Introduction
Breast cancer is the most common carcinoma among
Chinese women. It is estimated that there will be an increasing
number of new breast cancer patients in China (1). Radiation therapy may have a
transformative impact on the treatment of breast cancer, because it
allows women with early stage disease to maintain integrity of
their body and those with advanced disease to have relief from
suffering (2). Fisher et al
(3) proved that breast-conserving
surgery with radiation has the same therapeutic effect as
mastectomy after their 20-year radomized trial in 1851 women with
invasive breast tumors.
Although radiation therapy is a powerful anticancer
modality, radiation-induced stress response and gene expression
with an adaptive resistance may severely compromise the
effectiveness of radiation (4).
Mechanisms leading to radioresistance are diverse and still poorly
defined. Many investigations have indicated that breast cancer stem
cells evolve resistance to radiation due to intrinsic and extrinsic
mechanisms, genetic mutations and epigenetic modifications
(5).
Low-dose ionizing radiation (LDIR) is proved having
hormesis (6,7) and adoptive effect (8), which is quite different from
high-dose ionizing radiation (HDIR). Many investigations, incuding
ours, have indicated that LDIR stimulates the proliferation of
normal cells, such as rat mesenchymal stem cells, mouse bone marrow
hematopoietic progenitor cells and several human normal cell lines
(9,10), but LDIR does not induce
proliferation of tumor cells (11). Therefore, LDIR has been considering
as a promising assistant method of clinical radiotherapy.
p53 plays a key role in the process of radiation
response, controlling the activation of DNA repair and cell
apoptosis pathways after acute radiation injury (12,13).
Since more than half of all tumors harbor p53 gene mutation
and deletion (14), it is
necessary to investigate the biological behavior of these cells
with abnormal p53 gene after LDIR. Our previous study
demonstrated that 75 mGy LDIR inhibits the proliferation of
p53null type human prostate tumor cells. In this
investigation, we further illustrate how breast cancer cells with
p53 mutation responded to LDIR. We hope that the present
study provides a valuable theoretical reference and supplement to
the radiation treatment of breast cancer.
Materials and methods
Cell cultivation and treatments
The human breast cancer cell line MDA-MB-231 and
normal breast fibroblast cell line Hs 578Bst were purchased from
the American Type Culture Collection (ATCC; Manassas, VA, USA);
human embryonic kidney cell line 293T was kept in our laboratory.
MDA-MB-231 and 293T cells was maintained in Dulbecco's modified
eagle's media (DMEM; Life Technology, Shanghai, China) supplemented
with 10% fetal bovine serum (FBS; Hyclone Laboratories, Beijing,
China) and 1% antibiotics (penicillin/streptomycin; Invitrogen,
Carlsbad, CA, USA). Hs 578Bst cells were maintained in DMEM/F12
supplemented with 10% FBS and 30 ng/ml epidermal growth factor
(EGF; Invitrogen). The cells were cultured at 37°C in a humidified
incubator with a constant air flow of 5% CO2.
In order to inhibit the function of ATM, 10
µM KU55933 (Selleck Chemicals, Co., Ltd., Shanghai, China)
dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich, Shanghai,
China) was added to the cell culture media 2 h prior to
irradiation. Media containing the chemical inhibitors were replaced
with fresh medium immediately after the irradiation.
Lentiviral expression of wtP53 and p53
siRNA
The full-length 1.2 kb human p53 gene coding
sequence (CDS) was amplified from the P53-expressing plasmid of
pcDNA3.1(-)-P53 kept in our laboratory. The PCR primers contain the
XbaI and EcoRV restriction sites. The primers
sequence are as follows. Forward,
5′-ATAGAAGATTCTAGATGTACAGCCGCCACCATGGAGGAGCCGCAGTCAGAT-3′ and
reverse, 5′-TCAGTCTGAG TCAGGCCCTTTAATGAGATATCGTCGACGAAT-3′. The pCR
product was gel-purified, cut by restriction enzymes and ligated
into the pCMV-copGFP/Puro vector constructed in our laboratory. The
sense sequence of p53-siRNA and scrambled siRNA were
CACCATCCACTACAACTACAT (15) and
GGATTTCGAGTCGTCTTAA. The siRNA fragment was synthesized by PCR and
cloned into pGreenPuro vector (SBI, Los Angeles, CA, USA) via
restriction sites of BamHI and EcoRI. Both p53
overexpression and shRNA clones were confirmed by sequencing and
then packaged in 293T packing cells using the method described in
our laboratory (16). At 48 h
post-transduction, cell clones were selected by 2 µg/ml
puromycin (Invivogen, Inc., San Diego, CA, USA). After 7 days of
puromycin-selection, stable transduction cells were used for
LDIR.
Irradiation strategy
Monolayer cells were ionizing irradiated at the dose
of 12.5 mGy/min by X-RAD 320 (Precision X-RAD; Precision X-Ray,
North Branford, CT, USA). The total dose was 50, 100, 150 or 200
mGy. After LDIR, culture media were replaced and cells were
harvested immediately or continually cultured until the next step
of the experiment was carried out. Control groups were treated
similarly except for irradiation.
