Introduction
Pancreatic cancer has no early symptoms and is
generally silent until the disease is advanced. During the
progression of pancreatic carcinomas, cancer cells frequently
released from the surface of the tumor can adhere to and invade
tissues and organs in the peritoneal cavity. Patients have a median
survival of 4–8 months after the diagnosis of pancreatic cancer,
partly because of the advanced stage of the disease at the time of
diagnosis and start of treatment (1–3).
Notably, peritoneal dissemination is a frequent occurrence in
pancreatic cancer, which is associated with a poor prognosis. It is
therefore necessary to discover new therapies or therapeutic
combinations that control peritoneal dissemination in order to
significantly alter the poor outcome of this deadly disease. The
anti-metabolite agent gemcitabine is currently used to treat
pancreatic cancer (4,5). While gemcitabine has shown a
significant benefit in clinical applications, its ability to treat
pancreatic cancer is limited. Furthermore, the more recently
approved chemotherapy combinations folic acid, fluorouracil,
irinotecan, and oxaliplatin (FOLFIRINOX) and nab-paclitaxel plus
gemcitabine only modestly improve survival in advanced pancreatic
cancer (6–9). Therefore, new therapeutic approaches
need to be investigated to improve the treatment of this deadly
neoplasm.
Hepatocyte growth factor (HGF) was originally
identified as a liver mitogen and fibroblast-derived epithelial
motility factor, and it is the only physiological ligand for the
MET receptor tyrosine kinase (RTK) (10). Both levels of HGF and MET are
elevated in multiple cancers, including pancreatic ductal
adenocarcinoma (PDAC) (11), and
are associated with increased tumor cell invasion, distant
metastases, and a poor prognosis (10,12).
An important process in cancer-cell metastasis is the conversion of
epithelial cells to a migratory phenotype, a phenomenon known as
epithelial-mesenchymal transition (EMT) (13,14).
HGF-induced scattering of Madin-Darby canine kidney cells is a
routinely used model of EMT, in which actin cytoskeletal
rearrangement is known to be dependent on Rho family GTPases
(15,16). A model of HGF-induced EMT has been
developed using the human prostate cancer cell line DU145.
HGF-induced scattering of DU145 cells was dependent on the activity
of Rho family GTPases and on a phosphatidylinositide 3-kinase
pathway (17).
Crizotinib is a small-molecule inhibitor that is
selective for MET as well as anaplastic lymphoma kinase (ALK) and
ROS1 (18–21). It has been shown to inhibit cell
proliferation, migration, and invasion of several tumor cell lines
in vitro and it has also displayed significant antitumor
activity in xenograft mouse models (22–24).
In order to elucidate the mechanism of crizotinib on
the inhibition of PDAC progression, we evaluated the effect of the
drug on cell proliferation and invasion in vitro and
peritoneal dissemination in vivo. Our results showed that
crizotinib may be a potent drug for treating peritoneal
dissemination of PDAC by inhibiting cancer cell proliferation and
invasion.
Materials and methods
Cancer cell lines, animals, and
ethics
AsPC-1, BxPC-3, H-48-N, KP-2, KP-3, MIAPaCa-2,
Panc-1 and Suit-2 are cell lines that were derived from a human
PDAC. AsPC-1, BxPC-3, H-48-N, MIAPaCa-2 and Panc-1 were obtained
from American Type Culture Collection (Manassas, VA, USA). KP-2,
KP-3 and Suit-2 were obtained from the Japanese Collection of
Research Bioresources Cell Bank (Osaka, Japan). The human prostate
cancer cell line PC3, non-small cell lung cancer cell line A549 and
breast cancer cell line MCF-7 were obtained from American Type
Culture Collection. All cell lines were cultured in RPMI-1640
medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% FBS
and 50 U/ml penicillin and 50 μg/ml streptomycin in a
humidified atmosphere under 5% CO2 at 37°C. Male nude
mice (BALB/c nu/nu), 8 weeks of age (Clea Japan, Tokyo, Japan) were
kept under specific pathogen-free conditions. This study was
approved by the Institutional Animal Care and Use Committee
(permission no. 26-02) and carried out according to the National
Kyushu Cancer Center Animal Experimentation Regulation.
Drug sensitivity test
Human PDAC were seeded into 96-well plates
(1.25×103 cells/50 μl) and allowed to attach for
24 h. Cells were treated with crizotinib (0.03–10 μM) (LC
Laboratories, Woburn, MA, USA) for 24, 48 and 72 h in 100 μl
medium. At the end of drug exposure, 20 μl of
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
(MTS) reagent (Promega, Madison, WI, USA) was added and cells were
incubated in humidified 5% CO2 atmosphere. After 2 h,
spectrophotometric readings were taken for each sample according to
the manufacturer's instructions. Cell growth inhibition was
expressed as a percentage of the absorbance of control cultures
measured at 492 nm with a microplate reader. The 50% inhibitory
concentration of cell growth (IC50) was calculated by a
sigmoidal dose-response curve (GraphPad PRISM, San Diego, CA,
USA).
Reverse transcription and quantitative
real-time polymerase chain reaction (PCR)
Reverse transcription was performed using the
ThermoScript RT-PCR system (Invitrogen) according to the
manufacturer's instructions. Quantitative real-time PCR (qRT-PCR)
was performed using the ABI PRISM 7000 Sequence Detection system
(Applied Biosystems, Foster City, CA, USA) and SYBR Premix Ex Taq
(Takara Bio, Otsu, Shiga, Japan). The human gene-specific primers
were 5′-TACAGGGGCACTGTCAATACC-3′ and 5′-GGATACTGAGAATCCCAACGC-3′
for HGF, 5′-GCCCTCTGGAAGGTACATTGC-3′ and
5′-GAGCACTGTCCAACCATGCTT-3′ for ALK, 5′-TGGTGCAGAGGAGCAATGG-3′ and
5′-CATTCTGGATGGGTGTTTCCG-3′ for MET, 5′-CCACATAATCTGAGTGAACCGTG-3′
and 5′-CGCTGCTACAGCCAACCTC-3′ for ROS1, and
5′-CATGTACGTTGCTATCCAGGC-3′ and 5′-CTCCTTAATGTCACGCACGAT-3′ for
β-actin.
