Introduction
Colorectal cancer (CRC) is one of the most
aggressive malignancies worldwide. Twenty percent of primary
colorectal cancer diagnoses reveal remote metastases. Available
therapies still offer a poor prognosis and patients have a <10%
five-year survival rate (1). As
evidenced by genetic modifications, environmental impacts, diet and
lifestyles, the underlying mechanisms of CRC have been intensively
studied.
Dysregulation of cell energetics was just counted in
as one of the cancer hallmarks. Most cancers have a high request
for nutrients and energy to support their rapid growth. Almost a
century ago, the Nobel laureate Otto Warburg discovered more
lactate (indicating higher glycolysis) was secreted in cancer cells
than normal cells under aerobic conditions (2). This finding was later termed as the
'Warburg effect', indicating the hypothesis that even equipped with
normal mitochondria, cancer cells still relied on both
mitochondrial oxidative phosphorylation (OXPHOS) and glycolysis to
sustain survival and growth (3).
Mitochondria are important players in the
maintenance of cellular homeostasis, as they generate ATP via
OXPHOS. Mitochondrial metabolism occurs during the tricarboxylic
acid (TCA) cycle. Electron transport chain (ETC) has, therefore,
become a crucial focus of cancer treatment. In addition, TCA cycle
provides intermediates for amino acid and lipid synthesis. The
amplified demand for energy supplement in cancer cells endows
targeting ETC a promising strategy to restrict cancer progression.
Compounds that target the mitochondria have drawn worldwide
attention. For example, metformin, a diabetes drug, obstructed
mitochondrial complex I and exhibited impressing effects on various
cancer models (4).
As a metabolic sensor in energy homeostasis,
AMP-activated protein kinase (AMPK) is important for regulating
mitochondrial content and maintaining glucose homeostasis. In
metabolically active cancer cells, impaired mitochondrial OXPHOS
would lead to AMPK activation. AMPKα can be phosphorylated by
upstream kinases, including calcium/calmodulin-dependent protein
kinase 2 (CaMKK2), liver kinase B1 and TAK1 (5). Reactive oxygen species
(ROS)-dependent increases in cytosolic calcium activated AMPK
signaling via CAMKK2 (6). One of
the most important consequences of AMPK activation was mTORC1
inactivation. AMPK/mTOR activation has been demonstrated to be
involved in mesenchymal stem cells promoted-colorectal cancer
progression (7). Moreover, mTOR
signaling was greatly provoked in glandular elements of CRC and
colorectal adenomas with advanced intraepithelial neoplasia
(8).
Other than inhibiting TCA, blunting aerobic
glycolysis is also a promising approach to selectively inhibit
cancer cells which mainly rely on this pathway. Enzymes involved in
the glycolytic pathway served as substrates for anticancer therapy.
Right-sided colon tumors showed significantly higher glucose
transporter 1 (GLUT1) mRNA levels, which is a rate-limiting
transporter for glucose uptake (8). Hexokinase II (HKII) catalyzes the
ATP-dependent phosphorylation of glucose. High HKII and low
p-pyruvate dehydrogenase expression in the invasive lesions of CRC
tumors is predictive of tumor aggressiveness (9,10).
Recently, several potent glycolytic inhibitors have been developed,
such as 2-deoxy-D-glucose (2DG) and 3-bromopyruvate (3BP). 3BP is
an inhibitor of HKII (11,12).
The mitochondrial damage induced by caulerpin, a
Caulerpa spp. algal pigment, was first mentioned by Liu
et al (13), who suggested
that mitochondrial respiration can be intervened by caulerpin via
suppressing ETC complex I of breast cancer cells in hypoxia.
Recently, the study group of Ferramosca et al found that
caulerpin selectively repressed the activity of complex II in rat
liver mitochondria (14). The
observations that caulerpin suppresses HIF-1 activation and
displays cytotoxicity to human dermal fibroblasts support further
exploration into the anticancer mechanism of caulerpin. Recent
studies showed that glycolysis inhibitors stimulated cell death and
sensitized cancer cells to chemotherapeutic agents in various types
of cancers (15,16). To date, there is no published
literature showing that glycolysis inhibitors sensitized colorectal
cells to caulerpin.
Although caulerpin inhibited mitochondrial
respiration, whether disturbing complex I would abolish cells
proliferation remained obscure. Moreover, activation of the energy
sensor AMPK can promote glycolysis to replenish the loss of ATP.
Herein, our study aimed to explore the intracellular molecular
mechanism involved in caulerpin-modulated energy metabolism in CRC
cell lines and further examine whether the combination of caulerpin
and glycolysis inhibitors will orchestrate the stimulation of cell
death both in vitro and in vivo.
Materials and methods
Chemical and reagents
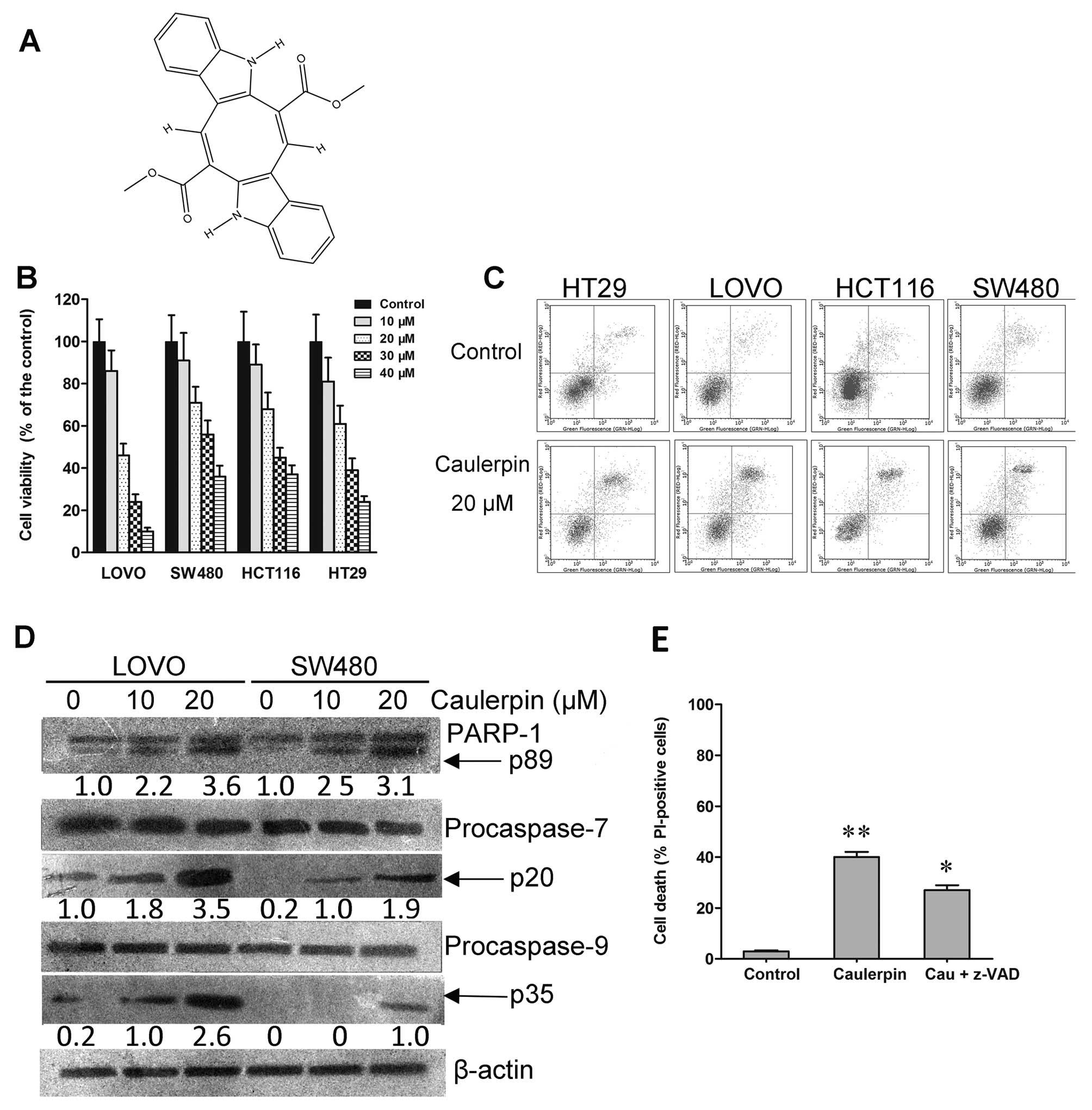
Caulerpin (Fig. 1A)
of 99% purity was purchased from Yuanye Pharmaceutics (Shanghai,
China). A stock solution of metformin was made at a concentration
of 100 mM in DMSO and stored at −20°C. KN93, 2DG and 3BP were
obtained from Sigma (Shanghai, China). Antibodies against PARP,
p-p70S6K, p70S6K, p-S6, S6, p-4EBP1, 4EBP1, AMPKα1, AMPKα2 and
β-actin were purchased from Santa Cruz Biotechnology, Inc. (Santa
Cruz, CA, USA). AMPKα, p-AMPKα (Thr172), p-ACC (Ser79), ACC,
procaspase-7, procaspase-9 antibodies were purchased from Cell
Signaling Technology (Denver, MA, USA). Horseradish
peroxidase-labeled secondary antibodies were obtained from Promega
(Madison, WI, USA).