Cell proliferation assay
Approximately 3×103 MDA-MB-231 cells and
Hs 578Bst cells were seeded in a 96-well plate 24 h before LDIR,
and then irradiated with 50, 100, 150 or 200 mGy of X-rays. After
the LDIR, cells were transferred to the incubator and cultured for
another 24, 48 or 72 h. Cell proliferation assays were determined
by WST-1 cell proliferation reagent (Roche Diagnostics, Shanghai,
China). According to the manufacturer's instructions, 20 µl
WST-1 reagent was added to 200 µl cell culture medium and
incubated in the dark for 2 h. Then, the absorbance of 450 nm and
630 nm were measured by microplate reader (Bioteck, Co., Ltd.,
Beijing, China). Final OD (optical density) was designated as
OD450-OD630-ODblank.
Cell cycle assay
Approximately 2×106 irradiated MDA-MB-231
cells or Hs 578Bst cells were harvested and washed by 1 ml cold
phosphate-buffered saline (PBS; Thermo Fisher Scientific, Beijing,
China) twice to remove the residual trypsin and serum. Cells were
resuspended in 1 ml fixation solution (300 µl PBS and 700
µl ethanol). After incubated at 4°C for 4 h, cells were
centrifuged at 1000 rpm for 5 min and fixation solution was
removed. After washed twice with 1 ml PBS, cells were pelleted and
suspended in 0.5 ml propidium iodide (PI; Sigma-Aldrich) staining
solution [50 µg/ml PI, 20 µg/ml RNase A (Takara Bio,
Dalian, China) and 0.2% Triton X-100 (Sigma-Aldrich)] and incubated
in the dark at 37°C for 30 min. Cell suspensions were filtered
through a 400-mesh sieve before analyzed by a BD FACSCalibur flow
cytometer (Becton-Dickinson, Sparks, MD, USA).
Protein extraction and western blot
analysis
Cell total protein was extracted with RIPA buffer
(Beyotime Institute of Biotechnology, Shanghai, China) supplemented
with cocktail protease inhibitor (Roche), and the protein
concentration was determined by BCA protein assay kit (Beyotime
Institute of Biotechnology).
Approximately 5–40 mg total protein was separated by