Immunoblot analysis
Anti-MET, p-MET (Tyr1234/1235), AKT, p-AKT (Ser473),
MAPK, and p-MAPK (Tyr202/204) antibodies were obtained from Cell
Signaling Technology (Beverly, MA, USA), and anti-β-actin antibody
was obtained from Biovision (Mountain View, CA, USA). Protein
samples were separated by sodium dodecyl sulfate-polyacrylamide gel
electro phoresis and then transferred to polyvinylidene difluo-ride
membranes (Millipore, Billerica, MA, USA). Following blocking, the
membrane was blotted with the appropriate antibody, and
subsequently, horseradish peroxi dase-conjugated anti-mouse or
anti-rabbit IgG (Santa Cruz Biotechnology, Dallas, TX, USA) was
applied. The final signal was revealed by ECL chemiluminescence (GE
Healthcare Bio-sciences, Pittsburgh, PA, USA). Digital images were
analyzed with ImageJ software to measure the density of each band
without a saturated signal.
RNA interference
Silencer Select Pre-designed siRNAs designed to
target MET (si-MET), ROS1 (si-ROS1), and ALK (si-ALK), and negative
controls designed not to target any known human gene (si-NC) were
purchased from Thermo Fisher Scientific (Waltham, MA, USA). For the
silencing assay, we transfected 1×106 of human PDAC
cells with 15 μl of stock Silencer Select siRNA duplexes (5
μM) using Lipofectamine RNAiMAX solution (Thermo Fisher
Scientific) in a 100-mm diameter culture dish. We harvested total
RNA from transfected cells 48 h after transfection to perform gene
expression profiling.
Cell invasion assays
Transwell cell invasion was evaluated using a
24-well chemotaxis chamber with membranes with 8-μm pores
(BD Biosciences, Franklin Lakes, NJ, USA). RNA interference for
Suit-2 cells was done 24 h before seeding into the upper chambers
as needed. Next, Suit-2 cells were incubated in serum-free culture
medium with either DMSO (solvent) or crizotinib (0.1 or 1
μM) for 24 h, transferred to the upper chambers
(2.5×105 cells/500 μl) and allowed to migrate
through Matrigel-coated (8.7 mg/ml) membranes for 24 h. The lower
chambers were filled with culture medium containing 10% FBS,
without or with HGF (50 ng/ml), and with the same concentrations of
crizotinib as in the upper chambers. Non-migrated cells were wiped
off with a cotton swab, the filter was stained with Diff-Quik stain
solution (Siemens, Munich, Germany), and the number of remaining
cells was counted under a microscope.
Rho pull-down assay
The Rho pull-down assay was performed using a Rho
activation assay kit according to the manufacturer's instructions
(Cytoskeleton, Denver, CO, USA). Briefly, cells
(3×105/ml) were cultured under serum-free conditions
without or with crizotinib (0.1 and 1 μM) for 24 h. After
incubation, the cells were stimulated with HGF (50 ng/ml) for 60
min and lysed in Mg2+ lysis buffer. Equal volumes of
cell lysates were incubated with Rhotekin-RBD beads. Bound RhoA
proteins were detected by western blotting using a monoclonal
antibody against RhoA. Western blotting of the total amount of RhoA
in cell lysates was performed for comparison with Rho activity
(level of GTP-bound Rho) in the same samples.
In vivo peritoneal dissemination model of
pancreatic cancer
Five-week-old male nude mice (BALB-cAJcl-nu/nu, Clea
Japan) were housed in filtered-air, laminar-flow cabinets and were