Cell culture
The human colorectal cancer cell lines HCT116 and
HT29 were obtained from ATCC. LOVO and SW480 were obtained from the
Cell Bank of the Shanghai Institute of Biochemistry and Cell
Biology, Shanghai, China (http://www.cellbank.org.cn). All cell lines were
cultured in DMEM medium supplemented with 10% fetal bovine serum,
100 U/ml penicillin and 100 µg/ml streptomycin and
maintained in a humidified atmosphere at 37°C with 5%
CO2. For glucose deprivation experiments, medium was
replaced with DMEM containing non-glucose (Invitrogen, Carlsbad,
CA, USA).
Cell viability assay
The effects of drug treatment on the viability of
colorectal cell lines were assessed with a cell counting kit-8 from
Sigma (17), as described by the
manufacturer. Briefly, 2×104 cells were seeded into
96-well plates. After treatment, CCK-8 solution was added to each
well and cells were further incubated at 37°C for 2 h. The
absorption was measured using a multiskan spectrum microplate
reader at 450 nm. All the experiments were repeated at least three
times.
Apoptosis detection
Briefly, the cells were treated with caulerpin for
24 h. Cells (1×106) were centrifuged and harvested
(18). Next, the cells were
suspended in binding buffer, containing Annexin V-FITC and PI and
then incubated for 15 min in the dark at room temperature. The
stained cells were analyzed using a FACSCanto flow cytometer (BD
Biosciences, Flanklin Lakes, NJ, USA).
Mitochondrial membrane potential (MMP)
and ROS determination
Briefly, LOVO and SW480 cells were seeded in a
12-well plate at ~80% confluence. After receiving indicated
treatment, cells were washed twice with phosphate-buffered saline
(PBS) and incubated at 37°C for 30 min in buffer solution
containing lipophilic cation JC-1 (KeyGen, Nanjing, China) or DCFDA
(Invitrogen), cells were re-suspended and fluorescence was measured
using a FACSCalibur (BD Biosciences). Two fluorescence parameters,
channel 1 (green fluorescence, FL1) at 530 nm and channel 2 (red
fluorescence, FL2) at 590 nm, were used to measure MMP (19). Bright green fluorescence produced
by DCFDA means high level of ROS (20).
Western blot analysis
Cells following different treatments were lysed with
RIPA buffer. Protein concentration was determined using a Coomassie
blue staining method (21). An
equal amount of protein (50 µg) was separated on 10%
SDS-PAGE and transferred to PVDF membranes. The membranes were
incubated with primary antibodies in 5% BSA at 4°C overnight. The
membranes were washed and incubated with HRP-conjugated secondary
antibody for 1 h at room temperature. The immunoreactive bands were
detected with an ECL kit (Beyotime Institute of Biotechnology,
Shanghai, China).
Metabolic analysis
Rat liver mitochondria (0.3 mg protein/ml) and
saponin-permeabilized or intact cells (5×106) were
incubated with caulerpin for indicated time. The oxygen consumption
rate (OCR) and extracellular acidification rate (ECAR) were
measured using an XFe-96 Extracellular Flux Analyzer (Seahorse
Bioscience, North Billerica, MA, USA). After attachment to XFe cell
culture plates, cells were washed with XF assay medium and
equilibrated for 1 h. ECAR were determined by the supplement of
2DG. ATP content was examined from OCR differences from basal rate
after adding oligomycin. Different substrates were added to
evaluate mitochondrial respiration when respiratory complex I to IV
was stimulated respectively. Bioenergetic profiling to distinguish
the site within the ETC controlled by caulerpin was performed as
previously described (22).
Measurements were normalized by total protein content with Lowry
assay.
Generation of AMPKα1-knockout cell lines
with a CRISPR/Cas9 system
Two single guide RNAs (sgRNA) targeting unique
sequences of the AMPKα1 gene were designed and cloned into the
pSpCas9 (BB)-2A-Puro (PX459) V2.0 (Plasmid #62988; Addgene)
plasmid, which contains a puromycin resistance cassette. sgRNAs
were as follows: 5′-CACCGTACATTCTGGGTGACACGCT-3′ and
5′-AAACAGCGTGTCACCCAGAATGTAC-3′. After transfection, the cells were
cultured in a medium containing puromycin for 48 h (23). Cells resistant to the drug were
then selected by FACS for clonal expansion. Gene deficiency in LOVO
cells was examined by western blotting with an anti-AMPKα1
antibody.
RNA interference
Scramble siRNA, siRNA targeting AMPKα2 and siRNA
transfection reagent were purchased from Invitrogen (Carlsbad, CA,
USA). Cells were transfected with Lipofectamine 2000 (Invitrogen)
according to the instruction (24).
In vivo tumor growth assay
Four to 6-week-old athymic mice, which were obtained
from SLAC Lab Animal Center of Shanghai (Shanghai, China), were
housed under standard conditions. This experiment was conducted in
accordance with the guidelines issued by the State Food and Drug
Administration (SFDA of China). The experiments were approved by
the Institutional Animal Care and Use Committee of Ningbo No. 2
Hospital, Ningbo, China. Cells (1×107) in culture medium
were injected subcutaneously into the right flank of each mouse
(25). Tumor size was monitored by
using micrometer calipers every other day for a 30-days period and
calculated with a formula: tumor volume (mm3) =
width2 × length/2. When tumor volume reached ~75
mm3, nude mice were divided into three groups (6
mice/group): saline control, caulerpin (30 mg/kg, in saline with
0.2% CMC-Na), 3BP (15 mg/kg, in saline) or combination group. The
oral gavage was executed every other day. It began on day 1, and
finished on day 30. At the end of the experiments, all the mice
were euthanized. Tumor tissues were used for immunohistochemistry
assay.
Immunohistochemistry
Paraffin-embedded sections were heat-fixed,
deparaffinized, and rehydrated through a graded alcohol series
(100, 95, 85 and 75%). Antigen retrieval was performed with citrate
buffer and treated with 3% H2O2 to block
endogenous peroxidase activity prior to the treatment with primary
antibodies (anti-PCNA, 1:100; anti-mTOR, 1:200). The tissues were
then incubated with the secondary biotinylated antispecies
antibody. Counterstaining was performed using hematoxylin (26).
Statistical analysis
Data are expressed as means ± SD. Statistical
significance were performed using one-way ANOVA followed by
unpaired Student's t-test. For comparison of multiple samples, the
Tukey-Kramer test was used. All data were analyzed with GraphPad
PRISM 5 (GraphPad Software, Inc., La Jolla, CA, USA).
Results
Caulerpin inhibits cell viability in
colorectal cell lines
In the present study, we examined the anticancer
property of caulerpin in various colorectal cancer cell lines
(HT29, LOVO, HCT116 and SW480). A growth inhibitory effect was
observed in the four colorectal cell lines after 48 h exposure,
with IC50 values ranging from 20 to 31 µM
(Fig. 1B). Among them, LOVO cells
were most sensitive, with 50% inhibition rate at the concentration
of 20 µM, while SW480 cells were least sensitive to
caulerpin. As shown in Fig. 1C,
caulerpin induced apoptosis to colorectal cells, though to a
different extent. Caulerpin augmented the activities of PARP-1,
caspase-9 and caspase-7 in LOVO cells, as indicated by their
cleavage (Fig. 1D), but the
broad-range caspase inhibitor z-VAD-fmk only partially prevented
cell death, signifying that other caspase-independent signaling was
engaged in the action of caulerpin (Fig. 1E).
