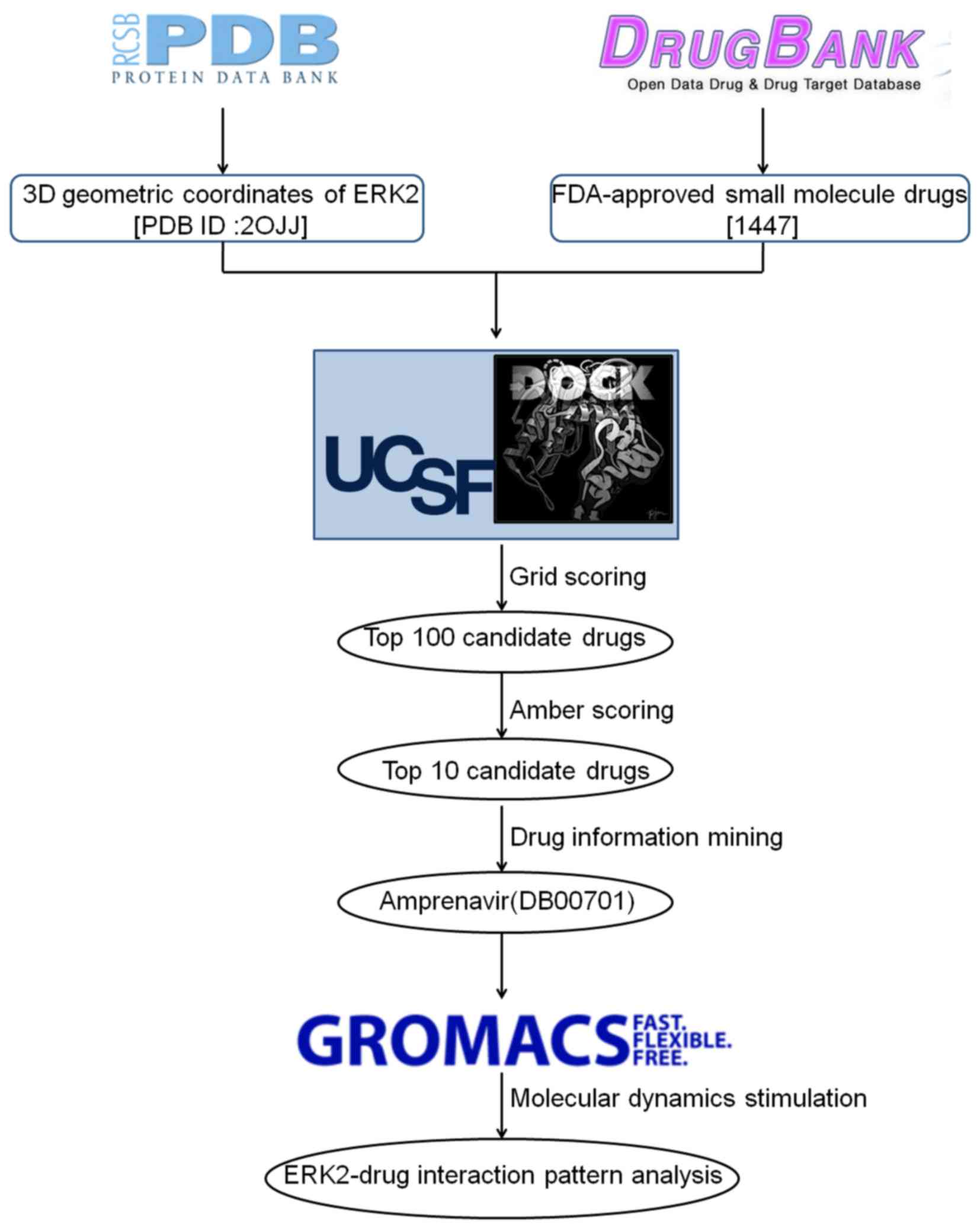
Introduction
Despite substantial advances in cancer therapeutics,
breast cancer remains a fundamental public health burden worldwide
(1,2). One of the most appealing
chemotherapies against breast cancer is inducing the expression of
Bim, a pro-apoptotic Bcl-2 homology (BH)-3-only protein within
Bcl-2 family (3), to mediate
apoptotic cancer cell death. However, Bim-targeted therapies may
lead to acquired chemoresistance and tumor selectivity (2,4) and
thus limiting their clinical application. In addition to
upregulated levels of BimEL (an isoform of Bim protein),
multiple breast cancer cells have recently been established to
express elevated phosphorylated forms of BimEL (such as
phospho-Ser69), which jointly determine the fate of breast cancer
cells (5). Although
BimEL is well-known for its apoptosis-inducing activity,
the fact that constitutively overexpressed BimEL do not
kill cancer cells suggests a potential pro-survival function of
phosphorylated BimEL. Studies demonstrate that
extracellular signal-regulated kinase 1/2 (ERK1/2)-mediated
phosphorylation of BimEL at Ser69 could enhance the
proteosomal degradation of BimEL (6) or reduce its sequestration with
pro-survival molecules, such as Mcl-1, Bcl-xL and Bcl-2 (7), and thus it promotes cancer cell
survival. Therefore, identification of compounds that can inhibit
ERK1/2 kinase activity is of considerably therapeutic interest for
breast cancer.
In silico drug repositioning by screening
existing approved drugs for previously unknown indications is
therapeutically essential and efficient for drug discovery
(8–10). The main method for computational
drug repositioning is molecular docking screening, which aimed at
predicting and stimulating the direct physical interactions between
existing drugs and novel therapeutic targets (11,12).
As only one X-ray structure of ERK1 has been reported (13) while several high-resolution crystal
structures of ERK2 in complex with selective inhibitors are
available now, the search of FDA-approved drugs against ERK2 to
screen novel ERK2 inhibitors become feasible.
In the present study, firstly we screened 1447
FDA-approved small molecule drugs and identified amprenavir as a
potential ATP-competitive inhibitor of ERK2. Subsequently, we found
amprenavir could inhibit the kinase activity of ERK2 and induce
apoptosis in MCF-7 human breast cancer cells by inhibiting
ERK2-mediated phosphorylation of BimEL at Ser69.
Furthermore, we confirmed the anti-proliferative and
apoptosis-inducing activities of amprenavir in vivo. Taken
together, these findings reveal the possible new therapeutic use of
amprenavir and provide a paradigm of drug repositioning to maximize
the additional therapeutic advantages of existing approved
drugs.
Materials and methods
Molecular docking screening
The initial 3D structure of ERK2 (PDB ID: 2OJJ)
(14) was downloaded from RCSB
Protein Data Bank (PDB) (http://www.pdb.org/pdb/). After removing ligand and
solvent molecules, hydrogens and standard charges were assigned to
each atom with UCSF Chimera (15),
and the inhibitor-binding cluster was generated using program
sphere_selector (16). The
screening library containing 1447 FDA-approved small molecule drugs
was constructed from DrugBank (version 3.0, http://www.drugbank.ca/). After removing established
drugs targeting ERK2, the remaining drugs were subjected to
OpenBabel toolbox (http://open-babel.org/) (17) to convert the 3D MOL2 format, which
were then uploaded to the ZINC database (http://zinc.docking.org/) (18) for structure editing. Docking screen
was conducted by UCSF DOCK6.3 program (19) with amber force field. Firstly,
flexible-ligand docking with grid scoring was performed, then,
topmost poses of the pre-generated top 100 low-scored drugs were
subjected to amber scoring for further refinement.
Molecular dynamics (MD) simulations
MD simulations were performed by GROMACS package
(version 4.5) (20). Topmost
structures of ERK2-amprenavir complex from previous docking
screening were used as the initial conformations. Protein topology
was constructed by GROMACS accessory pdb2gmx and ligand topology
was processed by PRODRG2 server (21) with GROMOS96 43a1 forcefield
(22). After two phases of
equilibrations, a 5000 ps MD simulation with the time step of 2 fs
was performed. The resulting trajectory files were viewed and
analyzed using VMD software (22,23)
and UCSF Chimera (15).
Chemicals
Amprenavir purchased from Santa Cruz Biotechnology,
Inc. (Santa Cruz, CA, USA) was dissolved in dimethyl sulfoxide
(DMSO) to a stock concentration of 1 mM and stored at −20°C. All
other chemical reagents with the highest purity available were
purchased from Sigma-Aldrich.
Cell culture and cytotoxicity assay
Human breast cancer MCF-7 cells were incubated in
Dulbecco's modified Eagle's medium (DMEM) supplemented with 10%
fetal bovine serum (FBS) (both from Gibco, Grand Island, NY, USA),
100 U/ml penicillin and 100 mg/ml streptomycin (both from
Invitrogen, Carlsbad, CA, USA), and were maintained at 37°C and 5%
CO2 in a humidified atmosphere.
Prior to drug treatment, MCF-7 cells were seeded in
a 96-well plate for 24 h. Serial doses of amprenavir were then
added and cells were allowed to grow for additional 24 or 48 h.
Cytotoxic effects were measured by MTT assay with a
spectrophotometer (Model 3550 Microplate Reader; Bio-Rad, Hercules,
CA, USA) and the cell growth inhibition rate was calculated as
follows:
Cell growth inhibitiry rate(%)=A540(DMSO)−A540(Amprenavir)A540(DMSO)−A540(Blank)×100%
Cell morphology observation and
staining
MCF-7 cells were treated with various concentrations
of amprenavir (0, 50, 100, 150 and 200μM) and then observed
by a phase-contrast microscope (Leica, Wetzlar, Germany). In
addition, Hoechst 33258 staining was applied and cells were
analyzed by a fluorescence microscope (Leica).
