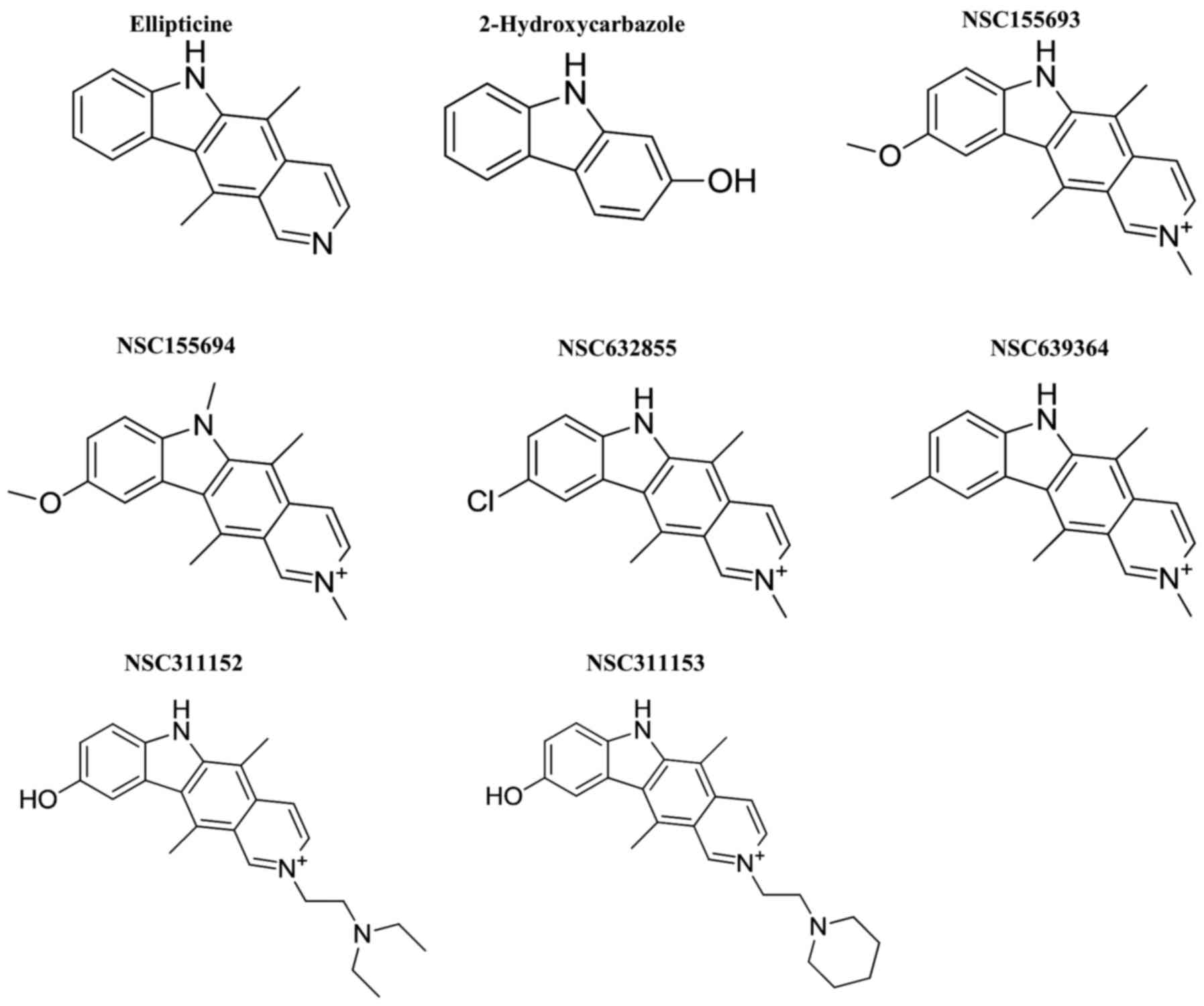
Introduction
The RET proto-oncogene, which is a key
component in the development of enteric nervous system, encodes a
trans-membrane receptor-type tyrosine kinase (1,2). The
domain organization of the RET protein includes a ligand-binding
extracellular domain, a transmembrane domain and an intracellular
tyrosine kinase domain (3-5). The ligands for RET receptor have been
identified as the members of glial cell line derived neurotrophic
factor (GDNF) family that activate this protein through the
interaction with glycosyl-phosphatidylinositol linked GFR-α
co-receptors (6,7). These co-receptors mediate RET
homodimerization that leads to trans-autophosphorylation of the
tyrosine residues present in the intracellular kinase domain
thereby activating its function (8). Upon activation, the phosphorylated
tyrosine residues act as binding sites for many adaptor molecules
that trigger a cascade of intracellular signaling pathways
(9). The major mitogenic signaling
pathways include the Raf/MEK/ERK and PI3K/Akt/mTOR pathways that
contribute to cell proliferation and survival (10-12).