4–8% (for ATM) or 5–12% (for other proteins) SDS polyacrylamide gel
electrophoresis (SDS-PAGE), and then was electrophoretically
transferred to PVDF membranes (0.45 µm; Millipore,
Billerica, MA, USA) and blocked at 37°C for 1 h with 5% skim milk
in TBST [TBS (10 mmol/l Tris pH 7.5, 150 mmol/l naCl) containing
0.1% Tween-20 (Sigma-Aldrich)]. Afterwards, membranes were
incubated in monoclonal antibodies against CDK4 (1:1,500; Cell
Signaling Technology, Beijing, China), CDK6 (1:1,500; Santa Cruz
Biotechnology, Santa Cruz, CA, USA), cyclin D1 (1:1,500; Abcam,
Shanghai, China), P53 (1:1,500; DO7, Santa Cruz Biotechnology), p21
(1:1,000, C-19; Santa Cruz Biotechnology), ATM (1:1,500, Y170; Cell
Signaling Technology) and β-actin (1:3,000; Santa Cruz
Biotechnology) at 4°C overnight. After washed for 3×5 min with
TBST, membranes were incubated with HRP conjugated goat anti-mouse
or goat anti-rabbit second antibodies (1:3,000; ZSGB-BIO, Beijing,
China) at 37°C for 1 h. Washed further 3 times, the immunocomplexes
were detected with the enhanced chemiluminescence system (ECL;
Thermo Fisher Scientific) and X-ray film (Kodak, Beijing, China).
Protein expression levels were determined semi-quantitatively by
densitometric analysis with Quantity One software (Bio-Rad
Laboratories).
Statistical analysis
All data and results were calculated from at least
three replicate measurements and were presented as means ± SD. The
significance was determined by the Student's t-test using the SPSS
20.0 (IBM). P<0.05 was considered statistically significant
(*P<0.05; **P<0.01; NS, not
significant).
Results
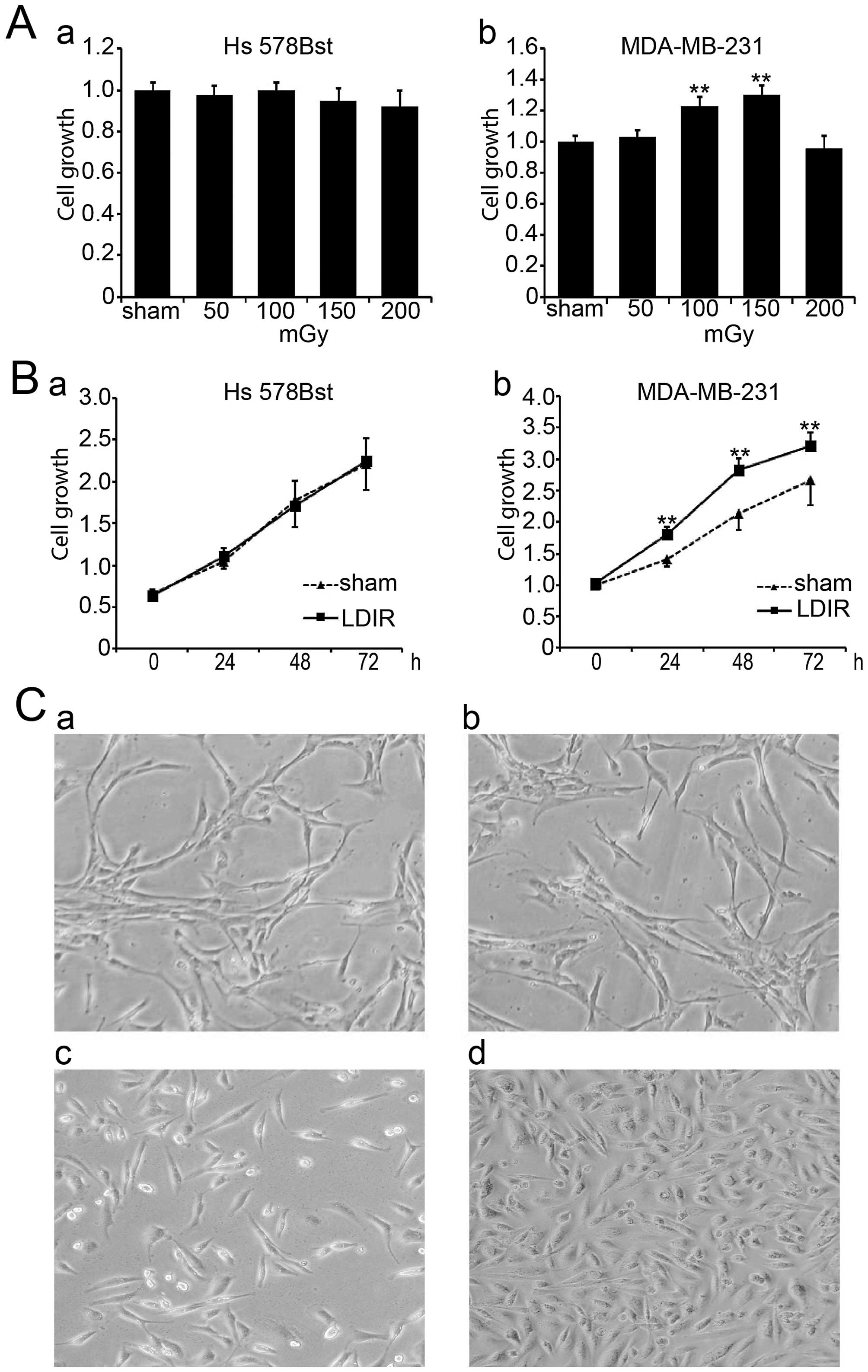
LDIR induces differential cell growth in
breast cancer MDA-MB-231 and normal Hs 578Bst cells
In order to investigate the function of LDIR on
breast cancer MDA-MB-231 and normal Hs 578Bst cells, we set up four
different doses of LDIR including 50, 100, 150 and 200 mGy. Cell
