Introduction
Glioma is one of the most common and aggressive
human malignancies worldwide (1).
The poor prognosis of glioma is largely due to the deregulation of
intercellular signaling pathways, including Notch and retinoic acid
pathways, and the Hedgehog (Hh) signaling pathway, which may
promote glioma progression by conferring cell proliferation and
survival advantages through several mechanisms (2).
Hh signaling transduction is initiated by the
binding of Hh proteins (sonic Hh, Shh; Indian Hh, Ihh; and Desert
Hh, Dhh) to the 12-pass transmembrane protein Patched (PTCH), which
abrogates the repressive activity of PTCH, allowing the 7-pass
transmembrane protein Smoothened (Smo) to transduce the signal to
the nucleus. Specifically, Smo promotes nuclear translocation of
the 5-zinc-finger transcription factors glioma-associated oncogenes
(Glis) and subsequently activates target gene transcription
(3,4). There are three members in the Gli
family, Gli1, Gli2, and Gli3. Gli1 and Gli2 primarily act as
transcriptional activators, whereas Gli3 acts as a transcriptional
repressor in the Hh signaling pathway (5). Gene targeting studies in mice have
demonstrated that Gli2 and Gli3 are the primary mediators of Hh
signaling and are essential for embryogenesis. Loss of Gli2 is
embryonically lethal, whereas Gli1 is dispensable for animal
development (6,7).
In humans, the Hh signaling pathway is critical for
embryonic development and adult homeostasis, and Hh signaling
activity normally ceases after embryogenesis. However, aberrant
activation of the Hh signaling cascade has been shown to be
associated with oncogenesis and maintenance of the malignant
phenotype in multiple types of human cancers (8,9).
Increasing evidence shows that excessive activating mutations in
the Smo gene, loss of function mutations in the PTCH gene, or
amplification of Glis cause the majority of human cancers (10). RNA analysis of clinical samples
found upregulated expression of Gli1 and PTCH1 in glioma tissues
(11), and vismodegib treatment
reduced Gli1 expression concomitant with the induction of apoptosis
and cell cycle arrest (12).
Blockade of Hh signaling by cyclopamine (13), Gli-ANTagonist 61 (GANT61) (14) or Smo shRNA (15) inhibits cell proliferation and
suppresses tumor formation. These observations indicate that
deregulated Hh signaling is correlated with rapid growth of human
glioma cells. Nevertheless, the mechanisms by which Hh signaling
promotes tumor growth need to be further elucidated.
MicroRNAs (miRNAs) are a class of short,
single-stranded endogenous non-coding RNAs (approximately 20–22
nucleotides in length) that post-transcriptionally control gene
expression via either translational repression and/or mRNA
degradation in multicellular eukaryotes (16). Computational and biological
analyses estimate that approximately 30% of all genes and the
majority of genetic pathways are subject to regulation by multiple
miRNAs (17). By targeting
multiple transcripts, miRNAs play important roles in a wide array
of biological processes, including development, differentiation,
cell proliferation, apoptosis, and metabolism.
It is noteworthy that as a regulatory element, miRNA
itself often acts as downstream effector of transcription factors
including p53, HIF-1, and c-myc, which have been verified to
regulate the expression of several miRNAs (18). Although the diversity and abundance
of miRNAs seem to be regulated by several transcription factors and
mediate gene expression in any given cancer type, to our knowledge
there has been no definitive description of miRNAs whose expression
is regulated by Hh signaling in a transcriptional manner. In
addition, the functional consequences of such regulation are
ambiguous.
In this study, we performed a set of experiments to
elucidate the molecular mechanisms by which the Hh signaling
pathway regulates cancer cell proliferation and tumor growth. Our
findings indicate that inhibition of Hh signaling suppresses cell
proliferation, at least in part, via the Gli2/miR-124/AURKA axis in
human glioma cells.
Materials and methods
Reagents, antibodies and constructs
Primary antibodies were purchased from Millipore
(GAPDH, mAb374) and Cell Signaling Technology (Aurka, 4718). A
PrimeScript™ RT reagent kit with gDNA Eraser,
SYBR®Premix Ex Taq™ II and a One Step PrimeScript miRNA
cDNA Synthesis kit were purchased from Takara (Tokyo, Japan). The
miR-124 inhibitor and control oligonucleotides were purchased from
Ribobio (Guangzhou, China) and prepared as 50 µM stock
solutions using RNase-free H2O. Lipofectamine 2000 was
obtained from Invitrogen Life Technologies (Carlsbad, CA, USA). The
working concentrations for small molecular inhibitors and chemicals
included GANT61 (20 µM, G9048, Sigma) and DMSO (0231,
Amresco), which was used as the solvent for the inhibitors and as
the vehicle control. Human AURKA (NM_000689) was subcloned between
EcoRI and XhoI sites in pcDNA-Flag3.0 (BD Biosciences
Clontech, Palo Alto, CA, USA) in-frame downstream of the Flag
epitope. shRNA plasmids that separately suppressed the expression
of Gli1 (NM_005269.2) and Gli2 (NM_005270) were generated using a
BlOCK-iT™ Pol II miR RNAi Expression Vector kit (K4936-00,
Invitrogen Life Technologies). The oligonucleotide sequences for
the shRNA constructs are listed in Table I. The authenticity of all
constructs was verified by DNA sequencing.
 | Table IInterference sequences. |
Table I
Interference sequences.
| Genes | Target site | Target
sequence |
|---|
| sh-Gli1-720 | 720 to 740 |
5′-TTCATACACAGATTCAGGCTC-3′ |
| sh-Gli1-1863 | 1863 to 1883 |
5′-TTCATACACAGATTCAGGCTC-3′ |
| sh-Gli1-2255 | 2255 to 2275 |
5′-AAGACCTATCCGATCCAGCGG-3′ |
| sh-Gli2-233 | 233 to 253 |
5′-AATGGTACCTTCCTTCCTGGT-3′ |
| sh-Gli2-1127 | 1127 to 1147 |
5′-TGGCCTGAAACGATGTCATC-3′ |
| sh-Gli2-2058 | 2058 to 2078 |
5′-TGTGAATGGCGACAGGGTTGA-3′ |
Cell culture and transfection
Human glioma cell lines (H4 and U87) and a human
embryonic kidney cell line (HEK293T) were purchased from the
American Type Culture Collection (ATCC, Manassas, VA, USA) and
cultured in DMEM containing 10% fetal bovine serum (Gibco,
Carlsbad, CA, USA), 50 mg/ml penicillin (100 U/ml) and streptomycin
(100 µg/ml) at 37°C in a humidified atmosphere of 5%
CO2 in air. All cell lines used tested negative for
mycoplasma and used in less than 2 months when the experiments were
performed. Transient transfection was performed with Lipofectamine
2000 (Invitrogen Life Technologies) according to the manufacturer's
instructions.
Gene expression profiling by microarray
analysis and real-time PCR
Total RNA, including miRNA, was isolated using
TRIzol (Ambion, Austin, TX, USA). The integrity of the total RNA
was analyzed by gel electrophoresis. Then, 200 ng of the isolated
total RNA was labeled using an Illumina Total Prep-96 RNA
Amplification kit (PN:4393543, Ambion Life Technologies, Grand
Island, NY, USA), and 750 ng of cRNA was generated and hybridized
into a Human HT-12 V4 BeadChip. Then, the BeadChip was washed and
stained as per the Illumina protocol and scanned on an iScan
(Illumina, San Diego, CA, USA). Data analysis was performed with
Genespring GX 12.0 Software (Agilent Technology, Inc., Santa Clara,
CA, USA). Raw data were filtered by percentile (lower cut-off: 20).
An unpaired t-test was used to identify significant (P<0.05)
gene expression changes with multiple testing correction
(Benjamini-Hochberg) to control the false discovery rate and obtain
statistically reliable results.
