Introduction
Nasopharyngeal carcinoma (NPC) is a relatively rare
malignancy worldwide, yet it is the most common cancer in some
endemic areas, including north Africa, south-eastern Asia and a
number of provinces in south-eastern China (1). Despite great progress in the systemic
treatment, including primarily radiotherapy, chemotherapy or
chemotherapy in combination with radiotherapy due to its intricate
anatomical location, against stage I and II NPC over the past
years, the 5-year survival rates of more than half of the patients
with advanced NPC is still <50% (2). Extensive studies have indicated that
uncontrolled proliferation ability of cancer cells is the most
common momentum responsible for the progression of NPC, where
multiple cell cycle-related genes have been implicated in the
proliferation of tumor cells (3,4).
Therefore, better understanding of the molecular mechanisms
contributing to the uncontrolled proliferation of cancer cells will
facilitate to improve the survival rate of NPC patients.
Normal cell growth and metabolism are under precise
control of cell cycle in which protein kinase complexes composed of
cyclins and cyclin-dependent kinase (CDK) play an important role
and determine a cell's progression in a sequential fashion
(5). From the molecular
perspective, cyclins act as the regulatory subunits of an activated
heterodimer and CDKs functions as the catalytic subunits, which
orchestrates coordinated entry into the S phase of the cell cycle
(5). Different cyclin-CDK
combinations specifically determine the downstream targeted
proteins that in turn promote the expression of cyclins and enzymes
required for DNA replication (6).
Previous studies showed that disregulation of the cell cycle
components may cause the tumor cell to multiply uncontrollably,
which finally leads to tumor formation (7). Importantly, therapies targeting CDK
inhibitor present a favorable prospect in the treatment of a
variety of cancers. O'Leary et al reported that phase III
trials investigating palbociclib in patients with advanced-stage
estrogen receptor-positive breast cancer have demonstrated a
substantial improvement in progression-free survival, with a
well-tolerated toxicity profile (8); furthermore, Kumar and colleagues
demonstrated a single agent activity of dinaciclib, a novel potent
small non-selective inhibitor of CDK1, CDK2, CDK5 and CDK9, in
relapsed myeloma (9). These
studies indicated that therapy targeting cell cycle related
proteins exhibits broad-ranging efficacy in many cancer and could
serve as a primary therapeutic avenue in the treatment of
cancers.
MicroRNAs (miRNAs) are a diverse group of small
non-coding RNAs composed of 19–25 nucleotides and mechanistically
function by binding to the 3′-untranslated region of downstream
mRNAs, leading to mRNA degradation or repression of translation
(10,11). A growing body of evidence has
demonstrated that miRNAs not only play crucial roles in many
biological processes including proliferation, differentiation, cell
cycle and apoptosis (11), but
also regulate the progression and metastasis in various types of
tumors (12–18). In recent studies, abnormal
expression of miRNAs has been reported to be broadly implicated in
the pathogenesis of NPC (19,20).
Furthermore, several lines of evidence indicated that miRNAs have
been reported to be critical regulators of cell proliferation in
multiple human cancers, including NPC (21–23).
For example, Zhao et al reported that miR-3188 regulates
nasopharyngeal carcinoma proliferation through a FOXO1-modulated
positive feedback loop with mTOR-p-PI3K/AKT-c-JUN (24); moreover, a study from He and
colleagues showed that miR-16 targeting fibroblast growth factor 2
inhibited NPC cell proliferation through PI3K/AKT and MAPK
signaling pathways (25). Notably,
numerous studies indicated that miRNAs regulated cancer cell
proliferation via directly targeting single or several cell
cycle-related genes, including cyclin D, cyclin E and
cyclin-dependent kinase (CDK), which promoted the unlimited
proliferation of cancer cells (26,27).
Therefore, the above results imply that dysregulation of miRNAs
promote the NPC cells proliferation, which contributes to the
progression and recurrence of NPC.
In this study, we found that miR-150 expression is
markedly decreased in NPC tissues and cells. Moreover, upregulation
of miR-150 suppresses, while silencing miR-150 promotes
nasopharyngeal carcinoma cell proliferation and cell cycle in
vitro, as well as tumorigenesis in vivo. Our results
further demonstrated that CCND1, CCND2, CDK2 and CCNE2 are the
direct targets of miR-150; moreover, they mediate the regulation of
miR-150 in NPC cells proliferation and cell cycle. Therefore, our
results demonstrate that miR-150 inhibits nasopharyngeal carcinoma
cells proliferation and cell cycle by directly targeting CCND1,
CCND2, CDK2 and CCNE2 and indicate that miR-150 play a
tumor-suppressive role by suppressing proliferation and cell cycle
in nasopharyngeal carcinoma.
Materials and methods
Cell lines and cell culture
The human nasopharyngeal carcinoma cell lines CNE1,
CNE2, C666-1, HNE1, HNE2, HONE1, SUNE1 and 5–8F were obtained from
Department of Biochemistry and Molecular Biology, Guangdong Medical
College (Zhanjiang, China) and were cultured in RPMI-1640 medium
(Life Technologies, Carlsbad, CA, USA) supplemented with penicillin
G (100 U/ml), streptomycin (100 mg/ml) and 10% fetal bovine serum
(FBS, Life Technologies). NP69 is an immortalized nasopharyngeal
epithelium cell line which was cultured in defined keratinocyte
serum-free medium supplemented with specific growth factors (Life
Technologies). 293T cells were cultured in Dulbecco's modified
Eagle's medium (DMEM, Life Technologies). All cells were incubated
at 37°C in a humidified atmosphere with 5% CO2 and were
routinely subcultured using 0.25% (w/v)
trypsin-ethylenedi-aminetetraacetic acid solution.
Patients and tumor tissues
Paired tumor specimens and the matched adjacent
normal tissues from 8 confirmed NPC patients were obtained from the
Affiliated Hospital of Guangdong Medical College between January
2014 and December 2014. Tissue specimens were obtained by fiber
optic nasopharyngoscopy directly on the tumor growth site and on
the adjacent side with observed normal mucosal morphology for the
corresponding non-tumor pair. Informed consent was obtained from
the patients involved, and ethics approval for this study was
obtained from the Institutional Research Ethics Committee. The
median age was 48 years and the age range from 32 to 72 years. The
histologic subtype of 8 NPC includes 7 undifferentiated
non-keratinizing and 1 differentiated non-keratinizing. The EBV
status of all 8 NPC patients were positive. The patient information
is summarized in Table I.
 | Table IThe basic information of 8 NPC
patients for miR-150 expression analysis. |
Table I
The basic information of 8 NPC
patients for miR-150 expression analysis.
| Cases (n) | Percentage (%) |
|---|
| Gender | | |
| Male | 5 |
62.5 |
| Female | 3 |
37.5 |
| Age | | |
| <50 | 5 |
62.5 |
| ≥50 | 3 |
37.5 |
| Histologic
subtype | | |
| Undifferentiated
non-keratinizing | 7 |
87.5 |
| Differentiated
non-keratinizing | 1 |
12.5 |
| EBV status | | |
| Positive | 8 | 100.0 |
| Negative | 0 |
0.0 |
RNA extraction, reverse transcription,
and real-time PCR
Total RNA from tissues or cells were extracted using
TRIzol (Life Technologies) according to the manufacturer's
instructions. Messenger RNA (mRNA) and miRNA were polyadenylated
using a poly-A polymerase-based First-Strand Synthesis kit (Takara,
Dalian, China) and reverse transcription (RT) of total mRNA was
performed using a PrimeScript RT Reagent kit (Takara) according to
the manufacturer's protocol. Complementary DNA (cDNA) was amplified
and quantified on ABI 7500HT system (Applied Biosystems, Foster
City, CA, USA) using SYBR Green I (Applied Biosystems). Table II lists the primers used in the
reactions. Real-time PCR was performed according to a standard
method, as described previously (28). Primers for U6 (no. MQP-0202) and
miR-150 (no. miRQ0000451) were synthesized and purified by RiboBio
(Guangzhou, China). U6 or glyceraldehyde-3-phosphate dehydrogenase
(GAPDH) was used as endogenous controls for miRNA or mRNA,
respective. Relative fold expressions were calculated with the
comparative threshold cycle (2−∆∆Ct) method.
| Table IIThe primers used in the reactions for
real-time RT-PCR. |
Table II
The primers used in the reactions for
real-time RT-PCR.
| Gene | Sequence
(5′-3′) |
|---|
| CCND1-up |
GCCCTCGGTGTCCTACTTC |
| CCND1-dn |
CTCCTCCTCGCACTTCTGTT |
| CCND2-up |
GGTCGGGTTTTCAATCACAC |
| CCND2-dn |
CCTCTTCACCTCCCTTCAACT |
| CCND3-up |
TCCTCTCCCATTGTCCCTCT |
| CCND3-dn |
CCACCAGCCTAAACCTTGC |
| CDK4-up |
GTCTGGTTGTTTGTTGTGTGC |
| CDK4-dn |
CGGTGTGTTTGTTGTTTGTCC |
| CDK6-up |
GTCTGGTTGTTTGTTGTGTGC |
| CDK6-dn |
CGGTGTGTTTGTTGTTTGTCC |
| CCNE1-up |
CGGTATATGGCGACACAAGA |
| CCNE1-dn |
ACATACGCAAACTGGTGCAA |
| CCNE2-up |
AGGAAAACTACCCAGGATGTCA |
| CCNE2-dn |
ATCAGGCAAAGGTGAAGGATTA |
| CDK2-up |
CTGCTTCCTGTTGGCTCTTTCT |
| CDK2-dn |
CTTTGTTTCTGCCTTCTCTCCT |
| GAPDH-up |
GCACCGTCAAGGCTGAGAAC |
| GAPDH-dn |
TGGTGAAGACGCCAGTGGA |
Plasmid, small interfering RNA and
transfection
The human miR-150 expression plasmid was generated
by cloning the genomic pre-miR-150 gene, with a 300-bp sequence on
each flanking side, into retroviral transfer plasmid pMSCV-puro
(Clontech Laboratories Inc., Tokyo, Japan) to generate plasmid
pMSCV-miR-150. pMSCV-miR-150 was cotransfected with the pIK
packaging plasmid into 293FT cells using the standard calcium
phosphate transfection method, as previously described (29). Thirty-six hours after the
cotransfection, supernatants were collected and incubated with
cells to be infected for 24 h in the presence of polybrene (2.5
µg/ml). After infection, puro-mycin (1.5 µg/ml) was
used to select stably transduced cells over a 10-day period. The
luciferase reporter system of pE2F-luc (Clontech) and pRb-luc
(Clontech) were used to examine the transcriptional activity of
E2Fs and the transcriptional repression capability of Rb,
respectively. The 3′-untranslated region (3′-UTR) regions of the
human CCND1, CCND2, CDK2 and CCNE2 were PCR-amplified from genomic
DNA and cloned into pmirGLO luciferase reporter vector (Promega,
Madison, WI, USA), and the list of primers used in clone reactions
is presented in Table III. The
miArrest plasmids for anti-miR150 and negative control plasmids
were constructed and cloned into pH1 plasmids by GeneChem
(Shanghai, China). Small interfering RNA (siRNA) for CCND1, CCND2,
CDK2 and CCNE2 knockdown were obtained from Santa Cruz (Dallas, TX,
USA). Transfection of siRNAs and plasmids was performed using
Lipofectamine 3000 (Life Technologies) according to the
manufacturer's instructions.