Cell cycle and apoptosis analysis
MCF-7 cells treated with amprenavir or PBS for 24 h
and 48 h were harvested, and cells at different phases of cell
cycle were measured by FACScan flow cytometer (Becton-Dickinson,
Franklin Lakes, NJ, USA). Annexin V (AV)/propidium iodide (PI)
staining assays were performed using AV-fluorescein isothiocyanate
(FITC) apoptosis detection kit (BD Pharmingen, San Diego, CA, USA),
and different phases of apoptotic cells were measured by FACScan
flow cytometer.
Western blot analysis
MCF-7 cells were treated with DMSO or amprenavir for
indicated time periods. Then, both adherent and floating cells were
collected and western blot analysis was conducted as previously
described (24). Primary
antibodies and horseradish peroxidase-conjugated secondary antibody
were purchased from Zymed (San Francisco, CA, USA).
Expression and purification of
recombinant proteins
Plasmids for ERK2/pT/pY, ERK2/T183A/pY and
ERK2/pT/Y185F containing His6-tag were kindly provided by Dr Yi
Wang (Nationwide Children's Hospital, USA). Plasmids were
transformed into E. coli BL21 cells and protein expression
was achieved by inducing cells with 0.4 mM
isopropyl-b-D-thiogalactopyranoside (IPTG) at 22°C for 4 h. Then
proteins were isolated by Ni-nitrilotriacetic acid (NTA)-agarose
chromatography (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA)
and quantified by NanoDrop spectrophotometer (Life Science).
Small interfering RNA (siRNA)
transfection
MCF-7 cells were transfected with siRNAs against
intracellular ERK2 and BimEL using Lipofectamine 2000
(Invitrogen) according to the manufacturer's instructions.
Transfection efficiency was identified by western blotting and
cells were used for further experiments 24 h later.
DNA constructs and mutagenesis
PCR-amplified hemagglutinin (HA)-tagged ERK2 and
Flag-tagged BimEL were subcloned into pcDNA3.1 vector
between BamHI and XbaI, respectively. pcDNA
3.1-ERK2/T183A/pY, ERK2/pT/Y 185F and pcDNA 3.1-BimEL
(S69A), BimEL (S69E) were made using the QuikChange
site-directed mutagenesis kit (Stratagene).
Transient expression, stable transfection
and immunoprecipitation
Transient expression and immunoprecipitation were
conducted as previously described (25). Briefly, MCF-7 cells were plated at
a density of 4×105/well of a 6-well plate 16 h prior to
transfection. Cells were transiently transfected with the indicated
plasmids using Expressfect Transfection reagents (Denville
Scientific). Forty-eight hours after transfection, cell cultures
were cultivated for another 2 weeks in the presence of active G418
(400 μg/ml) (Gibco). Antibiotic-resistant colonies that
stably expressed indicated proteins were picked, pooled, identified
and expanded for further experiments. After 24–48 h transfection
with the indicated recombinants, MCF-7 cells were exposed to
various doses of amprenavir for another 24 h. Then the indicated
proteins were pulled down with the Pierce HA (or Flag) Tag IP/Co-IP
kit (Thermo Scientific) according to the manufacturer's
instructions.
In vitro kinase assays
Bacterially purified or immunoprecipitated wild-type
and mutant ERK2s were incubated for 20 min at 30°C in kinase buffer
(10 mM Tris-HCl (pH 7.4), 5 mM MnCl2 and 1 mM
dithiothreitol) containing 20 μM ATP and 10 μCi
[γ-32P]ATP (Perkin-Elmer) with myelin basic protein
(MBP) or BimEL as a substrate. The final concentrations
of amprenavir were 0–200 μM. Reactions were stopped with an
equal volume of 2X SDS-PAGE loading buffer and then heated to
100°C. Proteins were isolated by SDS-PAGE and transferred onto
polyvinylidene difluoride (PVDF) membranes. Then 32P
incorporation into substrate was detected by exposing the membrane
to X-ray film.
In vivo kinase assays
After knockdown of intracellular ERK2 and
BimEL, wild-type and mutant ERK2 and BimEL
plasmids were transfected into MCF-7 cells. The stably transfected
cells were metabolically labeled with 32P for 4 h and
then treated with amprenavir (150 μM) or 0.01% DMSO for 24
h. ERK2-BimEL complex was isolated and 32P
incorporation into BimEL was then detected as described
previously (26). Western blot
analysis with specific anti-phospho antibodies was conducted to
analyze the phosphorylation of BimEL by ERK2.
Xenograft studies
Male C57BL/6J nude mice (4–6 weeks old, weighing
18–22 g) were subcutaneously injected with MCF-7 cells, when tumors
became palpable, mice were randomly divided into the following
three groups (8 mice/group): vehicle control (normal saline),
amprenavir (45 mg/kg/day, q1d) and cisplatin (5 mg/kg/day, q7d).
The dosage of amprenavir used here was determined by our
preliminary studies (data not shown). Amprenavir and cisplatin were
intraperitoneally administered into mice, and the drug treatments
lasted for 2 weeks. Tumor volume was measured by caliper according
to formula (0.5 × long-axis × short-axis × short-axis). Mice were
sacrificed by cervical dislocation every three days and tumors were
weighed. All animals were handled according to guidelines of the
Institutional Animal Care and Use Committee of the Sichuan
University in China (IACUC no. 20100318).
Immunohistochemistry staining
After two weeks of treatment, mice were sacrificed
by cervical dislocation, and whole tumor tissues were harvested.
Tumor tissues were fixed in 10% neutral buffered formalin for 24 h
at room temperature, dehydrated, embedded in paraffin and
sectioned. Tumor tissue sections were then processed for
immunohistochemical staining with anti-proliferating cell nuclear
antigen (PCNA) antibody and anti-cleaved caspase-3 antibody (both
from CapitalBio Corporation). Apoptosis in tumor tissues were
detected by terminal deoxynucleotidyl transferase-mediated dUTP
nick-end labeling (TUNEL) staining using the Roche Fluorescence
DeadEnd kit following the manufacturer's instructions (27). Representative images of each
section were captured using a fluorescence microscopy (Nikon
Eclipse 80i, Nikon, Tokyo, Japan).
Statistical analysis
Data on mouse body weight changes and the
therapeutic efficacy (cell viability, tumor volume and tumor
weight) were expressed as mean ± SD from at least three independent
experiments. Statistical significance was determined by Student's
t-test and two-way analysis of variance (ANOVA) with P<0.05 were
considered as statistically significant, and P<0.01 highly
significant.
Results
In silico identification of amprenavir as
a novel ERK2 inhibitor
Grid scoring and amber scoring were applied to
predict novel approved drugs targeting ERK2 among the 1447
FDA-approved small molecule drugs, and MD simulations were then
launched to stimulate the interaction pattern between ERK2 and the
predicted drug (Fig. 1). After two
rounds of docking screening, amprenavir appears to be the topmost
low-scored drug potential as a candidate ERK2 inhibitor with the
grid score and amber score of −62.397369 and −46.427078,
respectively. According to the data available in DrugBank,
amprenavir is an HIV-1 protease inhibitor approved for the
treatment of HIV infection, and has not been documented yet to
target ERK2.