Real-time PCR analysis of the expression of all mRNA
and miRNA was analyzed using a SYBR Green kit (Takara) according to
the manufacturer's instructions. Briefly, for the detection of
mRNA, 1 µg of total RNA was used to generate cDNA via
reverse transcription using a PrimeScript RT reagent kit with gDNA
Eraser. Then, PCR was performed using a SYBR Premix Ex Taq II. For
the detection and estimation of miRNAs, we employed a real-time PCR
method that involves the formation of miRNA-specific cDNA from
total RNA (10 ng) using a specific primer. A diluted reverse
transcription product, which is a miRNA-specific cDNA was used for
each real-time PCR reaction. The primers for PCR are shown in
Table II. All experiments were
performed using the ABI StepOnePlus™ Real-Time qPCR System (Applied
Biosystems Inc., Carlsbad, CA, USA), miRNA and mRNA values were
normalized to two endogenous controls, U6 and GAPDH, respectively.
The 2−ΔΔCT method was used to calculate the expression
ratios.
| Table IIPrimer sequences for real-time
PCR. |
Table II
Primer sequences for real-time
PCR.
| Genes | Forward primer (5′
to 3′) | Reverse primer (5′
to 3′) |
|---|
| Gli1 |
5′-TCCTACCAGAGTCCCAAGTT-3′ |
5′-CCCTATGTGAAGCCCTATTT-3′ |
| Gli2 |
5′-CTGTGGGTTAGGGATGGACTG-3′ |
5′-GTAAAGTGGGTGGACGTTGCA-3′ |
| AURKA |
5′-GGAATATGCACCACTTGGAACA-3′ |
5′-TAAGACAGGGCATTTGCCAAT-3′ |
| GAPDH |
5′-GAAGGTGAAGGTCGGAGTC-3′ |
5′-GAAGATGGTGATGGGATTTC-3′ |
| miR-301b |
5′-GCAGTGCAATGATATTGTCAAAGC-3′ | Uni-miR PCR
Primer |
| miR-302d |
5′-GCTAAGTGCTTCCATGTTTGAGTGT-3′ | Uni-miR PCR
Primer |
| miR-519a |
5′-GCAAAGTGCATCCTTTTAGAGTGT-3′ | Uni-miR PCR
Primer |
| miR-335 |
5′-GCTCAAGAGCAATAACGAAAAATGT-3′ | Uni-miR PCR
Primer |
| miR-122 |
5′-GTGGAGTGTGACAATGGTGTTTG-3′ | Uni-miR PCR
Primer |
Bioinformatic analysis
Identification of transcription factor binding sites
in the pre-miR-124 promoter was performed using Cisgenome 2.0
software to identify putative transcription factors that could
potentially bind and regulate the expression of miR-124. The motif
resembling the known Gli binding site was CTGGGTGGTC (11). A 10 kb region in the promoter of
pri-miR-124 was used for the transcription factor analysis. The
public databases TargetScan, PicTar, and Miranda were used to
identify putative miRNA seed matching sequences in the 3′-UTR of
AURKA.
Chromatin immunoprecipitation
A chromatin immunoprecipitation (CHIP) analysis was
performed to detect the occupation of the Gli2 transcription factor
on the putative regions of miR-124. In brief, H4 cells were
cultured in a 10-cm dish until reaching approximately 90%
confluence. After discarding the original medium, H4 cells were
crosslinked with 5 ml of PBS containing 1% formaldehyde at room
temperature for 10 min with gentle shaking. Then, DNA was sonicated
into a range of 200–1000 base pairs in size using a Bioruptor
Sonicator (Diagenode) for five cycles of 3 sec on/3 sec off. The
extracts were pre-cleared in BSA-blocked protein A beads and
incubated with anti-Gli2 or IgG control overnight at 4°C. After
being washed, DNA was eluted and reverse cross-linked overnight at
65°C and then purified and amplified by PCR. The primers for PCR
are shown in Table III.
| Table IIIPrimer sequences for CHIP. |
Table III
Primer sequences for CHIP.
| Genes | Forward primer (5′
to 3′) | Reverse primer (5′
to 3′) |
|---|
| BS1 |
5′-TACAGAGGGATCTGTTGGGAGT-3′ |
5′-TGGCCTTACCTACAAAATGGG-3′ |
| BS2 |
5′-AGGCTGGTTTCAAACTCCTG-3′ |
5′-TAGTGTCTAGGCTGGGTGC-3′ |
| BS3 |
5′-AGGGAAATGATTCCAAGCC-3′ |
5′-CTGGGAAGTTCTGAATGTTTG-3′ |
| BS4 |
5′-GAACTTCCCAGTCTAAACAGC-3′ |
5′-GGCTTAGGGATTGCTACAAC-3′ |
| BS5 |
5′-CGCTTCCAACCTCCTCTTG-3′ |
5′-GGGCTGGTCTTGAACTCCT-3′ |
| BS6 |
5′-GCTGGGAACTGTAGTCTTGC-3′ |
5′-GCCACTGGAGGTAGTGATT-3′ |
| BS7 |
5′-TTCTTCCCAGCAGAGTCAAG-3′ |
5′-TAATACCTCGCAAAGCATGG-3′ |
Luciferase assay
The wild-type (WT) AURKA-3′-UTR was amplified by PCR
from human cDNA using the primers (forward) 5′-CAA GCT TCA CAT CAG
GTG GAT GGA GAG AC-3′ and (reverse) 5′-GAG CTC GGC AGG GGA AAG CTG
TAG GAA T-3′. The mutant-type (Mut) AURKA-3′ UTR was amplified
using the primers (forward) 5′-CAA GCT TCA CAT CAG GTG GAT GGA GAG
AC-3′ and (reverse) 5′-GAG CTC GGC AGG GGT ATG GTC TAG GAA T-3′.
Then, the cDNA fragments were inserted into a pGL3 Vector using the
SacI and HindIII sites.
HEK293T cells were cultured in 24-well plates and
co-transfected with pGL3.0 vectors containing either the WT or
mutated AURKA-3′-UTR vectors and miR-124 expression plasmid or
control plasmid using Lipofectamine 2000. After 48 h, cells were
lysed, and luciferase assays were performed using a dual luciferase
reporter assay kit (Promega, Madison, WI, USA). Renilla
luciferase activity was used to normalize the transfection
efficiency. Three independent experiments were performed in
triplicate.
Western blotting
Cells were washed with chilled PBS and harvested by
trypsinization. Then, cells were lysed in lysis buffer at 4°C for
30 min and centrifuged (12,000 rpm, 15 min at 4°C) to collect the
supernatant. Protein concentrations were determined by the BCA
method using Pierce™ BCA Protein Assay kit (Thermo Scientific,
Rockford, IL, USA) according to the manufacturer's instructions.
Subsequently, the lysates were separated on SDS-PAGE gels and
immunoblotted using standard procedures. The primary antibodies
used were anti-AURKA (Abcam, 1:1000) and anti-GAPDH (Millipore,
1:2000). Finally, immunostaining was visualized using Kodak X-ray
film, which was subsequently scanned with an Epson 1680 scanner.
Quantitative analysis was performed on scanned images of blots
using ImageJ software.
Cell viability and colony formation
assays
Cell viability assays were performed as previously
described (19). H4 cells
(~5×103 per well) were seeded into a 96-well plate and
cultured for 72 h. Then, MTT solution was added and cells were
incubated for 4 h. The remaining MTT formazan crystals were
solubilized in DMSO, and the absorbance was measured at OD490. For
the colony formation assays, H4 cells (~3×103 per well)
were equivalently plated in 6-well plates in DMEM with 10% FBS.
Then, the cells were cultured, and the medium was changed every 5
days. Cells were cultured for up to 12 days. Then, the cells were
washed with PBS, fixed with 4% paraformaldehyde and stained with
0.1% crystal violet. Dishes were graphed and positive colonies
containing more than 50 cells were counted under a microscope.
Colony-formation rates were then calculated.