proliferation was assayed by WST-1 24 h post-LDIR. Compared to the
sham-irradiated group, we found that 100 and 150 mGy doses of LDIR
significantly promoted the growth of MDA-MB-231 cells (Fig. 1A-b; P<0.01), and that the 150
mGy irradiation had the most significant promotion effect. In
contrast, none of the irradiation doses affected the growth of Hs
578Bst cells (Fig. 1A-a).
We compared the cell growth in two cell lines at
different time-points following 150 mGy LDIR. Cell proliferation
was determined by WST-1 at 0, 24, 48 and 72 h post-LDIR.
Apparently, MDA-MB-231 cells showed an accelerated proliferation 24
h post-LDIR (Fig. 1B-b;
P<0.01). However, normal breast Hs 578Bst cells showed no
obvious change after 150 mGy irradiation compared with the sham
control group (Fig. 1B-a;
P>0.05). Cell morphology also showed the differential speed of
cell growth between MDA-MB-231 cells and Hs 578Bst cells after LDIR
(Fig. 1C).
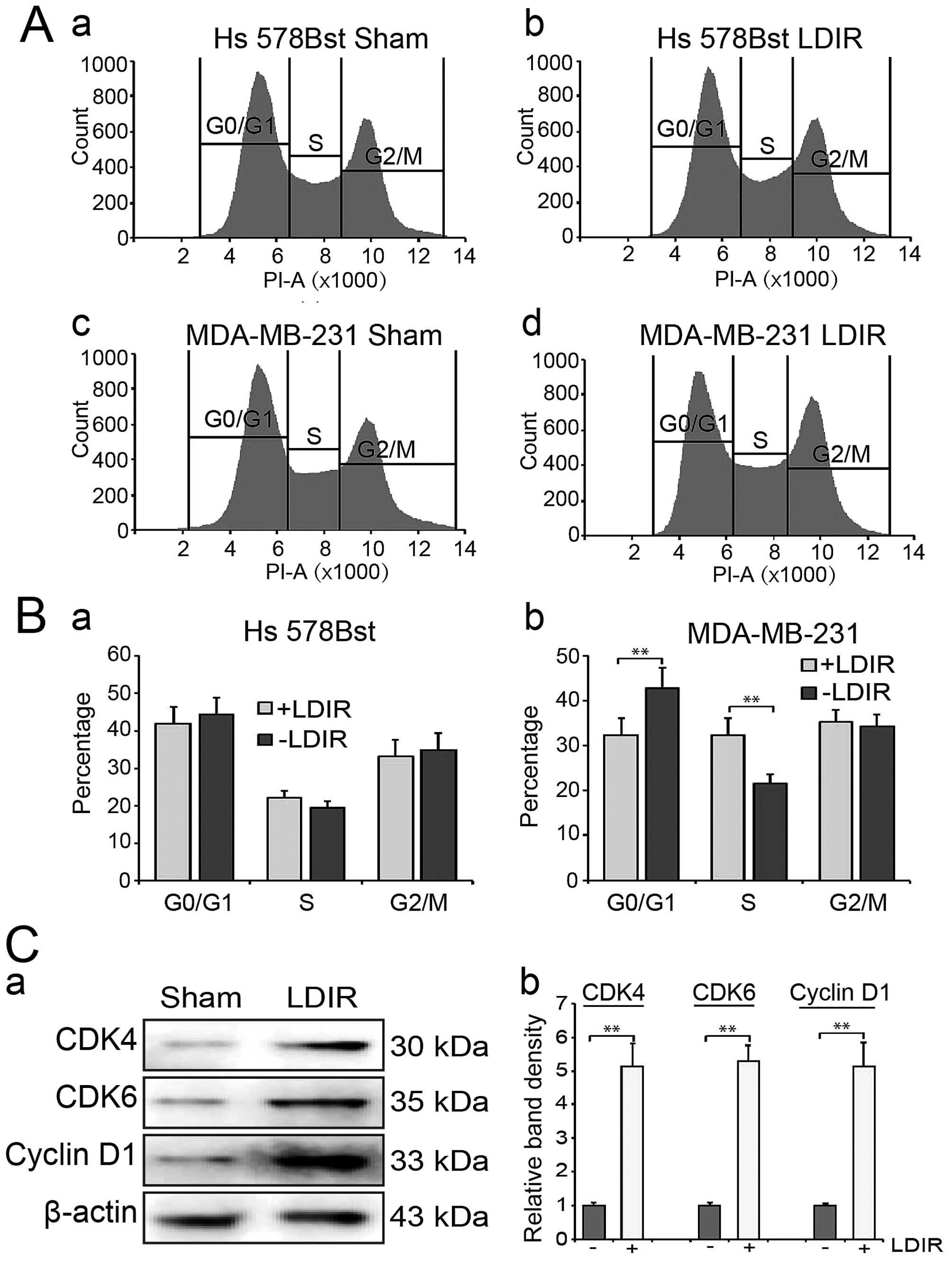
LDIR accelerates entry into S phase in
MDA-MB-231 cells
We then used flow cytometry to analyze cell cycle
distribution at 24 h post-LDIR. We found that LDIR may increase S
phase cell percentage in MDA-MB-231 cells compared to the sham
group (Fig. 2A-c and -d). In
contrast, we did not observe any significant change of cell cycle
distribution in Hs 578Bst cells after LDIR (Fig. 2A-a and -b). Quantitation data
showed that in MDA-MB-231 cells, the S phase percentage was
increased to 1.5-fold (32.4 vs. 21.6%, P<0.01; Fig. 2B).
Activation of cyclin dependent
kinases
We further detected the expression of G1/S-phase
related cyclins and cyclin-dependent kinases (CDKs). The expression
of cyclin D1, cyclin E, CDK2, CDK4 and CDK6 was detected by western
blotting at 24 h post-150 mGy LDIR. We observed a significant
increment in cyclin-dependent kinases following LDIR (Fig. 2C-a). Quantitation analyses showed
that the expression of CDK4, CDK6 and cyclin D1 significantly