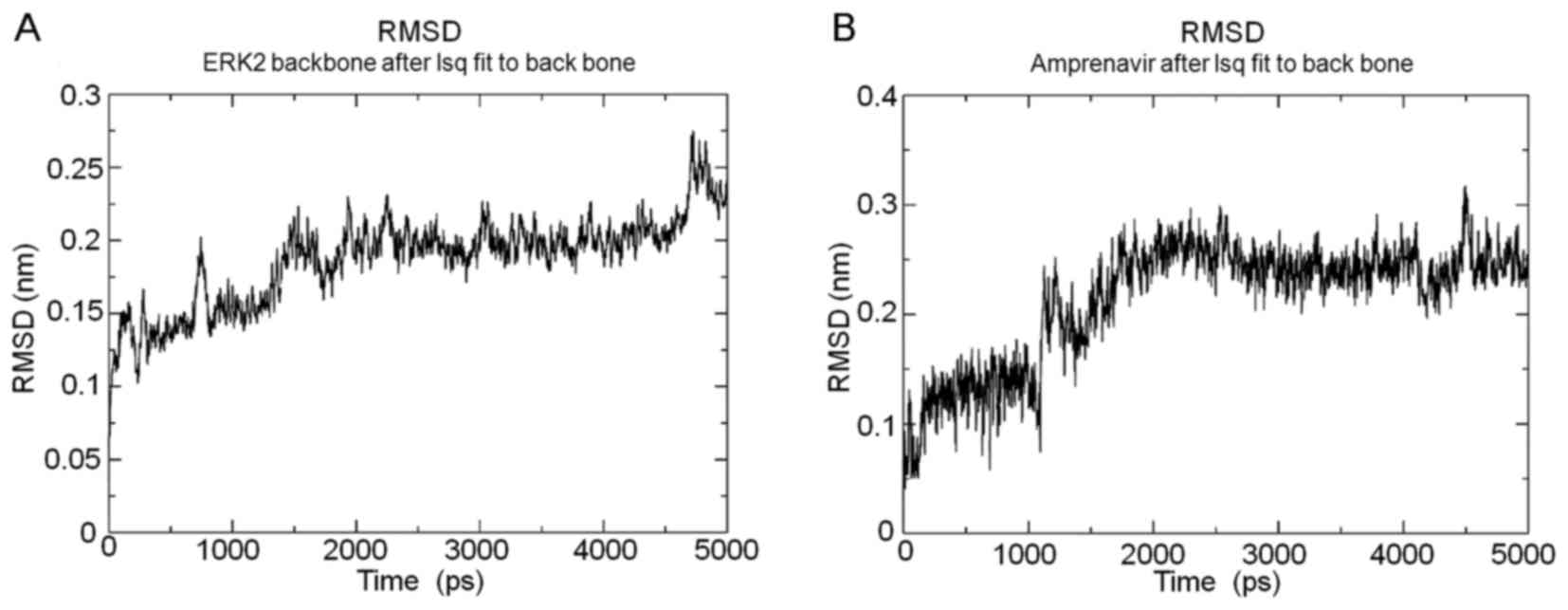
Subsequently, MD simulations were conducted to
evaluate the interaction stabilities and mechanisms between ERK2
and amprenavir, and the root mean square deviations (RMSDs) of ERK2
backbone and amprenavir over the 5000 ps simulation process shown
in Fig. 2, respectively. The
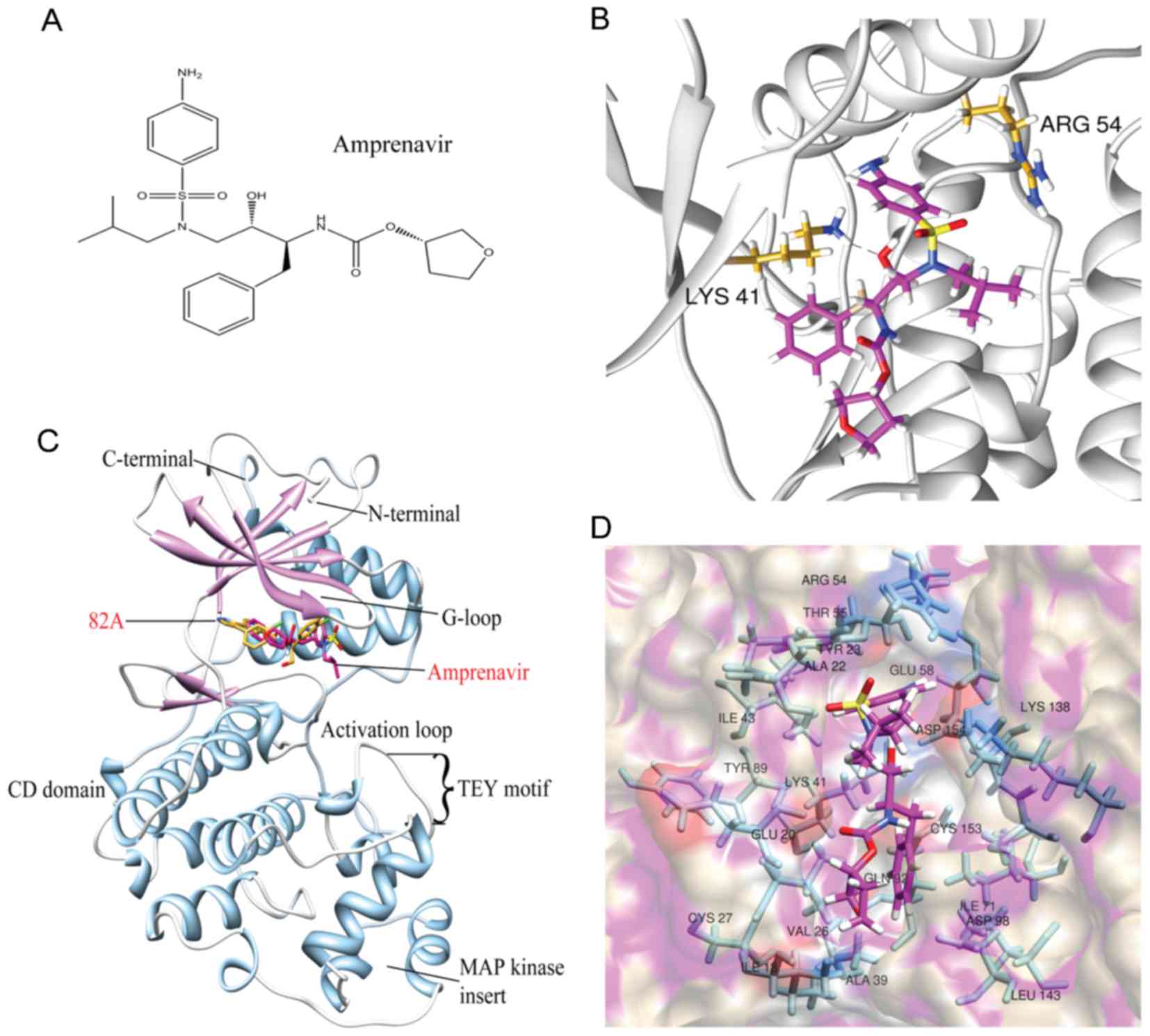
chemical structure of amprenavir is illustrated in Fig. 3A. Overall, amprenavir binds to the
hinge point between two domains of ERK2, in the ATP-binding site
(Fig. 3B). Choosing the trajectory
when the RMSDs of ERK2-amprenavir complex kept stable for at least
1 ns as the representative snapshot, two hydrogen bonds were formed
between amprenavir and ERK2 with Arg54 and Lys41 as donors
(Fig. 3C). Moreover, amprenavir
was stabilized in the ATP-binding site via hydrophobic interactions
with the nonpolar residues of ERK2 (Ile18, Ala22, Val26, Ala39,
Ile43, Ile71 and Leu143) (Fig.
3D). These findings reveal that hydrogen bonds and van der
Waals contacts can synergistically contribute to the stabilization
of amprenavir in the ATP-binding site of ERK2.
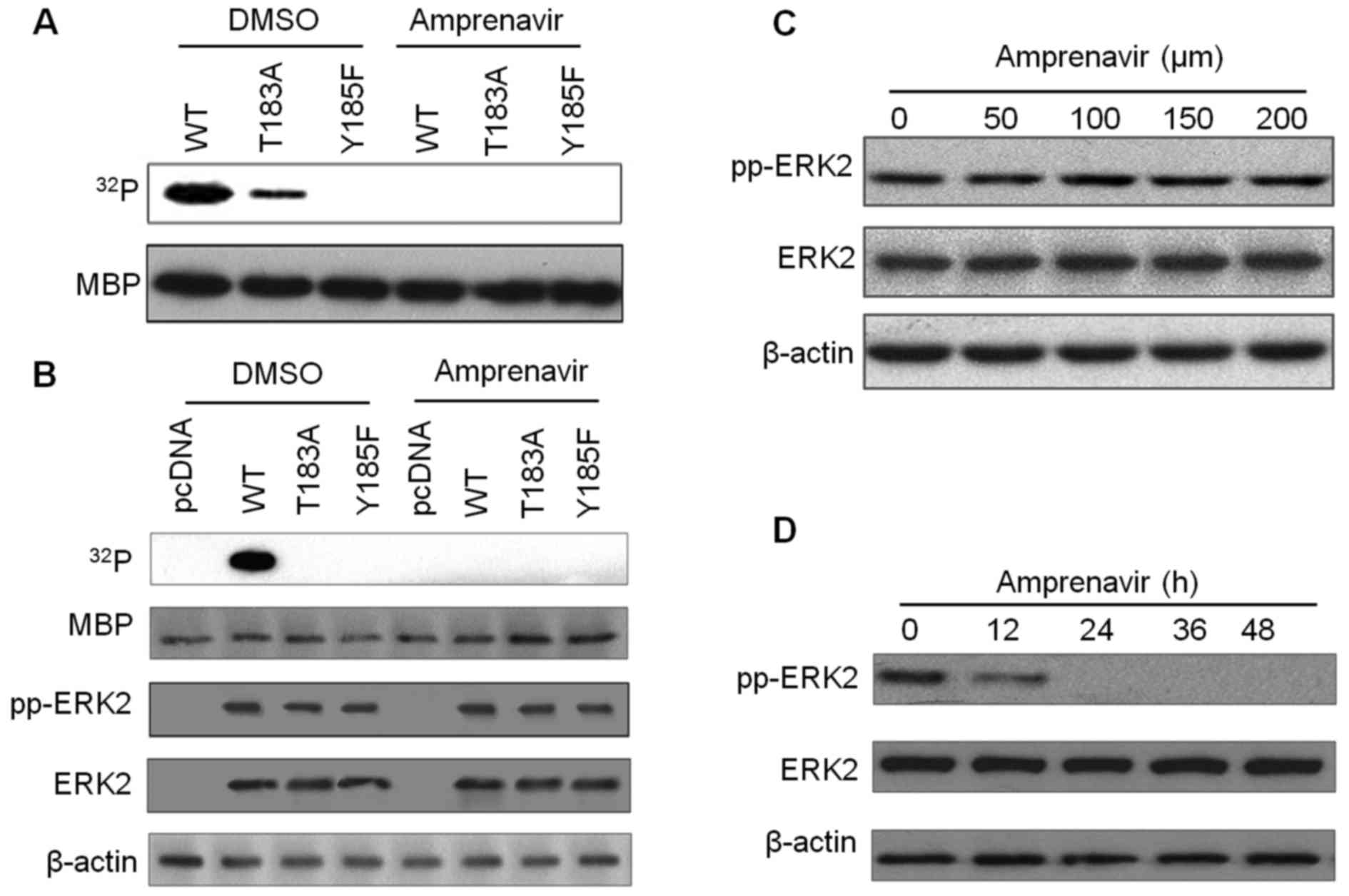
Amprenavir inhibits the kinase activity
of ERK2
To determine the inhibitory effects of amprenavir on
the kinase activity of ERK2, kinase assays with MBP as a substrate
were performed (Fig. 4). In the
kinase system with bacterially purified recombinant ERK2s, exposure
to 150 μM amprenavir for 24 h completely blocked the
phosphorylation of MBP by ERK2/pTpY and ERK2/T183A/pY (Fig. 4A). Additionally, in the kinase
system with ERK2s immunoprecipitated from transfected MCF-7 cells,
MBP was significantly phosphorylated by dually phosphorylated
ERK2/pTpY in diluted DMSO, whereas this phosphorylation phenomenon
disappeared after 150 μM amprenavir treatment (Fig. 4B), confirming that amprenavir could
potently inhibit the kinase activity of ERK2. We then examined if
amprenavir could exert an influence on ERK2 activation by detecting
total ERK2 and fully activated ERK2 in MCF-7 cells. As shown in
Fig. 4C and D, increasing doses of
amprenavir did not affect the activation of ERK2, whereas exposure
to 150 μM amprenavir for 0 to 48 h inhibited the activation
of ERK2 to some extent. These findings demonstrated that amprenavir
may inhibit the catalytic activity of ERK2 while having little
inhibitory effect on its activation.
