Introduction
Vasculogenesis/angiogenesis is an essential process
for embryonic and postnatal development, wound healing and also
endometrial angiogenesis during the menstrual cycle in women.
Angiogenesis is also vital for the growth and dissemination of
solid tumours (1,2). Tumour-associated angiogenesis is
pivotal for a solid tumour to grow beyond a certain size (2–3 mm)
as it can be restricted by interspatial diffusion of nutrients. The
newly formed vasculature also provides a pathway for cancer cells
to spread to other parts of the body. Angiogenesis is affected by
multiple factors and various cells in the tumour microenvironment
(3).
Angiogenesis can be induced or promoted by
pro-angiogenic factors, such as VEGF, FGF, TNF-α and HGF which can
be produced by cancer cells, stromal cells, inflammatory cells and
endothelial cells (4–7). For example, VEGF binds to two
tyrosine kinase receptors, VEGFR1 and VEGFR2. VEGFR1 is expressed
by hematopoietic and vascular endothelial cells and acts as a
negative regulator of angiogenesis, whilst VEGFR2 is mainly
expressed by vascular endothelial cells and is crucial for
vasculogenesis leading to the formation of primary vascular plexus
(8–10). Tie-2 receptor and its ligands,
angiopoietins, are also important for angiogenesis. Angiopoietin-2
has been demonstrated as a VEGF negative regulator in several
cancers (11,12). In addition to their pro-angiogenic
effect, FGF and HGF can also directly promote proliferation,
migration and invasion of cancer cells (13,14).
Protein tyrosine phosphatases (PTPs) are involved in
regulation of cellular functions by coordinating signal
transduction through dephosphorylation of certain signalling
molecules. PTPs have also been indicated as important regulators in
tumorigenesis and angiogenesis (15–18).
Receptor-like protein tyrosine phosphatase beta (PTPRB, also known
as vascular endothelial VE-PTP), for example, plays a crucial role
in the angiogenesis of breast cancer by regulating several
signalling pathways such as the Tie-2 pathway (19). Blocking PTPRB with a small
inhibitor, AKB-9778, reduced tumour growth and metastases of breast
cancer (20). Furthermore, Src
homology 2 domain-containing protein tyrosine phosphatase-1 (SHP-1)
suppresses angiogenesis by inhibiting the VEGF signalling in
microvascular endothelial cells (21).
To date, aberrant expression of receptor-like
protein tyrosine phosphatase kappa (PTPRK) has been observed in
glioma, lymphoma, prostate and breast cancer (22–26).
However, the role played by PTPRK in angiogenesis remains largely
unknown. The present study aimed to investigate the role played by
this molecule in angiogenesis, in particular VEGF and FGF-promoted
angiogenesis.
Materials and methods
Cell lines and cells culture
HECV (human endothelial vascular cell line) cells
were purchased from Interlab (Naples, Italy); PANC-1, MSA-MB-231
and MRC-5 cells were purchased from the American Type Culture
Collection (ATCC; Manassas, VA, USA) and HT115 cells were purchased
from the European Collection of Authenticated Cell Cultures (ECACC;
Salisbury, UK). Cells were routinely cultured with Dulbecco's
modified Eagle's medium (DMEM) containing 10% fetal calf serum
(FCS) and antibiotics at 37°C with 5% CO2. PCR primers
were designed using Primer-3 and synthesised by Sigma-Aldrich
(Dorset, UK) and the sequences are provided in Table I.
 | Table IPrimer sequences used in the present
study. |
Table I
Primer sequences used in the present
study.
| Gene | Forward primers
(5′–3′) | Reverse primer
(5′–3′) |
|---|
| GAPDH |
GGCTGCTTTTAACTCTGGTA |
GACTGTGGTCATGAGTCCTT |
| GAPDH (Q-PCR) |
CTGAGTACGTCGTGGAGTC |
ACTGAACCTGACCGTACACAGAGATGACCCTTTTG |
| PTPRK |
AATTACAATTGATGGGGAGA |
CCACTTTTCCACCTGAAGTA |
| PTPRK (Q-PCR) |
AATTACAATTGATGGGGAGA |
ACTGAACCTGACCGTACATATTGTGTGACGATGAAAGC |
Reverse transcription-PCR
Total RNA extraction from cells was performed using
Tri reagent (Sigma-Aldrich). Following reverse transcription, PCR
was carried out using GoTaq DNA olymerase (Promega, Southampton,
UK). Reactions were carried out with the following process: 94°C
for 5 min, 30 cycles of 94°C for 30 sec, 55°C for 30 sec and 72°C
for 30 sec, followed by a final extension of 7 min at 72°C. PCR
products were separated on a 1.5% agarose gel and photographed
after staining with SYBR Safe DNA dye (Invitrogen, Paisley,
UK).
Real-time quantitative PCR
The level of PTPRK transcripts in HECV cells was
determined using a real-time quantitative PCR, based on a
previously reported method (27).
The reaction was carried out on an iCycler iQ™ (Bio-Rad
Laboratories, Hempstead, UK) which is equipped with an optical unit
that allows real-time detection of 96 reactions. The reaction
conditions were: 94°C for 12 min, 100 cycles of 94°C for 15 sec,
55°C for 35 sec (the data capture step) and 72°C for 15 sec. The
levels of the transcripts were generated from an internal standard
that was simultaneously amplified.
Construction of ribozyme transgene
targeting human PTPRK and the establishment of corresponding stable
transfectants
Anti-human PTPRK hammerhead ribozymes were designed
based on the secondary structure of the gene transcript and
generated using the Zuker RNA Mfold program (28). The ribozymes were synthesized and
then cloned into a pEF6/V5-His TOPO vector (Invitrogen). The
verified ribo-zyme transgenes and empty plasmids were transfected
into HECV (HECVPTPRKkd and HECVpEF) cells,
respectively using an Easyjet Plus electroporator (Equibio, Kent,
UK). After a period of selection with 5 μg/ml blasticidin
(up to 10 days), the verified transfectants were cultured in
maintenance medium containing 0.5 μg/ml blasticidin. Primer
sequences of the ribozymes were
5′-CTGCAGTTTGCTCTTTTTTACAATTAATATCTGATGAGTCCGTGAGGA-3′ and
5′-ACTAGTTCATCCTCCTTCTCCTAGTTGTTTCGTCCTCACGGACT-3′.
In vitro cell growth assay
HECV cells (3,000 cells/well) were plated into two
identical 96-well plates. Cells were fixed in 4% formalin after 24
and 72 h of culture. The cells were then stained with 0.5% (w/v)
crystal violet. Following washing, stained crystal violet was
extracted with 10% (v/v) acetic acid (29). Absorbance was then determined at a
wavelength of 540 nm using an EL×800 spectrophotometer (BioTek
Instruments, Inc., Swindon, UK). Growth rate of day 3 (%) =
absorbance of day 3/absorbance of day 1 × 100.
In vitro cell-matrix adhesion
Cells (20,000/well) were seeded to each well of a
96-well plate which was pre-coated with Matrigel (5 μg/well)
(BD Biosciences, Oxford, UK). After 40 min of incubation, the
non-adherent cells were washed off using balanced salt solution
(BSS; comprising 137 mM NaCl, 2.6 mM KCl, 1.7 mM
Na2HPO4 and 8.0 mM
KH2PO4 and adjusting the pH to 7.4 with 1 M
NaOH). The remaining cells were fixed with formalin and were
stained with crystal violet. The number of adherent cells was then
counted under a microscope.