Statistical analysis
The statistical significance between two groups was
calculated by unpaired Student's t-test using SPSS 16.0 software.
For experiments involving more than one group for comparison, ANOVA
was used with a suitable post hoc test. All data are expressed as
the mean ± SD for experiments performed at least three times.
Differences were considered significant at P<0.05 or
P<0.01.
Results
Inhibition of Hh signaling results in
miRNAome alteration
To identify miRNAs potentially regulated by Hh
signaling, we treated H4 cells with GANT61, a specific inhibitor of
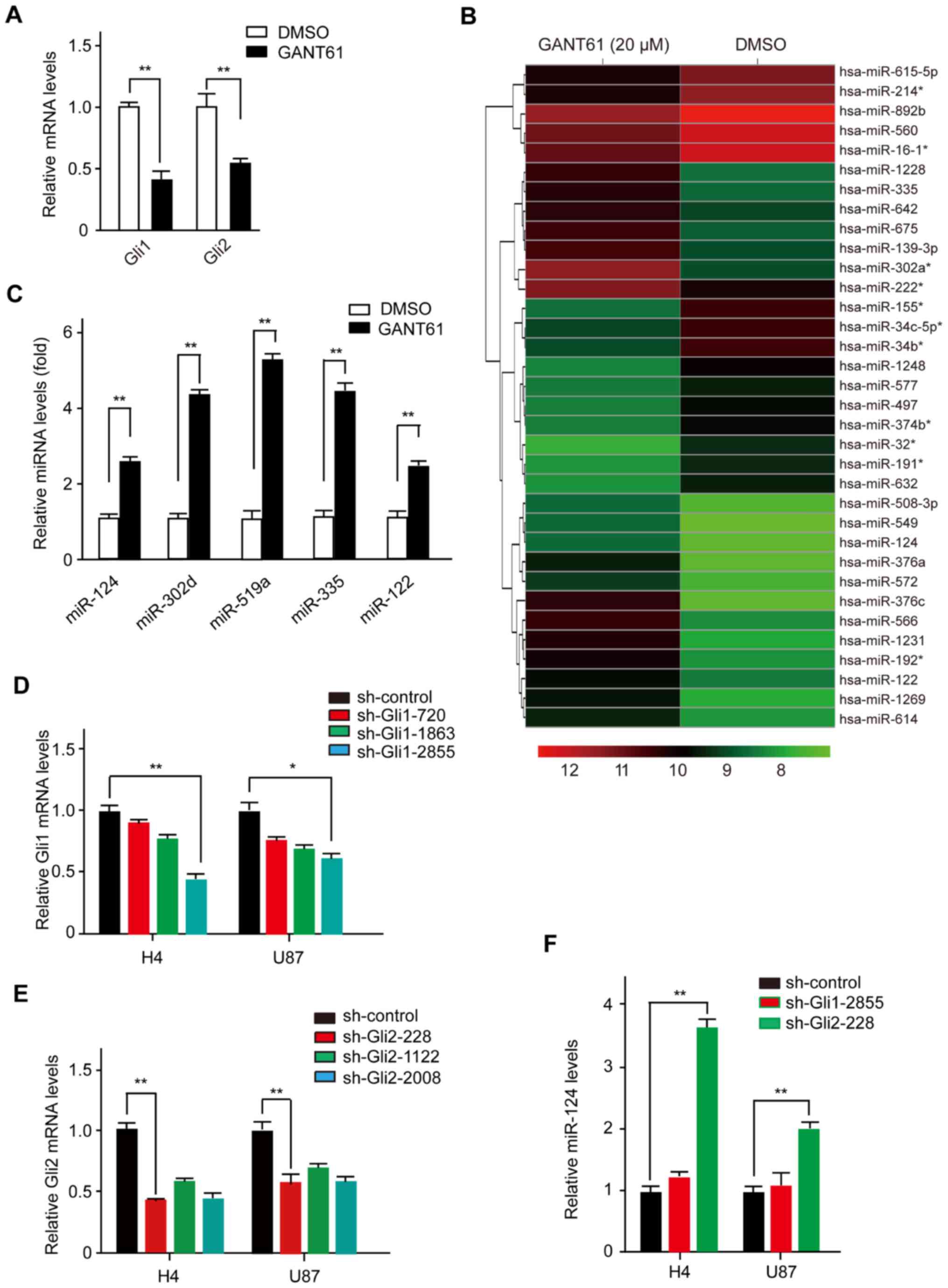
Gli1 and Gli2 (14). As expected,
GANT61 inhibited the expression of Gli1 and Gli2 (Fig. 1A). Subsequently, we purified total
RNA species from H4 cells that were treated with DMSO (Vehicle) or
GANT61 (20 µM, 48 h) and then labeled miRNAs with
fluorescent dyes and hybridized them to an oligonucleotide array
representing known miRNAs. As shown in Fig. 1B, upon efficient blockade of the Hh
signaling pathway by GANT61, a total of 34 miRNAs were
significantly overexpressed or underexpressed (Fig. 1B). To confirm the microarray-based
observations described above, we validated several mature miRNAs
using real-time PCR with stem-loop primers (Fig. 1C). As expected, several mature
miRNAs (i.e., miR-124, miR-302d, miR-519a, miR-335 and miR-122)
were induced by GANT61 treatment in the tested cell lines, which
was consistent with the microarray data. Together, our results
suggest that deregulation of Hh signaling may be involved in the
regulation of miRNA generation. Similar to other cancers, there is
a characteristic miRNA expression pattern in human glioma cells
(20). In particular, miR-124,
whose mature sequences are conserved from Caenorhabditis
elegans to humans, is one of the most deficient miRNAs in
glioma tissue compared with normal brain tissue (20,21).
In addition, glioma-associated loss of normal brain-enriched
miR-124 enhances stem-like traits and the invasiveness of glioma
cells (22). Therefore, we decided
to direct our attention toward miR-124, given that miR-124 may have
critical roles in human glioma tumorigenesis and progression.
In vertebrates, the Gli family of transcription
factors, specifically Gli1 and Gli2, mediates the Hh signaling
pathway by regulating the transcription of target genes. Their
cooperative roles are vital in Hh signaling, while their specific
roles have only been partially defined. To interrogate which one of
the Glis influences miR-124 biogenesis, we transfected H4 cells
with either a Gli1-shRNA or Gli2-shRNA plasmid. We found that
sh-Gli1-2855 and sh-Gli2-228 were more efficient in knocking down
endogenous Gli1 and Gli2 expression (Fig. 1D and E). miR-124 was only repressed
after cells were transfected with Gli2-shRNA plasmid, while
Gli1-shRNA had little effect on miR-124 expression in H4 cells.
Similar results were also observed in U87 cells (Fig. 1F). Taken together, these results
reveal a previously unrecognized function of Hh signaling in miRNA
biogenesis, in which Gli2 negatively regulates the expression of
miR-124 in human glioma cells.