increased by 5.1-, 5.3- and 5.1-fold, respectively (Fig. 2C-b; P<0.01). However, there was
no significant changes in cyclin E and CDK2 following irradiation
(data not shown).
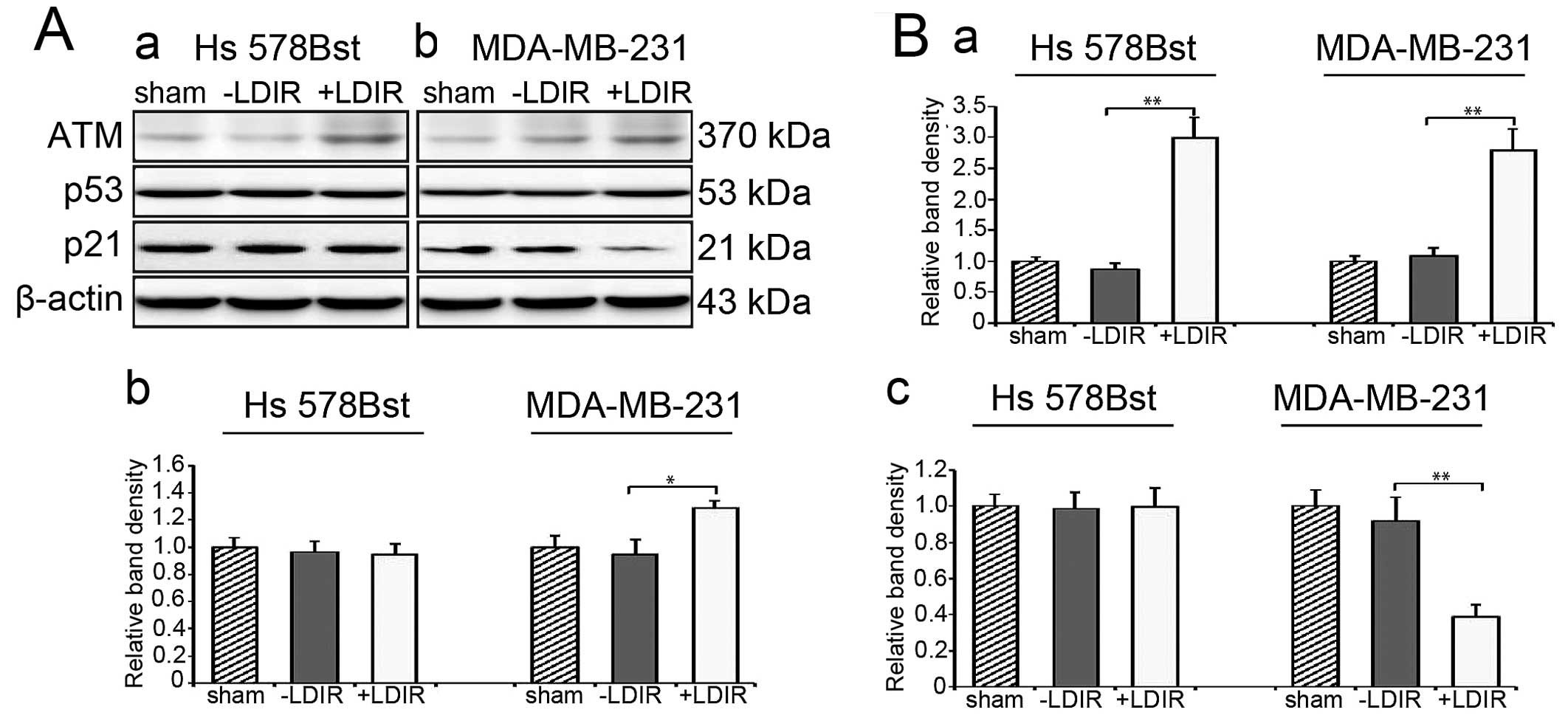
LDIR specifically activates the
ATM/p53/p21 pathway in breast cancer cells
We have demonstrated that ATM/p53/p21 pathway is a
critical LDIR-associated pathway leading to cell cycle and cell
proliferation change especially in p53 abnormal cancer cells
(unpublished data). We first checked the ATM level at 4 h
post-LDIR. We found that the expression of ATM in both Hs 578Bst
and MDA-MB-231 cells was upregulated by 150 mGy LDIR at 4 h
post-LDIR. In Hs 578Bst cells, the expression of ATM was
upregulated by 3-fold and in MDA-MB-231 cells, it was activated by
2.8-fold (Fig. 3A and B-a;
P<0.01). Then we examined the expression of P53. Western blot
results revealed that the expression of P53 increased by 1.3-fold
at 4 h post-LDIR in MDA-MB-231 cells (Fig. 3A and B-b; P<0.05). We also
quantitated the expression of p21 after LDIR. In MDA-MB-231 cells,
we observed a significant downregulation of p21 by 0.4-fold after
LDIR (Fig. 3A and B-c; P<0.01).
However, the LDIR-induced P53 and p21 changes were absent in normal
Hs 578Bst cells.
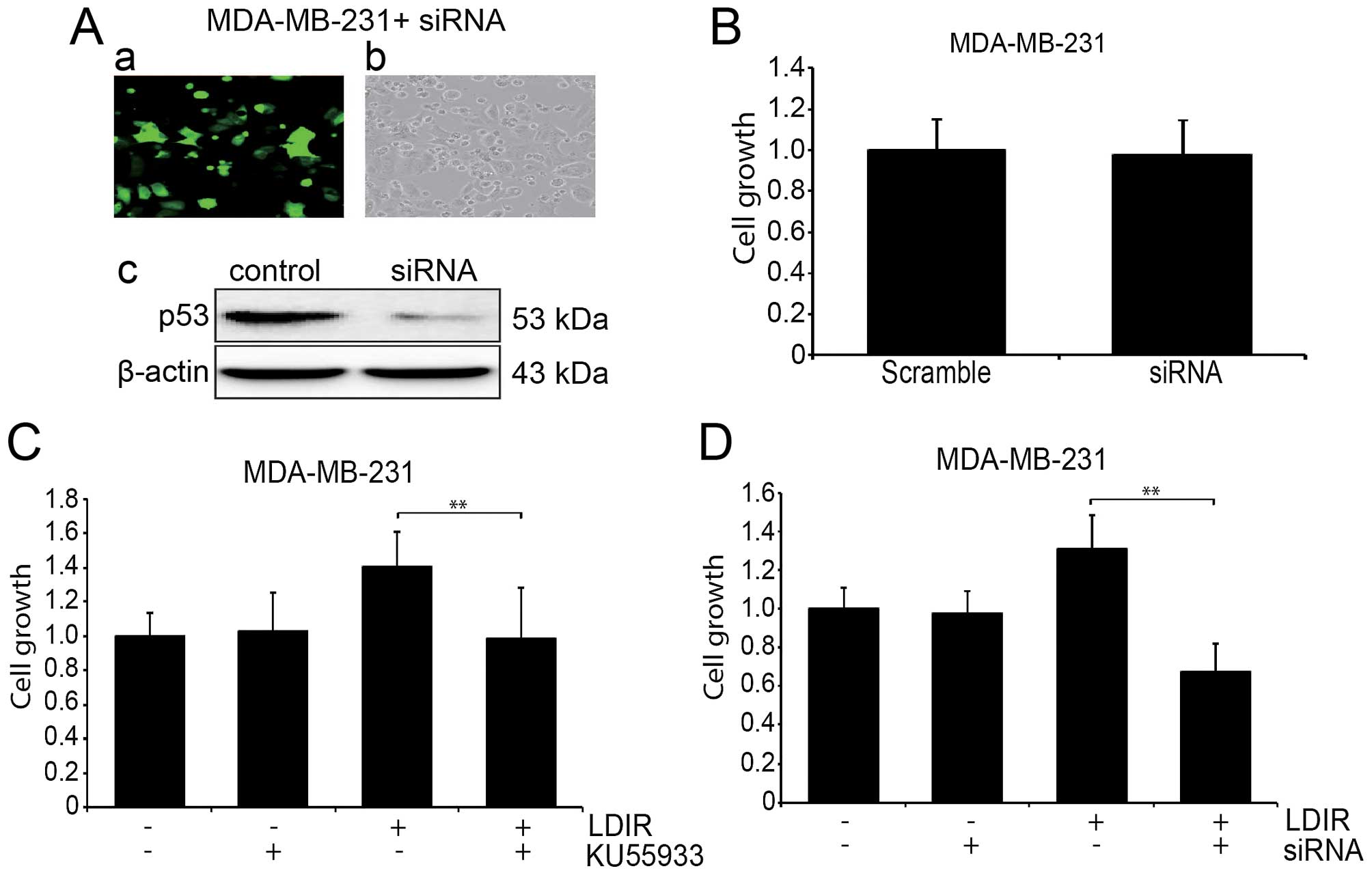
ATM inhibitor and P53 siRNA interferes
LDIR-induced cell proliferation in MDA-MB-231 cells
To further address the role of the ATM/p53/p21
pathway in the LDIR-induced cell proliferation change, we used
KU55933 to block the function of ATM in MDA-MB-231 cells and
measured the cell proliferation at 24 h post-LDIR. We found that
the LDIR induced proliferation in the MDA-MB-231 cells was
abolished after the treatment of KU55933 (Fig. 4C-a; P<0.05). We also used
lentiviral p53 shRNA to knock down the expression of mtP53 in the
MDA-MB-231 cells (Fig. 4A), and
the LDIR induced proliferation was abolished, (Fig. 4C-b; P<0.05).