manipulated using aseptic procedures. To prepare the in vivo
peritoneal dissemination model, Suit-2 cells were injected i.p. as
a cell suspension into nude mice (1×106 cells in 200
μl PBS per animal). This model using Suit-2 is not only
simple and reproducible but also has characteristics that resemble
those of human pancreatic cancer. The treatment regimens started on
the day of tumor inoculation and continued for 3 weeks. Crizotinib
was delivered using a vehicle (sterile saline with 0.5%
methylcellulose suspension) and given by oral gavage every day. The
daily dose of crizotinib used was 50 mg/kg/d (18–20).
At the end of the treatment period, the mice were sacrificed. The
volume of ascites was measured and tumor tissue was excised,
weighed, fixed in 10% neutral buffered formalin, and embedded in
paraffin. Paraffin sections (5 μm) were stained with
hematoxylin and eosin. Blood samples were collected from the left
heart ventricle and assayed for serum CA19-9.
Statistical analysis
Results are reported as the mean ± SD of triplicates
unless otherwise stated. Group comparisons were performed using
one-way analysis of variance (ANOVA) followed by an unpaired
Student's t-test. Significantly differentially expressed genes were
analyzed by Spearman's rank correlation. Differences between groups
were considered statistically significant at P<0.05. Tests were
done in triplicates unless otherwise noted.
Results
Cytotoxic effect of crizotinib on PDAC
cells
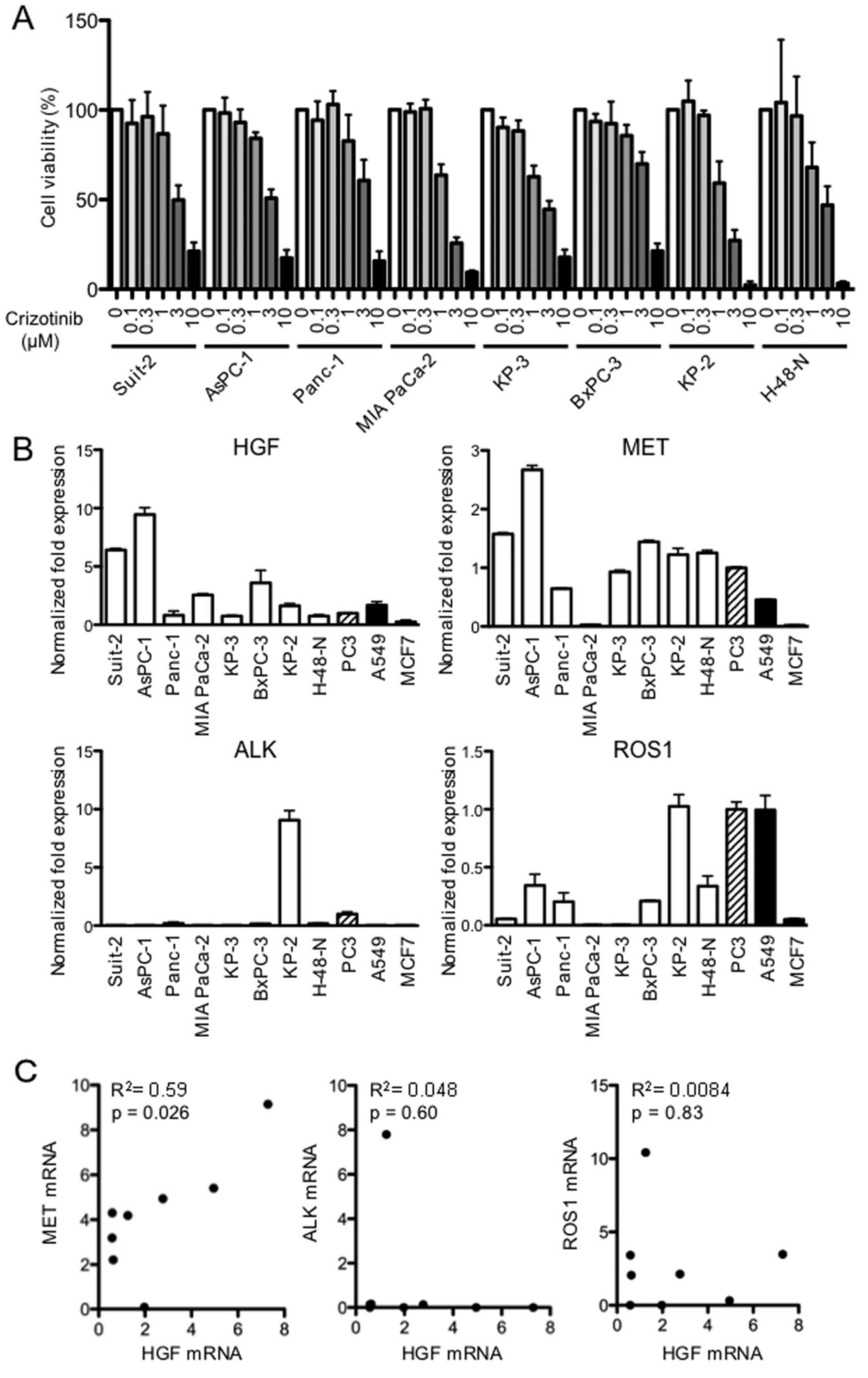
The effect of crizotinib on PDAC cell growth was
examined in vitro using the MTS assay. Treatment with
crizotinib (0.1–10 μM) resulted in a dose-dependent
reduction of cell growth after 48 h of treatment (Fig. 1A) and the IC50 was
calculated to be in the range of 1.4–4.3 μM (Table I). Next, we examined the mRNA
expression levels of HGF, MET, ALK and ROS1, which are the known
targets of crizotinib. Most of the PDAC cells expressed substantial
levels of HGF and MET mRNA compared with the levels expressed by
the human prostate cancer cell line PC3 used as a control cell line
(Fig. 1B). Furthermore, the mRNA
expression of HGF and MET were positively correlated in the PDAC
cells (P=0.026). However, the mRNA expression of HGF and ALK, or
HGF and ROS1 were not correlated in the same PDAC cells (Fig. 1C).
 | Table ICrizotinib inhibition
(IC50) of the growth of PDAC cells. |
Table I
Crizotinib inhibition
(IC50) of the growth of PDAC cells.
| Cell lines | IC50
(μM) |
|---|
| Suit-2 | 3.4±0.9 |
| AsPC-1 | 3.2±0.3 |
| Panc-1 | 3.5±1.0 |
| MIA PaCa-2 | 1.5±0.3 |
| KP-3 | 2.1±0.5 |
| BxPC-3 | 4.3±0.6 |
| KP-2 | 1.4±0.5 |
| H-48-N | 2.1±0.8 |
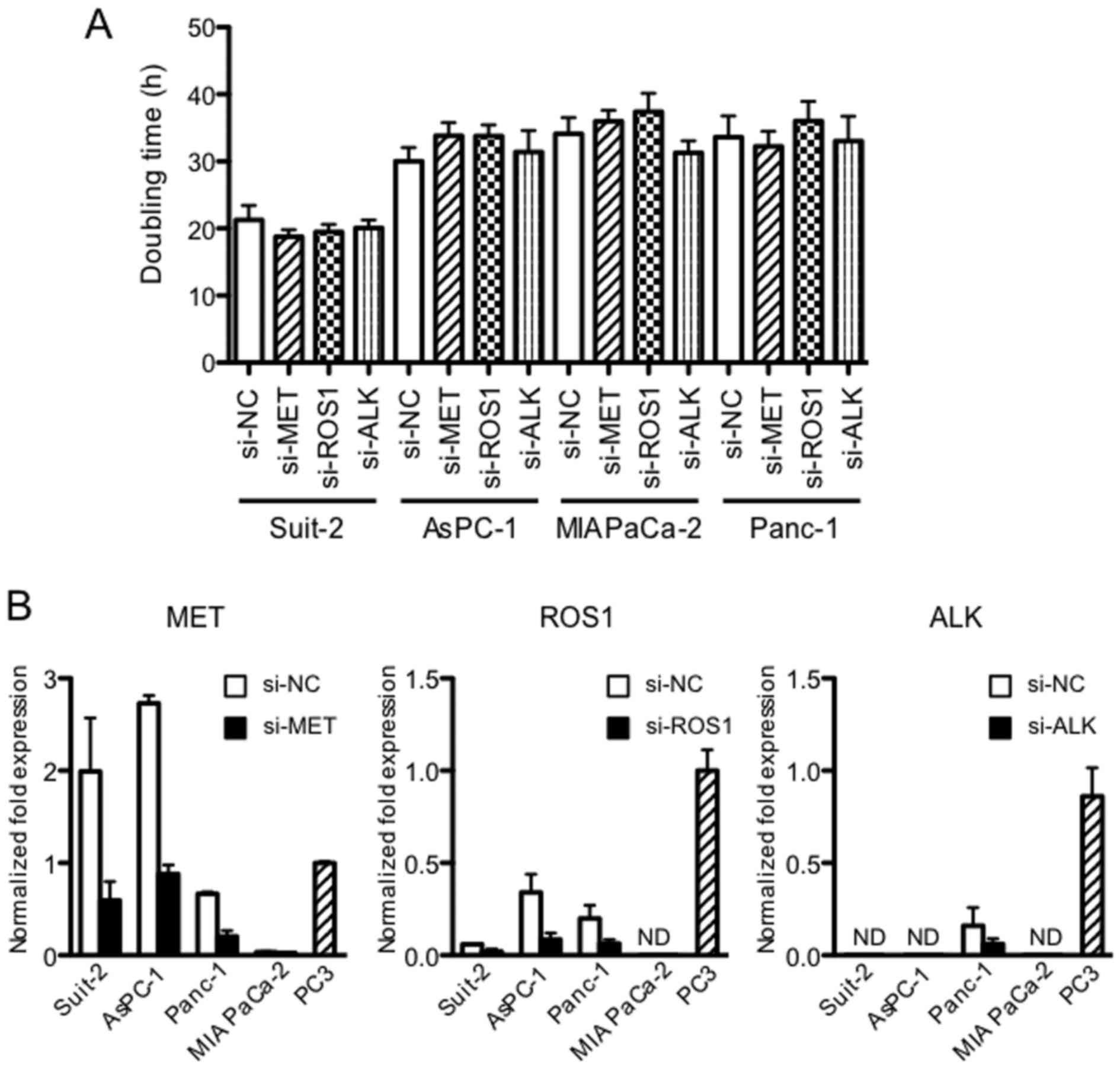
Downregulation of MET, ROS1 and ALK
kinases by siRNA does not affect the proliferation of PDAC cell
lines
Because dysregulated RTKs induce tumor growth and
metastasis, we tested the effect of MET, ROS1 and ALK
downregulation on proliferation of the PDAC cells. It was found
that the proliferation of four PDAC cell lines, assessed by
doubling time, was unchanged by the downregulation of MET, ROS1 and
ALK (Fig. 2A). Expression of MET,
ROS1 and ALK measured at 48 h after siRNA transfection was reduced
by 70–80% (P<0.05) (Fig.