Expression of miR-124 is regulated by
Gli2
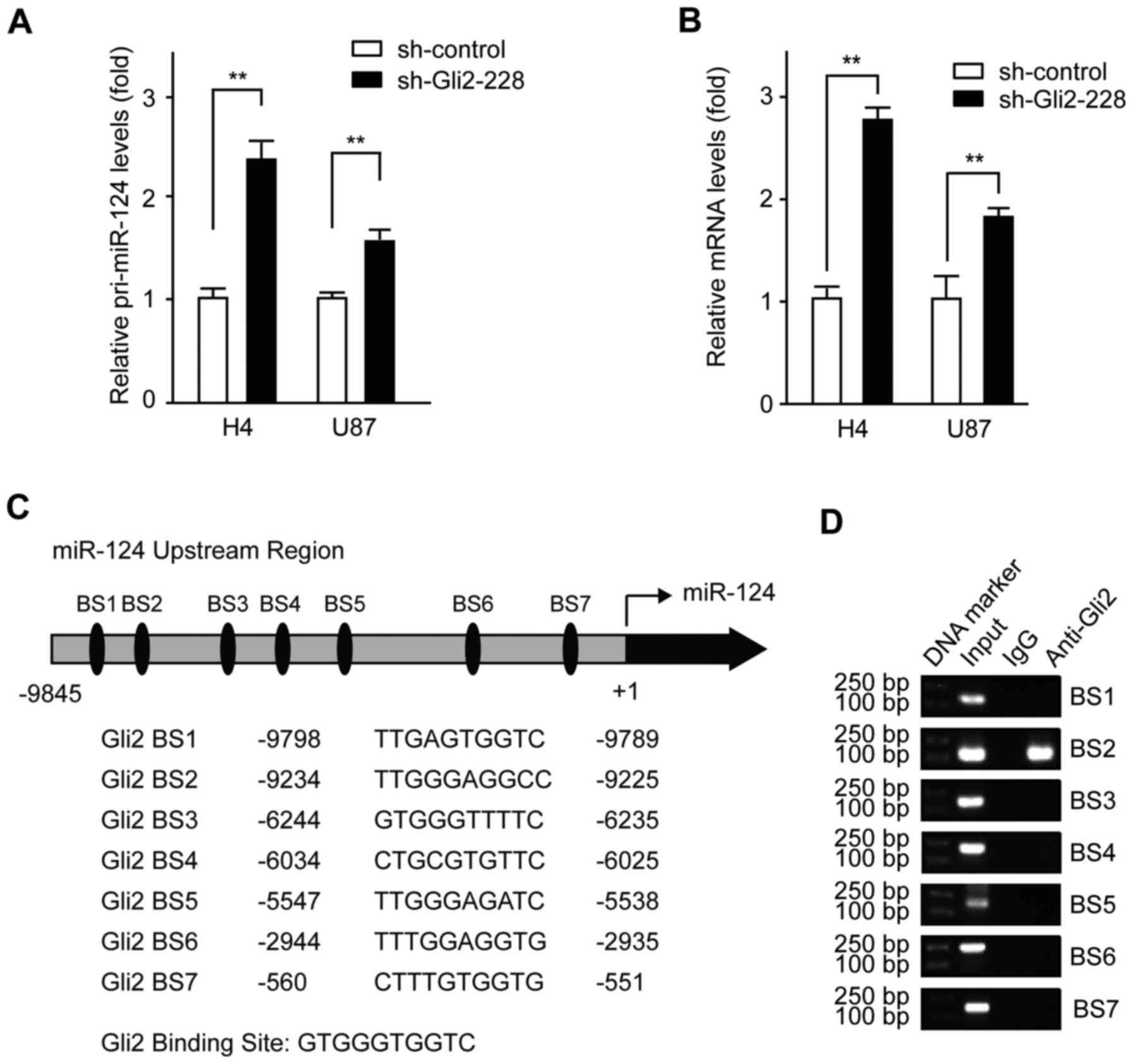
To investigate the molecular mechanism by which Gli2
orchestrates miR-124 expression, we measured the expression level
of pri-miR-124 and pre-miR-124 following Gli2 knockdown in H4
cells. As shown in Fig. 2A,
pri-miR-124 was upregulated after Gli2 depletion in H4 cells and
U87 cells, suggesting a transcriptional level of regulation.
Moreover, an increase in pre-miR-124 was also observed in both H4
cells and U87 cells when transfected with shRNA-Gli2-228 plasmid
(Fig. 2B). Thus, we suspected that
Gli2 might regulate the transcription of miR-124 in glioma
cells.
Next, in order to further illustrate the mechanism,
a search for putative Gli2 binding sites, using Cisgenome 2.0,
identified seven putative Gli2-binding DNA elements (BS1: +9789 ~
+9798, BS2: +9255 ~ +9234, BS3: +6235 ~ +6244, BS4: +6025 ~ +6034,
BS5: +5538 ~ +5547, BS6: +2935 ~ +2944, and BS2: +551 ~ +560)
located upstream of the transcriptional start site of miR-124
(Fig. 2C). Subsequently, we
conducted a set of ChIP assays in H4 cells and determined that Gli2
binds to BS2, but not to any of the other binding sites, in the
miR-124 upstream sequence (Fig.
2D). These findings suggest that Gli2 may be at least partially
responsible for the miR-124 downregulation observed in glioma
cells.
AURKA is a direct target of miR-124
To elaborate the functional consequences of the
Gli2-mediated inhibition of miR-124 expression, we analyzed the
target genes of miR-124. Noteworthy, based on the bioinformatic
analysis of potential miR-124 targets (www.miRNA.org), we determined that miR-124 may
interact with the 3′-UTR region of AURKA (Fig. 3A). AURKA expression promotes
centrosome maturation and separation, which plays multiple roles in
cancer development (23). To
examine whether miR-124 can regulate AURKA expression, a plasmid
construct was used to upregulate the expression of miR-124 in
glioma cells. The miR-124 expression level was upregulated in H4
cells transfected with the miR-124 expression plasmid. Then, we
analyzed the expression levels of AURKA mRNA, and the results
indicated that AURKA mRNA was decreased by ectopic expression of
miR-124 (Fig. 3B and C).
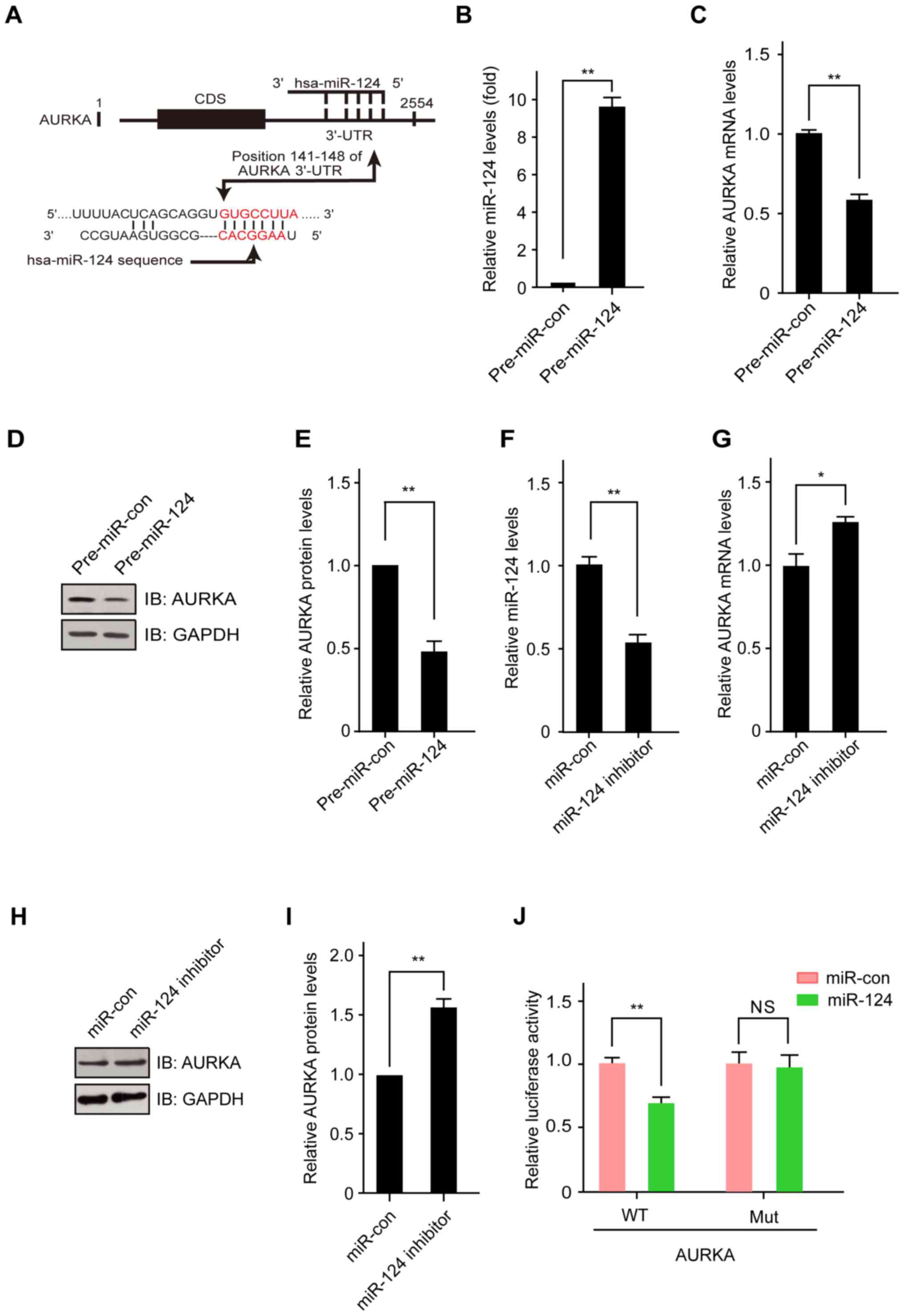 | Figure 3AURKA is a direct target of miR-124.