Wild-type P53 interferes LDIR-induced
cell proliferation in MDA-MB-231 cells
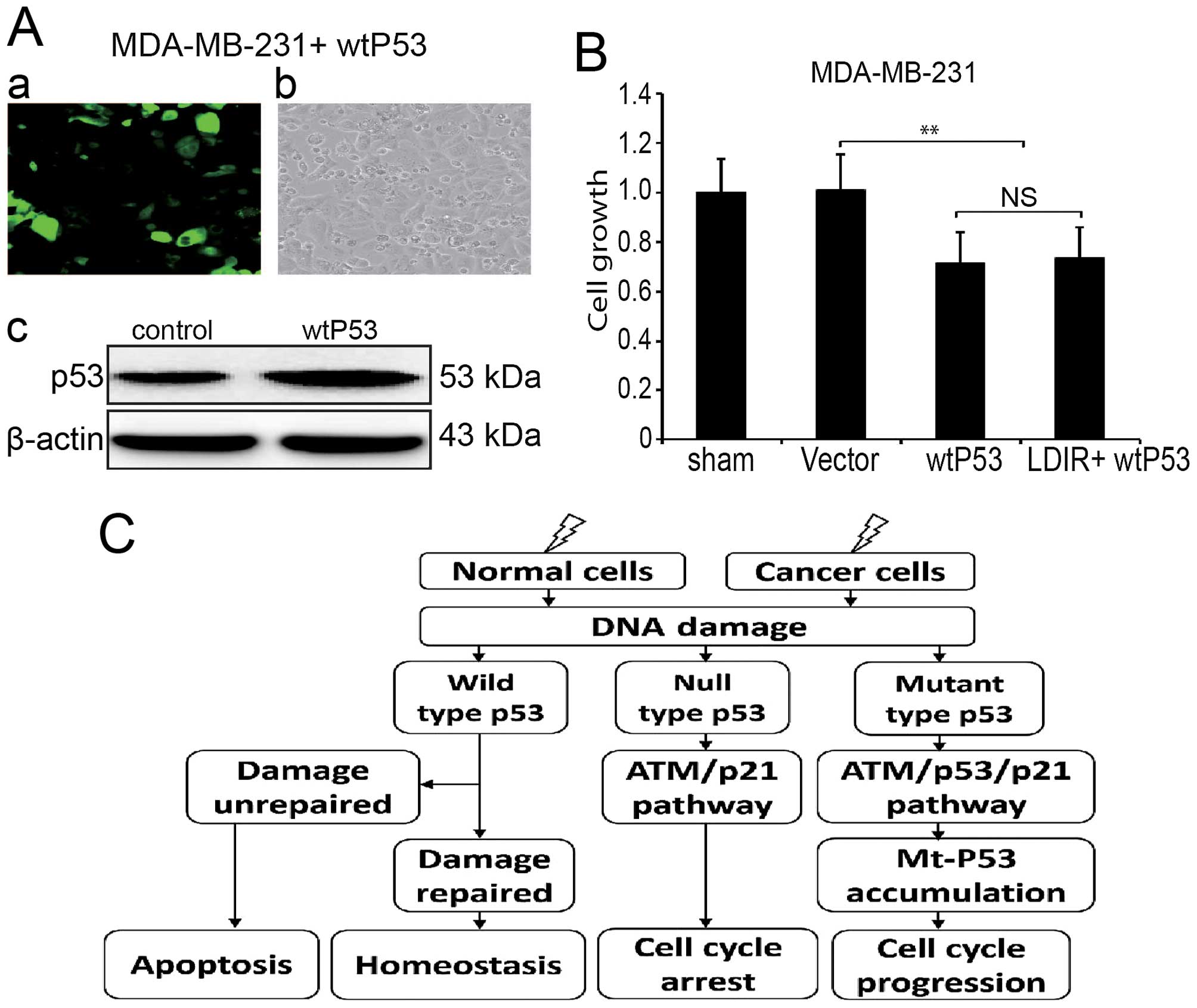
Since MDA-MB-231 expresses mtP53, we introduced
wtP53 by transducing MDA-MB-231 cells with lentivirus (Fig. 5A). We investigated the
proliferation of wtP53 transduced MDA-MB-231 cells as well as cells
irradiated by 150 mGy LDIR. We found that the LDIR-induced cell
proliferation was also abolished (Fig.