2B).
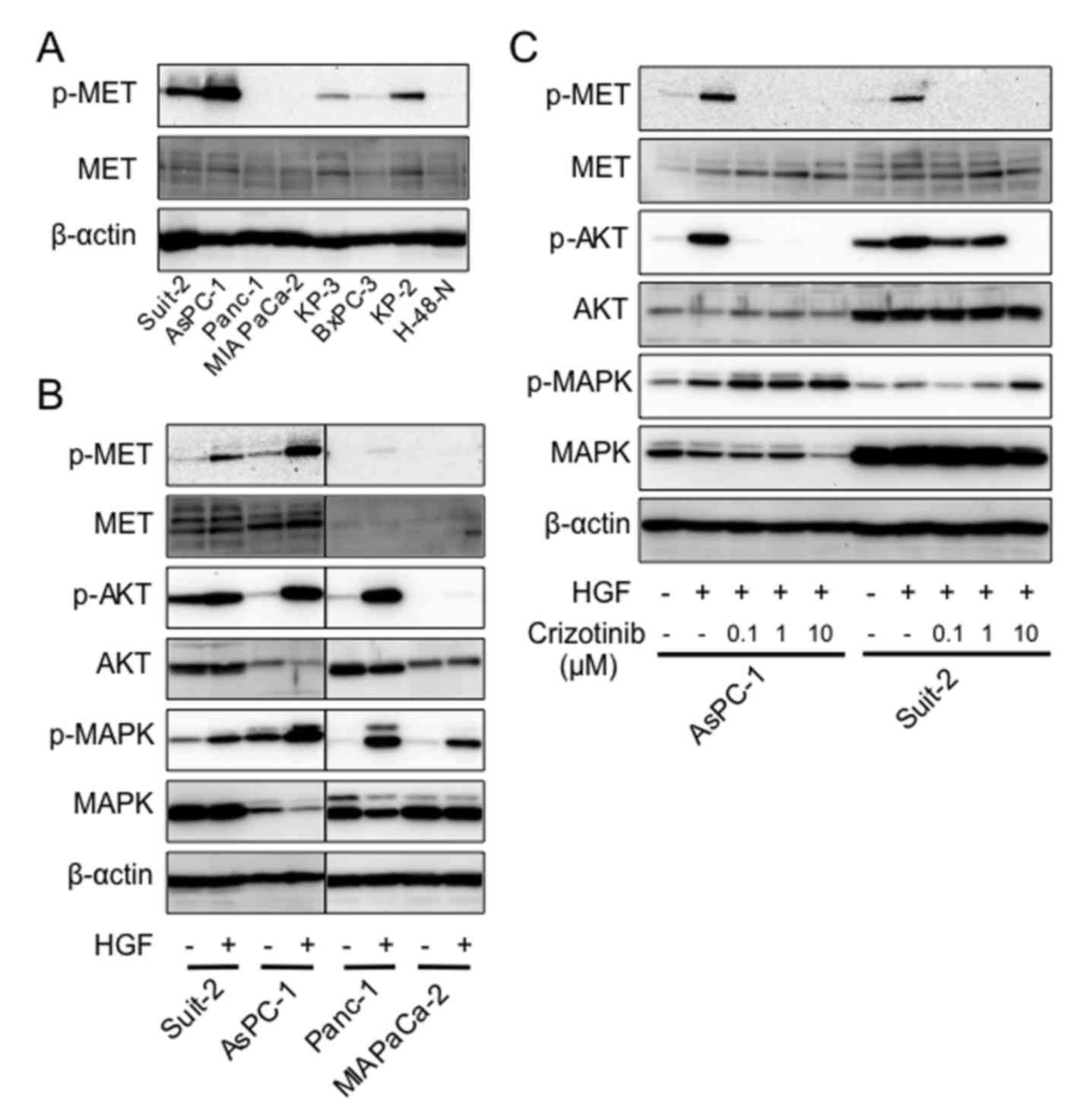
Phosphorylation of key regulatory factors
in HGF/MET signaling in PDAC cells
As a considerable amount of HGF and MET mRNA was
expressed in PDAC cells (Fig. 1B),
we then examined the phosphorylation of the key regulatory factors
in HGF/MET signaling. Phosphorylation of MET was higher in AsPC-1
and Suit-2 than in the other PDAC cell lines under normal culture
conditions (Fig. 3A). We next
examined whether the phosphorylation of MET was induced by the
addition of HGF in PDAC cells. Results showed that phosphorylation
was strongly induced in Suit-2 and AsPC-1, slightly induced in
Panc-1, and not induced in MIA PaCa-2 (Fig. 3B). We next examined whether
HGF-induced phosphorylation of the key regulatory factors in
HGF/MET signaling was inhibited by crizotinib in AsPC-1 and Suit-2.
Phosphorylation of MET was inhibited at the concentration of 0.1
μM in both cell lines, and phosphorylation of AKT was
inhibited at 0.1 μM and 10 μM of crizotinib in AsPC-1
and Suit-2, respectively. However, phosphorylation of MAPK was not
inhibited in both cell lines (Fig.
3C).