(A) miR-124 sequences and the predicted miR-124 binding sites in
the 3′-UTR of human AURKA. (B) Overexpression of miR-124 in H4
cells. H4 cells were transfected with control plasmid or miR-124
expression plasmid for 48 h, and then, miR-124 levels were detected
by real-time PCR. (C) miR-124 expression downregulates the mRNA
level of AURKA in H4 cells. H4 cells were transfected with control
plasmid or miR-124 expression plasmid for 48 h, and then, AURKA
mRNA levels were detected with real-time PCR. (D) miR-124
expression downregulates the protein level of AURKA in H4 cells.
Cells were transfected with control plasmid or miR-124 expression
plasmid for 48 h, and then, AURKA protein levels were detected by
western blotting. (E) Downregulation of the expression of miR-124
in H4 cells. Cells were treated with control oligonucleotides or
miR-124 inhibitor for 48 h, and then, miR-124 levels were detected
with real-time PCR. (F) Downregulation of the expression of miR-124
in H4 cells. Cells were treated with control oligonucleotides or
miR-124 inhibitor for 48 h, and then, AURKA mRNA levels were
detected with real-time PCR. (G) Downregulation of the expression
of miR-124 in H4 cells. Cells were treated with control
oligonucleotides or miR-124 inhibitor for 48 h, and then, AURKA
protein levels were detected by western blotting. (H,I,J) The
miR-124 binding site in the human AURKA 3′-UTR mediates the
repression of luciferase activity in HEK-293T cells. luciferase
reporter constructs containing the wild-type or mutant human AURKA
3′-UTR were fused to the 3′-end of the firefly luciferase gene.
Then, the AURKA 3′-UTR luciferase plasmid was transfected into
HEK293T cells together with or without the miR-124 expression
plasmid. The relative luciferase activity was measured 48 h after
transfection with a dual luciferase assay. The values shown are the
means ± SD for triplicate samples. *P<0.05;
**P<0.01; NS, no significance. |
In addition, overexpression of miR-124 significantly
decreased the expression of AURKA protein in H4 cells (Fig. 3D and E). In contrast, the loss of
miR-124 in H4 cells transfected with the miR-124-inhibitor led to
the increased expression of AURKA mRNA and protein when compared to
cells transfected with the control plasmids (Fig. 3F–I). To further confirm that AURKA
was directly targeted and regulated by miR-124, luciferase reporter
genes with the AURKA 3′-UTR and a mutant counterpart, mutated at
the miR-124 binding regions, were co-transfected with the miR-124
expression plasmid or control plasmid into HEK293T cells. The
luciferase reporter assay showed that overexpression of miR-124
significantly inhibited the luciferase activity of AURKA with the
wild-type 3′-UTR but not with the mutant 3′-UTR (Fig. 3J). These findings demonstrated that
AURKA is a direct target gene of miR-124.
Gli2 regulates the expression of AURKA
through miR-124
The results described above show that Hh signaling
inhibits the expression of miR-124, and miR-124 downregulates the
expression level of AURKA (Figs. 2
and 3). In addition, our previous
microarray data showed that AURKA was poorly expressed in the
GANT61 group compared with the control group in H4 cells (24). We next investigated if AURKA can be
directly regulated by Gli2. It is worth noting that we did not
detect any Gli2 occupancy in the analysis of the AURKA binding
sites (data not shown), which indicates that AURKA is presumably
not a direct transcriptional target of Hh signaling but can be
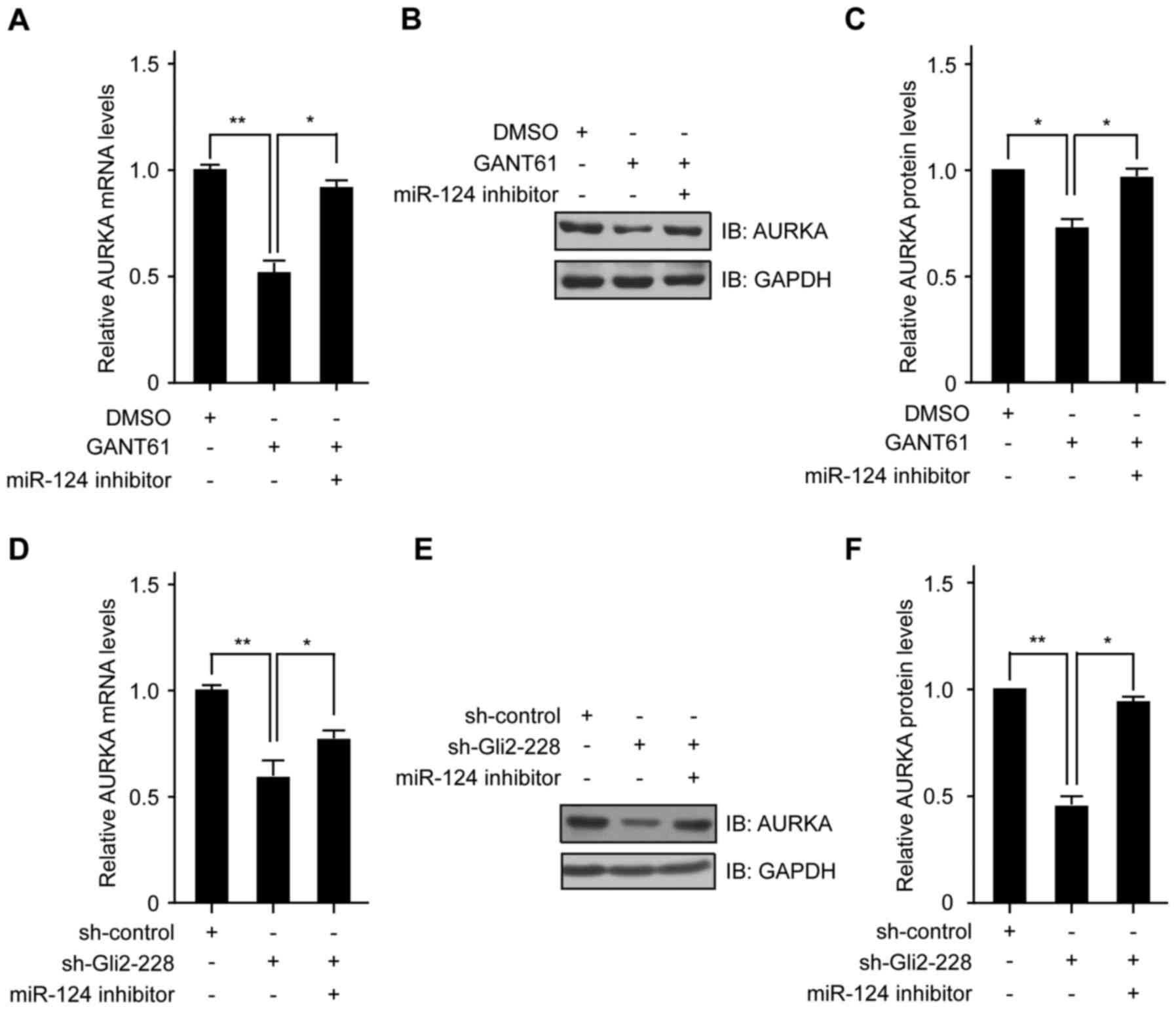
regulated by Hh signaling via miR-124. To validate our hypothesis,
we transfected the miR-124 inhibitor into H4 cells, accompanied by
GANT61.