In vitro migration/wounding assay
Cells (200,000/well) were seeded into a 24-well
plate and allowed to reach confluence. The cell monolayer was
scratched using a fine gauge needle to create an artificial wound
of ~200 μm in width (30).
Images were taken at 0.25, 1, 2, 3 and 4 h after wounding.
Migration distances were measured using ImageJ software (National
Institutes of Health, Bethesda, MD, USA).
Cell spreading assay
Cells (20,000/well) were seeded into a 96-well plate
which was pre-coated with Matrigel (5 μg/well) (BD
Biosciences) and the cells were fixed after an incubation of up to
4 h. The fixed cells were stained with fluorescein phalloidin
(F432; Life Technologies, Carlsbad, CA, USA) and DAPI (10236276001;
Roche Applied Science, Basel, Switzerland). Images were taken using
a Leica fluorescence microscope (Leica DM IL LED). Cell spreading
was measured using ImageJ software (National Institutes of
Health).
Tubule formation assay
Cells (40,000/well) were seeded into a 96-well plate
which was pre-coated with Matrigel (500 μg/well). The cells
were fixed with formalin after a 4-h incubation and photographed
immediately using a microscope. The sum of tubule perimeter was
measured using ImageJ software.
Electric cell-substrate impedance sensing
(ECIS)
An ECIS 9600 model instrument and 96W1E arrays
(Applied Biophysics, Inc., Troy, NY, USA) was also used for
migration assays in the study, as previously reported (31). HECVpEF and
HECVPTPRKkd cells were seeded at 40,000 cells/well in
200 μl DMEM medium alone or supplemented with 10 ng/ml FGF
(F0291; Sigma-Aldrich) or 10 ng/ml VEGF (293-VE; R&D Systems,
Abingdon, UK), with small inhibitors; 5 μM Akt inhibitor
(Akt124005; Millipore UK, Ltd., Watford, UK), 50 nM c-Src inhibitor
Src I1 (Tocris Bioscience, Bristol, UK), 5 nM PI3K inhibitor
(wortmannin; Tocris Bioscience) and 5 nM PLCg inhibitor (STK870702;
Vitas-M Laboratory, Ltd., Apeldoorn, The Netherlands),
respectively. The resistance was measured at 30 kHZ for 5 h after
electrical wounding, and data was analysed using an ECIS-9600
software package.
Western blot analysis
Equal amounts of protein were separated using
SDS-PAGE and blotted onto nitrocellulose membranes (SC-3724; Santa
Cruz Biotechnology, Heidelberg, Germany). Proteins were then probed
with the primary antibodies and corresponding peroxidase-conjugated
secondary antibodies. Protein bands were visualised using a
chemiluminescence detection kit (Luminata; Millipore) and
photographed using UVItec imager (Uvitec Printing Ink Co., Inc.,
Lodi, NJ, USA). Antibodies for GAPDH (sc-32233), PTPRK (sc-28906),
c-Src (sc-5266), PLCγ (sc-81) and Akt1 (sc-1618) were purchased
from Santa Cruz Biotechnology. Antibodies for FAK and Paxillin were
obtained from BD Biosciences.
Statistical analysis
Statistical analysis was performed using SigmaPlot
11 (SPSS, Inc., Chicago, IL, USA). Data were calculated as the mean
± SD, the Student's t-test was used for normally distributed data
and one-way ANOVA was used for multiple group comparison. Each
assay was performed three times. P<0.05 was considered
statistically significant.
Results
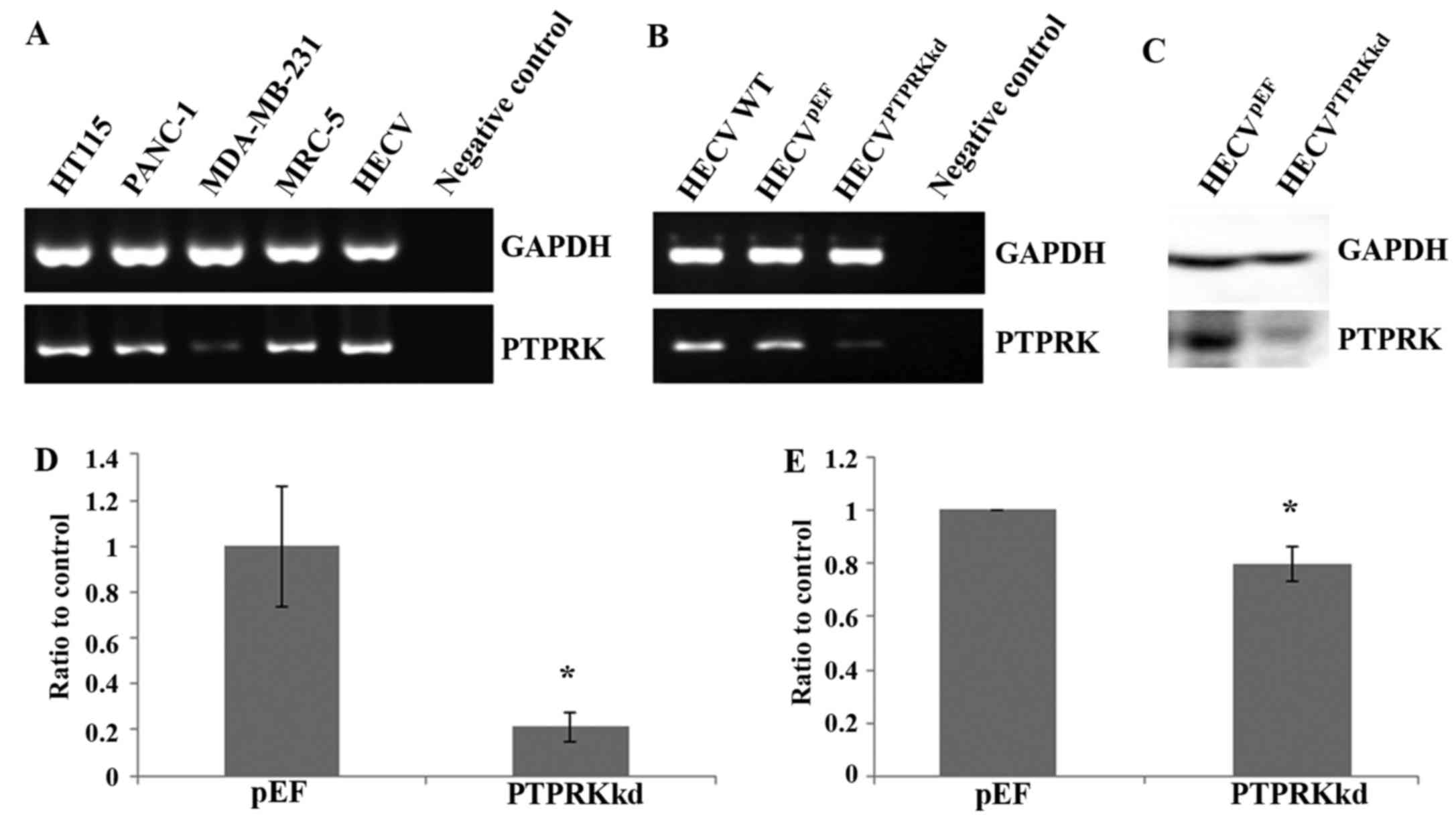
Knockdown of PTPRK in vascular
endothelial cells using anti-PTPRK ribozyme
The expression of PTPRK in the vascular endothelial
cell line HECV, was first determined using conventional PCR with a
comparison to its expression in several cancer cell lines including
colorectal cancer (HT115), pancreatic cancer (PANC-1) and breast
cancer (MDA-MB-231) and also a fibroblast cell line (MRC-5)
(Fig. 1A). All these cell lines
express PTPRK though levels had subtle variations.