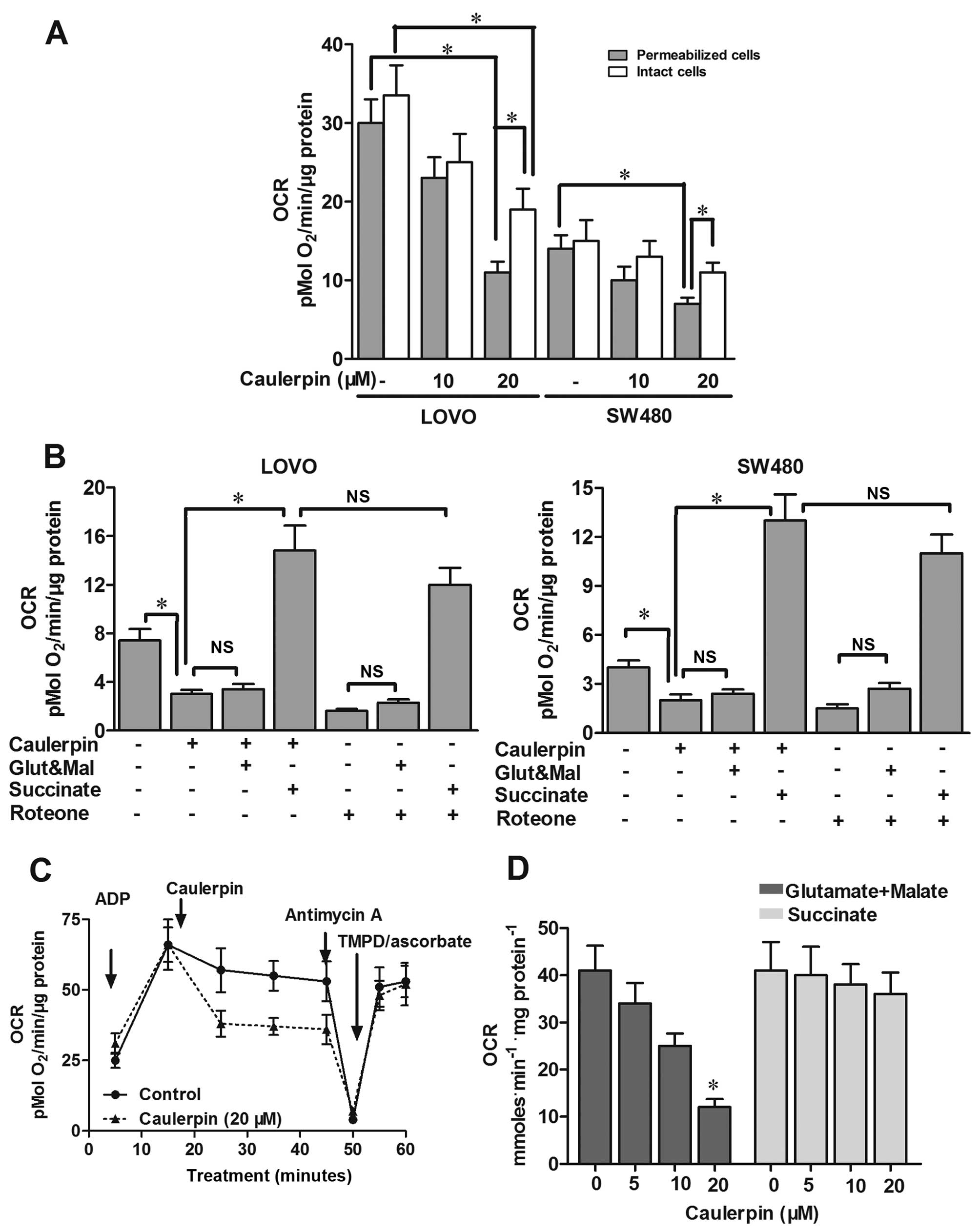
Caulerpin inhibits OXPHOS and disturbs
mitochondrial function via the inhibition of mitochondrial complex
I
Previous findings illustrated that caulerpin
impaired mitochondrial ETC in cisplatin-resistant C13 cells and
human breast cancer T47D cells (13,14),
but its effects on oxygen consumption have not been fully
addressed. Additionally, whether the emergence of an energy stress
correlates with the anticancer activity of caulerpin in colorectal
cells needs to be explored. In the present study, oxygen
consumption was inhibited by ~60% in SW480 and LOVO cells after
caulerpin treatment. The phenomenon was more obvious in
permeabilized cells than intact cells (Fig. 2A). LOVO cells pretreated with
caulerpin (20 µM, 2 h) were then permeabilized with saponin.
Although malate and glutamate (ETC complex I substrates) failed to
restore caulerpin-triggered downregulation of mitochondrial
respiration in LOVO cells, complex II substrate succinate rescued
mitochondrial respiration in LOVO cells pretreated with caulerpin,
indicating that caulerpin caused impairment to complex I (Fig. 2B). The inhibitory effect of
caulerpin on mitochondria respiration resembled that seen from
rotenone, a complex I inhibitor (Fig.
2B). Moreover, in the incubation media containing succinate,
respiration was suppressed by supplementation with complex III
inhibitor antimycin A, proving that the ETC still works. A
combination of TMPD/ascorbate (complex IV substrates) successfully
rebooted respiration perturbed by antimycin A (Fig. 2C). In mitochondria isolated from
rat liver, similar results were obtained. In the media containing
malate and glutamate, oxygen consumption was suppressed due to
caulerpin treatment, however, not in the presence of succinate
(Fig. 2D). The results
demonstrated that caulerpin brought about an intervention to
mitochondrial function, via inhibition of mitochondrial complex I
without interfering complexes II, III or IV.
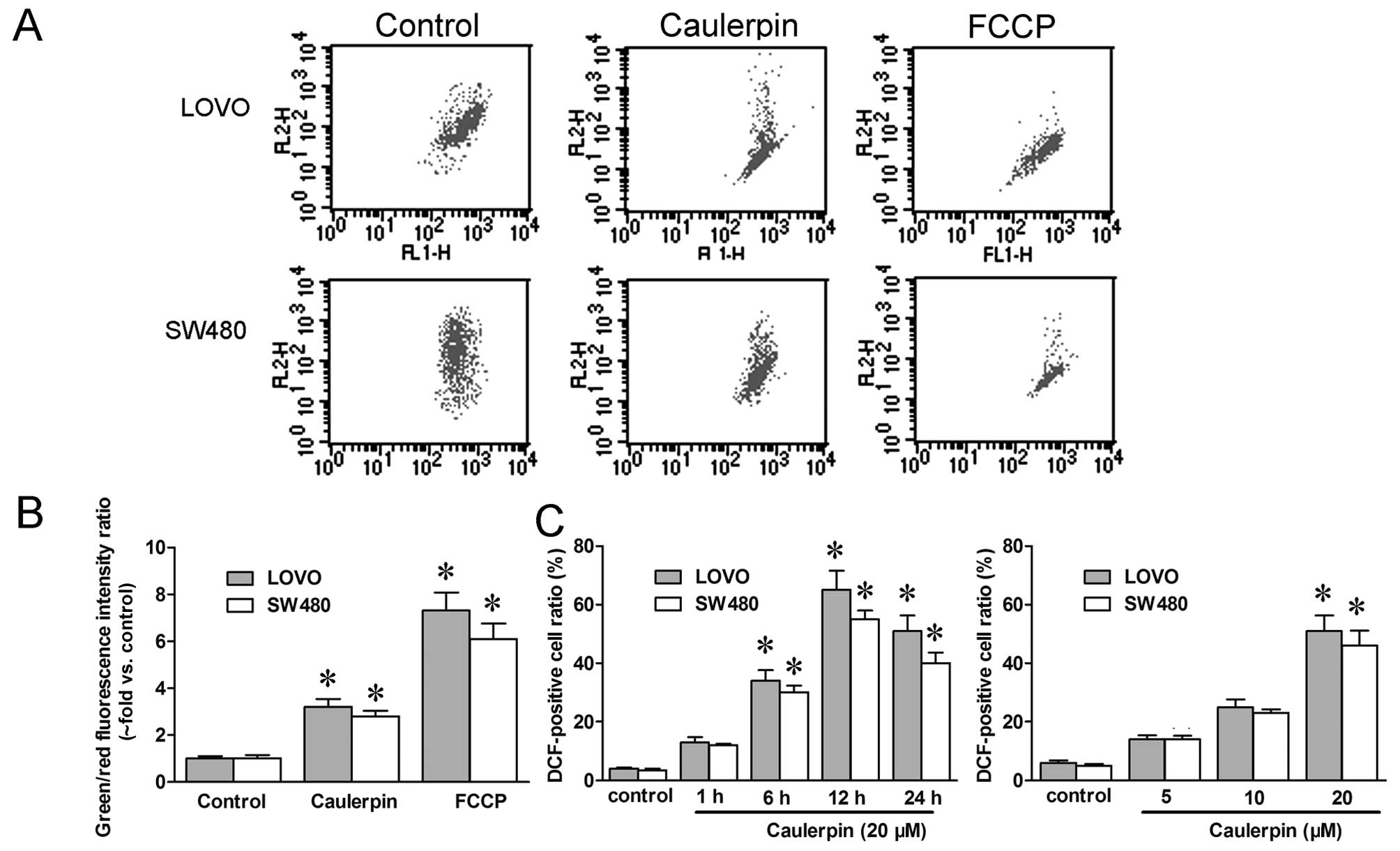
Caulerpin induces a drop in mitochondrial
membrane potential and a surge in ROS level
Since mitochondrial function was impaired by
caulerpin, we then examined mitochondrial membrane potential (ΔΨM)
and ROS with flow cytometry, repectively. JC-1 is a cationic dye
which accumulates in mitochondria in response to potential change,
and the rise in the green/red fluorescence intensity ratio reflects
mitochondrial depolarization. Herein, JC-1 fluorescent probes were
utilized to analyze ΔΨM. In Fig.