| Table IIIThe primers used in the reactions for
clone PCR. |
Table III
The primers used in the reactions for
clone PCR.
| Gene | Sequence
(5′-3′) |
|---|
| miR-150-up |
TAGGCGCCGGAATTACTCCCCTGGAGCCTGTTCA |
| miR-150-dn |
CTACCCGGTAGAATTGAGACGCCCCAACAATCAG |
| CCND1-3UTR-up |
TAGTTGTTTAAACGAGCATTTTGATACCAGAAGGGAAA |
| CCND1-3UTR-dn |
AGGTCGACTCTAGACTTGTCTTTTTGTCTTCTGCTGGA |
| CCND2-3UTR-up |
TAGTTGTTTAAACGAGTTCTGTGACATCCTGCTTCTT |
| CCND2-3UTR-dn |
AGGTCGACTCTAGACTACTTATCAGCACTTTCTAACATCC |
| CDK2-3UTR-up |
TAGTTGTTTAAACGAGCCCTAATCTCACCCTCTCCT |
| CDK2-3UTR-dn |
AGGTCGACTCTAGACCGTTAATAGCAAGAGCACTCAAGG |
| CCNE2-3UTR-up |
TAGTTGTTTAAACGAAATTCACCAAGATTGGGTAGAAC |
| CCNE2-3UTR-dn |
AGGTCGACTCTAGACAACAATGGGCTAAAAATAAACAGTA |
Cell counting kit-8 analysis and colony
formation assay
For cell counting kit-8 analysis, cells
(2×103) were seeded into 96-well plates and stained at
the indicated time-point with 100 µl cell counting kit-8
(CCK-8; Dojindo, Japan) dye for 2 h at 37°C, followed by the
absorbance measured at 450 nm, with 650 nm used as the reference
wavelength. For colony formation assay, cells (0.2×103)
were plated into 6-well plates and cultured for 10 days. Colonies
were then fixed for 15 min with 10% formaldehyde and stained with
1.0% crystal violet for 30 sec.
Anchorage-independent growth ability
assay
Cells (3×103) were suspended in 2 ml
complete medium plus 0.3% agar (Sigma-Aldrich, St. Louis, MO, USA).
The agar-cell mixture was plated as a top layer onto a bottom layer
comprising 0.6% complete medium agar mixture. After 14-day culture,
colony size was measured using an ocular micrometer and colonies
>0.1 mm in diameter were counted.
Cell cycle analysis
Pretreatment and staining was performed using Cell
Cycle Detection kit (KeyGen, China) according to the manufacturer's
instructions. Cells (5×105) were harvested by
trypsinization, washed in ice-cold phosphate-buffered saline (PBS)
and fixed in 75% ice-cold ethanol in PBS. Before staining, cells
were gently resuspended in cold PBS, and ribo-nuclease was added
into cell suspension tube incubated at 37°C for 30 min, followed by
incubation with propidium iodide (PI) for 20 min at room
temperature. Cell samples (2×104) were then analyzed by
FACSCanto II flow cytometer (Becton-Dickinson & Co., Franklin
Lakes, NJ, USA) and the data were analyzed using FlowJo 7.6
software (TreeStar Inc., Ashland, OR, USA).
Tumor xenografts
Four-week-old BALB/c-nu female mice weighing 15–20 g
were maintained in a standard pathogen-free environment where the
animals were housed in sterile cages under laminar flow hoods in a
20–26°C temperature controlled room with a 12-h light/dark cycle
and fed autoclaved chow and water. All experimental procedures were
approved by the Institutional Animal Care and Use Committee (IACUC)
of Guangdong Medical College. BALB/c-nu mice at 4–6 weeks of age
were randomly divided into four groups (n=5 per group) and
indicated CNE-2 cells (2×106) were inoculated
subcutaneously into the flanks of the nude mice. Tumor volume was
determined using an external caliper and calculated using the
equation (L x W2)/2. The mice were sacrificed by
inhaling CO2 on day 36 after inoculation and the tumors
were excised and subjected to pathologic examination.
Immunohistochemistry
The immunohistochemistry procedure and scoring of
Ki67 expression levels were performed as previously described
(12). The slides were incubated
overnight at 4°C in a humidified chamber with the rabbit anti-human
Ki67 antibody diluted 1:5,000 in PBS (no. ab16667. Abcam,
Cambridge, MA, USA).
Dual luciferase report experiments
Cells (5×105) were plated in 60-mm cell
culture dishes, at 60–80% confluence after 24 h of culture, and the
reporter constructs were transfected into cells using Lipofectamine
3000. After 12-h incubation, the transfection medium was replaced;
cells were harvested and washed with PBS, and lysed with passive
lysis buffer (Promega). The cell lysates were analyzed immediately
using Synergy™ 2 microplate system (BioTek, Winooski, VT, USA).
Luciferase and Renilla luciferase were measured using a
Dual-Luciferase Reporter assay system (Promega) according to the
manufacturer's instructions. The luciferase activity of each lysate
was normalized to Renilla luciferase activity. The relative
transcriptional activity was converted into fold induction above
the vehicle control value.
RNA immunoprecipitation
Cells (5×105) were plated in 60-mm cell
culture dishes, at 60–80% confluence after 24 h of culture, and the
pIRESneo-FLAG/HA-Ago2 plasma (10822; Addgene, Cambridge, MA, USA)
was cotransfected into cells using Lipofectamine 3000. After 48-h
transfection, cells were washed and lysed in
radioimmunoprecipitation buffer (Sigma-Aldrich) containing 10%
proteinase inhibitor cocktail (Sigma-Aldrich) and 1 mM
phenylmethylsulfonyl fluoride (Sigma-Aldrich). A fraction of the
whole cell lysate was used for RNA isolation, and the remaining
lysate was subjected to immunoprecipitation (IP) using an antibody
against Ago2 (Abcam) or immunoglobulin G (IgG) (Abcam). RNA from
whole cell lysates and RNA IP (RIP) fractions was extracted with
TRIzol (Life Technologies) according to the manufacturer's
instructions. The relative levels of mRNA were determined using
real-time RT-PCR as described above. The relative mRNA enrichment
in the RIP fractions was computed based on the ratio of relative
mRNA levels in the RIP fractions and the relative mRNA levels in
the whole cell lysates.
Western blotting
The proteins extracted from the cell lysates were
loaded with 50 µg in each lane, which was further separated
by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis
and transferred to polyvinylidene fluoride membranes (Millipore,
Billerica, MA, USA). The membranes were probed with antibodies
against CCND1 (no. 3300; dilution, 1:1,000), CCND2 (no. 3741;
dilution, 1:1,000), CDK2 (no. 2546; dilution, 1:1,000), and CCNE2
(no. 4132; dilution: 1:1,000) (Cell Signaling Technology, Beverly,
MA, USA) overnight at 4°C, and then incubated with horseradish
peroxidase-conjugated secondary antibodies (no. 7071; dilution,
1:3,000) (Cell Signaling Technology) for 1 h at room temperature.
Immune complexes were detected by enhanced chemiluminescence (Cell
Signaling Technology). α-tubulin (no. 12351; dilution, 1:1,000)
(Cell Signaling Technology) was used to correct for differences in
protein loading from the control and experimental groups.
Statistical analysis
All values are presented as means ± standard
deviation (SD). Significant differences were determined using SPSS
19.0 software (SPSS, Chicago, IL, USA). A paired Student's t-test
was used to analyze the paired control group (pMSCV-V or pH1-V) and
treatment group (miR-150 or anti-miR-150) of in vitro
experiments. An independent Student's t-test was used to analyze
the paired control group (pMSCV-V or pH1-V) and treatment group
(miR-150 or anti-miR-150) of in vivo experiments. Spearman's
correlation tests were used to evaluate the pairwise expression
correlation between miR-150 and targeted genes in NPC tissues.
P<0.05 was considered statistically significant.