Amprenavir inhibits proliferation and
induces apoptosis in MCF-7 cells
Given the implications of ERK2 signaling pathways in
tumor progression, we explored the antitumor effects of amprenavir
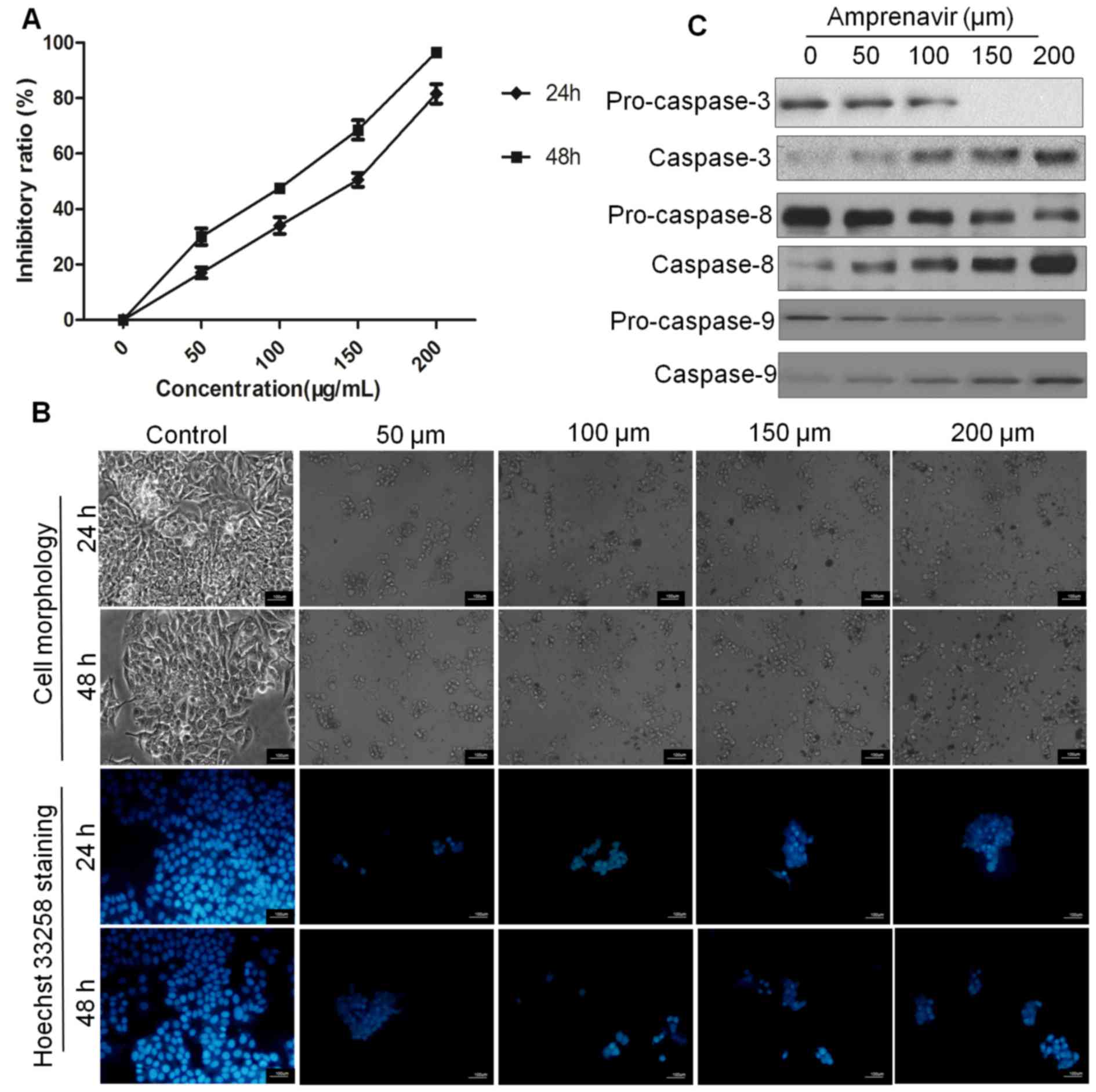
in MCF-7 cells. According to results of MTT assays, amprenavir
inhibited the proliferation of MCF-7 cells in a time-dependent and
dose-dependent manner (Fig. 5A),
and the IC50 values were ~150 and 105 μM for 24
and 48 h, respectively.
Then, cell and nuclear morphology were observed to
determine if apoptosis was involved in this anti-proliferative
activity. When observed by phase-contrast microscopy, MCF-7 cells
treated with amprenavir for 24 or 48 h showed typical morphological
characteristics of apoptosis (28), such as reduction in cell volume,
rounding up of cells, cell shrinkage and membrane blebbing.
Moreover, Hoechst 33258 staining of DMSO-treated MCF-7 cells
exhibited normal nuclear morphology and homogeneously stained
chromatin, whereas amprenavir-treated cells showed an apoptotic
nuclear morphology (29) with
shrunken, fragmented nuclei and brighter hypercondensed chromatin
(Fig. 5B).
When MCF-7 cells were treated with 150 μM for
different time periods, expression levels of pro-caspase-3, -8 and
-9 were decreased, whereas expression of caspase-3, -8 and -9 were
increased along with treatment time (Fig. 5C), indicating that amprenavir could
time-dependently activate caspase-3, -8 and -9 and thus induce
apoptosis in MCF-7 cells.
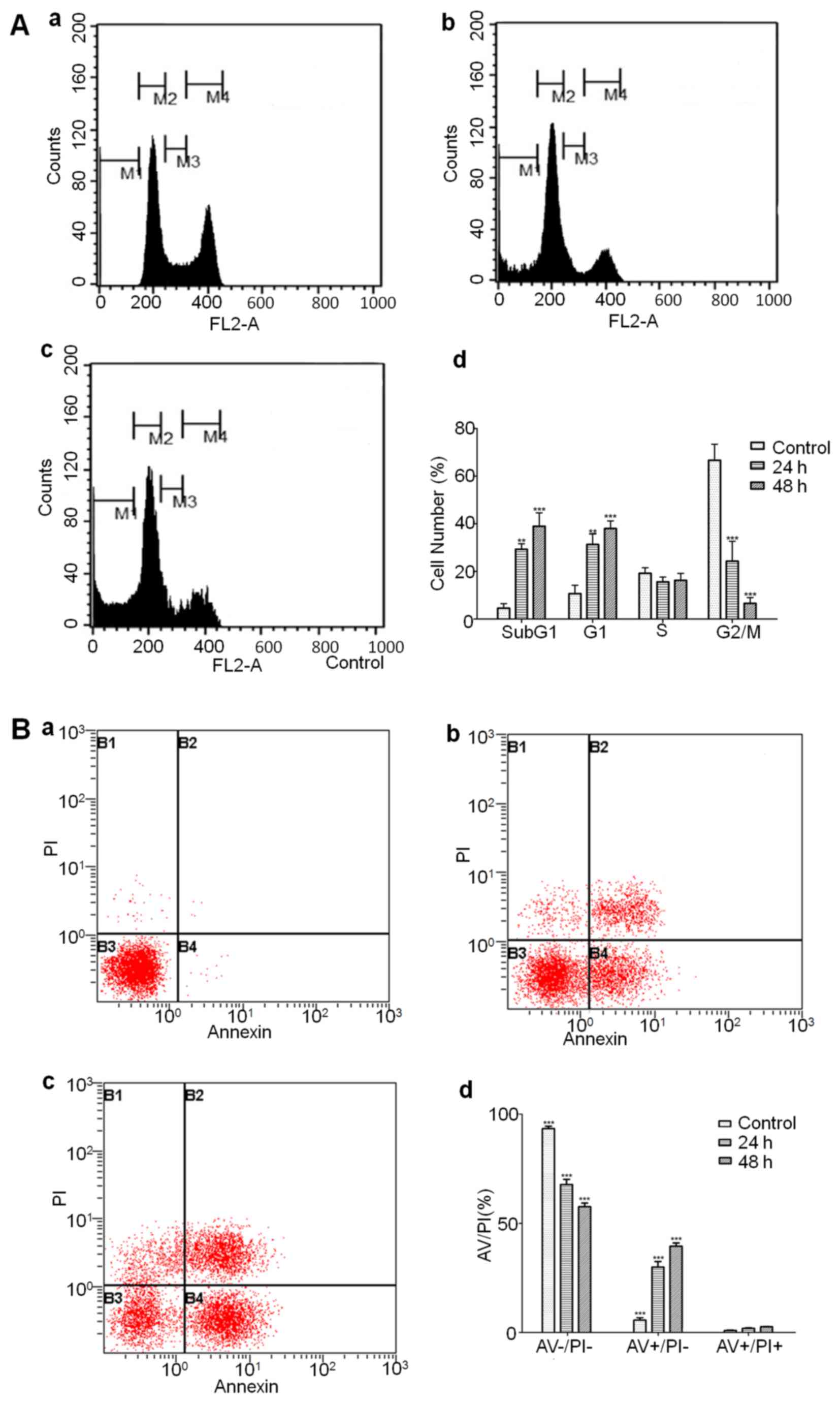
Flow cytometry to analyze cell cycle
arrest and detect different phases of apoptotic cells
Cell cycle analysis showed that amprenavir treatment
significantly upregulated the proportions of MCF-7 cells in sub-G1
and G1 phase, suggesting that amprenavir may trigger sub-G1 and G1
phase arrest along with the culturing time (Fig. 6A). AV/PI staining results showed
that amprenavir treatment reduced the number of live cells and
increased the number of early and late apoptotic cells, and
percentages of early apoptotic cells were significantly higher than
late apoptotic cells (Fig. 6B),
suggesting that amprenavir could effectively induce apoptosis in a
time-dependent manner.