3B, a significant advance in green/red fluorescence ratio
(almost 3-fold vs. control) was obtained in both LOVO and SW480
cells treated with caulerpin, pointing to a reduction in ΔΨM. FCCP,
an electron transport chain uncoupler, was applied as the positive
control. Likewise, intracellular ROS concentration was measured by
flow cytometry with the fluorescence dye DCF-DA. In a
concentration-dependent manner, caulerpin rapidly upregulated ROS
level at 1 h post-induction, and up to 24 h (Fig. 3C).
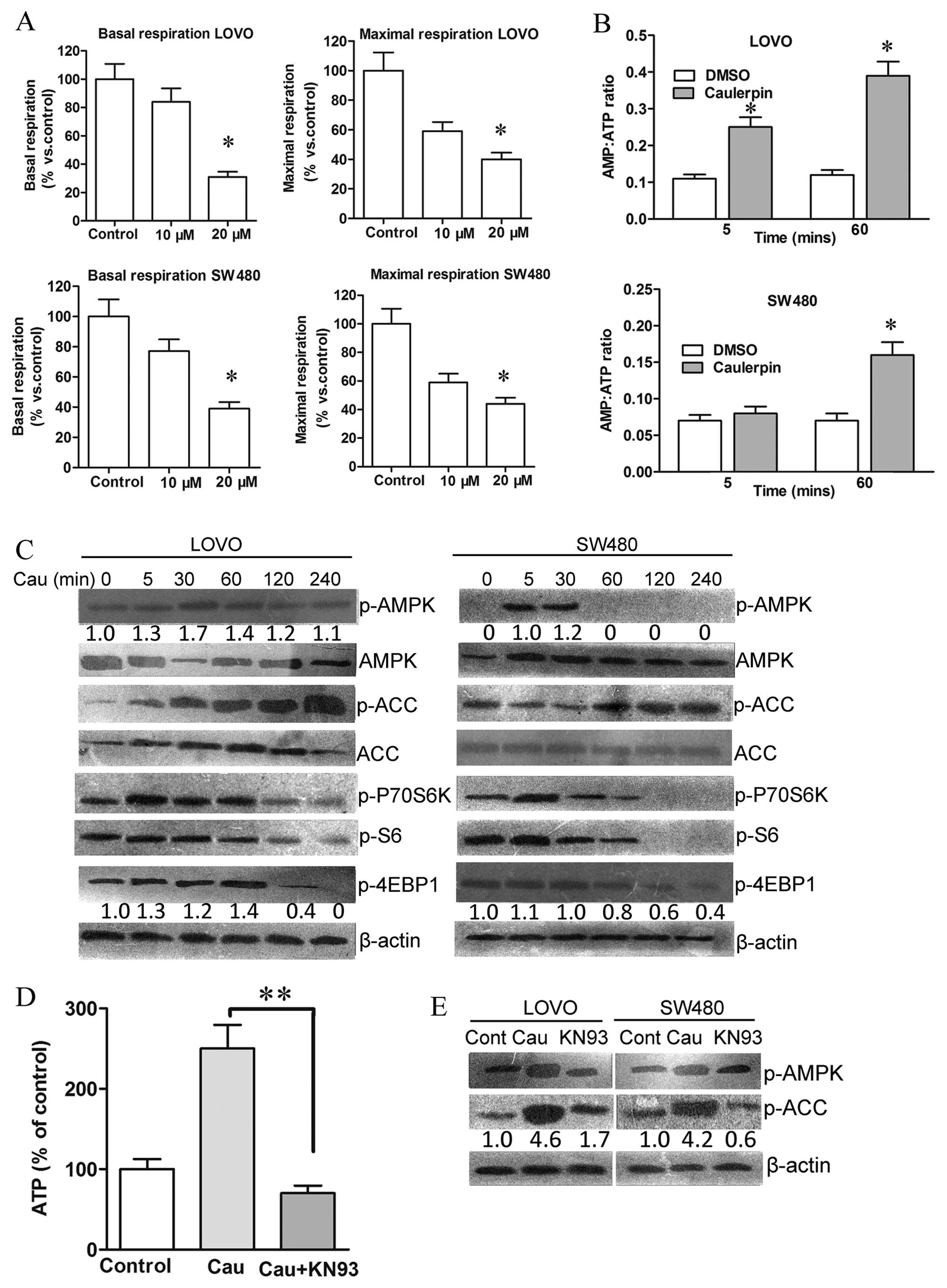
Caulerpin stimulates AMPK signaling and
inhibits mTORC1-4E-BP1 axis
The metabolic profile of colorectal cancer cells was
further examined with the Seahorse XF-e96 analyzer. Caulerpin
treatment inhibited both maximal and basal respiration (Fig. 4A), accompanied by an increment in
AMP/ATP ratio, which could be observed after 5 min and 1 h
incubation with caulerpin in LOVO cells (Fig. 4B). AMPK is a crucial energetic
sensor that is activated either by decreased cellular ATP levels or
by ROS elevation (27). Activated
AMPK will further suppress mTOR signaling for cellular adaptation
in a stress condition (28). In
the present study, caulerpin-triggered phosphorylation of AMPK and
the phosphorylation of the downstream target ACC were observed in
both cell lines, accompanied with inhibition of the downstream
mTORC1 targets 4E-BP1 and S6 (Fig.
4C).
As a kinase responsive to oxidative activation
upstream of AMPK, the essential role of CaMKII was explored to
uncover the mechanism of caulerpin-elicited AMPK activation
(29). As shown in Fig. 4D, the CaMKII inhibitor KN93 reduced
caulerpin-induced ATP levels in colorectal cells, and induced a
remarkable dephosphorylation of AMPK and ACC. These results
signified that caulerpin-stimulated AMPK activation was
CaMKII-dependent.
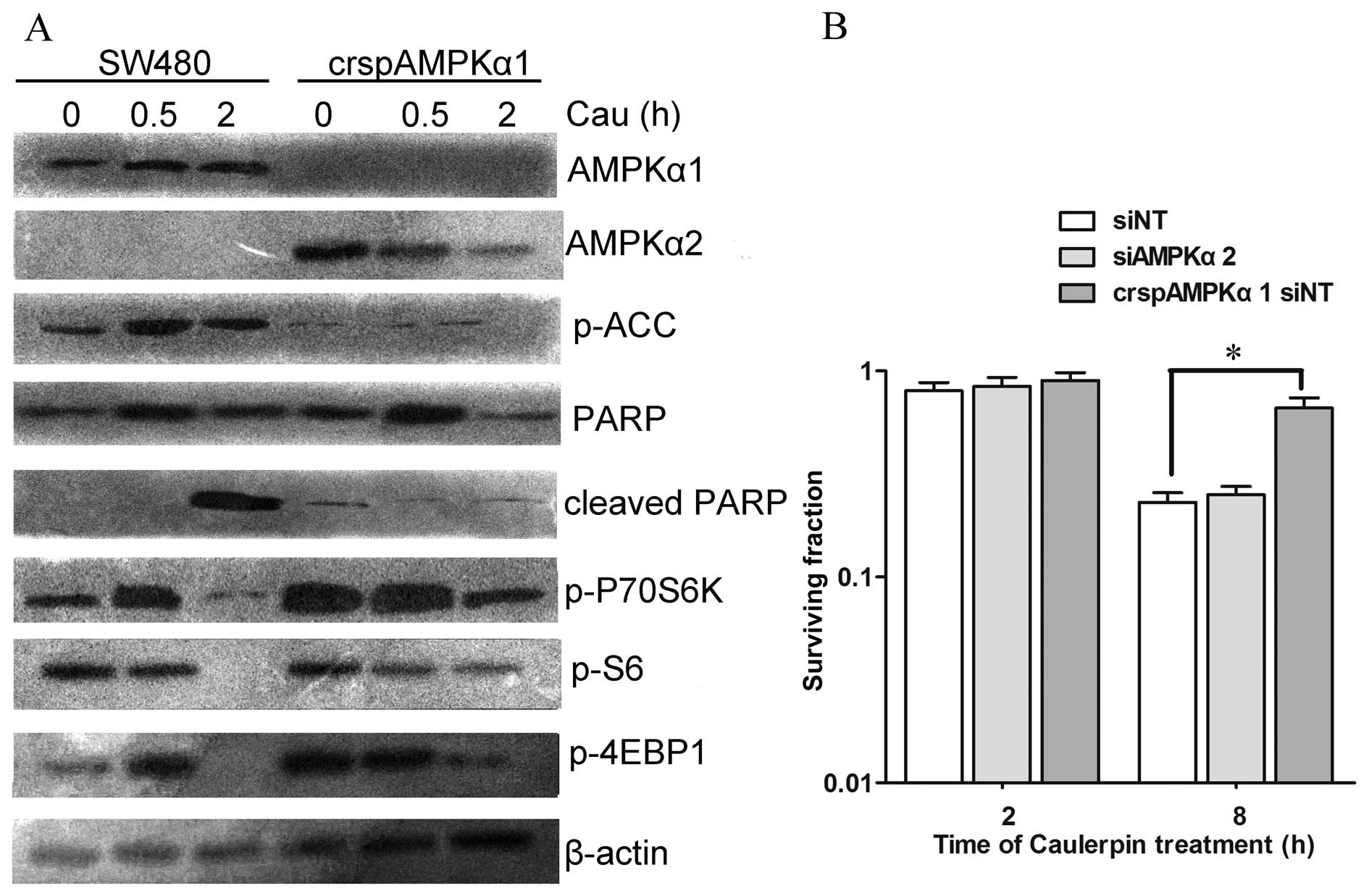
The role of AMPKα1 during caulerpin
treatment
To further dissect the effect of AMPK isoforms on
the viability of cells treated with caulerpin, the CRISPR-cas9
system was applied to knock out AMPKα1. crspAMPKα1 attenuated
caulerpin-induced inactivation of mTOR pathway. Loss of AMPKα1
generated a compensatory growth in AMPKα2 expression (Fig. 5A). CCK-8 assay displayed that knock
out of AMPKα1 protected cells against caulerpin-induced
cytotoxicity (2 and 8 h), while siAMPKα2 exhibited no significant
influence on cell death, which was induced by caulerpin treatment
(Fig. 5B).