Results
miR-150 is downregulated in
nasopharyngeal carcinoma tissues and cell lines
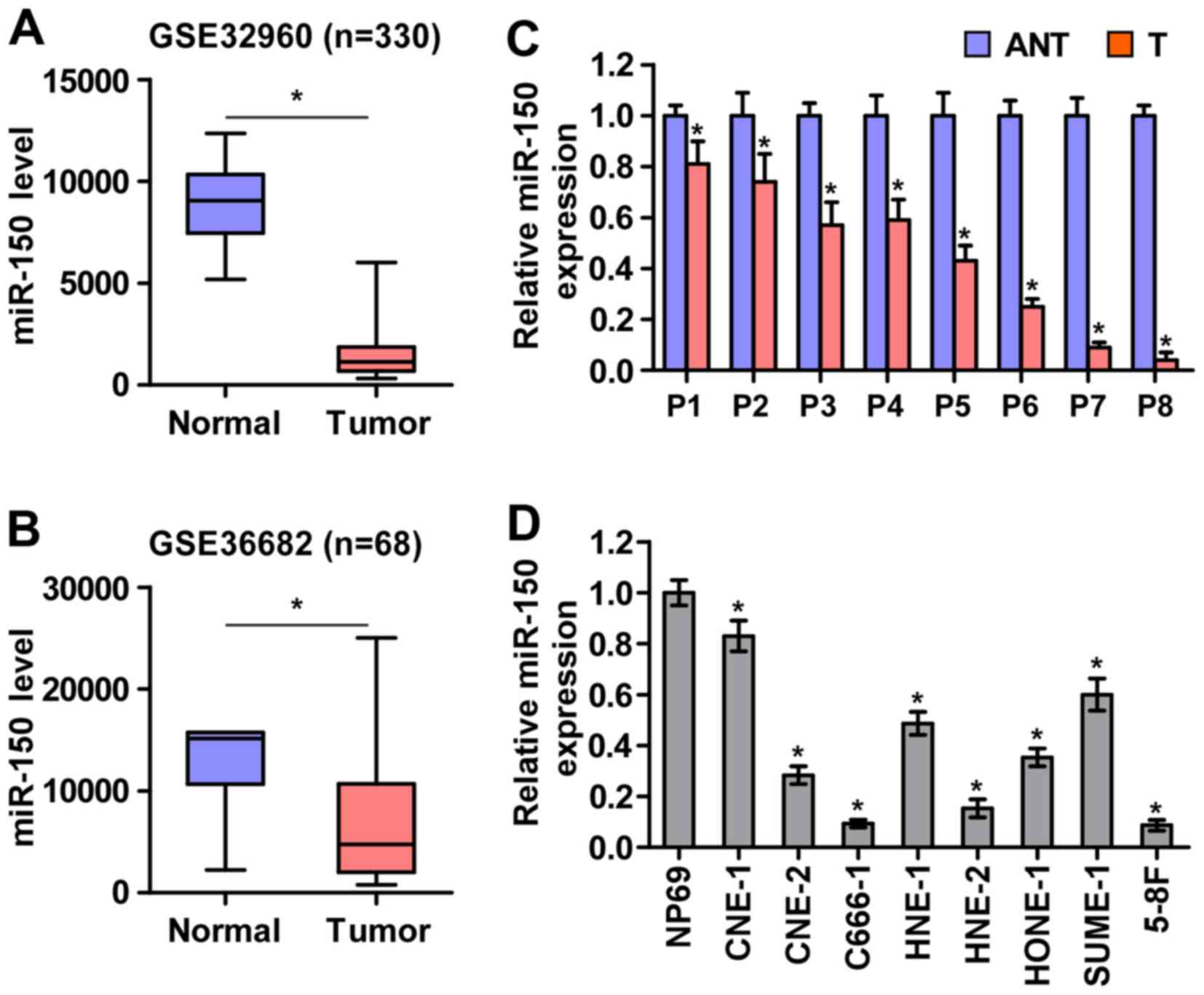
To screen the aberrant miRNA expression between NPC
tissues and normal nasopharyngeal tissues, two microarray-based
high-throughput datasets of NPC from GSE32960 (ftp://ftp.ncbi.nlm.nih.gov/geo/series/GSE32nnn/GSE32960/matrix/)
and GSE36682 (ftp://ftp.ncbi.nlm.nih.gov/geo/series/GSE36nnn/GSE36682/matrix/)
were analyzed and showed that miR-150 expression was downregulated
in NPC tissues compared with normal nasopharyngeal tissues
(Fig. 1A and B). To validate the
miR-150 expression in NPC tissues, real-time PCR was performed on
NPC clinical samples and cell lines. As shown in Fig. 1C and D, miR-150 expression was
differentially downregulated in the primary NPC tissues from 8
individual patients and NPC cell lines compared with that in the
matched adjacent normal tissues and immortalized nasopharyngeal
epithelium cell line (NP69), respectively. Therefore, the published
miRNA datasets and our results suggested that miR-150 is
downregulated in NPC tissues and cells.
Downregulation of miR-150 promotes NPC
cell proliferation in vitro
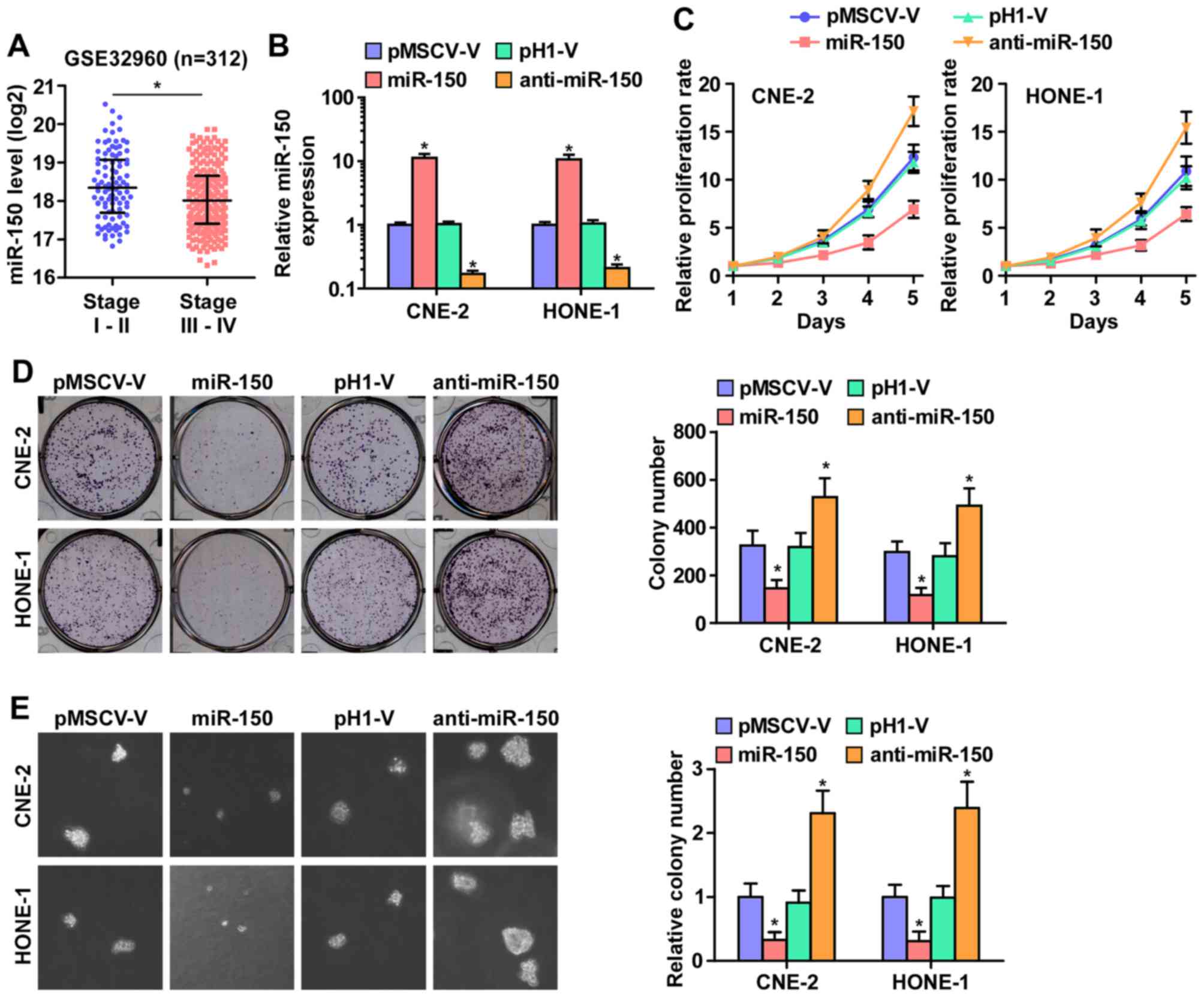
As downregulation of miR-150 was correlated with
clinical stage in NPC clinical tissues (Fig. 2A), we examined whether miR-150 is
involved in NPC cell proliferation. We first constructed
miR-150-expressing stably HONE1 and CNE2 cells via exogenously
overexpressing miR-150 and endogeneously silencing miR-150 via
virus transduction (Fig. 2B). The
reason why HONE1 and CNE2 cells were selected to study the
biological roles of miR-150 in NPC is that miR-150 expression
levels in CNE-2 and HONE1 cells were located in the intermediate
level in the miR-150 expression spectrum of all NPC cell lines when
we examined the miR-150 expression levels in 8 NPC cell lines.
Furthermore, CNE-2 and HONE1 cell lines were the most common cell
models in studying the proliferation and tumorigenesis of NPC in
vitro and in vivo assays (30,31).