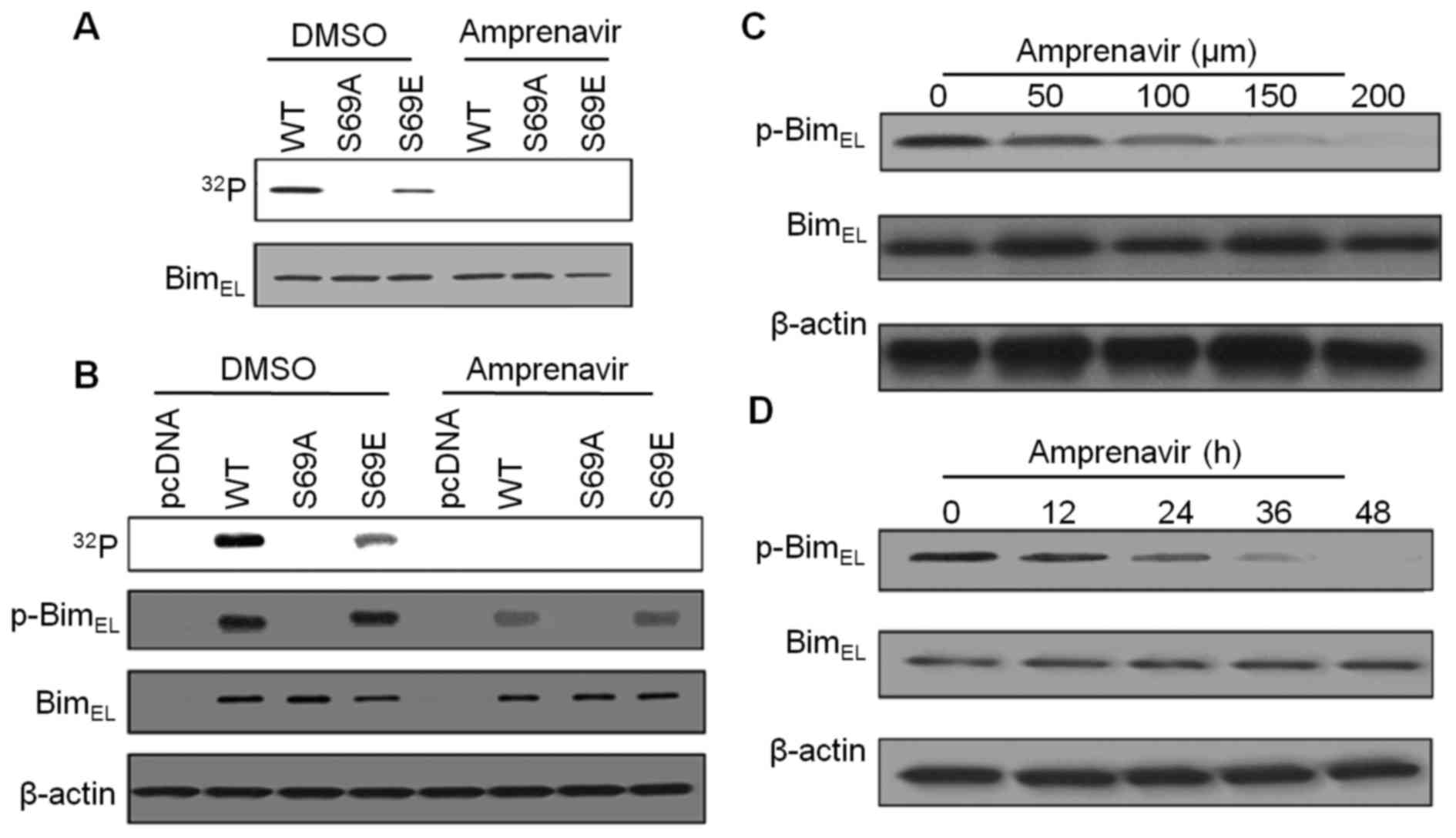
Amprenavir inhibits ERK2-mediated
phosphorylation of BimEL at Ser69
Phosphorylation of BimEL at S69 may
greatly facilitate ERK2-induced apoptosis, therefore, we performed
kinase assays to evaluate the inhibitory effects of amprenavir on
ERK2-mediated phosphorylation of BimEL at S69. As shown
in Fig. 7A, with bacterially
purified wild-type BimEL or BimEL mutant as a
substrate, strong phosphorylation of BimEL (WT) was
observed in diluted DMSO, whereas replacement of Ser69 with
glutamic acid (S69E) partially suppressed this phosphorylation
phenomenon, revealing the involvement of S69 in ERK2-mediated
BimEL phosphorylation. After 150 μM amprenavir
treatment, we failed to detect any phosphorylation of
BimEL (WT), BimEL (S69A) and BimEL
(S69E), demonstrating that amprenavir inhibited phosphorylation of
BimEL at S69. Similarly, using
32P-metabolically labeling MCF-7 cells cotransfected
with active ERK2 and indicated BimEL constructs, we
found that 150 μM amprenavir treatment could completely
interrupt the phosphorylation phenomena in BimEL (WT),
BimEL (S69A) and BimEL (S69E) (Fig. 7B), indicating that amprenavir
inhibited ERK2-mediated phosphorylation of BimEL at S69
in MCF-7 cells. As an alternative, specific antibody against
phospho-S69-BimEL was used. As shown in Fig. 7B, in DMSO group,
phospho-S69-BimEL could be detected in MCF-7 cells
transfected with BimEL (WT) and BimEL (S69E),
while the signals became significantly weaker after 150 μM
amprenavir treatment. Moreover, amprenavir dose- and
time-dependently reduced the expression levels of
phospho-S69-BimEL with the total amounts of
BimEL keeping constant (Fig. 7C and D). Taken together, these
results suggest that amprenavir could effectively inhibit
ERK2-mediated phosphorylation of BimEL at S69 without
affecting the expression levels of total BimEL in MCF-7
cells.