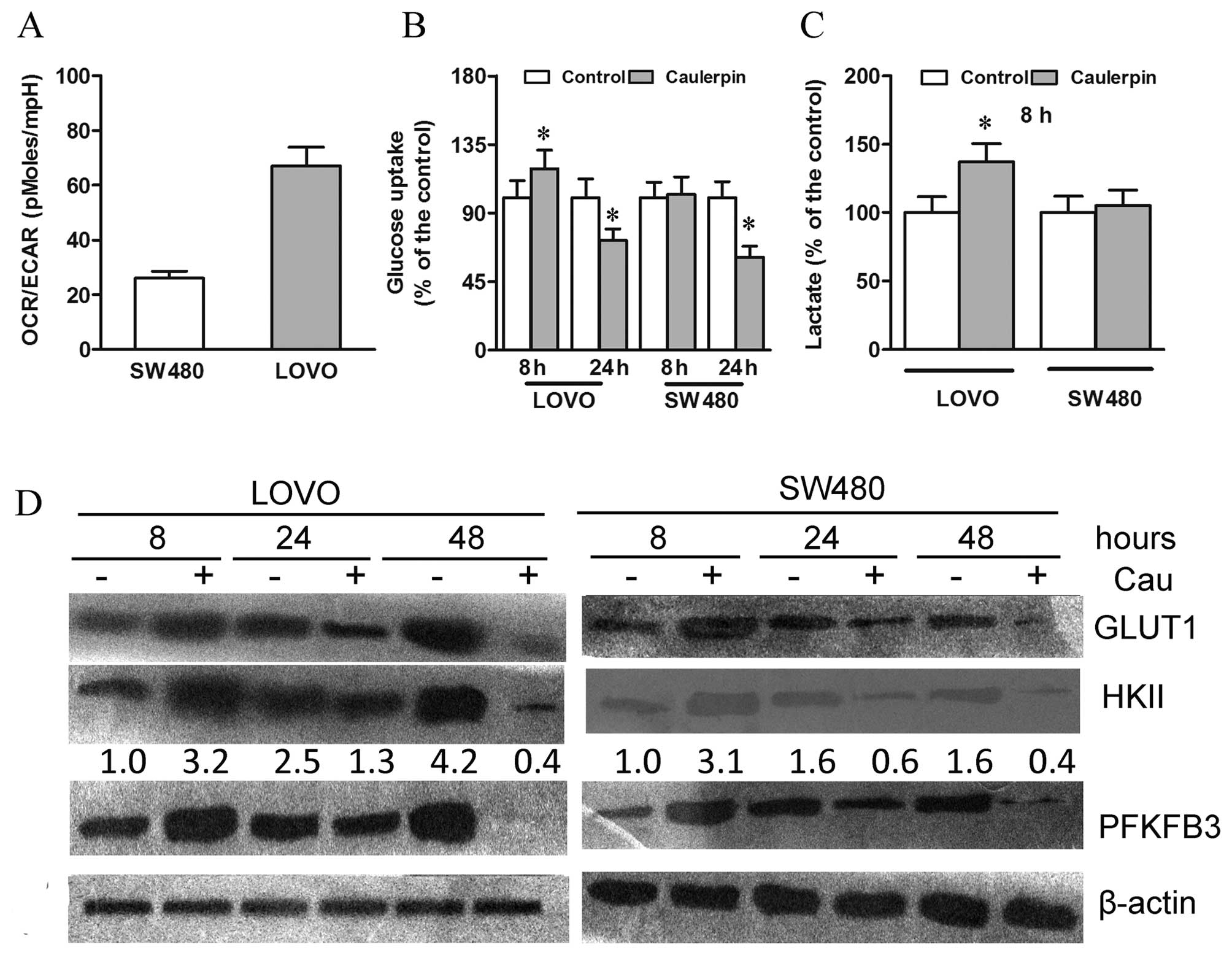
Caulerpin exhibits different early and
long-term effects on glycolysis
As a direct measure of ECAR, lactic acid was
secreted by cells and thus serving as a hint for glycolytic
activity. The baseline OCR/ECAR ratio was low in the SW480 cells
and high in the LOVO cells, indicating a higher intrinsic
glycolytic rate in SW480 cells than LOVO cells, which generate ATP
primarily through OXPHOS (Fig.
6A).
We further examined whether caulerpin-triggered
OXPHOS attenuation altered cell glucose metabolism. After 8 h
treatment with caulerpin, glycolysis in LOVO cells was strengthened
to compensate ATP loss, as revealed by significant increases in
glucose uptake and lactate production (Fig. 6B and C). Moreover, upregulation of
GLUT1, HKII, and 6-phosphofructo-2-kinase (PFKFB3) protein
expression was also observed (Fig.
6D). Nevertheless, a prolonged exposure to caulerpin,
unexpectedly, weakened glucose uptake in LOVO cells, as evidenced
by downregulation of GLUT1 level after 48 h of caulerpin incubation
(Fig. 6B and D). In the highly
glycolytic SW480 cells, 8 h treatment with caulerpin exerted no
significant effect on glucose metabolism; on the other hand, a
remarkable decline of GLUT1 protein expression was observed after
24 h and continued after 48 h. Therefore, in spite of the initial
attempt to adapt to the energy stress, long-term incubation with
caulerpin eventually elicited a notable impairment to glucose
metabolism.