CCK-8 assay was performed and the result indicated that miR-150
upregulation markedly decreased, while silencing miR-150 increased,
the cell proliferation rate of CNE2 and HONE1 cells (Fig. 2C). Furthermore, colony formation
assays and anchorage-independent growth assays revealed that
miR-150 downregulation increased the colony-generating capability
and anchorage-independent growth activity of CNE2 and HONE1 cells
(Fig. 2D and E). Moreover,
upregulated miR-150 drastically inhibited NPC cell growth, as
indicated by the decreased colony numbers (Fig. 2D and E). These results indicated
that miR-150 inhibits NPC cell proliferation ability in
vitro.
miR-150 inhibits nasopharyngeal carcinoma
cell tumorigenesis in vivo
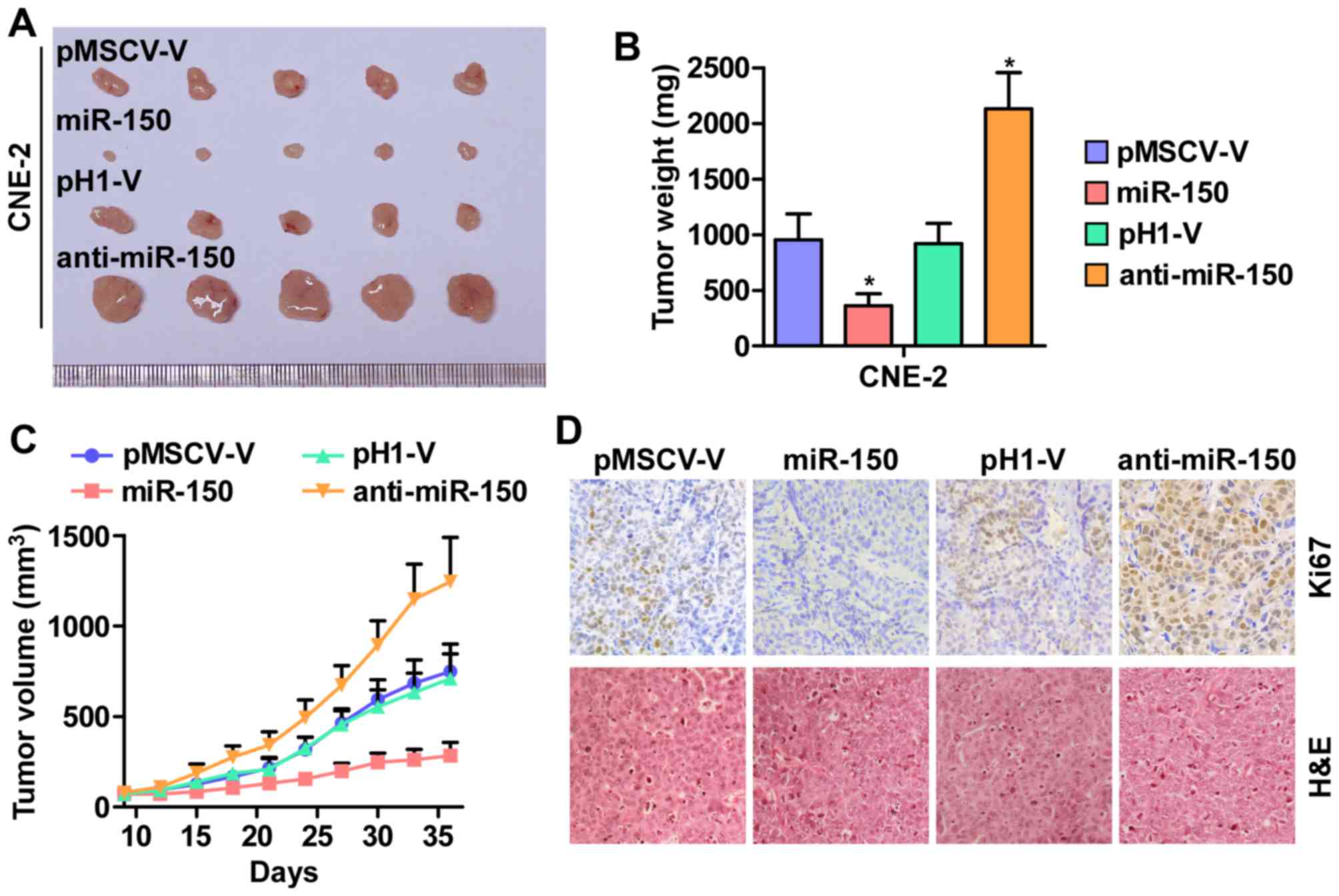
Furthermore, the effect of miR-150 on tumorigenesis
of human nasopharyngeal carcinoma CNE-2 cells was tested in
vivo. As shown in Fig. 3A–C,
tumor volumes and weight were increased significantly in the
miR-150-downregulation group compared with the control group. In
contrast, the tumors formed by the miR-150-overexpressing cells
were smaller and exhibited reduced tumor volume and weight compared
to the control group. The analysis of IHC and H&E staining
revealed that the miR-150-silencing tumor tissues displayed higher
Ki67 proliferation indexes, whereas the miR-150-overexpressing
tumor tissues exhibited reduced numbers of Ki67-positive cells
(Fig. 3D). Taken together, these
results indicated that miR-150 inhibits the tumorigenesis of NPC
cells in vivo.
miR-150 downregulation promotes cell
cycle progression of NPC cells
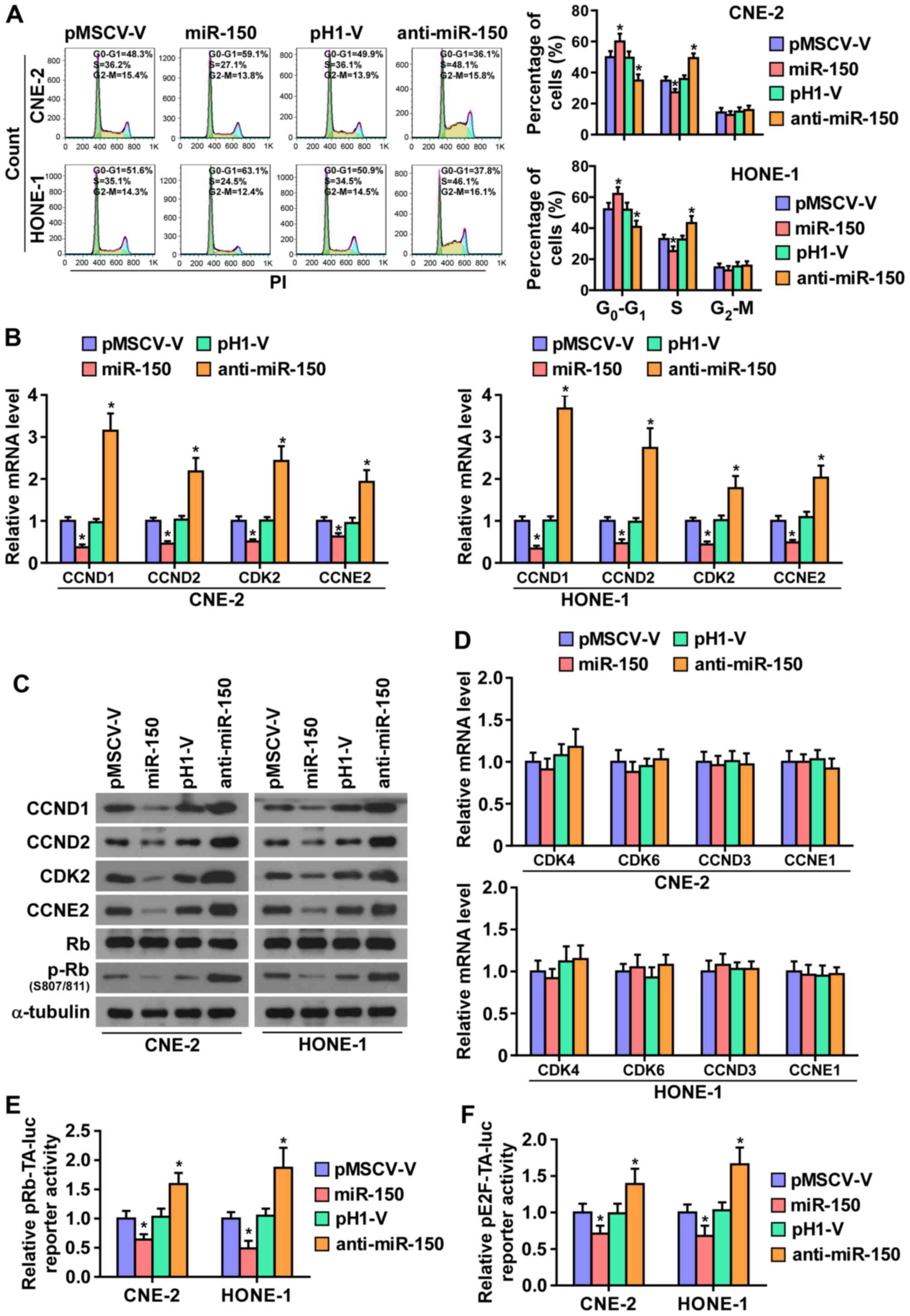
To further investigate the mechanism underlying the
miR-150-mediated inhibition of NPC cell proliferation, the cell
cycle progression was analyzed. As shown in Fig. 4A, flow cytometry showed that
miR-150 upregulation markedly decreased the percentage of cells in
the S phase and increased that of cells in the
G1/G0 phase, while silencing miR-150
increased the percentage of cells in the S phase and decreased that
of cells in the G1/G0 phase, suggesting that
upregulation miR-150 might induce G1/S arrest in NPC
cells. Furthermore, the expression levels of a number of critical
cell cycle regulators in G1/S checkpoint were detected.
As shown in Fig. 4B and C,
real-time PCR assays and western blotting revealed that
upregulating miR-150 decreased, while silencing miR-150 increased,
the expression levels of cyclin D1 (CCND1), cyclin D2 (CCND2),
cyclin-dependent kinase 2 (CDK2), and cyclin E2 (CCNE2) at both the
protein and mRNA levels. Whereas, the mRNA levels of
cyclin-dependent kinase 4 (CDK4), cyclin-dependent kinase 6 (CDK6),
cyclin D3 (CCND3), and cyclin E1 (CCNE1) were not affected by
miR-150 (Fig. 4D).
It has been well documented that progression of cell
cycle was promoted by E2F transcription factors. E2F can work with
Rb, a negative regulator of the cell cycle, in regulating
G1/S checkpoint. E2F activation or Rb inactivation can
induce entry of cells into S phase (32,33).