The expression of PTPRK was knocked down using
ribozyme transgenes targeting human PTPRK mRNA. Reduced mRNA
expression of PTPRK was seen in the cells which were transfected
with anti-PTPRK ribozyme transgenes using both conventional PCR
(Fig. 1B) and real-time
quantitative PCR (Fig. 1D). The
knockdown of PTPRK was further confirmed with western blot analysis
for its protein expression (Fig. 1C
and E).
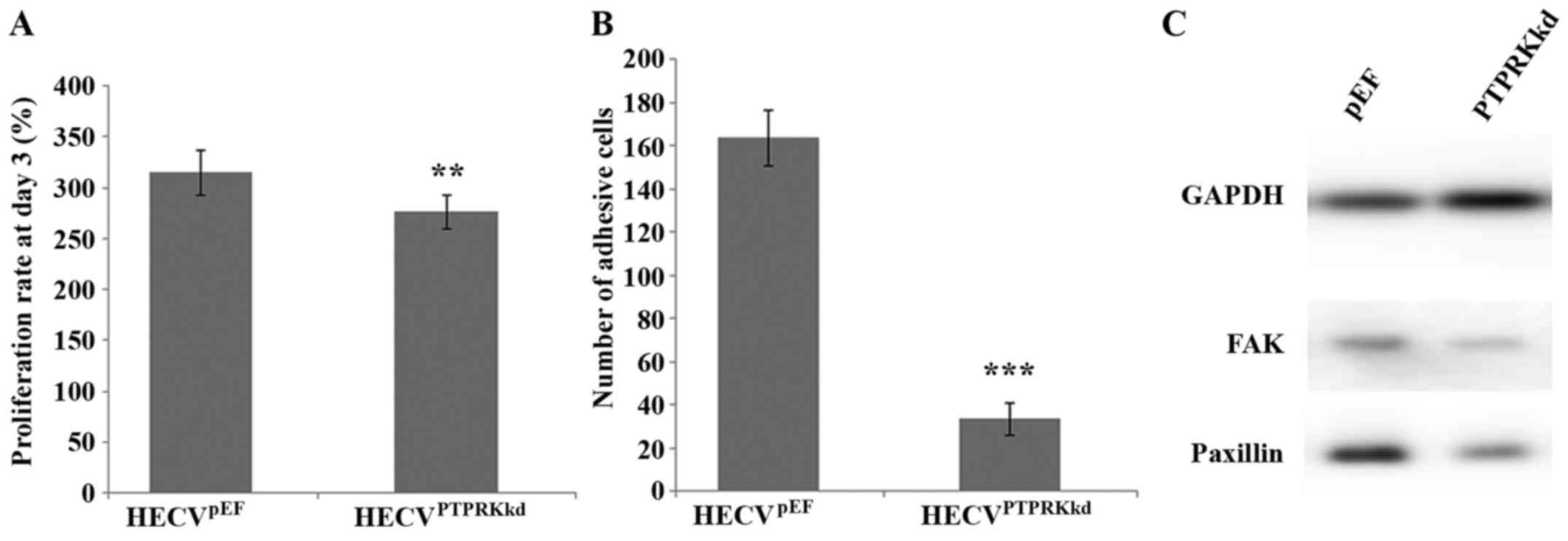
Effect of PTPRK knockdown on the
proliferation and cell-matrix adhesion of endothelial vascular
cells
After verification of the knockdown of PTPRK, we
also examined its influence on cell proliferation and cell-matrix
adhesion. Knockdown of PTPRK significantly reduced proliferation of
the vascular endothelial cells compared to the control cells
(276.27±16.37 vs. 314.77±21.93%; P<0.01) (Fig. 2A). PTPRK knockdown elicited a
significant influence on cell matrix adhesion. The number of
adhered HECVPTPRKkd cells (33.33±7.27) was much less
than the HECVpEF cells (163.50±12.96), P<0.001
(Fig. 2B). Furthermore, knockdown
of PTPRK resulted in a reduced expression of both FAK and paxillin
proteins (Fig. 2C).
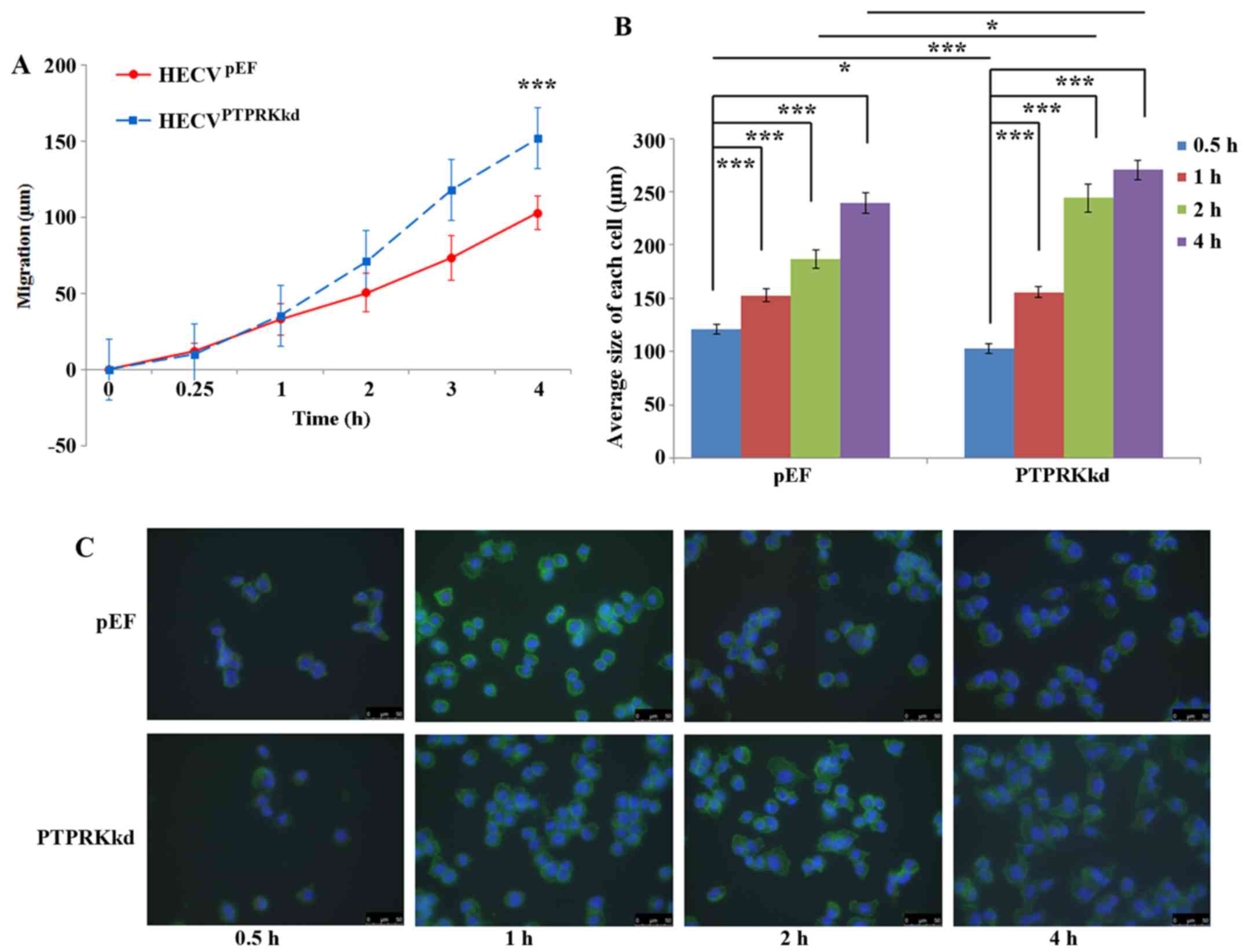
PTPRK and the migration and spreading of
endothelial vascular cells
PTPRK knockdown cells exhibited increased cell
motility compared with the control cells. After a 4-h incubation,
the distance that HECVPTPRKkd cells migrated was
152.02±18.98 μm compare with that of HECVpEF
cells (102.91±11.26 μm), P<0.001 (Fig. 3A). Cell spreading assays showed a
very interesting result. After a 30-min incubation, the size of
HECVpEF cells were found to be larger than
HECVPTPRKkd (120.98±4.66 vs. 102.82±4.25 μm;
P<0.05) which appeared to be in line with the inhibitory effect
on cell-matrix adhesion. Such an effect was diminished after 1-h