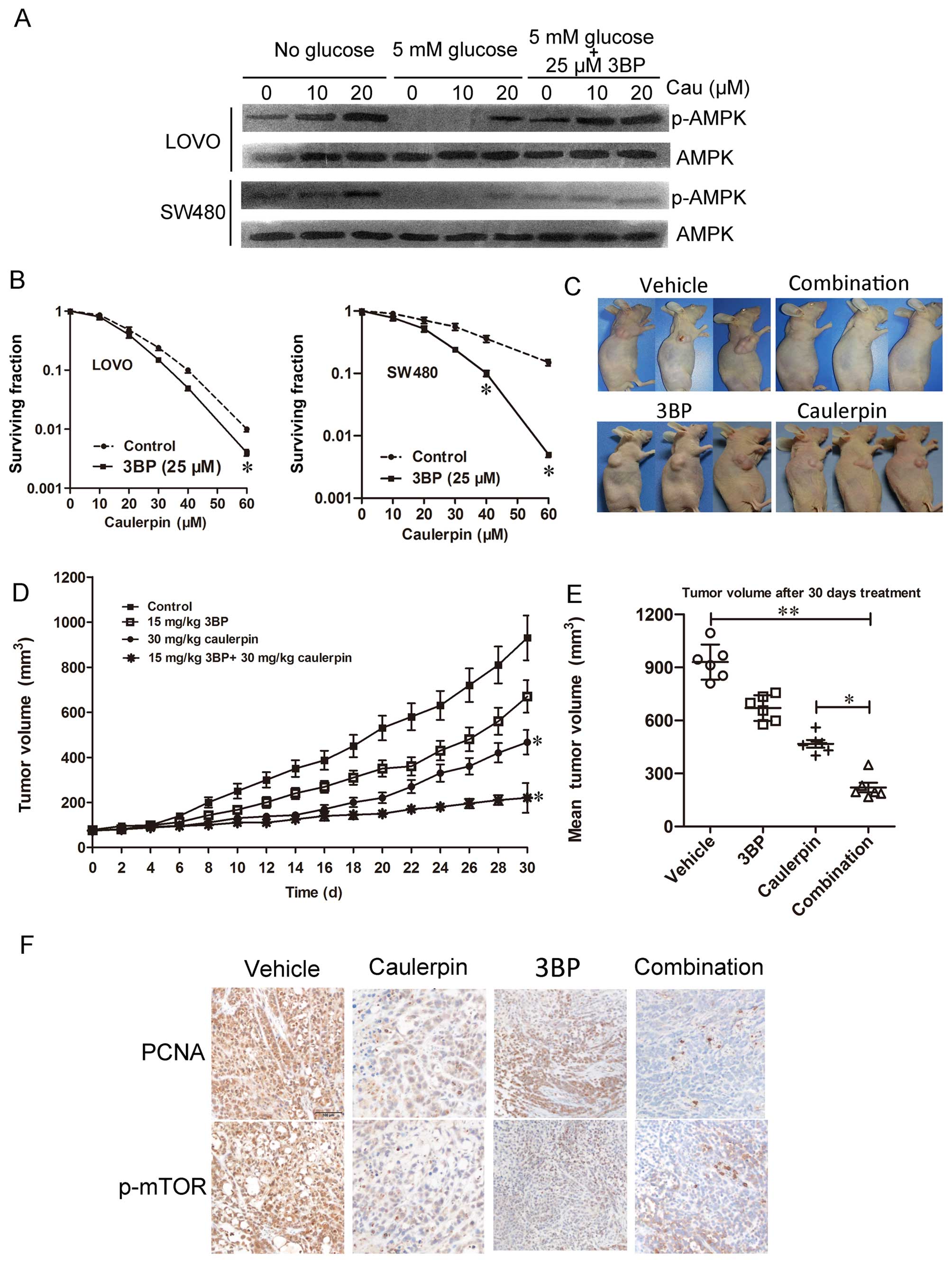
Synergistic effect of caulerpin combined
with glycolysis inhibitors
To further characterize the biochemical events
elicited by caulerpin and to examine its combination with the
glycolytic inhibitor, we investigated nutrient signaling events and
the AMPK kinase pathway. Glucose withdrawal promoted AMPK
activation in both cell lines. Interestingly, levels of p-AMPK is
higher in cells co-treated with caulerpin, 3BP (25 µM) and
glucose than cells treated with glucose only. These data emphasized
the key role of the glycolytic switch for compensating ATP
generation during caulerpin incubation (Fig. 7A). In Fig. 7B, in vitro CCK-8 assays were
applied to examine the cell viability. The IC50 of 3BP
is 41.2±1.6 and 38.9±1.6 µM for LOVO and SW480 cells,
respectively. As can be seen from the surviving factions, the
viability of cells treated with 3BP (25 µM) + caulerpin (60
µM) was significantly lower than cells treated with 3BP or
caulerpin alone. The combination index of 3BP (25 µM) +
caulerpin (60 µM) is 0.67 for LOVO cells and 0.21 for SW480
cells, suggesting a synergistic effect. When CI is >1, it means
antagonism. So we concluded that caulerpin plus 3BP may act
synergistically in inhibiting cellular proliferation in
vitro (Table I).
 | Table ICalcusyn analysis reveals synergistic
or antagonistic interactions between caulerpin and 3BP in CRC
cells. |
Table I
Calcusyn analysis reveals synergistic
or antagonistic interactions between caulerpin and 3BP in CRC
cells.
| Caulerpin
(µM) | 3BP
(µM) | Combination index
(CI)
|
|---|
| LOVO | SW480 |
|---|
| 20 | 25 | 1.43 | 1.38 |
| 40 | 25 | 1.02 | 0.68 |
| 60 | 25 | 0.67 | 0.21 |
To further assess the antitumor efficiency of the
combination in vivo, athymic nude mouse model bearing SW480
implanted xenografts were treated with control, 3BP, caulerpin, or
a combination of both agents. Tumor growth was slower in caulerpin
monotherapy group than the vehicle group. No significant difference
was observed in tumor size between mice treated with saline and
3BP. However, the combination of 3BP with caulerpin displayed
remarkable tumor regression (Fig.
7C). Tumor xenograft sections were subjected to
immunohistochemical analysis for p-mTOR and PCNA, which is an
indicator for cell proliferation. PCNA and p-mTOR was significantly
inhibited in combined treatment of 3BP with caulerpin (Fig. 7F).
Discussion
Cancer cell metabolism depends on a delicate balance
between oxidative phosphorylation and glycolysis. The former keeps
active to sustain energy needs required by the tumor, while the
latter extensively enlarged to afford the needs of an invasive and
rapid tumor growth (30).
Recently, new therapeutic approaches that target multiple
bioenergetics pathways have emerged. Interference with this unique
metabolic setting may provide novel opportunities for targeted
anticancer therapy.
Here, we focused on the energy regulating effect of
caulerpin, a mitochondrial ETC inhibitor. The obvious function
included the inhibition of OXPHOS, the loss of MMP, and the rise of
ROS in colorectal cancer cells. Although longtime treatment would
finally drive cells to death, a cell reprogramming towards
glycolysis largely attenuated the effect of caulerpin on cell death
in the initial few hours after caulerpin treatment. Given that
glycolysis triggered by caulerpin was a compensatory response, we
reasoned that combination with glycolysis inhibitors may reinforce
the anticancer effect of caulerpin. The marked increase in
caulerpin-induced AMPK activation by the glycolysis inhibitor 3BP
or by glucose deprivation evidently proves the above conclusion.
The enhancement of caulerpin sensitivity by 3BP was also confirmed
in the LOVO xenograft model, suggesting a key role of caulerpin in
energy metabolism-based combinational therapies.
As a crucial metabolic organelle during
carcinogenesis, mitochondria is regarded as a target for anticancer
therapy (4). Mostly, cancer cells
produce ATP via mitochondrial metabolism fueled by fatty acids and
amino acids (31). Furthermore,
mitochondrial oxidative metabolism is also required to sustain the
redox balance. Specifically, ATP generation associated with ROS
formation via complex I/III of the ETC (32). It is know that pro-tumorigenicity
is a key characteristic of ROS. When the generation of ROS
surpasses the intracellular clearance of ROS, cell-toxic oxidative
stress will be stimulated. ETC complex I, constitutes the entry
point of electrons and is an essential site for ROS generation
(33). Caulerpin has been reported
to reduce respiratory complex II activity and to exhibit
cytotoxicity to human dermal fibroblasts (14). In this study, caulerpin
considerably reduced oxygen consumption rate and ATP level in both
SW480 and LOVO cells, accompanied by an elevation in ROS.
Mitochondria isolated from rat liver have been selected as a system
in which mechanisms of bioenergetics are not altered (14). Therefore, liver mitochondrial and
LOVO cells were used to explore the complex interactions between
caulerpin and the organelle. Studies showed that complex I
inhibition by biguanides, such as phenformin and metformin,
suppressed cancer development in vitro and in vivo
(34,35). However, the effective inhibiting
concentrations of biguanides reached ~50 mM, with the
IC50 of caulerpin ranging from 20–31 µM. We found
that in the media containing malate and glutamate (substrates of
complex I), caulerpin treatment caused diminution of OCR, but
similar results were not obtained in the presence of succinate
(substrates of complex I), emphasizing the essential role of
complex I in the inhibitory effect of caulerpin.
Dynamic changes in mitochondria are closely
connected with post-translational modifications mediated by
metabolic sensors, such as SIRT1 and AMPK (36). Among them, AMPK acts as a
fuel-sensor that plays a vital role in manipulating bioenergetics
metabolism in both normal and malignant cells (37). Under conditions of metabolic
stress, activated AMPK will keep energy homeostasis by dampening
energy consuming (anabolic) pathways and strengthening energy
producing (catabolic) pathways to restore the loss of cellular ATP
and de-energization (38).
Caulerpin inhibited OXPHOS, accompanied by the decline of ATP
content and concomitant activation of the energy sensor AMPK. Once
activated, AMPK will promote glycolysis to replenish the loss of
ATP. In this study, in both LOVO and SW480 cells, the level of
p-AMPK was highest at 30 min after caulerpin treatment. We
hypothesized that OXPHOS was significantly inhibited by caulerpin,
and after 30 min, glycolysis was activated to some content to
counterbalance the loss of ATP. Therefore, the level of p-AMPK
increased during the initial 30 min and decreased since 60 min.
AMPK activation by caulerpin was dependent on CAMKK2, an upstream
effector of AMPK. In detail, AMPKα1 was demonstrated to mediate the
function of caulerpin, since crspAMPKα1 protected cells against
caulerpin-induced cytotoxicity and attenuated caulerpin-induced
activation of mTOR pathway. But siAMPKα2 failed to exhibit
impressive effect on cell death-induced by caulerpin treatment.
Likewise, AMPKα1-shRNA has been proved to improve mTORC1
phosphorylation in colorectal cancer cells (39). Herein, we concluded that AMPK
isoforms differentially regulated survival in response to caulerpin
treatment.