A real-time PCR assay showed that the AURKA mRNA
level was downregulated in the GANT61 group, while the
miR-124-inhibitor rescued the expression of AURKA mRNA (Fig. 4A). When the expression of miR-124
was prevented by transfection with the miR-124-inhibitor, the
inhibitory effect of GANT61 on AURKA protein could be alleviated
(Fig. 4B and C). In addition, we
inhibited Gli2 in H4 cells and then knocked down miR-124 expression
using the miR-124 inhibitor. We found that after induction of
conditional expression of Gli2, a significant reduction in AURKA
mRNA was observed in H4 cells. Of note, knockdown of miR-124
abrogated Gli2-dependent suppression of AURKA mRNA expression
(Fig. 4D). Moreover, the
expression of AURKA protein was also rescued by co-transfection
with the miR-124-inhibitor (Fig. 4E
and F). These data indicate that miR-124 acts as a downstream
effector of Gli2, and the repression of AURKA through Gli2
inhibition is mediated by miR-124.
Gli2 enhances glioma cell proliferation
via the miR-124/AURKA axis
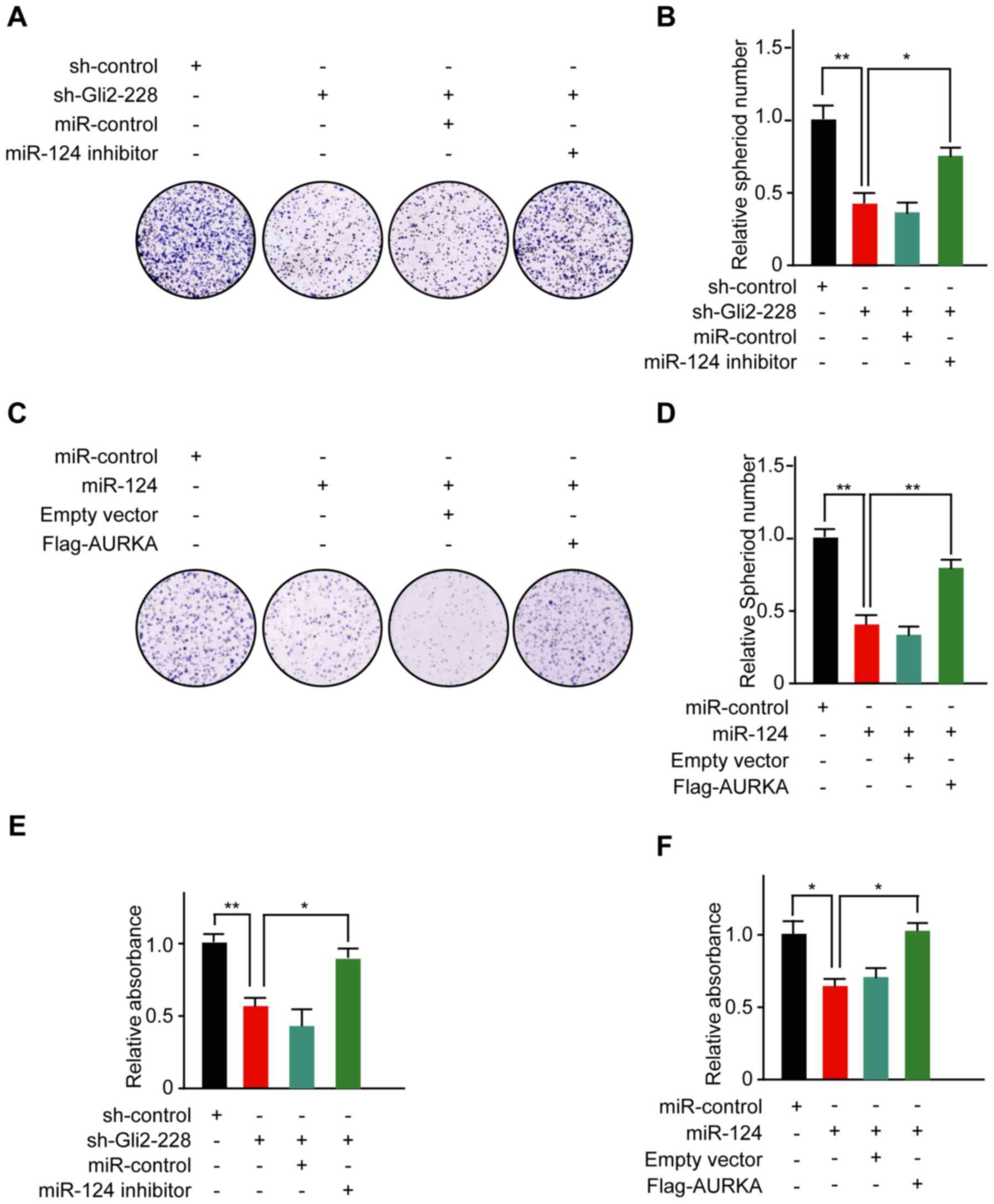
To illustrate the molecular mechanisms by which Hh
signaling regulates the proliferation process in glioma cells, we
inhibited the expression of Gli2 in H4 cells. Strikingly, the
number of colonies formed by H4 cells was significantly decreased
following Gli2 knockdown, while transfection with the miR-124
inhibitor rescued the proliferative ability of the cells (Fig. 5A and B). To functionally
characterize miR-124 in glioma cells, we upregulated miR-124 levels
by ectopically expressing miR-124 in H4 cells. Then, colony
formation assays were performed to assess the role of miR-124 in
cell proliferation. The cells transfected with miR-124 clearly grew
slower than in the control group, while co-transfection with the
AURKA construct upregulated the number of colonies in H4 cells
(Fig. 5C and D). In addition, an
MTT assay further supported the colony formation assay findings
(Fig. 5E and F). Collectively,
these results suggest that the Gli2/miR-124/AURKA axis can
influence the proliferation of human glioma cells.
Discussion
In this study, we determined that miR-124 acts as a
downstream effector of the Hh signaling pathway. Noteworthy, we
found that miR-124 potentially interacts with the 3′-UTR region of
AURKA. Further experiments showed that the Hh signaling pathway
regulated the expression of AURKA through miR-124, and
overexpression of miR-124 significantly decreased the expression of
AURKA and the proliferation of glioma cells. Our results suggest
that the Gli2/miR-124/AURKA axis is essential for the proliferation
and growth of human glioma cells.
The Gli transcription factors constitute the final
effectors of the Hh signaling pathway, which is frequently
hyperactivated in human cancers through multiple mechanisms. Hence,
targeting Gli may offer a highly effective therapeutic strategy for
the treatment of lethal tumors. Currently, there are multiple
studies aimed at assessing the efficacy of Gli inhibitors in
cancers. However, the downstream mechanisms initiated by Gli are
poorly understood, in part because relatively little is known about
the multiple specific genes directly regulated by Gli. Before the
discovery of noncoding RNAs, searches for transcription
factor-targeted genes were focused on protein-coding genes.
Intriguingly, it is worth noting that recently several
transcription factors have been discovered to regulate the
expression of miRNAs.