Thus, we hypothesized that miR-150 upregulation may suppress the
activity E2F-Rb complexes. As shown in Fig. 4C, the level of phosphorylation of
Rb (p-Rb) was markedly decreased in miR-150-overexpressing NPC
cells, while its expression level is increased in the
miR-150-silencing NPC cells. Moreover, the luciferase reporter
analysis shows that upregulating miR-150 enhanced, while
miR-150-silenced inhibited, the transcriptional repression
capability of Rb (Fig. 4E).
Moreover, the transcriptional activity of E2F was strongly
repressed by miR-150 overexpression, whereas silencing miR-150
increased the activity (Fig. 4F).
These results suggested that miR-150 inhibits the cell cycle
progression in NPC cells.
miR-150 suppresses cell cycle progression
by directly targeting CCND1, CCND2, CDK2 and CCNE2
It has been confirmed that miRNAs regulate gene
expression by targeting the 3′-UTR of mRNA, and the degradation of
target mRNA was mediated by RNA-induced silencing complex (RISC).
Therefore, we tested whether miR-150 mediated the RISC binding to
the mRNA of critical cell cycle regulators in G1/S
checkpoint using miRNP immunoprecipitation assay. As shown in
Fig. 5A and B, miR-150 directly
interacted with the mRNA of CCND1, CCND2, CDK2 and CCNE2, but did
not affect the mRNA of CDK4, CDK6, CCND3 and CCNE1. Furthermore,
publicly available algorithms (miRanda and TargetScan) were
employed and showed that CCND1, CCND2, CDK2 and CCNE2 are potential
targets of miR-150 (Fig. 5C). To
examine whether miR-150-mediated CCND1, CCND2, CDK2 and CCNE2
downregulation occurs through a miR-150-binding in the 3′-UTR of
CCND1, CCND2, CDK2 and CCNE2, the 3′-UTR of four genes were cloned
into pmirGLO luciferase reporter vectors. As predicted, miR-150
overexpression reduced, whereas anti-miR-150 increased, the
luciferase reporter activity of these four genes (Fig. 5D). Moreover, flow cytometry showed
that downregulated CCND1, CCND2, CDK2 and CCNE2 significantly
increased the percentage of NPC cells in G0 phase, while
the percentage of cells in S phase was decreased in
miR-150-silenced CNE2 and HONE1 cells (Fig. 5E). Taken together, these results
indicated that miR-150 retards the cell cycle progression via
directly targeting CCND1, CCND2, CDK2 and CCNE2, causing
G1/S phase arrest.
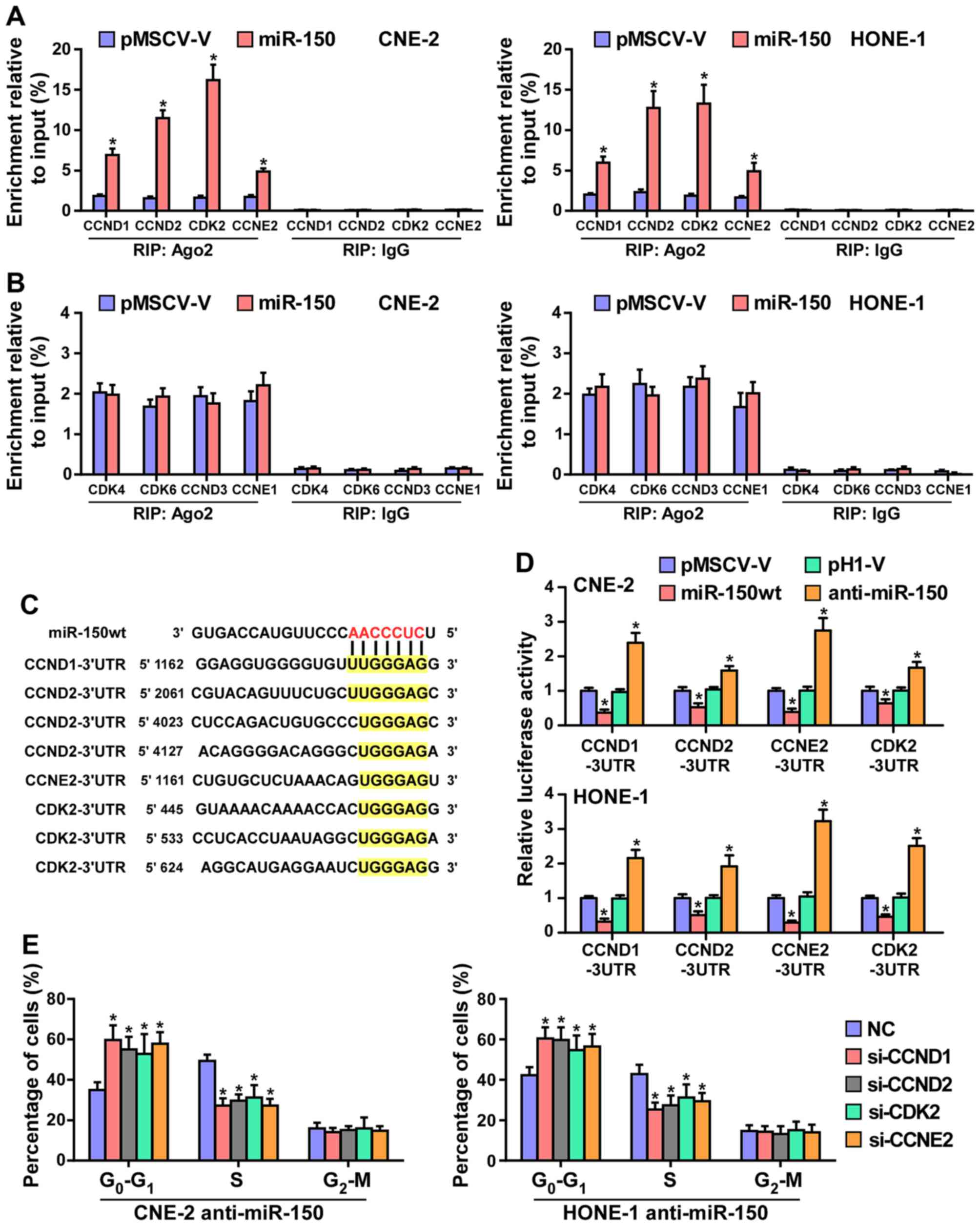 | Figure 5miR-150 inhibits the cell cycle
progression by directly targeting CCND1, CCND2, CDK2 and CCNE2. (A)
MiRNP IP (RIP) assay showing the association between miR-150 and
CCND1, CCND2, CDK2 and CCNE2 transcripts in CNE-2 and HONE1 cells.
Pulldown of IgG antibody served as the negative control. (B) RIP
analysis show that CDK4, CDK6, CCND3 and CCNE1 were not affected by
miR-150. RIP analysis, as assessed by immunoprecipitation of Ago2
in the indicated cells. IgG immunoprecipitation was used as a
negative control. *P<0.05. (C) Predicted miR-150
targeting sequence in 3′-UTRs of CCND1, CCND2, CDK2 and CCNE2. (D)
Luciferase assay of cells transfected with pmirGLO-3′-UTR reporter
of CCND1, CCND2, CDK2 and CCNE2 in miR-150 overexpressing and
silencing in CNE-2 and HONE1 cells, respectively. (E) Individual
silencing of CCND1, CCND2, CDK2 and CCNE2 increased the percentage
of NPC cells in G0 phase, while the percentage of cells
in S phase was decreased in miR-150-silencing cells. |
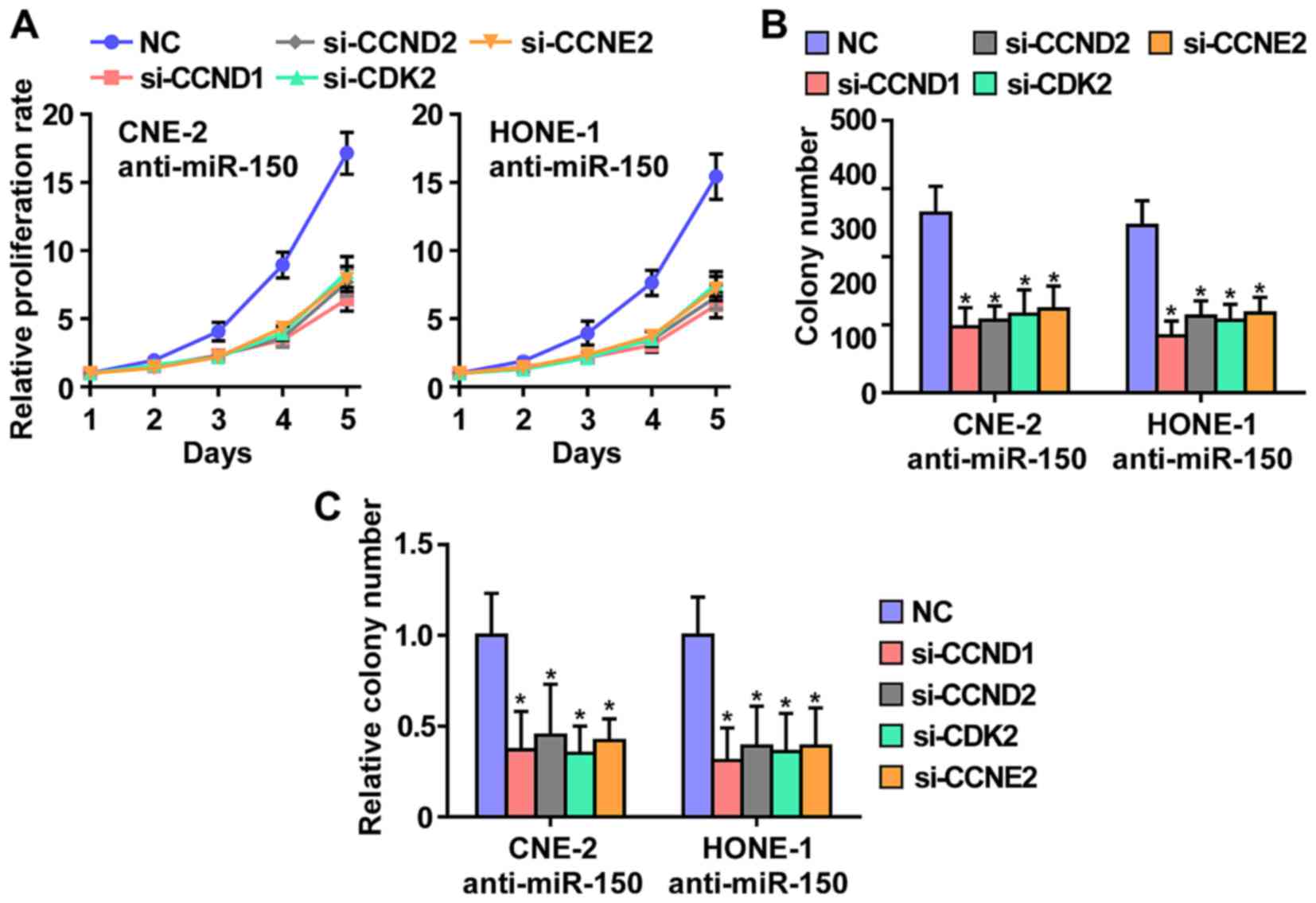
CCND1, CCND2, CDK2 and CCNE2 contribute
to miR-150 downregulation-induced NPC cell growth
To further investigate whether CCND1, CCND2, CDK2
and CCNE2 contribute to the phenotypes induced by
miR-150-downregulated in NPC cells, we used RNA interference to
knock down the CCND1, CCND2, CDK2 and CCNE2. As shown in Fig. 6, individual silencing of CCND1,
CCND2, CDK2 and CCNE2 significantly inhibited cell growth, cell
colony number and cell colony numbers on soft agar of anti-miR-150
CNE2 and HONE1 cells. These results indicated that miR-150 inhibits
NPC cells proliferation via silencing CCND1, CCND2, CDK2 and
CCNE2.