Under energy stress, activated AMPK can further
inhibit mTOR signaling for cellular adaptation in a stress
condition (28). Several groups
found that AMPK directly phosphorylated multiple components, such
as TSC2 in the mTORC1 pathway (40). Consistently, either AICAR (an AMPK
activator) or glucose withdrawal greatly attenuated mTORC1 activity
in TSC2-deficient cells (41). The
p70S6K and eIF4E-binding proteins (4E-BPs), which can be
phosphorylated by the mTOR complex, facilitate protein synthesis.
The p70S6K regulates sterol synthesis and switches glucose
metabolism from glycolysis to pentose phosphate pathway in cancer
cells (42). In the present study,
caulerpin triggered AMPK phosphorylation and further inhibited the
mTORC1/4E-BP1 axis.
Heightened glycolysis is a major cancer hallmark.
Firstly, we compared the basal metabolic profiles between LOVO and
SW480 CRC cells. OCR/ECAR ratio of LOVO cells was significantly
higher than that of SW480 cell, indicating that SW480 cells were
highly glycolytic, while LOVO was a less glycolytic cell line which
depended more on OXPHOS to produce ATP. This result could partially
explain why IC50 in LOVO cells is lower than SW480 cells
in response to caulerpin treatment, which was a potent inhibitor of
mitochondrial respiration. Moreover, mRNA of Transketolase-like
(TKTL)1, one of the key enzymes in glycolysis, was heterogeneously
expressed, with high level in SW480 cells but very low level in
LOVO cells (43,44). HKII is a rate-limiting enzyme in
glycolysis, thus giving rise to ATP decline and cell death. In
colon cancer cells, knockdown of HKII suppressed proliferation,
glycolysis, and lactate production under both normoxic and hypoxic
conditions (45). In this study,
distinguished effect was observed at different time-points after
caulerpin treatment. Glycolysis was enhanced in the initial hours
after treatment with caulerpin in LOVO cells rather than SW480
cells. However, long-term treatment of caulerpin considerably
inactivated mTOR pathway, thus causing cell death in both LOVO and
SW480 cells. GLUT1, HKII and PFKFB3 levels were weakened after 48 h
of caulerpin incubation.
Recently, combined treatment targeting both
glycolysis and mitochondria function emerges as an interesting
alternative for cancer therapy. Mitochondrial ETC blockers have
been reported to facilitate 2DG-induced oxidative stress and cell
killing effect in human colon carcinoma cells (46). In a phase I dose-escalation trial,
the recommended dose of 2DG in combination with weekly docetaxel is
63 mg/kg/day with tolerable adverse effects (47). The most noteworthy side effects at
this trial were reversible hyperglycemia, gastrointestinal bleeding
and reversible grade 3 QTc prolongation (47). 3BP is a small-molecule pyruvate
mimetic and anticancer drug, and its biological activity is
attributed to alkylation of free thiol groups on the cysteine
residues of proteins (48). 3BP
directly inhibits mitochondrial bound HKII. The combination of
caulerpin and 3BP retarded tumor growth in nude mice inoculated
with SW480 cells, along with a decrement of PCNA and mTOR
expression in IHC assays, showing the key role of AMPK/mTOR pathway
in the anticancer activity of caulerpin.
The present study provided a preliminary insight
into the potential use of caulerpin for the treatment of human
colorectal cancer. Only transplanted nude mice model with
xenografts was applied in our study to investigate the anticancer
efficiency of caulerpin plus 3BP. Orthotopic CRC model can also be
used in the future to verify the synergistical effect. In this
study, we demonstrated that caulerpin disturbed the mitochondrial
function mainly via inhibiting mTORC1-4E-BP1 axis. However,
knocking out of AMPKα1 did not lead to a total abolishment of
caulerpin-elicited cytotoxicity on LOVO cells. Moreover, the
broad-range caspase inhibitor z-VAD-fmk only partially prevented
cell death. Therefore, it is reasonable that other pathways are
also involved in the actions by caulerpin in CRC cells. Thus, it is
valuable to further uncover the interactions between AMPK signaling
and other possible pathways in mediating the effects of caulerpin
on colorectal carcinoma or other cancer models.
Acknowledgments
This study was supported by the Municipal key
disciplines of Ningbo (no. 2013001) and the Natural Science
Foundation of Ningbo (nos. 2014A610225 and 2016A610135).
Abbreviations:
|
CRC
|
colorectal cancer
|
|
OXPHOS
|
oxidative phosphorylation
|
|
ETC
|
electron transport chain
|
|
AMPK
|
AMP-activated protein kinase
|
|
CaMKK2
|
calcium/calmodulin-dependent protein
kinase 2
|
|
GLUT1
|
glucose transporter 1
|
|
HKII
|
hexokinase II
|
|
PFKFB3
|
6-phosphofructo-2-kinase
|
|
2DG
|
2-deoxy-D-glucose
|
|
3BP
|
3-bromopyruvate
|
|
MMP
|
mitochondrial membrane potential
|
|
ROS
|
reactive oxygen species
|
|
PBS
|
phosphate-buffered saline
|
|
OCR
|
oxygen consumption rate
|
|
ECAR
|
extracellular acidification rate
|
|
sgRNA
|
single guide RNA
|
References
|
1
|
Siegel R, Desantis C and Jemal A:
Colorectal cancer statistics, 2014. CA Cancer J Clin. 64:104–117.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Warburg O, Wind F and Negelein E: The
metabolism of tumors in the body. J Gen Physiol. 8:519–530. 1927.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ward PS and Thompson CB: Metabolic
reprogramming: A cancer hallmark even Warburg did not anticipate.
Cancer Cell. 21:297–308. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wheaton WW, Weinberg SE, Hamanaka RB,
Soberanes S, Sullivan LB, Anso E, Glasauer A, Dufour E, Mutlu GM,
Budigner GS, et al: Metformin inhibits mitochondrial complex I of
cancer cells to reduce tumorigenesis. eLife. 3:e022422014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Inoki K, Kim J and Guan KL: AMPK and mTOR
in cellular energy homeostasis and drug targets. Annu Rev Pharmacol
Toxicol. 52:381–400. 2012. View Article : Google Scholar
|
|
6
|
Sinha RA, Singh BK, Zhou J, Wu Y, Farah
BL, Ohba K, Lesmana R, Gooding J, Bay BH and Yen PM: Thyroid
hormone induction of mitochondrial activity is coupled to mitophagy
via ROS-AMPK-ULK1 signaling. Autophagy. 11:1341–1357. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wu XB, Liu Y, Wang GH, Xu X, Cai Y, Wang
HY, Li YQ, Meng HF, Dai F and Jin JD: Mesenchymal stem cells
promote colorectal cancer progression through AMPK/mTOR-mediated
NF-κB activation. Sci Rep. 6:214202016. View Article : Google Scholar
|
|
8
|
Zhang YJ, Dai Q, Sun DF, Xiong H, Tian XQ,
Gao FH, Xu MH, Chen GQ, Han ZG and Fang JY: mTOR signaling pathway
is a target for the treatment of colorectal cancer. Ann Surg Oncol.
16:2617–2628. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
He X, Lin X, Cai M, Zheng X, Lian L, Fan
D, Wu X, Lan P and Wang J: Overexpression of Hexokinase 1 as a poor
prognosticator in human colorectal cancer. Tumour Biol.
37:3887–3895. 2016. View Article : Google Scholar
|
|
10
|
Hamabe A, Yamamoto H, Konno M, Uemura M,
Nishimura J, Hata T, Takemasa I, Mizushima T, Nishida N, Kawamoto
K, et al: Combined evaluation of hexokinase 2 and phosphorylated
pyruvate dehydrogenase-E1α in invasive front lesions of colorectal
tumors predicts cancer metabolism and patient prognosis. Cancer
Sci. 105:1100–1108. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gandham SK, Talekar M, Singh A and Amiji
MM: Inhibition of hexokinase-2 with targeted liposomal
3-bromopyruvate in an ovarian tumor spheroid model of aerobic
glycolysis. Int J Nanomedicine. 10:4405–4423. 2015.PubMed/NCBI
|
|
12
|
Xintaropoulou C, Ward C, Wise A, Marston
H, Turnbull A and Langdon SP: A comparative analysis of inhibitors
of the glycolysis pathway in breast and ovarian cancer cell line
models. Oncotarget. 6:25677–25695. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu Y, Morgan JB, Coothankandaswamy V, Liu
R, Jekabsons MB, Mahdi F, Nagle DG and Zhou YD: The Caulerpa
pigment caulerpin inhibits HIF-1 activation and mitochondrial
respiration. J Nat Prod. 72:2104–2109. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ferramosca A, Conte A, Guerra F, Felline
S, Rimoli MG, Mollo E, Zara V and Terlizzi A: Metabolites from
invasive pests inhibit mitochondrial complex II: A potential
strategy for the treatment of human ovarian carcinoma? Biochem
Biophys Res Commun. 473:1133–1138. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Graham NA, Tahmasian M, Kohli B,
Komisopoulou E, Zhu M, Vivanco I, Teitell MA, Wu H, Ribas A, Lo RS,
et al: Glucose deprivation activates a metabolic and signaling
amplification loop leading to cell death. Mol Syst Biol. 8:5892012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Priebe A, Tan L, Wahl H, Kueck A, He G,
Kwok R, Opipari A and Liu JR: Glucose deprivation activates AMPK
and induces cell death through modulation of Akt in ovarian cancer
cells. Gynecol Oncol. 122:389–395. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen Y, Qing W, Sun M, Lv L, Guo D and
Jiang Y: Melatonin protects hepatocytes against bile acid-induced
mitochondrial oxidative stress via the AMPK-SIRT3-SOD2 pathway.