The central tumor suppressor p53 enhances the
transcriptional activity of the miR-34 family by binding to the
promoter of miR-34a (25). The
proto-oncogene c-myc encodes a transcription factor that regulates
cell proliferation and apoptosis, and a CHIP analysis has shown
that c-myc binds directly to the locus of a cluster of six miRNAs
on human chromosome 13 (26). In
addition, interactions between the Hh signaling pathway and miRNA
have been recently demonstrated by the discovery that several key
components of the Hh signaling pathway are regulated by miRNAs
(27–29). For example, in hepatic stellate
cells, the expression of Gli2 was markedly inhibited by miR-200a
(29). In addition, miR-324-5p
resulted in a reduction of Gli1 in MB cells (27). Moreover, miR-326 acts as a negative
modulator of the Hh signaling pathway by directly targeting Gli2
(30). These findings strongly
suggest that regulation of components of the Hh signaling pathway,
such as Glis, by miRNAs contributes to the functions of Hh
signaling.
In this study, we demonstrated that Gli2 can
directly modulate the expression of miR-124 by binding to one
binding site in the upstream region of the transcriptional start
site, thereby fine tuning the function of miR-124. Notably, most
studies have focused attention on protein-coding genes that can be
regulated by Hh signaling. Our findings suggest that, in addition
to many protein-coding genes, miRNAs can also be regulated by Gli2.
Our study raises the possibility that Gli2 functions as a global
modifier of gene expression through the regulation of miRNA
transcription. However, it is not known whether there are other
miRNAs that might be directly modulated by Gli2. Further
investigations will provide insight into how great a portion of the
pri-miRNAs are regulated by Gli2 to fully understand the regulatory
mechanism of Gli2 and miRNAs.
The biogenesis of miRNAs in mammalian systems is
composed of multiple steps, including transcription of primary
miRNA (pri-miRNA), cleavage of pri-miRNA to precursor miRNA
(pre-miRNA), nucleocytoplasmic transport of pre-miRNA and cleavage
of pre-miRNA to an miRNA duplex (31). Our findings suggested that miR-124
is transcriptionally regulated by Gli2 through binding of the
upstream region of the miR-124 transcription start site, which
contains a putative Gli2-binding element. This Gli2-mediated
transcriptional regulation of miR-124 is mediated through direct
binding of Gli2 to the upstream region of the transcriptional start
site for miR-124. These findings suggest that
transcription-dependent modulation of miRNA-124 biogenesis is
governed by Gli2. Apart from the important role of Gli2 as a
sequence-specific transcription factor, whether there is a
transcription-independent mechanism is unclear.
Consistent with Gli2 function, many signature
miRNAs, especially miR-124, are considered tumor-associated
molecules, and miR-124 expression is lost in diverse types of
tumors (32-34). miR-124 has previously been reported
to be downregulated in glioma. A significant difference was found
between glioma patients with a low miR-124 expression level, who
had distinctly shorter survival times, and patients with a high
miR-124 expression level (32).
miR-124 usually regulates its target genes at the
post-transcriptional level, and it is involved in multiple
biological processes, including proliferation and metastasis. In
renal clear cell carcinoma, miR-124 targets CAV1 and FlOT1 to
inhibit cell proliferation (35).
Through bioinformatic analysis and luciferase assays, we discovered
that miR-124 interacts with the 3′-UTR region of AURKA
(serine/threonine kinase, aurora kinase A). Moreover, AURKA was
downregulated by miR-124 overexpression and upregulated by miR-124
knockdown.
AURKA, also referred to as Aurora-2, BTAK, ARK1, and
STK15, maintains cell division by regulating centrosome separation,
bipolar spindle assembly, and chromosome segregation (36). AURKA dysfunction can cause
aneuploidy, mitotic arrest, genetic instability, poor histologic
differentiation, and poor prognosis in various types of cancers,
including colorectal, pancreatic, gastric, and breast cancers
(37). AURKA expression can
transform cells and drive tumor formation in mice (38). In addition, AURKA can block p53
function, thereby preventing cell apoptosis (39). Finally, AURKA has been shown to
cooperate with RAS to induce malignant transformation (40). AURKA is a target of several miRNAs
in various cancers. In non-small cell lung cancer, miR-32 can
suppress NSCLC by targeting AURKA (41). Furthermore, increased expression of
miR-25 downregulates the expression of the E3 ubiquitin ligase
FBXW7, resulting in elevated levels of AURKA (42). In this study, we identified miR-124
as a direct negative regulator of AURKA. miR-124 directly repressed
the expression of AURKA mRNA and protein through binding to one
binding site in the 3′-UTR of the human AURKA gene, thereby
negatively regulating AURKA functions. We have determined that
miR-124 influences glioma cell proliferation by targeting
AURKA.
In summary, our data indicate that aberrant
expression of miR-124 through Gli2 inhibition in glioma cells can
lead to the repression of AURKA, which can repress cell
proliferation in glioma cells. Our results highlight an additional
mechanism by which the Hh signaling pathway controls gene
expression and influences cancer progression, and they elucidate a
new mechanism through which the Hh signaling pathway regulates
glioma development.
Acknowledgments
This work was supported in part by grants from the
National Natural Science Foundation of China (31560314 to Q.L.) and
the Natural Science Foundation of Jiangxi Province (2016BAB204168
to Q.L.).
References
|
1
|
Sul J and Fine HA: Malignant gliomas: New
translational therapies. Mt Sinai J Med. 77:655–666. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Martín V, Herrera F, Carrera-Gonzalez P,
García-Santos G, Antolín I, Rodriguez-Blanco J and Rodriguez C:
Intracellular signaling pathways involved in the cell growth
inhibition of glioma cells by melatonin. Cancer Res. 66:1081–1088.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jiang J and Hui CC: Hedgehog signaling in
development and cancer. Dev Cell. 15:801–812. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ruiz i Altaba A: Gli proteins and Hedgehog
signaling: Development and cancer. Trends Genet. 15:418–425. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hui CC and Angers S: Gli proteins in
development and disease. Annu Rev Cell Dev Biol. 27:513–537. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bai CB, Auerbach W, Lee JS, Stephen D and
Joyner AL: Gli2, but not Gli1, is required for initial SHh
signaling and ectopic activation of the SHh pathway. Development.
129:4753–4761. 2002.PubMed/NCBI
|
|
7
|
Mo R, Freer AM, Zinyk DL, Crackower MA,
Michaud J, Heng HH, Chik KW, Shi XM, Tsui LC, Cheng SH, et al:
Specific and redundant functions of Gli2 and Gli3 zinc finger genes
in skeletal patterning and development. Development. 124:113–123.
1997.PubMed/NCBI
|
|
8
|
Brechbiel J, Miller-Moslin K and Adjei AA:
Crosstalk between hedgehog and other signaling pathways as a basis
for combination therapies in cancer. Cancer Treat Rev. 40:750–759.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Santoni M, Burattini L, Nabissi M, Morelli
MB, Berardi R, Santoni G and Cascinu S: Essential role of Gli
proteins in glioblastoma multiforme. Curr Protein Pept Sci.
14:133–140. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Karhadkar SS, Bova GS, Abdallah N, Dhara
S, Gardner D, Maitra A, Isaacs JT, Berman DM and Beachy PA:
Hedgehog signalling in prostate regeneration, neoplasia and
metastasis. Nature. 431:707–712. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee EY, Ji H, Ouyang Z, Zhou B, Ma W,
Vokes SA, McMahon AP, Wong WH and Scott MP: Hedgehog
pathway-regulated gene networks in cerebellum development and
tumorigenesis. Proc Natl Acad Sci USA. 107:9736–9741. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ferruzzi P, Mennillo F, De Rosa A,
Giordano C, Rossi M, Benedetti G, Magrini R, Pericot Mohr G,
Miragliotta V, Magnoni L, et al: In vitro and in vivo
characterization of a novel Hedgehog signaling antagonist in human
glioblastoma cell lines. Int J Cancer. 131:E33–E44. 2012.