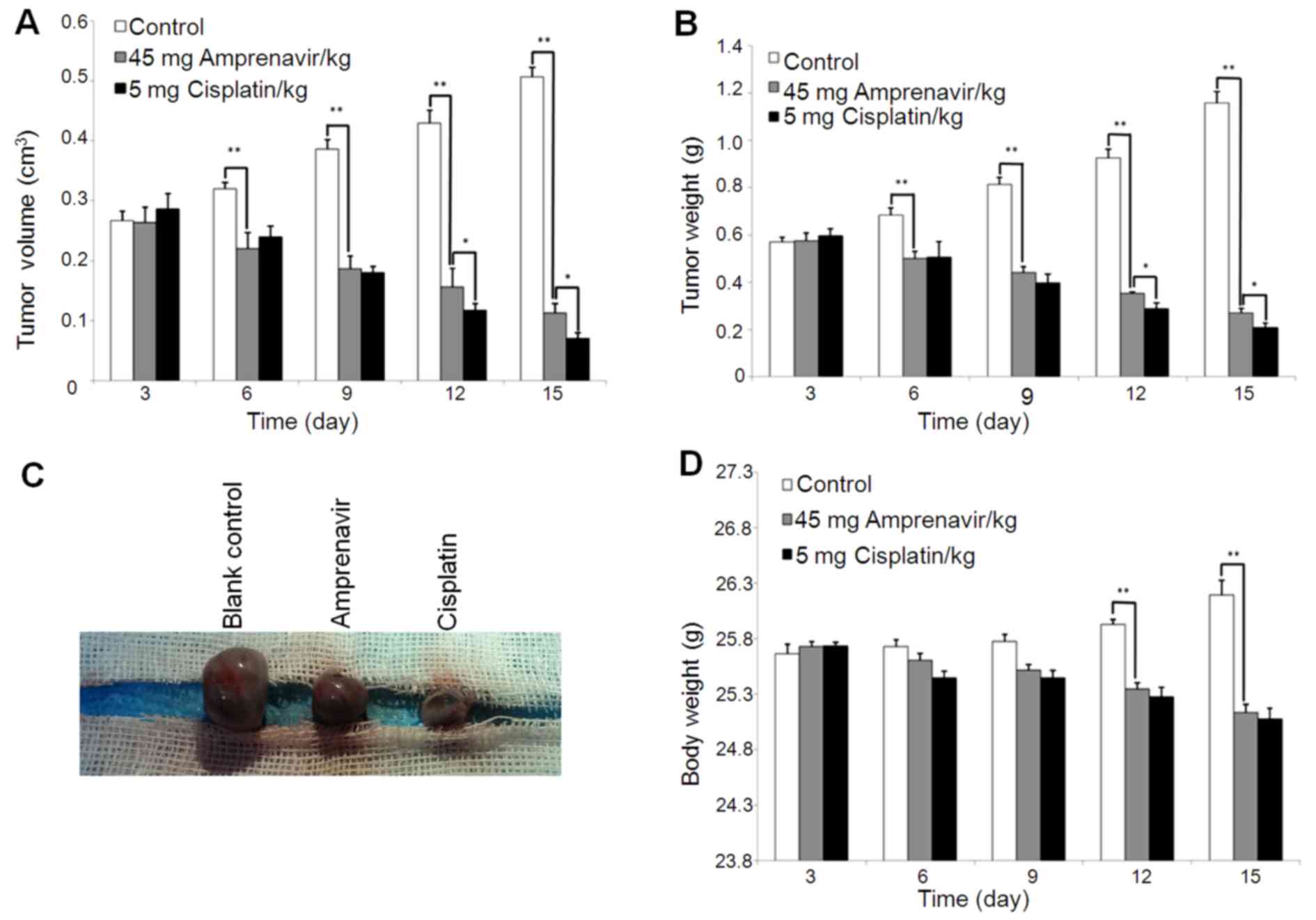
Antitumor effects and mechanisms of
amprenavir in MCF-7 xenograft models
MCF-7 xenograft models were constructed to establish
the antitumor effects and mechanisms of amprenavir in vivo
with the administration of DMSO and cisplatin as blank and positive
control, respectively. In general, administration of amprenavir
significantly inhibited the growth of MCF-7 tumors from day 6, and
the growth inhibitory effects exhibited a time-dependent manner
(P<0.001) (Fig. 8A–C). After 2
weeks of treatment, the average tumor volumes were inhibited by 78%
(vehicle vs. amprenavir; mean, 0.50 vs. 0.11 mm3;
difference, 0.39 mm3; P=0.000007) and tumor weights were
inhibited by 77% (vehicle vs. amprenavir; mean, 1.16 vs. 0.27 g;
difference, 0.89 g; P=0.000007). Of note, treated mice with
amprenavir for 9 days successfully suppressed tumor growth by ~50%.
In addition, the therapeutic efficiency of amprenavir and cisplatin
did not significantly differ from each other during the early time
periods of treatment. Regarding body weight change as an indication
of general toxicity (Fig. 8D),
average body weights of amprenavir and cisplatin-treated mice did
not display a significant difference during the early periods of
treatment, and amprenavir showed more moderate toxicity than
cisplatin after day 9. Collectively, daily injecting mice with 45
mg/kg/day amprenavir could effectively inhibit the growth of MCF-7
tumor xenografts, and for early periods of treatment, its
therapeutic efficiency was comparable to cisplatin, which is a
clinically widely used antitumor agent.
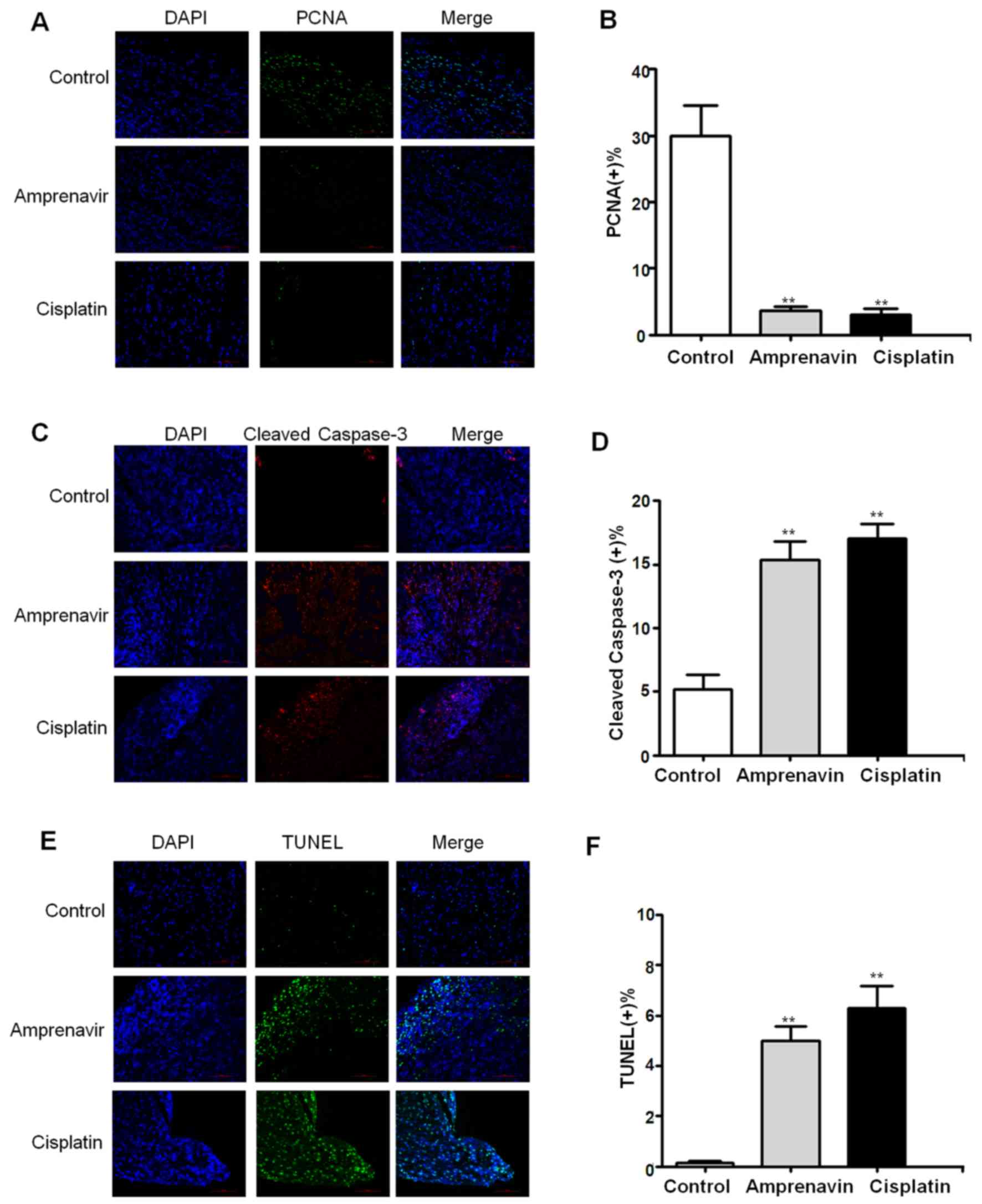
Immunohistochemical staining was then conducted to
validate the antitumor mechanisms of amprenavir in vivo.
Proliferation of MCF-7 xenografts in different treatment groups was
evaluated by the localization of PCNA (Fig. 9A), and 2 weeks of amprenavir
treatment showed significant anti-proliferative effects, which was
even comparable to that of cisplatin (Fig. 9B). Moreover, cleaved caspase-3
staining and TUNEL staining results indicated that apoptosis was
widely distributed in tumor specimens exposed to amprenavir
treatment for 2 weeks (Fig. 9C and
E). Statistical analysis confirmed a significant apoptosis
level in amprenavir-treated MCF-7 xenografts, which was only
slightly weaker than cisplatin-treated groups (Fig. 9D and F) (P<0.01).
Discussion
In the present study, for the purpose of
repositioning existing approved drugs for novel anticancer uses, we
launched a structure-based virtual screening of 1447 FDA-approved
small molecule drugs against ERK2. As a result, we identified
amprenavir, a HIV-1 protease inhibitor, as a potent kinase
inhibitor of ERK2, and this activity was further confirmed to
contribute to its anti-proliferative and apoptosis-inducing effects
in MCF-7 breast cancer cells both in vitro and in
vivo.
According to our in silico screening results,
among the top 10 drugs predicted to target ERK2, three drugs
(amprenavir, indinavir and saquinavir) are established HIV-1
protease inhibitors approved for the treatment of HIV infection
(data not shown). From the perspective of function, ERK2 has been
reported to play important roles in the pathogenesis of HIV
infection. For example, HIV-1 accessory protein Nef can act by
activating the Ras-Raf-MAPK1/2 pathways (24), stimulating the transcription factor
activating protein-1 through Hck and MAPK to increase CD4 T cell
ERK activity (30), and thus
affecting T cell activity, viral replication and viral infectivity
(31). Therefore, ERK2 may be a
latent target of HIV-1 proteases and its inhibition may facilitate
the treatment of HIV infection. From the perspective of structure,
although the backbone chains of HIV-1 protease and ERK2 hardly have
any superposition, their ligand-binding cavities overlap by a large
margin (data not shown), which may provide structural basis for the
sharing of identical small molecule inhibitors between these two
proteins.