incubation. However, an increased spreading was seen in the
HECVPTPRKkd cells after 2-h incubation, their size being
244.20±12.93 ImageJ units, ~30% bigger than the size
HECVpEF cells (186.76±8.29 ImageJ units; P<0.001). A
similar influence on the spreading was also observed after an
incubation of 4 h (Fig. 3B and
C).
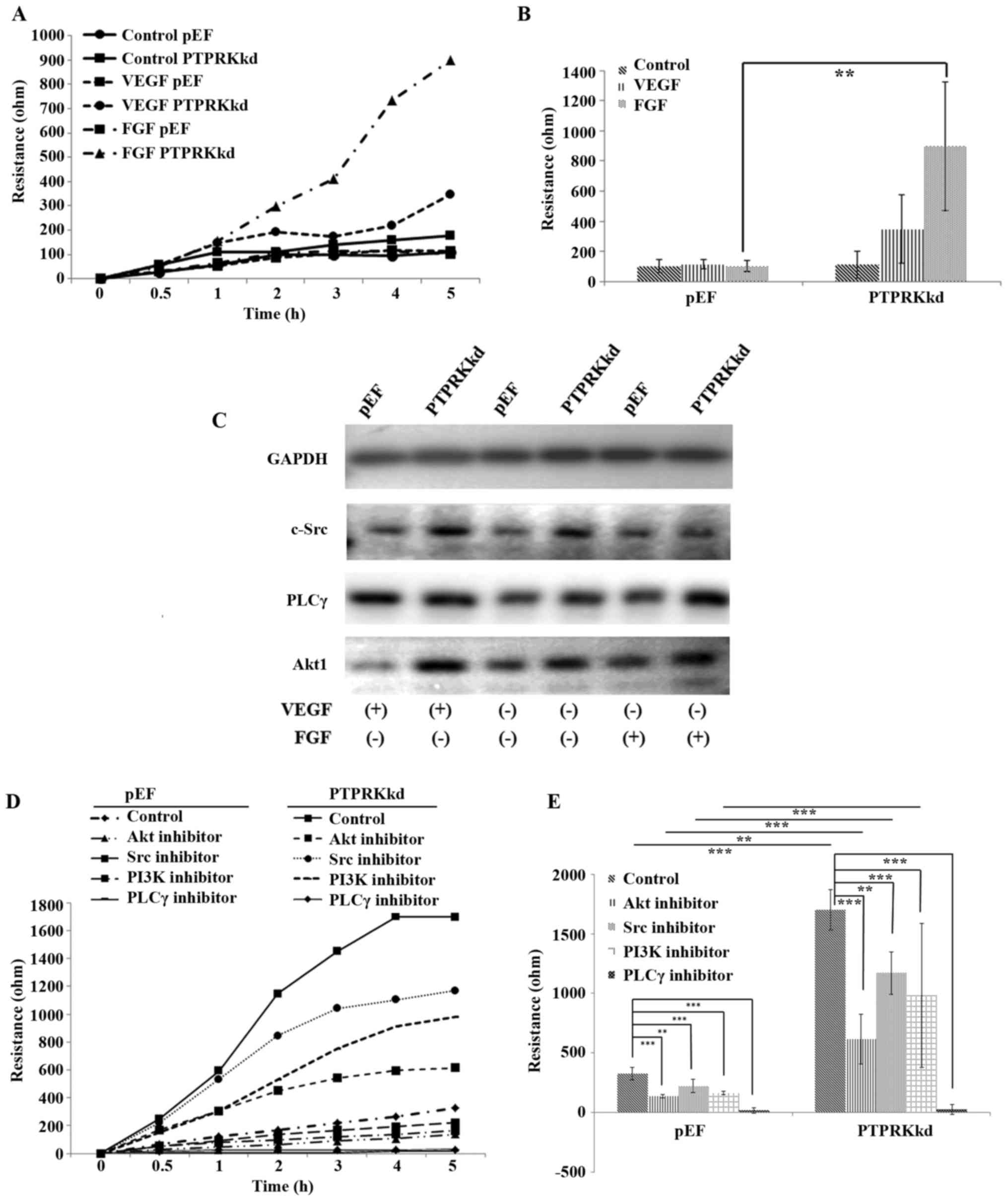
PTPRK in VEGF and FGF induced migration
of vascular endothelial cells
VEGF and FGF pathways play crucial roles for several
cell functions including vasculogenesis and angiogenesis (8,32).
To explore the involvement of PTPRK in VEGF and FGF induced
angiogenesis, we determined the migration of HECV cells with
additions of VEGF and FGF, respectively. Little effect on cell
migration was seen in the HECVpEF cells exposed to VEGF
(10 ng/ml) and FGF (10 ng/ml), respectively. However, the knockdown
of PTPRK conferred an increased sensitivity to these two
pro-angiogenic factors. HECVPTPRKkd cells were more
responsive to the treatment of both VEGF and FGF. A more marked
increase was seen in FGF treated HECVPTPRKkd cells
(Fig. 4A and B). We then examined
the expression of three key molecules, c-Src, PLCγ and Akt1 which
mediate signalling downstream of the respective receptors of VEGF
and FGF. To our surprise, an elevated protein level was seen for
Akt1 and c-Src in the HECVPTPRKkd cells, which was not
observed for the PLCγ. An enhanced expression of Akt1 was seen in
the PTPRK knockdown cells when they were exposed to the VEGF, while
FGF treated cells exhibited similar levels of Akt1 protein compared
with the untreated controls. A similar pattern and expression
levels of c-Src were seen in the cells treated with VEGF compared
with the untreated controls, while the FGF-treated
HECVPTPRKkd cells had a reduced expression of c-Src
which was down to a similar level of the HECVpEF cells.