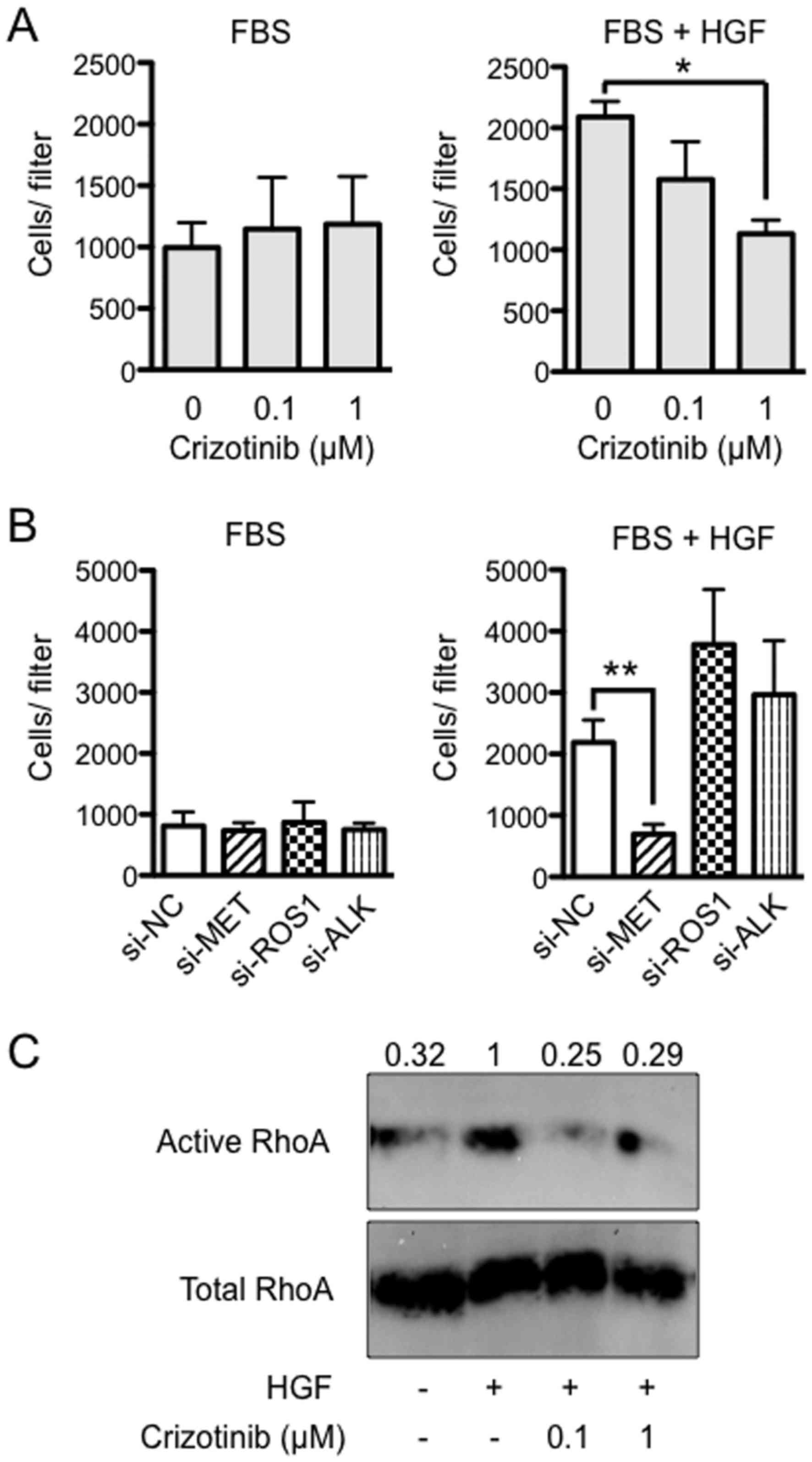
Importance of HGF/MET signaling in
invasion by Suit-2
To assess the effect of crizotinib on cell invasion,
Suit-2 cells were seeded into Matrigel chambers in serum-free
culture medium with either DMSO (solvent) or crizotinib (0.1 or 1
μM) and allowed to migrate into the medium in the lower
chamber containing 10% FBS and with the same concentrations of
crizotinib as in the upper chambers. It was found that there was no
significance between the groups (Fig.
4A, left). However, when medium was supplemented with 50 ng/ml
of HGF in the lower chambers, HGF-induced invasion was completely
blocked by 1 μM of crizotinib (P<0.05) (Fig. 4A, right). Next, to determine which
RTK was critical to the HGF-induced invasion, Suit-2 cells were
treated with MET, ROS1 and ALK siRNA, and allowed to migrate into
the medium containing FBS in the lower chamber. It was found that
there was no significant difference between the groups (Fig. 4B, left). However, when HGF was
added to FBS, HGF-induced invasion was not blocked by treatment
with NC, ROS1 and ALK siRNAs, while it was completely blocked by
treatment with MET siRNA (P<0.01) (Fig. 4B, right). Expression of MET, ROS1
and ALK measured at 48 h after siRNA transfection was reduced by
70–80% (P<0.05, data not shown). Next, to evaluate whether HGF
induces RhoA activity in Suit-2 cells, we used a pull-down assay
with the fusion protein GST-Rhotekin-RBD, which recognizes only
RhoA-GTP, the active form of RhoA. An increase in RhoA-GTP was
observed in Suit-2 treated for 1 h with HGF (50 ng/ml).
Furthermore, the activation of RhoA was suppressed by the addition
of crizotinib (0.1 or 1 μM) (Fig. 4C).
Crizotinib inhibits the peritoneal
dissemination of Suit-2 cells
To examine the effect of crizotinib on peritoneal
dissemination in vivo, we used a pancreatic cancer model
with i.p. carcinomatosis in nude mice. We started the
administration of crizotinib on the day of inoculation of cancer
cells. Preliminary experiments revealed that tumor-bearing mice
began to exhibit abdominal swelling with ascites ~2.5 weeks after
the inoculation of cancer cells and died with cachexia after the
fifth week without any treatment. Therefore, we sacrificed and
examined the mice 3 weeks after the inoculation of cancer cells.
Fig. 5A shows the effects of
crizotinib treatment on ascites formation. The mean volume of
ascites was significantly reduced (by ~60%) in the group given 50
mg/kg/d compared with the untreated group (1.2±1.7 vs. 3.3±1.0 ml;
P<0.01). The mean tumor weight on the peritoneum was
significantly reduced (by ~30%) in the treatment group given 50
mg/kg/d compared with the untreated group (0.67±0.22 vs. 0.91±0.19
g; P<0.05) (Fig. 5B). The
concentration of CA19-9, which is expressed by Suit-2, was examined
in the sera collected from the left heart ventricle (Fig. 5C). The mean concentration of CA19-9
was significantly reduced (by ~85%) in the group given 50 mg/kg/d
of crizotinib compared with the untreated group (4.0±6.1 vs.