Among varieties of in silico drug
repositioning strategies, the combination of molecular docking and
MD simulations, in which docking is first utilized to fast screen
of large libraries and MD simulations are then applied to optimize
the conformations of protein-drug complexes, is probably a rational
protocol to accelerate the virtual screening process (12). According to the binding mechanisms
analyzed by MD simulations (Fig. 3C
and D), some interactions between amprenavir and ERK2 are
conserved compared with other reported ERK2-inhibitor complexes.
For example, the hydrophobic interactions between amprenavir and
the non-polar groups Leu143 and Cys153 are consistent with the
published X-ray crystallographic data (32) and structure-based virtual screening
studies (33,34), suggesting the importance of these
two residues in accommodating an inhibitor in the ATP-binding site
of ERK2. Moreover, interactions between the terminal groups of
amprenavir with Lys41 and Asp154 in ERK2 have been established to
make ample space available to fit the ligand substituent (32–34),
and Gln92 has been demonstrated to provide structural basis for the
inhibitor selectivity of ERK2 (35,36).
Collectively, these conserved residues can form hydrogen bonds or
van der Waals interactions between amprenavir and ERK2, and further
contribute to the selectivity and stabilization of amprenavir in
the ATP-binding site of ERK2. Structural studies are needed to
validate the detailed structure-activity relationships between
amprenavir and ERK2, and improved therapeutic potency may be
achieved by modifying the drug's chemical features or examining
structurally similar compounds.
HIV-1 proteinase inhibitors, such as ritonavir,
saquinavir, atazanavir and nelfinavir, have demonstrated
broad-spectrum antitumor activities and diverse action mechanisms
(37–40). However, the antitumor potentials of
amprenavir have not yet been well-clarified. Amprenavir (agenerase,
GlaxoSmithKline) was approved by FDA for the treatment of HIV
infection in 1999. In 2003, Lexiva (fosamprenavir), a prodrug of
amprenavir, was approved and is now the preferred form to extend
the duration of available amprenavir and thus reducing the pill
burden in patients. The antitumor activity of amprenavir has been
validated in U87MG human glioblastoma cells (41) and Huh-7 hepatocarcinoma cells
(42), and the mechanisms may
involve the downregulation of vascular endothelial growth factor
(VEQF) and hypoxia-inducible factor-1 (HIF-1) expression. given the
implications of ERK2 in the regulation of VEGF and HIF-1, it is
reasonable to speculate that amprenavir may act by inhibiting
ERK2-mediated angiogenesis and thus suppress tumor development.
Additionally, a recent study revealed that amprenavir could
blockage the activation of matrix metalloproteinase-2 (MMP-2) and
inhibit the invasion of Huh-7 cells (42). Intriguingly, activation of ERK1/2
can contribute to the conversion of pro-MMP-2 to MMP-2 and then
activate this enzyme (41).
Therefore, inhibited ERK2-mediated activation of MMP-2 may also
contribute to the antitumor effects of amprenavir.
Inhibited ERK1/2 activity may be implicated in the
apoptotic cell death induced by several approved HIV-1 protease
inhibitors. For example, inhibition of ERK1/2 signaling by
ritonavir, nelfinavir and saquinavir could initiate growth arrest
and apoptosis in human multiple myeloma cells (37), and a pro-apoptoptic action has been
recorded in amprenavir-treated hepatocellular carcinoma xenograft
models (42), though the accurate
mechanisms remain unclear. Since phosphorylation of
BimEL Ser69 can enhance the proteosomal degradation of
BimEL (6) and reduce
its binding to pro-survival molecules, such as Mcl-1, Bcl-xL and
Bcl-2 (7), ERK2-mediated of
BimEL phosphorylation at Ser69 appears to be one of the
critical modulators of its pro-apoptotic function. Intriguingly, a
most recent research reported that in human mammary epithelial
cancer cells including MCF-7 cells, expression levels of
phosphorylated BimEL at Ser69 were elevated, and
pro-survival proteins Bcl-xL and Mcl-1, who sequester
BimEL in cancer cells, were also upregulated (5). Moreover, phosphorylation of
BimEL also participates in the apoptosis regulation in
chronic lymphocytic leukemia (CLL) cells and thus is of great
clinical significance in CLL (43). Our findings support a mechanism
that amprenavir could induce apoptotic response in MCF-7 cells
through the inhibition of ERK2-mediated BimEL
phosphorylation at Ser69 (Fig. 7),
suggesting the suppression of pro-survival, phosphorylated
BimEL as a potential strategy for anticancer
therapeutics.
Animal study results confirmed the tumor growth
inhibition and apoptosis induction effects of amprenavir in the
in vivo settings (Figs. 8
and 9). As an approved drug, the
pharmacokinetics of amprenavir has been extensively studied.
According to the information available in FDA, for adults, its
recommended capsule doses are 1,200 mg twice daily; for patients
<13 years of age or adolescents <50 kg, its recommended
capsule dosage is 20 mg/kg (22.5 mg/kg for oral solution) twice
daily or 15 mg/kg (17 mg/kg for oral solution) 3 times a day to a
maximum dose of 2,400 mg (2,800 mg for oral solution). In our in
vivo study, we adopted a daily dose of 45 mg/kg/day, which is
within the range of therapeutically recommended dosage of this
drug. These results demonstrate that amprenavir is expected to be a
promising anticancer drug under the established drug regimen. A
most recent study has reported that the combination of
amprenavir-doxorubicin could reach a significantly inhibitory
effects of tumor growth earlier than the two drugs individually
(42), suggesting the combined use
of amprenavir and other chemotherapeutic agents as a potential
strategy to increase antitumor efficacy and decrease
chemoresistance.
In conclusion, in the present study, we in
silico screened 1447 FDA-approved small molecule drugs against
ERK2 and identified amprenavir as a potential inhibitor of ERK2. We
confirmed its ability to inhibit ERK2 kinase activity and human
MCF-7 breast cancer cell proliferation, elucidated its unique
apoptosis-inducing mechanisms by inhibiting ERK2-mediated
phosphorylation of BimEL at Ser69, and validated its
anti-proliferative and apoptosis-inducing effects in MCF-7
xenograft models. These findings suggest amprenavir as a novel
modulator of ERK2 signaling pathway and provide insights into its
antitumor activities and mechanisms in MCF-7 cells both in
vitro and in vivo, which may reveal possible new uses of
this drug in anticancer treatment and supply a paradigm of
repositioning existing approved drugs for additional therapeutic
uses.
Acknowledgments
This study was supported in part by the Sichuan
Science Foundation (no. 2015JY0183), by Sichuan Health and Family
Planning Commission Funding (16ZD0253), the Researcher Foundation
of Sichuan Academy of Medical Science and Sichuan Provincial
People's Hospital and Sichuan Scientific Research Foundation of the
Returned Overseas Chinese Scholars. We are grateful to Rong Sun,
Bin Zhang, Huailong Xu, Lei Wu, Na An and Nan Zhou (Sichuan
University) for their kind help and useful suggestions on this
manuscript.
References
|
1
|
Giacinti L, Claudio PP, Lopez M and
Giordano A: Epigenetic information and estrogen receptor alpha
expression in breast cancer. Oncologist. 11:1–8. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
O'Connor L, Strasser A, O'Reilly LA,
Hausmann G, Adams JM, Cory S and Huang DC: Bim: A novel member of
the Bcl-2 family that promotes apoptosis. EMBO J. 17:384–395. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Akiyama T, Dass CR and Choong PF:
Bim-targeted cancer therapy: A link between drug action and
underlying molecular changes. Mol Cancer Ther. 8:3173–3180. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gogada R, Yadav N, Liu J, Tang S, Zhang D,
Schneider A, Seshadri A, Sun L, Aldaz CM, Tang DG, et al: Bim, a
proapoptotic protein, up-regulated via transcription factor
E2F1-dependent mechanism, functions as a prosurvival molecule in
cancer. J Biol Chem. 288:368–381. 2013. View Article : Google Scholar :
|
|
6
|
Luciano F, Jacquel A, Colosetti P, Herrant
M, Cagnol S, Pages G and Auberger P: Phosphorylation of Bim-EL by
Erk1/2 on serine 69 promotes its degradation via the proteasome
pathway and regulates its proapoptotic function. Oncogene.
22:6785–6793. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ewings KE, Hadfield-Moorhouse K, Wiggins
CM, Wickenden JA, Balmanno K, Gilley R, Degenhardt K, White E and
Cook SJ: ERK1/2-dependent phosphorylation of BimEL promotes its
rapid dissociation from Mcl-1 and Bcl-xL. EMBO J. 26:2856–2867.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang H, Qian DZ, Tan YS, Lee K, Gao P,
Ren YR, Rey S, Hammers H, Chang D, Pili R, et al: Digoxin and other
cardiac glycosides inhibit HIF-1alpha synthesis and block tumor
growth. Proc Natl Acad Sci USA. 105:19579–19586. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Miller SC, Huang R, Sakamuru S, Shukla SJ,
Attene-Ramos MS, Shinn P, Van Leer D, Leister W, Austin CP and Xia
M: Identification of known drugs that act as inhibitors of
NF-kappaB signaling and their mechanism of action. Biochem
Pharmacol. 79:1272–1280. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Biechele TL, Camp ND, Fass DM, Kulikauskas
RM, Robin NC, White BD, Taraska CM, Moore EC, Muster J, Karmacharya
R, et al: Chemical-genetic screen identifies riluzole as an
enhancer of Wnt/β-catenin signaling in melanoma. Chem Biol.