VEGF increased the protein level of PLCγ in both
HECVPTPRKkd and HECVpEF cells. In contrast to
the VEGF-elevated expression of PLCγ, an increased protein
expression of PLCγ was only seen in the PTPRK knockdown cells when
they were treated with FGF (Fig.
4C). Since an increased response to FGF was seen in the
migration of PTPRK knockdown cells, we treated cells with FGF with
additions of small inhibitors targeting c-Src, PLCγ, Akt and also
PI3K to verify their involvement in the FGF-promoted cell motility.
Four inhibitors suppressed the cell migration which were promoted
by FGF, however, only the PLCγ inhibitor repressed the migration of
HECVPTPRKkd cells to a level similar to the control
cells (Fig. 4D and E).
Involvement of PTPRK in pro-angiogenic
factor and cancer cell induced tubule formation of vascular
endothelial cells
An in vitro tubule formation assay was used
to assess the influence of PTPRK knockdown on the capability of
vascular endothelial cells to form new vasculature. Knockdown of
PTPRK resulted in a decrease of proliferation and cell-matrix
adhesion, a similar inhibitory effect was also seen in the tubule
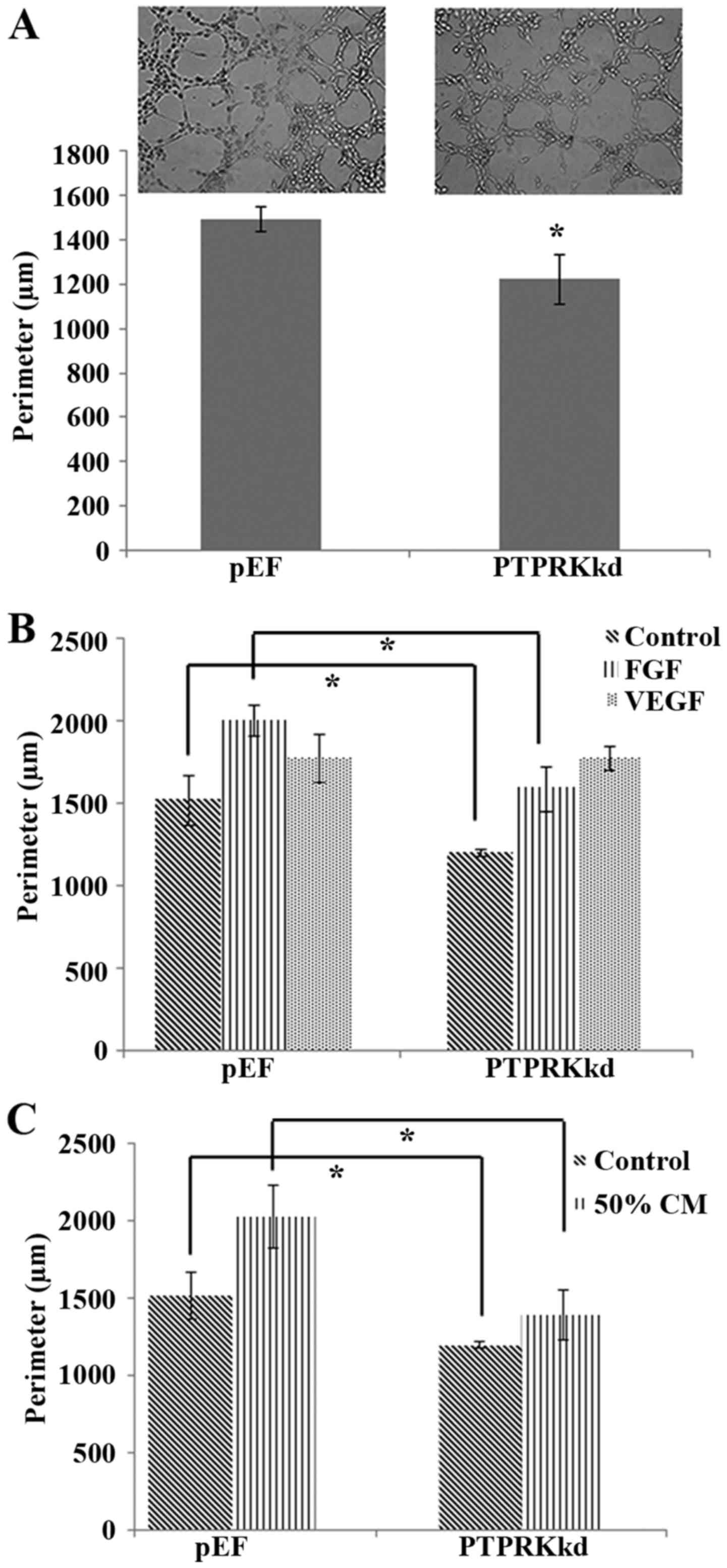
formation (Fig. 5A), though the
motility of endothelial cells was enhanced after the PTPRK
knockdown. We then investigated the proangiogenic factor, in
particular the VEGF and FGF-induced angiogenesis. The reduced
tubule formation in the PTPRK knockdown cells was diminished by an
exposure to VEGF (10 ng/ml) and the PTPRK knockdown cells appeared
to be more responsive to VEGF compared with the HECVpEF
cells but not to a significant level. However, an increased tubule
formation was seen in both HECVPTPRKkd and
HECVpEF cells which were treated with FGF (10 ng/ml)
(1588.92±134.61 vs. 2002.02±96.39 μm; P<0.05). However,
the PTPRK knockdown-inhibited tubule formation still existed when
the cells were treated with FGF (Fig.
5B).
To mimic the tumour associated angiogenesis, we
treated the endothelial cells with medium collected from breast
cancer cells (MDA-MB-231). We aimed to clarify the involvement of
PTPRK in cancer cell-induced angiogenesis. Cells incubated with 50%
medium collected from breast cancer showed similar result with FGF
treated cells, it promoted both tubule formation of
HECVPTPRKkd and HECVpEF cells but the PTPRK
knockdown-inhibited tubule formation was still present
(1392.87±157.59 vs. 2029.87±204.46 μm; P<0.01) (Fig. 5C).
Discussion
Our previous studies have shown that the expression
of PTPRK is reduced in the breast cancer which is associated with
poor prognosis of the disease (26). Knockdown of PTPRK in breast cancer
cells promoted their proliferation, adhesion, invasion and
migration. In contrast to the reduced expression of PTPRK observed
in the breast cancer, an increased PTPRK expression was seen in
prostate cancer (25). An
inhibitory effect on cellular functions was also seen in prostate
cancer cells following PTPRK knockdown. It suggests that PTPRK
plays different roles in different cancers which can be cancer
specific. It has been indicated that some PTPs participate in the
regulation of angiogenesis. For example, SHP-1 (PTPN6) has been
identified as an anti-angiogenic regulator via VEGFR2 signalling
pathway (21,33). VE-PTP (PTPRB) plays an important
role in angiogenesis by targeting the VEGFR2 (34,35)
and Tie2 pathways (36–39). However, to date, little is known
about the role of PTPRK in tumour-associated angiogenesis.