27.3±23.3 U/ml; P<0.05). At autopsy examination, tumors were
found on the surface of the peritoneum, diaphragm, intestines,
liver, spleen, pancreas, and kidney, with massive ascites in the
control group. Histological appearance of the tumor nests that
formed after dissemination of tumor cells in the peritoneum
(Fig. 5D) and to the pancreas
(Fig. 5F) and liver (Fig. 5G) from the untreated group showed
how the tumor extensively invaded the peritoneum.
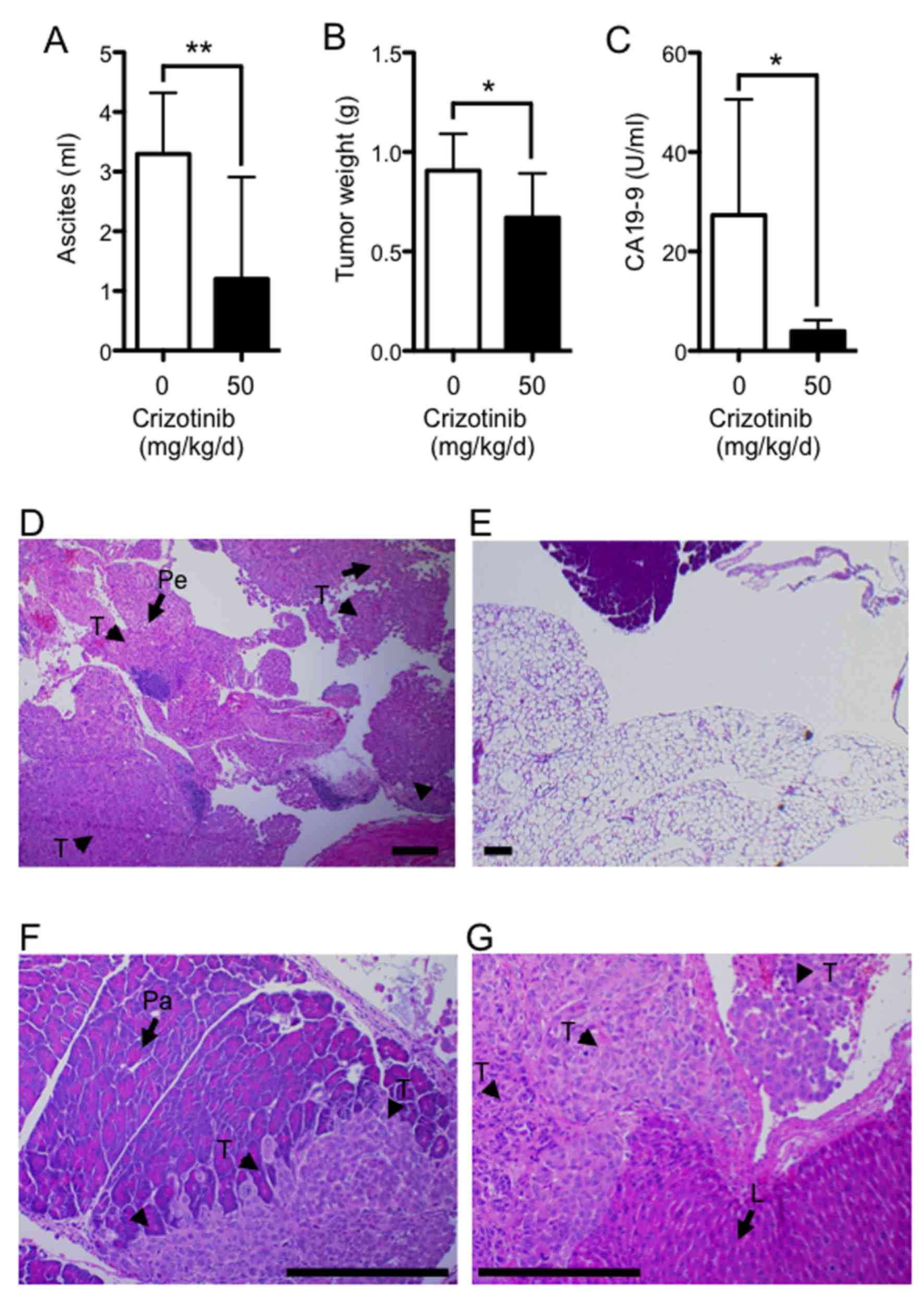 | Figure 5Crizotinib inhibits peritoneal
dissemination of Suit-2. Suit-2 cell suspension
(1×106/200 μl/mouse) was injected i.p. into nude
mice and treated with crizotinib (50 mg/kg) daily. The mice were
sacrificed at 21 days after implantation, and ascites, peritonea
and blood sera were collected. The volume of ascites (A), weight of
tumor that had disseminated on peritonea (B) and the concentrations
of CA19-9 (C) were measured (n=8). **P<0.01,
*P<0.05. Histologic appearance of disseminated tumor
nests in the peritoneum (D), pancreas (F) and liver (G) of
Suit-2-implanted nude mouse and peritoneum (E) of mouse treated
with crizotinib. Nude mice were sacrificed at 21 days after i.p.
injection of Suit-2 cells. Collected tissues were fixed, sectioned,
and stained with hematoxylin and eosin. T, tumor nest (arrowhead);
Pe, peritoneum (arrow); Pa, pancreas (arrow); L, liver (arrow);
bar, 200 μm in D–F; 800 μm in G. |
Discussion
Peritoneal dissemination frequently occurs in
pancreatic cancer, which is associated with a poor prognosis
(1–3). MET is associated with the progression
of pancreatic cancer (10–12); therefore, we evaluated the effect
of the MET inhibitor, crizotinib, on peritoneal dissemination of
pancreatic cancer. Crizotinib inhibited the growth of 8 pancreatic
cancer cell lines with the IC50 ranging from 1.4 to 4.3
μM. Invasion of a pancreatic cancer cell line, Suit-2, was
suppressed in vitro at a concentration of 1.0 μM,
which is sufficient for the inhibition of MET phosphorylation. This
effect on cell invasion was also recapitulated by the reduction of
MET expression in Suit-2 with siRNA. Crizotinib also inhibited RhoA
activation in addition to MET phosphorylation. We further evaluated
the effect of crizotinib on peritoneal dissemination of pancreatic
cancer in vivo. Crizotinib reduced tumor burden and ascites
accumulation due to peritoneal dissemination after inoculation of
Suit-2.