miR-150 levels clinically correlate with
CCND1, CCND2, CDK2 and CCNE2 expression in NPC
Finally, we investigated whether miR-150 correlated
with CCND1, CCND2, CDK2 and CCNE2 expression in NPC clinical
tissues. The rationale used to perform the correlation of miR-150
with CCND1, CCND2, CKD2 and CCNE2 expression in NPC tissues as
follows: we first examined the miR-150 expression and the mRNA
expression levels of CCND1, CCND2, CKD2 and CCNE2 via real-time PCR
in 8 NPC tissues. Relative fold expressions of miR-150, CCND1,
CCND2, CKD2 and CCNE2 in NPC tissue samples were calculated. Then,
one tissue sample was randomly selected and numbered as T1, other 7
tissue samples were numbered in order as T2, T3, T4, T5, T6, T7 and
T8. The expression levels of miR-150, CCND1, CCND2, CKD2 and CCNE2
in each NPC tissue were normalized by the corresponding miR-150,
CCND1, CCND2, CKD2 and CCNE2 expression levels in T1 NPC tissue.
The relative expression levels of miR-150, CCND1, CCND2, CKD2 and
CCNE2 after normalization were used to perform the correlation
analysis among miR-150 and CCND1, CCND2, CKD2 and CCNE2 expression.
This analysis method of clinical correlation was widely used in
multiple lines of studies (34,35).
The analysis of clinical correlation revealed that a significant
inverse correlation was found between miR-150 and CCND1 (r=−0.841;
P<0.05), CCND2 (r=−0.668; P<0.05), CDK2 (r=−0.799;
P<0.05), and CCNE2(r=−0.639; P<0.05) expression in NPC
(Fig. 7) in eight fresh NPC
tissues. Taken together, these results indicate that CCND1, CCND2,
CDK2 and CCNE2 expression negatively correlate with miR-150 levels
in NPC tissues.
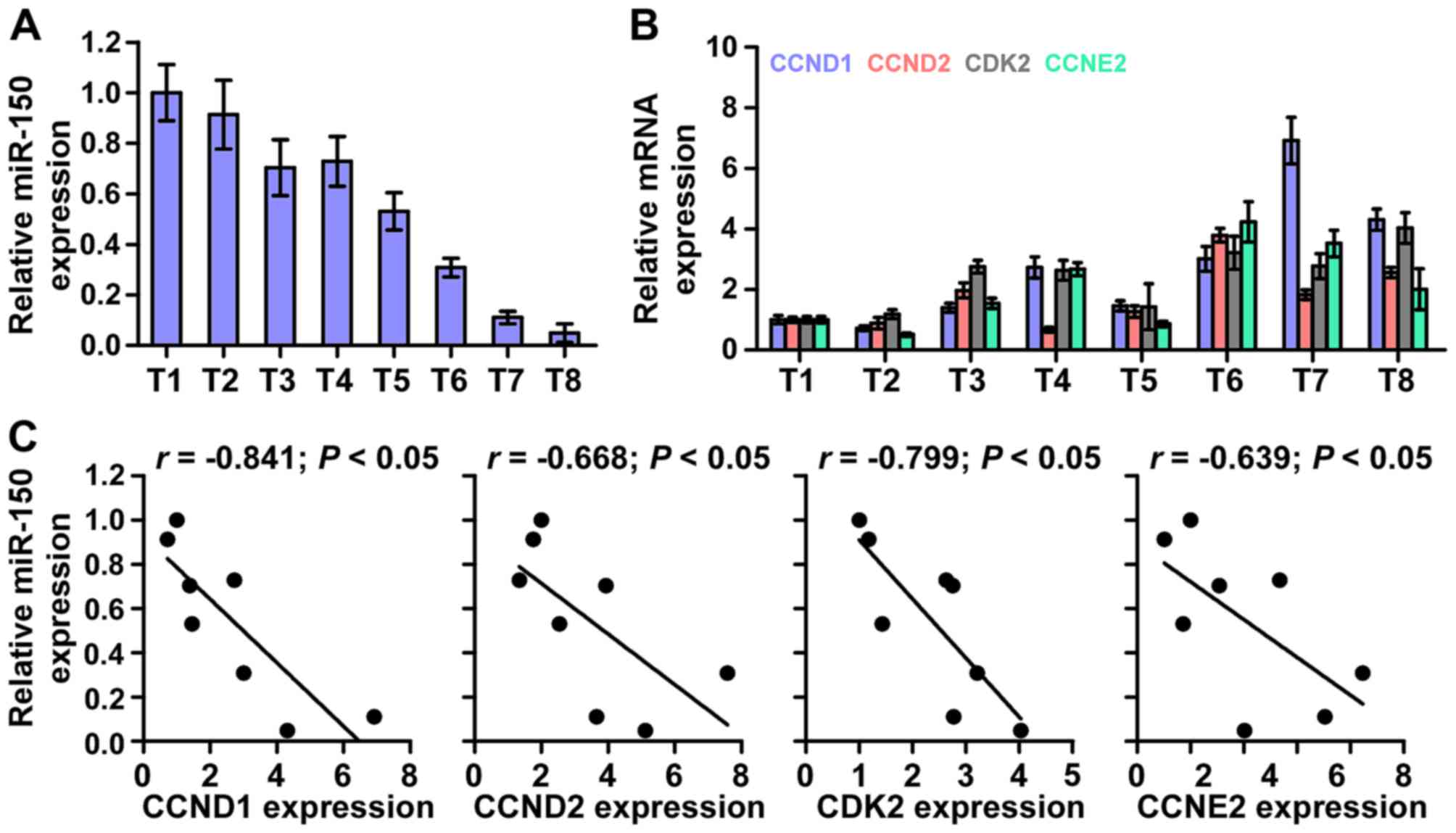 | Figure 7Clinical relevance of miR-150 with
CCND1, CCND2, CDK2 and CCNE2 in nasopharyngeal carcinoma tissues.
(A) miR-150 expression in eight freshly-frozen human nasopharyngeal
carcinoma tissues. Transcript levels were normalized by U6
expression. One tissue sample was randomly selected and numbered as
T1, other 7 tissue samples were numbered in order as T2, T3, T4,
T5, T6, T7 and T8. The expression levels of miR-150 in each NPC
tissue were normalized by the corresponding miR-150 expression in
T1 NPC tissue. The relative expression levels of miR-150 after
normalization were used to perform the correlation analysis among
miR-150 and CCND1, CCND2, CKD2 and CCNE2 expression. (B) Analysis
of CCND1, CCND2, CDK2 and CCNE2 expression in eight freshly-frozen
human nasopharyngeal carcinoma tissues. Transcript levels were
normalized by GAPDH expression. One tissue sample was randomly
selected and numbered as T1, other 7 tissue samples were numbered
in order as T2, T3, T4, T5, T6, T7 and T8. The expression levels of
CCND1, CCND2, CKD2 and CCNE2 in each NPC tissue were normalized by
the corresponding CCND1, CCND2, CKD2 and CCNE2 expression levels in
T1 NPC tissue. The relative expression levels of CCND1, CCND2, CKD2
and CCNE2 after normalization were used to perform the correlation
analysis among miR-150 and CCND1, CCND2, CKD2 and CCNE2 expression.
(C) Correlation between miR-150 levels and CCND1, CCND2, CDK2 and
CCNE2 expression in nasopharyngeal carcinoma tissues. |
Discussion
This study found that miR-150 expression is markedly
down-regulated in NPC tissues and cells, which is consistent with
the analysis of the results of publicly available NPC datasets.
Moreover, upregulation of miR-150 suppresses NPC cell proliferation
and cell cycle in vitro and tumorigenesis in vivo.
Conversely, silencing miR-150 displays the opposite effect on NPC
cells. Our results further demonstrated that CCND1, CCND2, CDK2 and
CCNE2 are the direct targets of miR-150 and importantly the
stimulatory effects of downregulating miR-150 on the proliferation
and cell cycle of NPC cells are reversed by individual silencing of
these target proteins. Therefore, these results present our
mechanistic understanding of miR-150-mediated tumor suppression in
NPC.