In the present study, we first confirmed expression
of PTPRK in a vascular endothelial cell lines (HECV). PTPRK was
found to be extensively expressed by a variety of different cell
lines, including vascular endothelial cells (HECV), colorectal
cancer (HT115), pancreatic cancer (PANC-1) and fibroblasts (MRC-5).
We then employed the anti-PTPRK ribozyme plasmid constructs to
establish a cell model for the present study. Knockdown of PTPRK
expression in the HECV cells resulted in a decreased proliferation
and cell-matrix adhesion. This is similar to the inhibitory effect
observed in the prostate cancer cells with knockdown of PTPRK
(25). In contrast, PTPRK
knockdown promoted migration of the endothelial cells which was
similar to the effect seen in breast cancer cells (26). Such contrasting effects on
different cellular functions of vascular endothelial cells suggest
that PTPRK elicits more complex functions by interacting with
different molecules.
Focal adhesion kinase (FAK)-paxillin signalling
pathway regulates many cellular functions such as cell survival and
migration; it has also been indicated that PTPs are involved in
this mechanism. For example, PTP-PEST interacts with paxillin and
Grb2 which are key players in the focal adhesion complex (40). DEP-1 (PTPRJ) inhibits cell
proliferation, formation of vinculin and paxillin-containing
adhesion plaques and also the activation of FAK (41). DEP-1 can interact with c-Src and
promotes cell adhesion through an activation of FAK and paxillin
(42). In the present study,
knockdown of PTPRK in human endothelia cells reduced cell-matrix
adhesion. To examine the involvement of FAK and paxillin in the
inhibitory effect on cell adhesion of HECV cells, we investigated
the protein expression using western blot analysis. The results
showed that protein levels of both FAK and paxillin were decreased
in the PTPRK knockdown cells. The reduced FAK and paxillin protein
levels are in line with the inhibitory effect on cell-matrix
adhesion. It suggests that PTPRK may play a role by either directly
mediating the adhesion and/or stabilising focal adhesion complex
which includes FAK and paxillin. However, the exact machinery
operated by PTPRK in regulation of cell adhesion requires further
investigation.
Moreover, knockdown of PTPRK promoted cell motility
in endothelial cells and cell spreading assay showed us that the
average cell size of PTPRK knockdown cells were smaller than
control cell after 30-min incubation. This is consistent with the
inhibitory effect of PTPRK knockdown on cell adhesion and also the
reduced FAK and paxillin proteins. The cellular spreading of HECV
cells following the initial adhesion was enhanced by the PTPRK
knockdown due to its effect on cell migration. Furthermore,
fibronectin induced endothelial cell migration was regulated by
Src-dependent phosphorylation of FGFR1 (43). Fibroblast induced
cell-contact-dependent colorectal cancer cell migration and
invasion via regulation of the FGF2-FGFRs-Src-αvβ5 pathway
(44). In migratory gliomatropic
neural stem cells, promotion of VEGFR2 expression resulted in
activation of VEGFR pathway downstream molecules such as PLCγ, FAK
and Akt (45). Our results showed
that c-Src and Akt were increased at their protein levels after the
knockdown of PTPRK. PLCγ appeared to be more involved in the FGF
induced effect. Along with c-Src, PLCγ and Akt, we also included
PI3K which is a key molecule in mediating signalling for FGF in our
experiment with small inhibitors. We treated the HECV cells with
FGF and small inhibitors targeting c-Src, PLCγ, Akt and PI3K. The
addition of small inhibitors showed that all four molecules play
important roles in cell migration and were involved in the PTPRK
knockdown promoted cell migration to various levels, in which PLCγ
tended to be vital for the migration. However, a more comprehensive
method is required to determine PTPRK-regulated protein
phosphorylation and signal transduction, for example, the Kinex™
antibody microarray (Kinexus Bioinformatics Corp., Vancouver,
Canada).
Studies have shown that certain PTPs contribute to
the inhibitory effect on cell proliferation, such as PTPN3 in
cholangiocarcinoma, PTPRM and PTPRK in breast cancer (26,46,47).
However, studies also showed that same PTPs may confer a favour to
the proliferation and inhibition of PTPRM in glioblastoma
multiforme cells has been shown to result in decreased cell growth
and survival (48). Furthermore,
our previous studies have also shown that knockdown of PTPRK in
prostate cancer cells reduces cell proliferation due to promotion
of apoptosis via JNK pathway (25). In the present study, we tried to
investigate which pathway was involved in the regulation of cell
growth in human endothelial cells. Little effect was seen in the
proliferation of HECV with addition of small inhibitors targeting
Src, Akt and also PI3K which is another key player downstream of
the respective receptors of VEGF and FGF (data not shown). The
mechanism underlying the inhibitor effect of proliferation is yet
to be elucidated.
A further in vitro tubule formation test
showed promotion of tubule formation triggered by the knockdown of
PTPRK, which could be the predominant effect of PTRPK knockdown on
angiogenesis unless it is further validated by ex vivo and
in vivo evidence. It has been reported that FGF and VEGF
pathways participate in the regulation of many cell function such
as cell motility and angiogenesis (49,50).
Reduction of PTP1B expression increased VEGF-induced migration and
proliferation of mouse heart microvascular endothelial cells and
FGF-induced proliferation of rat aortic smooth muscle cells
(51). SHP-2 was shown to
positively regulate endothelial cell motility and angiogenesis
in vitro and in vivo (52). To elucidate the involvement of
PTPRK in the pro-angiogenic factors-induced angiogenesis and also
the tumour-associated angiogenesis, we treated the HECV cells with
VEGF, FGF and also the conditioned medium from breast cancer cell
lines. The PTPRK knockdown HECV cells were more responsive to the
FGF in their migration suggesting a key role played by PTPRK in
suppression of FGF-induced cell migration. In the tubule formation,
PTPRK knockdown did not suppress the VEGF-induced tubule formation
though it exhibited inhibition on the tubule formation of the
untreated cells. In contrast, PTPRK knockdown cells tended to be
less responsive to the FGF treatment. Moreover, the PTPRK knockdown
cells were less responsive in their tubule formation by an exposure
to the conditioned medium from breast cancer cells. It suggests
that PTPRK bears inhibitory effect on the tubule formation by
suppressing pathways triggered by FGF and cancer cells. Therefore,
PTPRK may play a positive role in coordinating cancer cell induced
angiogenesis. Further investigation of targeting soluble factors,
such as VEGF and FGF released from cancer cells using neutralizing
antibodies will help to expand the current understanding of cancer
cell-regulated angiogenesis which may help to develop a novel
anti-angiogenic strategy.
In conclusion, PTPRK knockdown exhibited diverse
effects on different cellular functions of vascular endothelial
cells; inhibitory effect on cell proliferation, adhesion and tubule
formation, but a positive effect on cell migration. A positive
correlation in the expression between PTPRK and focal adhesion
complex (FAK and paxillin) contributes to the cell adhesion.