The effect of crizotinib on PDAC cell proliferation
was evaluated in vitro. The IC50 values were
calculated to be in the range of 1.4–4.3 μM, which was
comparable to the IC50 values of uveal melanoma cells in
the range of 0.75–2 μM (24), IC50 values of thyroid
cancer cells in the range of 2–3.5 μM, IC50 value
of MDA-MB-231 breast cancer cell (2.8 μM), and the
IC50 value of HT-29 colon cancer cell (2.6 μM)
(25). MET-negative MDA-MB-435
melanoma cells had a minimal response to crizotinib, not reaching
the IC50 value even in the presence of the maximal drug
dose (10 μM) (25). We then
examined the relationship between the expression of MET, ROS1 and
ALK and the effect of crizotinib on PDAC cell proliferation. To do
this, we chose highly MET-expressing Suit-2 and AsPC-1, and
marginally MET-expressing Panc-1 and MIA PaCa-2, as these 4 PDAC
cell lines showed similar IC50 values. While expression
of each RTK was downregulated by siRNA, the doubling time of each
cell line was not changed in any of the cases, suggesting that the
cell proliferation of PDAC cells was not controlled through only
one of the three RTKs targeted by crizotinib. At this point, since
no specific ligand for ALK or ROS1 has been found, it is impossible
to conduct an experiment to observe the effect of adding ligand to
PDAC cells expressing ALK or ROS1. If a specific ligand for them is
found in the future, it will lead to more accurate prediction of
the efficacy of crizotinib. However, it may actually be difficult
as it could vary depending on a variety of factors related to the
drug resistance mechanism of cancer cells.
To examine which signaling factors were activated by
the addition of HGF to PDAC cells, phosphorylation of MET, AKT and
MAPK was examined in the highly MET-expressing Suit-2 and AsPC-1,
and marginally MET-expressing Panc-1 and MIA PaCa-2. The
phosphorylation of MET, AKT and MAPK after the addition of HGF was
considerably induced in Suit-2 and AsPC-1. However, the
phosphorylation of MET after the addition of HGF was slightly or
not induced in Panc-1 or MIA PaCa-2, respectively. However, the
phosphorylation of AKT was considerably or slightly induced in
Panc-1 or MIA PaCa-2, respectively, and the phosphorylation of MAPK
was considerably induced in both cells. These data suggest that the
phosphorylation of AKT and MAPK may be induced through non-MET
signaling in marginally MET-expressing PDAC cells. Next, blocking
of HGF-induced phosphorylation of MET, AKT and MAPK by crizotinib
was examined in the highly MET-expressing Suit-2 and AsPC-1. The
phosphorylation of MET was blocked at a dose of 0.1 μM of
crizotinib in both cells, and the phosphorylation of AKT were
blocked at 0.1 μM and 10 μM of crizotinib in Suit-2
and AsPC-1, respectively. The phosphorylation of MAPK was not
blocked in either cell even at the maximal drug dose (10 μM)
of crizotinib, suggesting that the cell proliferation of highly
MET-expressing PDAC cells is controlled through HGF/MET/AKT rather
than HGF/MET/MAPK signaling. Taken together, our data suggest that
crizotinib inhibits the cell proliferation of PDAC cells,
regardless of MET expression and phosphorylation.
To explore the mechanism that controls the
peritoneal dissemination of PDAC cells, the effects of crizotinib
and downregulation of MET, ROS1 and ALK on the invasion of highly
MET-expressing Suit-2 were evaluated in vitro. Based on the
results, crizotinib was considered to inhibit the invasion of
Suit-2 induced by HGF/MET signaling. We then attempted to identify
key regulatory factors in HGF/MET signaling involved in the
invasion of Suit-2 cells. Since it is now well established that
invasion of cancer cells is induced by Ras-related GTPases
(especially RhoA) (12), the
activation of RhoA by the addition of HGF in Suit-2 was examined by
a pull-down assay with the fusion protein GST-Rhotekin-RBD. Indeed,
an increase in RhoA-GTP, the active form of RhoA, was observed in
Suit-2 cells that were treated for 1 h with HGF (50 ng/ml).
Moreover, the activation of RhoA by HGF was suppressed by the
addition of crizotinib (0.1 μM). Therefore, the invasion of
Suit-2 induced by HGF might be mediated by the activation of RhoA
and could be targeted by crizotinib. Furthermore, to examine the
effect of crizotinib on peritoneal dissemination in vivo, we
used a pancreatic cancer model with i.p. carcinomatosis in nude
mice. It was found that the volume of ascites, the tumor weight on
peritonea, and the concentration of CA19-9 in the untreated group
were significantly reduced in the group given 50 mg/kg/d of
crizotinib. Taken together, crizotinib may be a potent drug for
treating peritoneal dissemination of pancreatic cancer, possibly by
inhibiting cancer cell invasion through the inhibition of the
HGF/MET signaling and the activation of RhoA.
Several reports on the contribution of HGF/MET
signaling to proliferation, motility or metastasis of PDAC cells
were published (26,27). Treatment with the combination of
crizotinib and gemcitabine was shown to synergistically reduce
tumor growth and metastases in orthotopic PDAC models. In one
report, crizotinib was shown to inhibit metabolic inactivation of
gemcitabine in MET-driven pancreatic carcinoma (26). P21-activated kinase 1 (PAK1) was
shown to be a central node in PDAC cells, downstream of multiple
growth factor signaling pathways, including HGF/MET signaling. PAK1
inhibition blocks signaling to cytoskeletal effectors and tumor
cell motility driven by HGF/MET signaling. Inhibition of PAK1
attenuated in vivo tumor growth and metastasis in a model of
pancreatic adenocarcinoma (27).
In accordance with previous studies, our results indicate that the
phosphorylation of MET, AKT and MAPK by the addition of HGF was
considerably induced in highly MET-expressing PDAC cells. However,
the presence of non-MET-expressing PDAC cells urges us to
investigate the contribution of non-MET signaling in PDAC cell
proliferation or metastasis.
The following conclusions were drawn from the
present study. First, crizotinib has a potent inhibitory effect on
the proliferation of PDAC cells, and the effect might not be
mediated through one of the three known RTKs targeted by
crizotinib. Furthermore, the inhibitory effect was largely
unaffected by the status of MET expression and phosphorylation of
each PDAC cell line. Second, crizotinib has a potent inhibitory
effect on the invasion of highly MET-expressing PDAC cells through
the inhibition of HGF/MET signaling and RhoA activation. Finally,
crizotinib might be a potent drug for treating peritoneal
dissemination of highly MET-expressing pancreatic cancer, possibly
by inhibiting cancer cell proliferation and invasion.
Acknowledgments
This study was supported in part by the Grant-in-Aid
for Cancer Research (project no. 00054) from the Ministry of
Health, Labour, and Welfare of Japan (H.I.) and Grant-in-Aid for
Scientific Research (project no. 25462356) from Japan Society for
the Promotion of Science (S.T.).
