035. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cabanillas ME, Patel A, Danysh BP, Dadu R,
Kopetz S and Falchook G: BRAF inhibitors: Experience in thyroid
cancer and general review of toxicity. Horm Cancer. 6:21–36. 2015.
View Article : Google Scholar :
|