Reduced PTPRK expression enhanced FGF-induced migration, but
elicited inhibitory effects on the tubule formation that was
promoted by FGF and cancer cells. PTPRK tends to be less involved
in the VEGF-induced tubule formation. It suggests that PTPRK plays
diverse roles in coordinating angiogenesis which can be more
specific to certain pro-angiogenic factors.
Acknowledgments
The authors would like to thank for the support from
the Cancer Research Wales and Ser Cymru Welsh Life Science Research
Network. The authors would also like to thank Dr Sioned Owen and Dr
Andrew Sanders for their kind help in proof reading.
References
|
1
|
Tseng JC, Chen HF and Wu KJ: A twist tale
of cancer metastasis and tumor angiogenesis. Histol Histopathol.
30:1283–1294. 2015.PubMed/NCBI
|
|
2
|
Wang Z, Dabrosin C, Yin X, Fuster MM,
Arreola A, Rathmell WK, Generali D, Nagaraju GP, El-Rayes B,
Ribatti D, et al: Broad targeting of angiogenesis for cancer
prevention and therapy. Semin Cancer Biol. 35(Suppl): S224–S243.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Semenza GL: Cancer-stromal cell
interactions mediated by hypoxia-inducible factors promote
angiogenesis, lymphangio-genesis, and metastasis. Oncogene.
32:4057–4063. 2013. View Article : Google Scholar
|
|
4
|
Gacche RN and Meshram RJ: Angiogenic
factors as potential drug target: Efficacy and limitations of
anti-angiogenic therapy. Biochim Biophys Acta. 1846:161–179.
2014.PubMed/NCBI
|
|
5
|
Khan KA and Bicknell R: Anti-angiogenic
alternatives to VEGF blockade. Clin Exp Metastasis. 33:197–210.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Waters JP, Pober JS and Bradley JR: Tumour
necrosis factor and cancer. J Pathol. 230:241–248. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yadav L, Puri N, Rastogi V, Satpute P and
Sharma V: Tumour angiogenesis and angiogenic inhibitors: A review.
J Clin Diagn Res. 9:XE01–XE05. 2015.PubMed/NCBI
|
|
8
|
Ferrara N, Gerber HP and LeCouter J: The
biology of VEGF and its receptors. Nat Med. 9:669–676. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Koch S, Tugues S, Li X, Gualandi L and
Claesson-Welsh L: Signal transduction by vascular endothelial
growth factor receptors. Biochem J. 437:169–183. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Olsson AK, Dimberg A, Kreuger J and
Claesson-Welsh L: VEGF receptor signalling - in control of vascular
function. Nat Rev Mol Cell Biol. 7:359–371. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Eklund L and Olsen BR: Tie receptors and
their angiopoietin ligands are context-dependent regulators of
vascular remodeling. Exp Cell Res. 312:630–641. 2006. View Article : Google Scholar
|
|
12
|
Nakazawa Y, Kawano S, Matsui J, Funahashi
Y, Tohyama O, Muto H, Nakagawa T and Matsushima T: Multitargeting
strategy using lenvatinib and golvatinib: Maximizing
anti-angiogenesis activity in a preclinical cancer model. Cancer
Sci. 106:201–207. 2015. View Article : Google Scholar :
|
|
13
|
Turner N and Grose R: Fibroblast growth
factor signalling: From development to cancer. Nat Rev Cancer.
10:116–129. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jiang WG, Martin TA, Parr C, Davies G,
Matsumoto K and Nakamura T: Hepatocyte growth factor, its receptor,
and their potential value in cancer therapies. Crit Rev Oncol
Hematol. 53:35–69. 2005. View Article : Google Scholar
|
|
15
|
Du Y and Grandis JR: Receptor-type protein
tyrosine phosphatases in cancer. Chin J Cancer. 34:61–69. 2015.
View Article : Google Scholar :
|
|
16
|
Nikolaienko RM, Agyekum B and Bouyain S:
Receptor protein tyrosine phosphatases and cancer: New insights
from structural biology. Cell Adhes Migr. 6:356–364. 2012.
View Article : Google Scholar
|
|
17
|
Tautz L, Critton DA and Grotegut S:
Protein tyrosine phosphatases: Structure, function, and implication
in human disease. Methods Mol Biol. 1053:179–221. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhao S, Sedwick D and Wang Z: Genetic
alterations of protein tyrosine phosphatases in human cancers.
Oncogene. 34:3885–3894. 2015. View Article : Google Scholar :
|
|
19
|
Spring K, Fournier P, Lapointe L, Chabot
C, Roussy J, Pommey S, Stagg J and Royal I: The protein tyrosine
phosphatase DEP-1/PTPRJ promotes breast cancer cell invasion and
metastasis. Oncogene. 34:5536–5547. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Goel S, Gupta N, Walcott BP, Snuderl M,
Kesler CT, Kirkpatrick ND, Heishi T, Huang Y, Martin JD, Ager E, et
al: Effects of vascular-endothelial protein tyrosine phosphatase
inhibition on breast cancer vasculature and metastatic progression.
J Natl Cancer Inst. 105:1188–1201. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chu LY, Ramakrishnan DP and Silverstein
RL: Thrombospondin-1 modulates VEGF signaling via CD36 by
recruiting SHP-1 to VEGFR2 complex in microvascular endothelial
cells. Blood. 122:1822–1832. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Agarwal S, Al-Keilani MS, Alqudah MA,
Sibenaller ZA, Ryken TC and Assem M: Tumor derived mutations of
protein tyrosine phosphatase receptor type k affect its function
and alter sensitivity to chemotherapeutics in glioma. PLoS One.
8:e628522013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nakamura M, Kishi M, Sakaki T, Hashimoto
H, Nakase H, Shimada K, Ishida E and Konishi N: Novel tumor
suppressor loci on 6q22-23 in primary central nervous system
lymphomas. Cancer Res. 63:737–741. 2003.PubMed/NCBI
|
|
24
|
Stanford SM, Aleman Muench GR, Bartok B,
Sacchetti C, Kiosses WB, Sharma J, Maestre MF, Bottini M, Mustelin
T, Boyle DL, et al: TGFbeta responsive tyrosine phosphatase
promotes rheumatoid synovial fibroblast invasiveness. Ann Rheum
Dis. 7:295–302. 2014.
|
|
25
|
Sun PH, Ye L, Mason MD and Jiang WG:
Receptor-like protein tyrosine phosphatase κ negatively regulates
the apoptosis of prostate cancer cells via the JNK pathway. Int J
Oncol. 43:1560–1568. 2013.PubMed/NCBI
|
|
26
|
Sun PH, Ye L, Mason MD and Jiang WG:
Protein tyrosine phosphatase kappa (PTPRK) is a negative regulator
of adhesion and invasion of breast cancer cells, and associates
with poor prognosis of breast cancer. J Cancer Res Clin Oncol.
139:1129–1139. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jiang WG, Watkins G, Fodstad O,
Douglas-Jones A, Mokbel K and Mansel RE: Differential expression of
the CCN family members Cyr61, CTGF and Nov in human breast cancer.
Endocr Relat Cancer. 11:781–791. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zuker M: Mfold web server for nucleic acid
folding and hybridization prediction. Nucleic Acids Res.