Accumulating studies have indicated that several
individual components of the cell cycle machinery are targeted by
specific miRNAs and documented that the loss or gain of
miRNA-mediated cell cycle control contributed to malignancy in a
number of cancers. Several lines of evidence indicated that miRNAs
regulate classic cell cycle control pathways by directly targeting
the components of the cell cycle, including cyclins, CDKs, E2F
transcription factors and Cdk inhibitors (CKIs) (36). For example, a large body of
evidence indicated that upregulating let-7, a well-known tumor
suppressor as well as a critical regulator of the cell cycle,
induced a noted accumulation of G0 and G1
cell cycle stages. Microarray analysis and reporter assays revealed
that multiple genes implicated in promoting G1/S
transition, including CCND2, CDK6 and CDC25A, were the direct
targets of let-7 (37). Moreover,
Linsley and colleagues reported that miR-16 negatively regulated
cellular growth and cell cycle progression. Through microarray
profiling and functional screening, they found that the most
downregulated transcripts were highly enriched for cell cycle-
related genes, as indicated by gene ontology (GO) annotation,
indicating that miR-16 simultaneously regulates these targets that
may function in concert to control cell cycle progression (38). Therefore, these findings indicated
that the identified cell cycle-related target genes of these miRNAs
clarify well the molecular mechanisms by which miRNAs exert their
effects on cell cycle control. Furthermore, miR-150-mediated tumor
suppression via inhibiting cancer cell proliferation and cell cycle
progression has been well established in several cancer (39–42).
However, the expression level and biological role of miR-150 in NPC
lack evidence. In this study, we found that miR-150 expression was
decreased in NPC tissues and cells compared with normal NPC tissues
and cell lines. Upregulation of miR-150 suppressed, whereas miR-150
downregulation promoted, the proliferation and cell cycle of NPC
cells in vitro and tumorigenesis in vivo. To identify
the mechanisms by which miR-150 regulates the proliferation and
cell cycle pathways, we further examined the effects of miR-150 on
the cell cycle-related genes, including CCND1, CCND2, CCND3, CCNE1,
CCNE2, CDK2, CDK4 and CDK6. Of these, the mRNA and protein level of
CCND1, CCND2, CDK2 and CCNE2 were decreased in the
miR-150-overexpressing NPC cells and increased in the
miR-150-silencing NPC cells. The expression of the other four of
these genes were not influenced by miR-150. The analysis of the
publicly available algorithms (miRanda and TargetScan) revealed
that CCND1, CCND2, CDK2 and CCNE2 are the potential targets of
miR-150. Importantly, the stimulatory roles of silencing miR-150 in
the proliferation and cell cycle were attenuated by individual
knockdown of the targeted genes. Therefore, our results uncover a
novel mechanism responsible for the inhibitory effect of miR-150 on
the proliferation and cell cycle in NPC cells.
The majority of the previous studies showed that
miR-150 was downregulated in multiple human cancers, including
hepatocellular carcinoma, lymphoma, colorectal cancer, pancreatic
cancer which contributed to cancer cell proliferation, drug
resistance and metastasis via varing mechanisms (39,42–46).
Furthermore, the expression of miR-150 has also been reported to be
upregulated in non-small cell lung cancer, gastric cancer and
chronic lymphocytic leukemia (47–52).
These findings indicated that miR-150 functions as both an oncomiR
and tumor suppressive miRNA, depending on the tumor type. However,
the expression level and specific role of miR-150 in NPC remains
unknown. In this study, we found that miR-150 expression is
downregulated in NPC tissues and cells compared with normal NPC
tissues and cell lines. Upregulation of miR-150 suppressed, whereas
miR-150 downregulation promoted, the proliferation and cell cycle
of NPC cells in vitro and tumorigenesis in vivo.
Furthermore, the inhibitory role of miR-150 in the proliferation
and cell cycle progression in NPC were mediated by the cell
cycle-related genes, including CCND1, CCND2, CDK2 and CCNE2.
Intriguingly, Cao et al reported that miR-150 was
significantly upregulated in lung cancer clinical specimens and
high expression of miR-150 promoted the proliferation of lung
cancer cells by targeting SRC kinase signalling inhibitor 1
(49). This evidence suggests that
the complexity of the regulatory mechanisms by miR-150 might either
promote or inhibit cellular proliferation, is environment- and
tumor type-dependent.
In conclusion, this study revealed that
tumor-suppressive miR-150 inhibits the proliferation and cell cycle
progression via repressing cell cycle-related genes CCND1, CCND2,
CDK2 and CCNE2. Thus, improved understanding of the specific
mechanism of downregulation miR-150 in the pathogenesis of NPC will
increase our knowledge of nasopharyngeal carcinoma development,
which will help to develop new therapeutic measures against
nasopharyngeal carcinoma.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (81272434, 81500007), the Guangdong
Provincial Medical Research Program (A2016395), the Dongguan
Medical Research Program (no. 2016105101292), the Scientific and
Technological Project of Zhanjiang (2015B01043), the Surface
Cultivation Project of Guangdong Medical University (M2015010), and
the Open Program of Guangdong Provincial Key Laboratory of Medical
Molecular Diagnostics (FZZD201605).
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Qu C, Liang Z, Huang J, Zhao R, Su C, Wang
S, Wang X, Zhang R, Lee MH and Yang H: MiR-205 determines the
radioresistance of human nasopharyngeal carcinoma by directly
targeting PTEN. Cell Cycle. 11:785–796. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ai MD, Li LL, Zhao XR, Wu Y, Gong JP and
Cao Y: Regulation of survivin and CDK4 by Epstein-Barr virus
encoded latent membrane protein 1 in nasopharyngeal carcinoma cell
lines. Cell Res. 15:777–784. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhen Y, Fang W, Zhao M, Luo R, Liu Y, Fu
Q, Chen Y, Cheng C, Zhang Y and Liu Z:
miR-374a-CCND1-pPI3K/AKT-c-JUN feedback loop modulated by PDCD4
suppresses cell growth, metastasis, and sensitizes nasopharyngeal
carcinoma to cisplatin. Oncogene. 36:275–285. 2017. View Article : Google Scholar
|
|
5
|
Nigg EA: Cyclin-dependent protein kinases:
Key regulators of the eukaryotic cell cycle. BioEssays. 17:471–480.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Spellman PT, Sherlock G, Zhang MQ, Iyer
VR, Anders K, Eisen MB, Brown PO, Botstein D and Futcher B:
Comprehensive identification of cell cycle-regulated genes of the
yeast Saccharomyces cerevisiae by microarray hybridization. Mol
Biol Cell. 9:3273–3297. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Champeris Tsaniras S, Kanellakis N,
Symeonidou IE, Nikolopoulou P, Lygerou Z and Taraviras S: Licensing
of DNA replication, cancer, pluripotency and differentiation: An
interlinked world? Semin Cell Dev Biol. 30:174–180. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
O'Leary B, Finn RS and Turner NC: Treating
cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol.
13:417–430. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kumar SK, LaPlant B, Chng WJ, Zonder J,
Callander N, Fonseca R, Fruth B, Roy V, Erlichman C and Stewart AK;
Mayo Phase 2 Consortium: Dinaciclib, a novel CDK inhibitor,
demonstrates encouraging single-agent activity in patients with
relapsed multiple myeloma. Blood. 125:443–448. 2015. View Article : Google Scholar :
|
|
10
|
Ventura A and Jacks T: MicroRNAs and
cancer: Short RNAs go a long way. Cell. 136:586–591. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Khew-Goodall Y and Goodall GJ:
Myc-modulated miR-9 makes more metastases. Nat Cell Biol.
12:209–211. 2010.PubMed/NCBI
|
|
12
|
Ren D, Wang M, Guo W, Huang S, Wang Z,
Zhao X, Du H, Song L and Peng X: Double-negative feedback loop
between ZEB2 and miR-145 regulates epithelial-mesenchymal
transition and stem cell properties in prostate cancer cells. Cell
Tissue Res. 358:763–778. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Baranwal S and Alahari SK: miRNA control
of tumor cell invasion and metastasis. Int J Cancer. 126:1283–1290.
2010.
|
|
14
|
Ren D, Wang M, Guo W, Zhao X, Tu X, Huang
S, Zou X and Peng X: Wild-type p53 suppresses the
epithelial-mesenchymal transition and stemness in PC-3 prostate
cancer cells by modulating miR-145. Int J Oncol. 42:1473–1481.
2013.PubMed/NCBI
|
|
15
|
Garzon R, Calin GA and Croce CM: MicroRNAs
in cancer. Annu Rev Med. 60:167–179. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang X, Liu J, Zang D, Wu S, Liu A, Zhu
J, Wu G, Li J and Jiang L: Upregulation of miR-572
transcriptionally suppresses SOCS1 and p21 and contributes to human
ovarian cancer progression. Oncotarget. 6:15180–15193. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang M, Ren D, Guo W, Wang Z, Huang S, Du
H, Song L and Peng X: Loss of miR-100 enhances migration, invasion,
epithelial-mesenchymal transition and stemness properties in
prostate cancer cells through targeting Argonaute 2. Int J Oncol.
45:362–372. 2014.PubMed/NCBI
|
|
18
|
Huang S, Guo W, Tang Y, Ren D, Zou X and
Peng X: miR-143 and miR-145 inhibit stem cell characteristics of
PC-3 prostate cancer cells. Oncol Rep. 28:1831–1837.
2012.PubMed/NCBI
|
|
19
|
Cai L, Ye Y, Jiang Q, Chen Y, Lyu X, Li J,
Wang S, Liu T, Cai H, Yao K, et al: Epstein-Barr virus-encoded
microRNA BART1 induces tumour metastasis by regulating
PTEN-dependent pathways in nasopharyngeal carcinoma. Nat Commun.