View Article : Google Scholar
|
|
13
|
Von Hoff DD, LoRusso PM, Rudin CM, Reddy
JC, Yauch RL, Tibes R, Weiss GJ, Borad MJ, Hann CL, Brahmer JR, et
al: Inhibition of the hedgehog pathway in advanced basal-cell
carcinoma. N Engl J Med. 361:1164–1172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lauth M, Bergström A, Shimokawa T and
Toftgård R: Inhibition of GLI-mediated transcription and tumor cell
growth by small-molecule antagonists. Proc Natl Acad Sci USA.
104:8455–8460. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Varnat F, Duquet A, Malerba M, Zbinden M,
Mas C, Gervaz P and Ruiz i Altaba A: Human colon cancer epithelial
cells harbour active HEDGEHOG-GLI signalling that is essential for
tumour growth, recurrence, metastasis and stem cell survival and
expansion. EMBO Mol Med. 1:338–351. 2009. View Article : Google Scholar
|
|
16
|
Adams BD, Kasinski AL and Slack FJ:
Aberrant regulation and function of microRNAs in cancer. Curr Biol.
24:R762–R776. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lewis BP, Burge CB and Bartel DP:
Conserved seed pairing, often flanked by adenosines, indicates that
thousands of human genes are microRNA targets. Cell. 120:15–20.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Croce CM: Causes and consequences of
microRNA dysregulation in cancer. Nat Rev Genet. 10:704–714. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yan R, Peng X, Yuan X, Huang D, Chen J, Lu
Q, Lv N and Luo S: Suppression of growth and migration by blocking
the Hedgehog signaling pathway in gastric cancer cells. Cell Oncol
(Dordr). 36:421–435. 2013. View Article : Google Scholar
|
|
20
|
Ciafrè SA, Galardi S, Mangiola A, Ferracin
M, Liu CG, Sabatino G, Negrini M, Maira G, Croce CM and Farace MG:
Extensive modulation of a set of microRNAs in primary glioblastoma.
Biochem Biophys Res Commun. 334:1351–1358. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Silber J, Lim DA, Petritsch C, Persson AI,
Maunakea AK, Yu M, Vandenberg SR, Ginzinger DG, James CD, Costello
JF, et al: miR-124 and miR-137 inhibit proliferation of
glioblastoma multiforme cells and induce differentiation of brain
tumor stem cells. BMC Med. 6:142008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xia H, Cheung WK, Ng SS, Jiang X, Jiang S,
Sze J, Leung GK, Lu G, Chan DT, Bian XW, et al: loss of
brain-enriched miR-124 microRNA enhances stem-like traits and
invasiveness of glioma cells. J Biol Chem. 287:9962–9971. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Goldenson B and Crispino JD: The aurora
kinases in cell cycle and leukemia. Oncogene. 34:537–545. 2015.
View Article : Google Scholar
|
|
24
|
Tang X, Deng L, Chen Q, Wang Y, Xu R, Shi
C, Shao J, Hu G, Gao M, Rao H, et al: Inhibition of Hedgehog
signaling pathway impedes cancer cell proliferation by promotion of
autophagy. Eur J Cell Biol. 94:223–233. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Raver-Shapira N, Marciano E, Meiri E,
Spector Y, Rosenfeld N, Moskovits N, Bentwich Z and Oren M:
Transcriptional activation of miR-34a contributes to p53-mediated
apoptosis. Mol Cell. 26:731–743. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
O'Donnell KA, Wentzel EA, Zeller KI, Dang
CV and Mendell JT: c-Myc-regulated microRNAs modulate E2F1
expression. Nature. 435:839–843. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ferretti E, De Smaele E, Miele E, Laneve
P, Po A, Pelloni M, Paganelli A, Di Marcotullio L, Caffarelli E,
Screpanti I, et al: Concerted microRNA control of Hedgehog
signalling in cerebellar neuronal progenitor and tumour cells. EMBO
J. 27:2616–2627. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hyun J, Wang S, Kim J, Rao KM, Park SY,
Chung I, Ha CS, Kim SW, Yun YH and Jung Y: MicroRNA-378 limits
activation of hepatic stellate cells and liver fibrosis by
suppressing Gli3 expression. Nat Commun. 7:109932016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yu F, Zheng Y, Hong W, Chen B, Dong P and
Zheng J: MicroRNA-200a suppresses epithelial-to-mesenchymal
transition in rat hepatic stellate cells via GLI family zinc finger
2. Mol Med Rep. 12:8121–8128. 2015.PubMed/NCBI
|
|
30
|
Jiang Z, Cushing L, Ai X and Lü J: miR-326
is downstream of Sonic hedgehog signaling and regulates the
expression of Gli2 and smoothened. Am J Respir Cell Mol Biol.
51:273–283. 2014.PubMed/NCBI
|
|
31
|
Winter J, Jung S, Keller S, Gregory RI and
Diederichs S: Many roads to maturity: microRNA biogenesis pathways
and their regulation. Nat Cell Biol. 11:228–234. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen T, Wang XY, Li C and Xu SJ:
Downregulation of microRNA-124 predicts poor prognosis in glioma
patients. Neurol Sci. 36:131–135. 2015. View Article : Google Scholar
|
|
33
|
Hu CB, Li QL, Hu JF, Zhang Q, Xie JP and
Deng L: miR-124 inhibits growth and invasion of gastric cancer by
targeting ROCK1. Asian Pac J Cancer Prev. 15:6543–6546. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Peng XH, Huang HR, Lu J, Liu X, Zhao FP,
Zhang B, Lin SX, Wang L, Chen HH, Xu X, et al: MiR-124 suppresses
tumor growth and metastasis by targeting Foxq1 in nasopharyngeal
carcinoma. Mol Cancer. 13:1862014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Butz H, Szabó PM, Khella HW, Nofech-Mozes
R, Patocs A and Yousef GM: miRNA-target network reveals miR-124as a
key miRNA contributing to clear cell renal cell carcinoma
aggressive behaviour by targeting CAV1 and FLOT1. Oncotarget.
6:12543–12557. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Glover DM, Leibowitz MH, McLean DA and
Parry H: Mutations in aurora prevent centrosome separation leading
to the formation of monopolar spindles. Cell. 81:95–105. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gautschi O, Heighway J, Mack PC, Purnell
PR, Lara PN Jr and Gandara DR: Aurora kinases as anticancer drug
targets. Clin Cancer Res. 14:1639–1648. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bischoff JR, Anderson L, Zhu Y, Mossie K,
Ng L, Souza B, Schryver B, Flanagan P, Clairvoyant F, Ginther C, et
al: A homologue of Drosophila aurora kinase is oncogenic and
amplified in human colorectal cancers. EMBO J. 17:3052–3065. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Katayama H, Sasai K, Kawai H, Yuan ZM,
Bondaruk J, Suzuki F, Fujii S, Arlinghaus RB, Czerniak BA and Sen
S: Phosphorylation by aurora kinase A induces Mdm2-mediated
destabilization and inhibition of p53. Nat Genet. 36:55–62. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tatsuka M, Sato S, Kitajima S, Suto S,
Kawai H, Miyauchi M, Ogawa I, Maeda M, Ota T and Takata T:
Overexpression of Aurora-A potentiates HRAS-mediated oncogenic
transformation and is implicated in oral carcinogenesis. Oncogene.
24:1122–1127. 2005. View Article : Google Scholar
|
|
41
|
Ma ZL, Zhang BJ, Wang DT, Li X, Wei JL,
Zhao BT, Jin Y, Li YL and Jin YX: Tanshinones suppress AURKA
through up-regulation of miR-32 expression in non-small cell lung
cancer. Oncotarget. 6:20111–20120. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li Z, Sun Y, Chen X, Squires J,
Nowroozizadeh B, Liang C and Huang J: p53 mutation directs AURKA
overexpression via miR-25 and FBXW7 in prostatic small cell
neuroendocrine carcinoma. Mol Cancer Res. 13:584–591. 2015.
View Article : Google Scholar :
|