References
|
1
|
Kleeff J, Michalski C, Friess H and
Büchler MW: Pancreatic cancer: From bench to 5-year survival.
Pancreas. 33:111–118. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hidalgo M: Pancreatic cancer. N Engl J
Med. 362:1605–1617. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ryan DP, Hong TS and Bardeesy N:
Pancreatic adenocarcinoma. N Engl J Med. 371:1039–1049. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li D, Xie K, Wolff R and Abbruzzese JL:
Pancreatic cancer. Lancet. 363:1049–1057. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ueno H, Kiyosawa K and Kaniwa N:
Pharmacogenomics of gemcitabine: Can genetic studies lead to
tailor-made therapy? Br J Cancer. 97:145–151. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Conroy T, Desseigne F, Ychou M, Bouché O,
Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de
la Fouchardière C, et al Groupe Tumeurs Digestives of Unicancer;
PRODIGE Intergroup: FOLFIRINOX versus gemcitabine for metastatic
pancreatic cancer. N Engl J Med. 364:1817–1825. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Von Hoff DD, Ervin T, Arena FP, Chiorean
EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, et
al: Increased survival in pancreatic cancer with nab-paclitaxel
plus gemcitabine. N Engl J Med. 369:1691–1703. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Von Hoff DD, Goldstein D and Renschler MF:
Albumin-bound paclitaxel plus gemcitabine in pancreatic cancer. N
Engl J Med. 370:479–480. 2014.PubMed/NCBI
|
|
9
|
Suker M, Beumer BR, Sadot E, Marthey L,
Faris JE, Mellon EA, El-Rayes BF, Wang-Gillam A, Lacy J, Hosein PJ,
et al: FOLFIRINOX for locally advanced pancreatic cancer: A
systematic review and patient-level meta-analysis. Lancet Oncol.
17:801–810. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gherardi E, Birchmeier W, Birchmeier C and
Vande Woude G: Targeting MET in cancer: Rationale and progress. Nat
Rev Cancer. 12:89–103. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Di Renzo MF, Poulsom R, Olivero M,
Comoglio PM and Lemoine NR: Expression of the Met/hepatocyte growth
factor receptor in human pancreatic cancer. Cancer Res.
55:1129–1138. 1995.PubMed/NCBI
|
|
12
|
Birchmeier C, Birchmeier W, Gherardi E and
Vande Woude GF: Met, metastasis, motility and more. Nat Rev Mol
Cell Biol. 4:915–925. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kang Y and Massagué J:
Epithelial-mesenchymal transitions: Twist in development and
metastasis. Cell. 118:277–279. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gherardi E and Stoker M: Hepatocyte growth
factor - scatter factor: Mitogen, motogen, and met. Cancer Cells.
3:227–232. 1991.PubMed/NCBI
|
|
16
|
Ridley AJ, Comoglio PM and Hall A:
Regulation of scatter factor/hepatocyte growth factor responses by
Ras, Rac, and Rho in MDCK cells. Mol Cell Biol. 15:1110–1122. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wells CM, Ahmed T, Masters JR and Jones
GE: Rho family GTPases are activated during HGF-stimulated prostate
cancer-cell scattering. Cell Motil Cytoskeleton. 62:180–194. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zou HY, Li Q, Lee JH, Arango ME, McDonnell
SR, Yamazaki S, Koudriakova TB, Alton G, Cui JJ, Kung PP, et al: An
orally available small-molecule inhibitor of c-Met, PF-2341066,
exhibits cytoreductive antitumor efficacy through antiproliferative
and antiangiogenic mechanisms. Cancer Res. 67:4408–4417. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Christensen JG, Zou HY, Arango ME, Li Q,
Lee JH, McDonnell SR, Yamazaki S, Alton GR, Mroczkowski B and Los
G: Cytoreductive antitumor activity of PF-2341066, a novel
inhibitor of anaplastic lymphoma kinase and c-Met, in experimental
models of anaplastic large-cell lymphoma. Mol Cancer Ther.
6:3314–3322. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yamazaki S, Skaptason J, Romero D, Lee JH,
Zou HY, Christensen JG, Koup JR, Smith BJ and Koudriakova T:
Pharmacokinetic-pharmacodynamic modeling of biomarker response and
tumor growth inhibition to an orally available cMet kinase
inhibitor in human tumor xenograft mouse models. Drug Metab Dispos.
36:1267–1274. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Timofeevski SL, McTigue MA, Ryan K, Cui J,
Zou HY, Zhu JX, Chau F, Alton G, Karlicek S, Christensen JG, et al:
Enzymatic characterization of c-Met receptor tyrosine kinase
oncogenic mutants and kinetic studies with aminopyridine and
triazolopyrazine inhibitors. Biochemistry. 48:5339–5349. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sennino B, Ishiguro-Oonuma T, Wei Y,
Naylor RM, Williamson CW, Bhagwandin V, Tabruyn SP, You WK, Chapman
HA, Christensen JG, et al: Suppression of tumor invasion and
metastasis by concurrent inhibition of c-Met and VEGF signaling in
pancreatic neuroendocrine tumors. Cancer Discov. 2:270–287. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zheng X, He K, Zhang L and Yu J:
Crizotinib induces PUMA-dependent apoptosis in colon cancer cells.
Mol Cancer Ther. 12:777–786. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Surriga O, Rajasekhar VK, Ambrosini G,
Dogan Y, Huang R and Schwartz GK: Crizotinib, a c-Met inhibitor,
prevents metastasis in a metastatic uveal melanoma model. Mol
Cancer Ther. 12:2817–2826. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhou Y, Zhao C, Gery S, Braunstein GD,
Okamoto R, Alvarez R, Miles SA, Doan NB, Said JW, Gu J, et al:
Off-target effects of c-MET inhibitors on thyroid cancer cells. Mol
Cancer Ther. 13:134–143. 2014. View Article : Google Scholar
|
|
26
|
Avan A, Caretti V, Funel N, Galvani E,
Maftouh M, Honeywell RJ, Lagerweij T, Van Tellingen O, Campani D,
Fuchs D, et al: Crizotinib inhibits metabolic inactivation of
gemcitabine in c-Met-driven pancreatic carcinoma. Cancer Res.
73:6745–6756. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhou W, Jubb AM, Lyle K, Xiao Q, Ong CC,
Desai R, Fu L, Gnad F, Song Q, Haverty PM, et al: PAK1 mediates
pancreatic cancer cell migration and resistance to MET inhibition.
J Pathol. 234:502–513. 2014. View Article : Google Scholar : PubMed/NCBI
|