6:73532015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhen Y, Liu Z, Yang H, Yu X, Wu Q, Hua S,
Long X, Jiang Q, Song Y, Cheng C, et al: Tumor suppressor PDCD4
modulates miR-184-mediated direct suppression of C-MYC and BCL2
blocking cell growth and survival in nasopharyngeal carcinoma. Cell
Death Dis. 4:e8722013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Han Y, Meng F, Venter J, Wu N, Wan Y,
Standeford H, Francis H, Meininger C, Greene J Jr, Trzeciakowski
JP, et al: miR-34a-dependent overexpression of Per1 decreases
cholangio-carcinoma growth. J Hepatol. 64:1295–1304. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Aakula A, Kohonen P, Leivonen SK, Mäkelä
R, Hintsanen P, Mpindi JP, Martens-Uzunova E, Aittokallio T,
Jenster G, Perälä M, et al: Systematic identification of microRNAs
that impact on proliferation of prostate cancer cells and display
changed expression in tumor tissue. Eur Urol. 69:1120–1128. 2016.
View Article : Google Scholar
|
|
23
|
Qiu Z, Guo W, Wang Q, Chen Z, Huang S,
Zhao F, Yao M, Zhao Y and He X: MicroRNA-124 reduces the pentose
phosphate pathway and proliferation by targeting PRPS1 and RPIA
mRNAs in human colorectal cancer cells. Gastroenterology.
149:1587–1598.e11. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhao M, Luo R, Liu Y, Gao L, Fu Z, Fu Q,
Luo X, Chen Y, Deng X, Liang Z, et al: miR-3188 regulates
nasopharyngeal carcinoma proliferation and chemosensitivity through
a FOXO1-modulated positive feedback loop with mTOR-p-I3K/AKT-c-JUN.
Nat Commun. 7:113092016. View Article : Google Scholar
|
|
25
|
He Q, Ren X, Chen J, Li Y, Tang X, Wen X,
Yang X, Zhang J, Wang Y, Ma J, et al: miR-16 targets fibroblast
growth factor 2 to inhibit NPC cell proliferation and invasion via
PI3K/AKT and MAPK signaling pathways. Oncotarget. 7:3047–3058.
2016.
|
|
26
|
Jun GJ, Zhong GG and Ming ZS: miR-218
inhibits the proliferation of glioma U87 cells through the
inactivation of the CDK6/cyclin D1/p21(Cip1/Waf1) pathway. Oncol
Lett. 9:2743–2749. 2015.PubMed/NCBI
|
|
27
|
Schultz J, Lorenz P, Gross G, Ibrahim S
and Kunz M: MicroRNA let-7b targets important cell cycle molecules
in malignant melanoma cells and interferes with
anchorage-independent growth. Cell Res. 18:549–557. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Guo W, Ren D, Chen X, Tu X, Huang S, Wang
M, Song L, Zou X and Peng X: HEF1 promotes epithelial mesenchymal
transition and bone invasion in prostate cancer under the
regulation of microRNA-145. J Cell Biochem. 114:1606–1615. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hahn WC, Dessain SK, Brooks MW, King JE,
Elenbaas B, Sabatini DM, DeCaprio JA and Weinberg RA: Enumeration
of the simian virus 40 early region elements necessary for human
cell transformation. Mol Cell Biol. 22:2111–2123. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lee YM, Ting CM, Cheng YK, Fan TP, Wong
RN, Lung ML and Mak NK: Mechanisms of 2-methoxyestradiol-induced
apoptosis and G2/M cell-cycle arrest of nasopharyngeal carcinoma
cells. Cancer Lett. 268:295–307. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ghimire BR: Effects of zoledronic acid
(ZOL) on proliferation of nasopharyngeal carcinoma (NPC) cell lines
and synergistic effects of ZOL and cisplatin on NPC cell lines. J
Clin Oncol. 27:e135362009.
|
|
32
|
O'Donnell KA, Wentzel EA, Zeller KI, Dang
CV and Mendell JT: c-Myc-regulated microRNAs modulate E2F1
expression. Nature. 435:839–843. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sylvestre Y, De Guire V, Querido E,
Mukhopadhyay UK, Bourdeau V, Major F, Ferbeyre G and Chartrand P:
An E2F/miR-20a autoregulatory feedback loop. J Biol Chem.
282:2135–2143. 2007. View Article : Google Scholar
|
|
34
|
Lin C, Liu A, Zhu J, Zhang X, Wu G, Ren P,
Wu J, Li M, Li J and Song L: miR-508 sustains phosphoinositide
signalling and promotes aggressive phenotype of oesophageal
squamous cell carcinoma. Nat Commun. 5:46202014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cui Y, Ma W, Lei F, Li Q, Su Y, Lin X, Lin
C, Zhang X, Ye L, Wu S, et al: Prostate tumour overexpressed-1
promotes tumourigenicity in human breast cancer via activation of
Wnt/β-catenin signalling. J Pathol. 239:297–308. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chivukula RR and Mendell JT: Circular
reasoning: microRNAs and cell-cycle control. Trends Biochem Sci.
33:474–481. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Johnson CD, Esquela-Kerscher A, Stefani G,
Byrom M, Kelnar K, Ovcharenko D, Wilson M, Wang X, Shelton J,
Shingara J, et al: The let-7 microRNA represses cell proliferation
pathways in human cells. Cancer Res. 67:7713–7722. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Linsley PS, Schelter J, Burchard J,
Kibukawa M, Martin MM, Bartz SR, Johnson JM, Cummins JM, Raymond
CK, Dai H, et al: Transcripts targeted by the microRNA-16 family
cooperatively regulate cell cycle progression. Mol Cell Biol.
27:2240–2252. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Watanabe A, Tagawa H, Yamashita J, Teshima
K, Nara M, Iwamoto K, Kume M, Kameoka Y, Takahashi N, Nakagawa T,
et al: The role of microRNA-150 as a tumor suppressor in malignant
lymphoma. Leukemia. 25:1324–1334. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hornick NI, Doron B, Abdelhamed S, Huan J,
Harrington CA, Shen R, Cambronne XA, Chakkaramakkil Verghese S and
Kurre P: AML suppresses hematopoiesis by releasing exosomes that
contain microRNAs targeting c-MYB. Sci Signal. 9:ra882016.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Leoncini PP, Bertaina A, Papaioannou D,
Flotho C, Masetti R, Bresolin S, Menna G, Santoro N, Zecca M, Basso
G, et al: MicroRNA fingerprints in juvenile myelomonocytic leukemia
(JMML) identified miR-150-5p as a tumor suppressor and potential
target for treatment. Oncotarget. 7:55395–55408. 2016.PubMed/NCBI
|
|
42
|
Sun W, Zhang Z, Wang J, Shang R, Zhou L,
Wang X, Duan J, Ruan B, Gao Y, Dai B, et al: MicroRNA-150
suppresses cell proliferation and metastasis in hepatocellular
carcinoma by inhibiting the GAB1-ERK axis. Oncotarget.
7:11595–11608. 2016.PubMed/NCBI
|
|
43
|
Ito M, Teshima K, Ikeda S, Kitadate A,
Watanabe A, Nara M, Yamashita J, Ohshima K, Sawada K and Tagawa H:
MicroRNA-150 inhibits tumor invasion and metastasis by targeting
the chemokine receptor CCR6, in advanced cutaneous T-cell lymphoma.
Blood. 123:1499–1511. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ma Y, Zhang P, Wang F, Zhang H, Yang J,
Peng J, Liu W and Qin H: miR-150 as a potential biomarker
associated with prognosis and therapeutic outcome in colorectal
cancer. Gut. 61:1447–1453. 2012. View Article : Google Scholar
|
|
45
|
Srivastava SK, Bhardwaj A, Singh S, Arora
S, Wang B, Grizzle WE and Singh AP: MicroRNA-150 directly targets
MUC4 and suppresses growth and malignant behavior of pancreatic
cancer cells. Carcinogenesis. 32:1832–1839. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wuerkenbieke D, Wang J, Li Y and Ma C:
miRNA-150 down-regulation promotes pertuzumab resistance in ovarian
cancer cells via AKT activation. Arch Gynecol Obstet.
292:1109–1116. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yin QW, Sun XF, Yang GT, Li XB, Wu MS and
Zhao J: Increased expression of microRNA-150 is associated with
poor prognosis in non-small cell lung cancer. Int J Clin Exp
Pathol. 8:842–846. 2015.PubMed/NCBI
|
|
48
|
Wu Q, Jin H, Yang Z, Luo G, Lu Y, Li K,
Ren G, Su T, Pan Y, Feng B, et al: MiR-150 promotes gastric cancer
proliferation by negatively regulating the pro-apoptotic gene EGR2.
Biochem Biophys Res Commun. 392:340–345. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cao M, Hou D, Liang H, Gong F, Wang Y, Yan
X, Jiang X, Wang C, Zhang J, Zen K, et al: miR-150 promotes the
proliferation and migration of lung cancer cells by targeting SRC
kinase signalling inhibitor 1. Eur J Cancer. 50:1013–1024. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Papakonstantinou N, Ntoufa S,
Chartomatsidou E, Papadopoulos G, Hatzigeorgiou A, Anagnostopoulos
A, Chlichlia K, Ghia P, Muzio M, Belessi C, et al: Differential
microRNA profiles and their functional implications in different
immunogenetic subsets of chronic lymphocytic leukemia. Mol Med.
19:115–123. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yang M, Cui G, Ding M, Yang W, Liu Y, Dai
D and Chen L: miR-935 promotes gastric cancer cell proliferation by
targeting SOX7. Biomed Pharmacother. 79:153–158. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhang AX, Lu FQ, Yang YP, Ren XY, Li ZF
and Zhang W: MicroRNA-217 overexpression induces drug resistance
and invasion of breast cancer cells by targeting PTEN signaling.
Cell Biol Int. 2015.Epub ahead of print. View Article : Google Scholar
|