17:1177–1182. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dudley JT, Deshpande T and Butte AJ:
Exploiting drug-disease relationships for computational drug
repositioning. Brief Bioinform. 12:303–311. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Okimoto N, Futatsugi N, Fuji H, Suenaga A,
Morimoto G, Yanai R, Ohno Y, Narumi T and Taiji M: High-performance
drug discovery: Computational screening by combining docking and
molecular dynamics simulations. PLoS Comput Biol. 5:e10005282009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kinoshita T, Yoshida I, Nakae S, Okita K,
Gouda M, Matsubara M, Yokota K, Ishiguro H and Tada T: Crystal
structure of human mono-phosphorylated ERK1 at Tyr204. Biochem
Biophys Res Commun. 377:1123–1127. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Aronov AM, Baker C, Bemis GW, Cao J, Chen
G, Ford PJ, Germann UA, Green J, Hale MR, Jacobs M, et al: Flipped
out: Structure-guided design of selective pyrazolylpyrrole ERK
inhibitors. J Med Chem. 50:1280–1287. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rose PW, Bi C, Bluhm WF, Christie CH,
Dimitropoulos D, Dutta S, Green RK, Goodsell DS, Prlic A, Quesada
M, et al: The RCSB Protein Data Bank: New resources for research
and education. Nucleic Acids Res. 41:D475–D482. 2013. View Article : Google Scholar :
|
|
16
|
Pettersen EF, Goddard TD, Huang CC, Couch
GS, Greenblatt DM, Meng EC and Ferrin TE: UCSF Chimera - a
visualization system for exploratory research and analysis. J
Comput Chem. 25:1605–1612. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kuntz ID, Blaney JM, Oatley SJ, Langridge
R and Ferrin TE: A geometric approach to macromolecule-ligand
interactions. J Mol Biol. 161:269–288. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Knox C, Law V, Jewison T, Liu P, Ly S,
Frolkis A, Pon A, Banco K, Mak C, Neveu V, et al: DrugBank 3.0: A
comprehensive resource for 'omics' research on drugs. Nucleic Acids
Res. 39:D1035–D1041. 2011. View Article : Google Scholar
|
|
19
|
O'Boyle NM, Banck M, James CA, Morley C,
Vandermeersch T and Hutchison GR: Open Babel: An open chemical
toolbox. J Cheminform. 3:332011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Irwin JJ, Sterling T, Mysinger MM, Bolstad
ES and Coleman RG: ZINC: A free tool to discover chemistry for
biology. J Chem Inf Model. 52:1757–1768. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lang PT, Brozell SR, Mukherjee S,
Pettersen EF, Meng EC, Thomas V, Rizzo RC, Case DA, James TL and
Kuntz ID: DOCK 6: Combining techniques to model RNA-small molecule
complexes. RNA. 15:1219–1230. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Van Der Spoel D, Lindahl E, Hess B,
Groenhof G, Mark AE and Berendsen HJ: GROMACS: Fast, flexible, and
free. J Comput Chem. 26:1701–1718. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Schüttelkopf AW and van Aalten DM: PRODRG:
A tool for highthroughput crystallography of protein-ligand
complexes. Acta Crystallogr D Biol Crystallogr. 60:1355–1363. 2004.
View Article : Google Scholar
|
|
24
|
Li C, Chen J, Lu B, Shi Z, Wang H, Zhang
B, Zhao K, Qi W, Bao J and Wang Y: Molecular switch role of Akt in
Polygonatum odoratum lectin-induced apoptosis and autophagy in
human non-small cell lung cancer A549 cells. PLoS One.
9:e1015262014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shi W, Deng J, Tong R, Yang Y, He X, Lv J,
Wang H, Deng S, Qi P, Zhang D, et al: Molecular mechanisms
underlying mangiferininduced apoptosis and cell cycle arrest in
A549 human lung carcinoma cells. Mol Med Rep. 13:3423–3432.
2016.PubMed/NCBI
|
|
26
|
Wang Y, Liu Y, Wang H, Li C, Qi P and Bao
J: Agaricus bisporus lectins mediates islet β-cell proliferation
through regulation of cell cycle proteins. Exp Biol Med (Maywood).
237:287–296. 2012. View Article : Google Scholar
|
|
27
|
Jiagang D, Li C, Wang H, Hao E, Du Z, Bao
C, Lv J and Wang Y: Amygdalin mediates relieved atherosclerosis in
apolipoprotein E deficient mice through the induction of regulatory
T cells. Biochem Biophys Res Commun. 411:523–529. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang W, Zheng Y, Xia Y, Ji H, Chen X, Guo
F, Lyssiotis CA, Aldape K, Cantley LC and Lu Z: ERK1/2-dependent
phosphorylation and nuclear translocation of PKM2 promotes the
Warburg effect. Nat Cell Biol. 14:1295–1304. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang Y, Wang H, Liu Y, Li C, Qi P and Bao
J: Antihyperglycemic effect of ginsenoside Rh2 by inducing islet
beta-cell regeneration in mice. Horm Metab Res. 44:33–40. 2012.
View Article : Google Scholar
|
|
30
|
Yun J, Lv YG, Yao Q, Wang L, Li YP and Yi
J: Wortmannin inhibits proliferation and induces apoptosis of MCF-7
breast cancer cells. Eur J Gynaecol Oncol. 33:367–369.
2012.PubMed/NCBI
|
|
31
|
He JC, Husain M, Sunamoto M, D'Agati VD,
Klotman ME, Iyengar R and Klotman PE: Nef stimulates proliferation
of glomerular podocytes through activation of Src-dependent Stat3
and MAPK1,2 pathways. J Clin Invest. 114:643–651. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Biggs TE, Cooke SJ, Barton CH, Harris MP,
Saksela K and Mann DA: Induction of activator protein 1 (AP-1) in
macrophages by human immunodeficiency virus type-1 NEF is a
cell-typespecific response that requires both hck and MAPK
signaling events. J Mol Biol. 290:21–35. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Schrager JA, Der Minassian V and Marsh JW:
HIV Nef increases T cell ERK MAP kinase activity. J Biol Chem.
277:6137–6142. 2002. View Article : Google Scholar
|
|
34
|
Kinoshita T, Warizaya M, Ohori M, Sato K,
Neya M and Fujii T: Crystal structure of human ERK2 complexed with
a pyrazolo[3,4-c]pyridazine derivative. Bioorg Med Chem Lett.
16:55–58. 2006. View Article : Google Scholar
|
|
35
|
Park H, Bahn YJ, Jeong DG, Woo EJ, Kwon JS
and Ryu SE: Identification of novel inhibitors of extracellular
signal-regulated kinase 2 based on the structure-based virtual
screening. Bioorg Med Chem Lett. 18:5372–5376. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ohori M, Kinoshita T, Okubo M, Sato K,
Yamazaki A, Arakawa H, Nishimura S, Inamura N, Nakajima H, Neya M,
et al: Identification of a selective ERK inhibitor and structural
determination of the inhibitor-ERK2 complex. Biochem Biophys Res
Commun. 336:357–363. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Aronov AM, Tang Q, Martinez-Botella G,
Bemis GW, Cao J, Chen G, Ewing NP, Ford PJ, Germann UA, Green J, et
al: Structure-guided design of potent and selective
pyrimidylpyrrole inhibitors of extracellular signal-regulated
kinase (ERK) using conformational control. J Med Chem.
52:6362–6368. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang Z, Canagarajah BJ, Boehm JC, Kassisà
S, Cobb MH, Young PR, Abdel-Meguid S, Adams JL and Goldsmith EJ:
Structural basis of inhibitor selectivity in MAP kinases.
Structure. 6:1117–1128. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ikezoe T, Saito T, Bandobashi K, Yang Y,
Koeffler HP and Taguchi H: HIV-1 protease inhibitor induces growth
arrest and apoptosis of human multiple myeloma cells via
inactivation of signal transducer and activator of transcription 3
and extracellular signal-regulated kinase 1/2. Mol Cancer Ther.
3:473–479. 2004.PubMed/NCBI
|
|
40
|
Pyrko P, Kardosh A, Wang W, Xiong W,
Schönthal AH and Chen TC: HIV-1 protease inhibitors nelfinavir and
atazanavir induce malignant glioma death by triggering endoplasmic
reticulum stress. Cancer Res. 67:10920–10928. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Pajonk F, Himmelsbach J, Riess K, Sommer A
and McBride WH: The human immunodeficiency virus (HIV)-1 protease
inhibitor saquinavir inhibits proteasome function and causes
apoptosis and radiosensitization in non-HIV-associated human cancer
cells. Cancer Res. 62:5230–5235. 2002.PubMed/NCBI
|
|
42
|
Gills JJ1, Lopiccolo J, Tsurutani J,
Shoemaker RH, Best CJ, Abu-Asab MS, Borojerdi J, Warfel NA, Gardner
ER, Danish M, et al: Nelfinavir, A lead HIV protease inhibitor, is
a broadspectrum, anticancer agent that induces endoplasmic
reticulum stress, autophagy, and apoptosis in vitro and in vivo.
Clin Cancer Res. 13:5183–5194. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Esposito V, Verdina A, Manente L, Spugnini
EP, Viglietti R, Parrella R, Pagliano P, Parrella G, Galati R, De
Luca A, et al: Amprenavir inhibits the migration in human
hepatocarcinoma cell and the growth of xenografts. J Cell Physiol.
228:640–645. 2013. View Article : Google Scholar
|