5B).
Discussion
The linear no-threshold (LNT) model assumes that
even very low doses of ionizing radiation may generate detrimental
effects on human health (17).
However, it is indicated that although ionizing radiation can harm
cells, such injurious effects are not linear with the radiation
dose mainly due to different cellular mechanisms to adapt or die
(18,19). Besides, there is increasing
evidence showing that radiation below certain doses could be
protective (20,21).
LDIR is usually defined as ≤0.2 Gy at low linear
energy transfer (LET) or ≤0.05 Gy at high LET (22). In general, biological effects of
LDIR can be summarized as 'hormesis', 'adaptive response (AR)',
'bystander effects', 'hyper-radiosensitivity (HRS)' and 'induced
radioresistance (IRR)' (23).
These biological effects of LDIR, especially hormesis and AR, have
been investigated for the potential applicability in the treatment
of cancer due to its suppressive effect on cancer induction, growth
and metastasis (24). However, the
health risks associated with LDIR are still non-negligible, and
owing to a lack of understanding of the mechanisms underlying the
response of LDIR, the potential applicability of LDIR in clinical
anticancer therapy remains controversial. Therefore, it is
worthwhile to further clarify and provide more investigation
evidence of LDIR on molecular level.
Our previous study elucidated that ATM/p53/p21 is an
important responding pathway to LDIR in human prostate cells
(unpublished data). Because of a lack of normal p53 function, the
proliferation of PC-3 cells was suppressed by LDIR. In the present
study, we chose MDA-MB-231 cells to investigate the biological
effect of LDIR on breast cancer. MDA-MB-231 cells have a mutation
on p53 gene at exon 8 (25). We investigated the cell
proliferation after 50–200 mGy LDIR. MDA-MB-231 cells showed its
radiation sensitivity at 100 and 150 mGy LDIR at the dose rate of
12.5 mGy/min, but the normal breast fibroblast cell line Hs 578Bst
did not respond to LDIR. In our series of studies, we have
investigated the LDIR biological effects on several cancer cell
lines, such as K562, HL-60, NCI-H446, BEL7402, U251, HCT-8 and HeLa
(9–11). However, we found LDIR has no
obvious effect on these cancer cell lines. It is noteworthy that in
these investigations, we did not focus on the genetic backgrounds
of these cells, until we found that the proliferation of PC-3 cells
were inhibited by LDIR.
When the cell cycle distribution was analyzed, we
found LDIR accelerates the cell cycle entry into S phase in
MDA-MB-231 cells, and the expression of CDK4, CDK6 and cyclin D1
increases. When we further investigated the molecular mechanism
during this process, we found that ATM/p53/p21 pathway was also
activated by LDIR in MDA-MB-231 cells. Although ATM in both
MDA-MB-231 cells and Hs 578Bst cells was activated by LDIR, only
the mtP53 protein in MDA-MB-231 cells was shown to be
increased.
The p53, as a tumor suppressor gene, is
crucial in the protection against DNA damage and other forms of
physiological stress primarily by inducing cell cycle arrest or
apoptosis (26). Mutation of p53,
however, inactivates these growth regulatory functions and causes a
loss of tumor suppressor activity. It has been reported that
overexpression of mtP53 proteins in p53null cells
resulted in enhancement of plating efficiency and tumorigenicity
(27,28). Stable expression of p53 plays a
central role in LDIR activated ATM/p53/p21 pathway, affecting cell
proliferation and cell cycle. In normal cells, radiation-induced
DNA damage can be repaired by p53 and cells maintain homeostasis;
in breast cancer MDA-MB-231 cells, our results indicated that the
ATM/p53/p21 pathway was also activated by LDIR. However, since
mtP53 has an increased half-life (29), the fast growth of MDA-MB-231 cells
can be ascribed to the abnormal accumulation of mtP53 (Fig. 5C).
Of importance, different cells and organs may
respond differently to the same kind of radiation even at the same
dose level. Differential genetic backgrounds of cells and tumor
microenvironment also influence the activity of signaling pathways.
Therefore, further investigation directed toward the tumor niche
should be carried out to elucidate the biological effect of
LDIR.
Acknowledgments
The present study was supported by grants from the
National Science Foundation of China (81302380, to D.H.Y), and the
Jilin Provincial Science and Technology Department
(20140520017JH).
References
|
1
|
GLOBOCAN: Estimated Cancer Incidence,
Mortality and Prevalence Worldwide in 2012. http://globocan.iarc.fr/Default.aspx.
Access date: June 10, 2016.
|
|
2
|
Rodin D, Knaul FM, Lui TY and
Gospodarowicz M: Radiotherapy for breast cancer: The predictable
consequences of an unmet need. Breast. 29:120–122. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fisher B, Anderson S, Bryant J, Margolese
RG, Deutsch M, Fisher ER, Jeong JH and Wolmark N: Twenty-year
follow-up of a randomized trial comparing total mastectomy,
lumpectomy, and lumpectomy plus irradiation for the treatment of
invasive breast cancer. N Engl J Med. 347:1233–1241. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kim JJ and Tannock IF: Repopulation of
cancer cells during therapy: An important cause of treatment
failure. Nat Rev Cancer. 5:516–525. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Peitzsch C, Kurth I, Kunz-Schughart L,
Baumann M and Dubrovska A: Discovery of the cancer stem cell
related determinants of radioresistance. Radiother Oncol.