31:3406–3415. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jiang WG, Davies G, Martin TA, Parr C,
Watkins G, Mason MD, Mokbel K and Mansel RE: Targeting matrilysin
and its impact on tumor growth in vivo: The potential implications
in breast cancer therapy. Clin Cancer Res. 11:6012–6019. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jiang WG, Hiscox SE, Parr C, Martin TA,
Matsumoto K, Nakamura T and Mansel RE: Antagonistic effect of NK4,
a novel hepatocyte growth factor variant, on in vitro angiogenesis
of human vascular endothelial cells. Clin Cancer Res. 5:3695–3703.
1999.PubMed/NCBI
|
|
31
|
Jiang WG, Martin TA, Lewis-Russell JM,
Douglas-Jones A, Ye L and Mansel RE: Eplinalpha expression in human
breast cancer, the impact on cellular migration and clinical
outcome. Mol Cancer. 7:712008. View Article : Google Scholar
|
|
32
|
Raju R, Palapetta SM, Sandhya VK, Sahu A,
Alipoor A, Balakrishnan L, Advani J, George B, Kini KR, Geetha NP,
et al: A Network Map of FGF-1/FGFR Signaling System. J Signal
Transduct. 2014:9629622014.PubMed/NCBI
|
|
33
|
Chang YF, Hsu YF, Chiu PT, Huang WJ, Huang
SW, Ou G, Sheu JR and Hsu MJ: WMJ-S-001, a novel aliphatic
hydroxamate derivative, exhibits anti-angiogenic activities via
Src-homology-2-domain-containing protein tyrosine phosphatase 1.
Oncotarget. 6:85–100. 2015.
|
|
34
|
Mellberg S, Dimberg A, Bahram F, Hayashi
M, Rennel E, Ameur A, Westholm JO, Larsson E, Lindahl P, Cross MJ,
et al: Transcriptional profiling reveals a critical role for
tyrosine phosphatase VE-PTP in regulation of VEGFR2 activity and
endothelial cell morphogenesis. FASEB J. 23:1490–1502. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hayashi M, Majumdar A, Li X, Adler J, Sun
Z, Vertuani S, Hellberg C, Mellberg S, Koch S, Dimberg A, et al:
VE-PTP regulates VEGFR2 activity in stalk cells to establish
endothelial cell polarity and lumen formation. Nat Commun.
4:16722013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fachinger G, Deutsch U and Risau W:
Functional interaction of vascular endothelial-protein-tyrosine
phosphatase with the angiopoietin receptor Tie-2. Oncogene.
18:5948–5953. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shen J, Frye M, Lee BL, Reinardy JL,
McClung JM, Ding K, Kojima M, Xia H, Seidel C, Lima e Silva R, et
al: Targeting VE-PTP activates TIE2 and stabilizes the ocular
vasculature. J Clin Invest. 124:4564–4576. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Winderlich M, Keller L, Cagna G, Broermann
A, Kamenyeva O, Kiefer F, Deutsch U, Nottebaum AF and Vestweber D:
VE-PTP controls blood vessel development by balancing Tie-2
activity. J Cell Biol. 185:657–671. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bäumer S, Keller L, Holtmann A, Funke R,
August B, Gamp A, Wolburg H, Wolburg-Buchholz K, Deutsch U and
Vestweber D: Vascular endothelial cell-specific phosphotyrosine
phosphatase (VE-PTP) activity is required for blood vessel
development. Blood. 107:4754–4762. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sirois J, Côté JF, Charest A, Uetani N,
Bourdeau A, Duncan SA, Daniels E and Tremblay ML: Essential
function of PTP-PEST during mouse embryonic vascularization,
mesenchyme formation, neurogenesis and early liver development.
Mech Dev. 123:869–880. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kellie S, Craggs G, Bird IN and Jones GE:
The tyrosine phosphatase DEP-1 induces cytoskeletal rearrangements,
aberrant cell-substratum interactions and a reduction in cell
proliferation. J Cell Sci. 117:609–618. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kaur H, Burden-Gulley SM, Phillips-Mason
PJ, Basilion JP, Sloan AE and Brady-Kalnay SM: Protein tyrosine
phosphatase mu regulates glioblastoma cell growth and survival in
vivo. Neuro Oncol. 14:561–573. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zou L, Cao S, Kang N, Huebert RC and Shah
VH: Fibronectin induces endothelial cell migration through β1
integrin and Src-dependent phosphorylation of fibroblast growth
factor receptor-1 at tyrosines 653/654 and 766. J Biol Chem.
287:7190–7202. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Knuchel S, Anderle P, Werfelli P,
Diamantis E and Rüegg C: Fibroblast surface-associated FGF-2
promotes contact-dependent colorectal cancer cell migration and
invasion through FGFR-SRC signaling and integrin αvβ5-mediated
adhesion. Oncotarget. 6:14300–14317. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Alexiades NG, Auffinger B, Kim CK, Hasan
T, Lee G, Deheeger M, Tobias AL, Kim J, Balyasnikova I, Lesniak MS,
et al: MMP14 as a novel downstream target of VEGFR2 in migratory
gliomatropic neural stem cells. Stem Cell Res (Amst). 15:598–607.
2015. View Article : Google Scholar
|
|
46
|
Dominguez MG, Hughes VC, Pan L, Simmons M,
Daly C, Anderson K, Noguera-Troise I, Murphy AJ, Valenzuela DM,
Davis S, et al: Vascular endothelial tyrosine phosphatase
(VE-PTP)-null mice undergo vasculogenesis but die embryonically
because of defects in angiogenesis. Proc Natl Acad Sci USA.
104:3243–3248. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Gao Q, Zhao YJ, Wang XY, Guo WJ, Gao S,
Wei L, Shi JY, Shi GM, Wang ZC, Zhang YN, et al: Activating
mutations in PTPN3 promote cholangiocarcinoma cell proliferation
and migration and are associated with tumor recurrence in patients.
Gastroenterology. 146:1397–1407. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sun PH, Ye L, Mason MD and Jiang WG:
Protein tyrosine phosphatase μ (PTP μ or PTPRM), a negative
regulator of proliferation and invasion of breast cancer cells, is
associated with disease prognosis. PLoS One. 7:e501832012.
View Article : Google Scholar
|
|
49
|
Choi HJ, Armaiz Pena GN, Pradeep S, Cho
MS, Coleman RL and Sood AK: Anti-vascular therapies in ovarian
cancer: Moving beyond anti-VEGF approaches. Cancer Metastasis Rev.
34:19–40. 2015. View Article : Google Scholar :
|
|
50
|
Zhao Y and Adjei AA: Targeting
angiogenesis in cancer therapy: Moving beyond vascular endothelial
growth factor. Oncologist. 20:660–673. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Besnier M, Galaup A, Nicol L, Henry JP,
Coquerel D, Gueret A, Mulder P, Brakenhielm E, Thuillez C, Germain
S, et al: Enhanced angiogenesis and increased cardiac perfusion
after myocardial infarction in protein tyrosine phosphatase
1B-deficient mice. FASEB J. 28:3351–3361. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Mannell H and Krotz F: SHP-2 regulates
growth factor dependent vascular signalling and function. Mini Rev
Med Chem. 14:471–483. 2014. View Article : Google Scholar
|