Free Radic Res. 49:1275–1284. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang SY, Wei YH, Shieh DB, Lin LL, Cheng
SP, Wang PW and Chuang JH: 2-Deoxy-d-Glucose can complement
doxorubicin and sorafenib to suppress the growth of papillary
thyroid carcinoma cells. PLoS One. 10:e01309592015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wei L, Zhou Y, Dai Q, Qiao C, Zhao L, Hui
H, Lu N and Guo QL: Oroxylin A induces dissociation of hexokinase
II from the mitochondria and inhibits glycolysis by SIRT3-mediated
deacetylation of cyclophilin D in breast carcinoma. Cell Death Dis.
4:e6012013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jeong SM, Lee J, Finley LW, Schmidt PJ,
Fleming MD and Haigis MC: SIRT3 regulates cellular iron metabolism
and cancer growth by repressing iron regulatory protein 1.
Oncogene. 34:2115–2124. 2015. View Article : Google Scholar
|
|
21
|
Song J, Feng L, Zhong R, Xia Z, Zhang L,
Cui L, Yan H, Jia X and Zhang Z: Icariside II inhibits the EMT of
NSCLC cells in inflammatory microenvironment via down-regulation of
Akt/NF-κB signaling pathway. Mol Carcinog. Feb 9–2016.Epub ahead of
print.
|
|
22
|
Liu Y, Veena CK, Morgan JB, Mohammed KA,
Jekabsons MB, Nagle DG and Zhou YD: Methylalpinum isoflavone
inhibits hypoxia-inducible factor-1 (HIF-1) activation by
simultaneously targeting multiple pathways. J Biol Chem.
284:5859–5868. 2009. View Article : Google Scholar :
|
|
23
|
Bonifati S, Daly MB, St Gelais C, Kim SH,
Hollenbaugh JA, Shepard C, Kennedy EM, Kim DH, Schinazi RF, Kim B,
et al: SAMHD1 controls cell cycle status, apoptosis and HIV-1
infection in monocytic THP-1 cells. Virology. 495:92–100. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhou X, Chen M, Zeng X, Yang J, Deng H, Yi
L and Mi MT: Resveratrol regulates mitochondrial reactive oxygen
species homeostasis through Sirt3 signaling pathway in human
vascular endothelial cells. Cell Death Dis. 5:e15762014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang W, Tong D, Liu F, Li D, Li J, Cheng
X and Wang Z: RPS7 inhibits colorectal cancer growth via decreasing
HIF-1α-mediated glycolysis. Oncotarget. 7:5800–5814.
2016.PubMed/NCBI
|
|
26
|
Yamada C, Aikawa T, Okuno E, Miyagawa K,
Amano K, Takahata S, Kimata M, Okura M, Iida S and Kogo M: TGF-β in
jaw tumor fluids induces RANKL expression in stromal fibroblasts.
Int J Oncol. 49:499–508. 2016.PubMed/NCBI
|
|
27
|
Zhang M, Dong Y, Xu J, Xie Z, Wu Y, Song
P, Guzman M, Wu J and Zou MH: Thromboxane receptor activates the
AMP-activated protein kinase in vascular smooth muscle cells via
hydrogen peroxide. Circ Res. 102:328–337. 2008. View Article : Google Scholar
|
|
28
|
Hardie DG, Ross FA and Hawley SA: AMPK: A
nutrient and energy sensor that maintains energy homeostasis. Nat
Rev Mol Cell Biol. 13:251–262. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Luczak ED and Anderson ME: CaMKII
oxidative activation and the pathogenesis of cardiac disease. J Mol
Cell Cardiol. 73:112–116. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dang CV: Links between metabolism and
cancer. Genes Dev. 26:877–890. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hensley CT, Wasti AT and DeBerardinis RJ:
Glutamine and cancer: Cell biology, physiology, and clinical
opportunities. J Clin Invest. 123:3678–3684. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Willems PH, Rossignol R, Dieteren CE,
Murphy MP and Koopman WJ: Redox homeostasis and mitochondrial
dynamics. Cell Metab. 22:207–218. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Murphy MP: How mitochondria produce
reactive oxygen species. Biochem J. 417:1–13. 2009. View Article : Google Scholar
|
|
34
|
Pollak M: Overcoming drug development
bottlenecks with repurposing: Repurposing biguanides to target
energy metabolism for cancer treatment. Nat Med. 20:591–593. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Birsoy K, Possemato R, Lorbeer FK,
Bayraktar EC, Thiru P, Yucel B, Wang T, Chen WW, Clish CB and
Sabatini DM: Metabolic determinants of cancer cell sensitivity to
glucose limitation and biguanides. Nature. 508:108–112. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Duarte FV, Amorim JA, Palmeira CM and Rolo
AP: Regulation of mitochondrial function and its impact in
metabolic stress. Curr Med Chem. 22:2468–2479. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shackelford DB and Shaw RJ: The LKB1-AMPK
pathway: Metabolism and growth control in tumour suppression. Nat
Rev Cancer. 9:563–575. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fennema E, Rivron N, Rouwkema J, van
Blitterswijk C and de Boer J: Spheroid culture as a tool for
creating 3D complex tissues. Trends Biotechnol. 31:108–115. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lu PH, Chen MB, Ji C, Li WT, Wei MX and Wu
MH: Aqueous Oldenlandia diffusa extracts inhibits colorectal cancer
cells via activating AMP-activated protein kinase signalings.
Oncotarget. Jun 13–2016.Epub ahead of print.
|
|
40
|
Inoki K, Zhu T and Guan KL: TSC2 mediates
cellular energy response to control cell growth and survival. Cell.
115:577–590. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wolff NC, Vega-Rubin-de-Celis S, Xie XJ,
Castrillon DH, Kabbani W and Brugarolas J: Cell-type-dependent
regulation of mTORC1 by REDD1 and the tumor suppressors TSC1/TSC2
and LKB1 in response to hypoxia. Mol Cell Biol. 31:1870–1884. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Düvel K, Yecies JL, Menon S, Raman P,
Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S,
et al: Activation of a metabolic gene regulatory network downstream
of mTOR complex 1. Mol Cell. 39:171–183. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fanciulli M, Bruno T, Giovannelli A,
Gentile FP, Di Padova M, Rubiu O and Floridi A: Energy metabolism
of human LoVo colon carcinoma cells: Correlation to drug resistance
and influence of lonidamine. Clin Cancer Res. 6:1590–1597.
2000.PubMed/NCBI
|
|
44
|
Bentz S, Cee A, Endlicher E, Wojtal KA,
Naami A, Pesch T, Lang S, Schubert P, Fried M, Weber A, et al:
Hypoxia induces the expression of transketolase-like 1 in human
colorectal cancer. Digestion. 88:182–192. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Qin Y, Cheng C, Lu H and Wang Y: miR-4458
suppresses glycolysis and lactate production by directly targeting
hexokinase2 in colon cancer cells. Biochem Biophys Res Commun.
469:37–43. 2016. View Article : Google Scholar
|
|
46
|
Fath MA, Diers AR, Aykin-Burns N, Simons
AL, Hua L and Spitz DR: Mitochondrial electron transport chain
blockers enhance 2-deoxy-D-glucose induced oxidative stress and
cell killing in human colon carcinoma cells. Cancer Biol Ther.
8:1228–1236. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Raez LE, Papadopoulos K, Ricart AD,
Chiorean EG, Dipaola RS, Stein MN, Rocha Lima CM, Schlesselman JJ,
Tolba K, Langmuir VK, et al: A phase I dose-escalation trial of
2-deoxy-D-glucose alone or combined with docetaxel in patients with
advanced solid tumors. Cancer Chemother Pharmacol. 71:523–530.
2013. View Article : Google Scholar
|
|
48
|
Ko YH, Verhoeven HA, Lee MJ, Corbin DJ,
Vogl TJ and Pedersen PL: A translational study 'case report' on the
small molecule 'energy blocker' 3-bromopyruvate (3BP) as a potent
anticancer agent: From bench side to bedside. J Bioenerg Biomembr.
44:163–170. 2012. View Article : Google Scholar : PubMed/NCBI
|