108:378–387. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Luckey TD: physiological benefits from low
levels of ionizing radiation. Health Phys. 43:771–789. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Feinendegen LE: Evidence for beneficial
low level radiation effects and radiation hormesis. Br J Radiol.
78:3–7. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Olivieri G, Bodycote J and Wolff S:
Adaptive response of human lymphocytes to low concentrations of
radioactive thymidine. Science. 223:594–597. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li W, Wang G, Cui J, Xue L and Cai L:
Low-dose radiation (LDR) induces hematopoietic hormesis:
LDR-induced mobilization of hematopoietic progenitor cells into
peripheral blood circulation. Exp Hematol. 32:1088–1096. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liang X, So YH, Cui J, Ma K, Xu X, Zhao Y,
Cai L and Li W: The low-dose ionizing radiation stimulates cell
proliferation via activation of the MAPK/ERK pathway in rat
cultured mesenchymal stem cells. J Radiat Res (Tokyo). 52:380–386.
2011. View Article : Google Scholar
|
|
11
|
Jiang H, Xu Y, Li W, Ma K, Cai L and Wang
G: Low-dose radiation does not induce proliferation in tumor cells
in vitro and in vivo. Radiat Res. 170:477–487. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee CL, Blum JM and Kirsch DG: Role of p53
in regulating tissue response to radiation by mechanisms
independent of apoptosis. Transl Cancer Res. 2:412–421. 2013.
|
|
13
|
Menon V and Povirk L: Involvement of p53
in the repair of DNA double strand breaks: Multifaceted roles of
p53 in homologous recombination repair (HRR) and non-homologous end
joining (NHEJ). Subcell Biochem. 85:321–336. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Freed-Pastor WA and Prives C: Mutant p53:
One name, many proteins. Genes Dev. 26:1268–1286. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim JS, Lee C, Bonifant CL, Ressom H and
Waldman T: Activation of p53-dependent growth suppression in human
cells by mutations in PTEN or PIK3CA. Mol Cell Biol. 27:662–677.
2007. View Article : Google Scholar :
|
|
16
|
Zhang H, Zeitz MJ, Wang H, Niu B, Ge S, Li
W, Cui J, Wang G, Qian G, Higgins MJ, et al: Long noncoding
RNA-mediated intrachromosomal interactions promote imprinting at
the Kcnq1 locus. J Cell Biol. 204:61–75. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lindell B and Sowby D: The 1958 UNSCEAR
report. J Radiol Prot. 28:277–282. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kadhim M, Salomaa S, Wright E, Hildebrandt
G, Belyakov OV, Prise KM and Little MP: Non-targeted effects of
ionising radiation - implications for low dose risk. Mutat Res.
752:84–98. 2013. View Article : Google Scholar
|
|
19
|
Stankevicins L, Almeida da Silva AP,
Ventura Dos Passos F, Dos Santos Ferreira E, Menks Ribeiro MC, G
David M, J Pires E, Ferreira-Machado SC, Vassetzky Y, de Almeida
CE, et al: MiR-34a is up-regulated in response to low dose, low
energy X-ray induced DNA damage in breast cells. Radiat Oncol.
8:2312013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lehrer S and Rosenzweig KE: Lung cancer
hormesis in high impact states where nuclear testing occurred. Clin
Lung Cancer. 16:152–155. 2015. View Article : Google Scholar
|
|
21
|
Dobrzynski L, Fornalski KW and Feinendegen
LE: Cancer mortality among people living in areas with various
levels of natural background radiation. Dose Response.
13:15593258155923912015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mettler FA, Sinclair WK, Anspaugh L,
Edington C, Harley JH, Ricks RC, Selby PB, Webster EW and Wyckoff
HO: The 1986 and 1988 UNSCEAR (United nations Scientific Committee
on the Effects of Atomic Radiation) reports: Findings and
implications. Health Phys. 58:241–250. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang G, Li W, Jiang H, Liang X, Zhao Y, Yu
D, Zhou L, Wang G, Tian H, Han F, et al: Low-dose radiation may be
a novel approach to enhance the effectiveness of cancer
therapeutics. Int J Cancer. 139:2157–2168. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu SZ: On radiation hormesis expressed in
the immune system. Crit Rev Toxicol. 33:431–441. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bartek J, Iggo R, Gannon J and Lane DP:
Genetic and immunochemical analysis of mutant p53 in human breast
cancer cell lines. Oncogene. 5:893–899. 1990.PubMed/NCBI
|
|
26
|
Cadwell C and Zambetti GP: The effects of
wild-type p53 tumor suppressor activity and mutant p53
gain-of-function on cell growth. Gene. 277:15–30. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shaulsky G, Goldfinger N and Rotter V:
Alterations in tumor development in vivo mediated by expression of
wild type or mutant p53 proteins. Cancer Res. 51:5232–5237.
1991.PubMed/NCBI
|
|
28
|
Dittmer D, Pati S, Zambetti G, Chu S,
Teresky AK, Moore M, Finlay C and Levine AJ: Gain of function
mutations in p53. Nat Genet. 4:42–46. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yan W, Zhang Y, Zhang J, Liu S, Cho SJ and
Chen X: Mutant p53 protein is targeted by arsenic for degradation
and plays a role in arsenic-mediated growth suppression. J Biol
Chem. 286:17478–17486. 2011. View Article : Google Scholar : PubMed/NCBI
|