Activating RET germline mutations have been
identified as the key cause for the pathogenesis of medullary
thyroid carcinoma (MTC), which is a part of multiple endocrine
neoplasia type 2 (MEN2) syndrome (13,14).
Patients with MEN2 harbor several mutations in the RET protein that
leads to ligand independent phosphorylation and activation of the
receptor thereby resulting in the constitutive signaling of
intracellular pathways (15,16).
Unlike other differentiated thyroid cancers such as papillary
thyroid carcinoma (PTC) and follicular thyroid carcinoma (FTC), MTC
is poorly-differentiated and hence it metastasizes rapidly to
distant organs like bones, lungs and liver (17,18).
Moreover, most MTC patients have tumor invasion at the time of
diagnosis that hampers the effectiveness of standard therapies such
as surgical resection of the thyroid gland and external beam
radiation (18). Hence, a systemic
targeted therapy has gained considerable clinical interest for the
treatment of advanced and progressive MTC (19). Owing to the oncogenic potential of
RET in the development of MTC, it has been regarded as an ideal
molecular target for therapeutic intervention (19,20).
Although several approaches have been developed including the use
of kinase inhibitors and small interfering RNA (siRNA) to abrogate
the RET kinase activity and its expression, developing a specific
RET inhibitor still remains challenging (20-22).
Our previous study clearly showed that the transcriptional
inhibition of the RET gene by targeting its promoter region
could be a promising strategy for MTC specific therapy (23,24).
The transcriptional activation of the RET
gene is regulated by the presence of polypurine (guanine) and
polypyrimidine (cytosine) tract in the proximal promoter region
(25,26). This upstream core promoter region
serves as the binding site for RNA Pol II, Sp1 and other
transcriptional factors that are responsible for the basal promoter
activity (27). Our previous
studies have clearly demonstrated that the G-rich sequences present
within the RET promoter region are highly dynamic in nature,
adopting non-B-DNA conformations such as G-quadruplex structures
under negative supercoiling conditions (28-30).
These secondary structures are four-stranded intramolecular folding
of a single-stranded DNA, which is formed by the stacking of two or
more G-tetrads (31). Each
G-tetrad comprises of four guanines that are arranged in a square
planar conformation and are interconnected through Hoogsteen
hydrogen bonding (31,32). The G-quadruplex structure on the
promoter region is believed to serve as a silencer element for the
RET transcription through the sequestration of the
transcriptional factor binding sites (23). In our previous study, we have
clearly demonstrated that the stabilization of the G-quadruplex
structure formed on the RET promoter region by a
small-molecule, berberine interfered with the binding of SP1 and
RNA Pol II thereby silencing the transcription of this gene
(23,24).
In continuation of our previous study, the main
objective of the present study was to discover new drug-like
small-molecules that have clinical implications as a potent
suppressor of RET gene through the stabilization of the
promoter G-quadruplex structure. Hence, we repurposed ellipticine
and its structural derivatives as potential G-quadruplex
stabilizing agents and also exerting transcriptional inhibitory
effect on the RET gene in medullary thyroid carcinoma (MTC)
derived TT cell line. Ellipticine is a natural alkaloid, which has
been demonstrated to have therapeutic benefits in different types
of cancers (33,34). The anti-neoplastic activity of this
molecule has been attributed to its ability to intercalate with DNA
and/or to inhibit the topoisomerase II activity (35,36).
However, recent studies have revealed that ellipticine also binds
and stabilizes the G-quadruplex structures formed on the telomere
region and the promoter region of c-Myc oncogene in
vivo (37,38). In order to identify promising lead
compounds for the present study, we explored for different
structural derivatives of ellip-ticine from the NCI/DTP open
chemical repository (Fig. 1). This
led us to the identification of NSC311153 and NSC311152 as lead
compounds that silence the RET gene expression by targeting
the G-quadruplex structure formed on the promoter region of this
gene. We also investigated the cellular effects mediated by these
compounds in TT cells and its in vivo antitumor efficacy
using MTC xenograft models to further understand the therapeutic
implications in targeting the RET transcription through the
promoter G-quadruplex.
Materials and methods
Chemicals
Ellipticine was obtained from Santa Cruz
Biotechnology (SC-200878; Santa Cruz, CA, USA). NSC311153 and other
structural analogs were kindly provided from the U.S. NCI/DTP Open
Chemical Repository. All the compounds were dissolved in dimethyl
sulfoxide (DMSO) at a final concentration of 10 mg/ml.
Materials
The 5′-FAM labelled RET-WT
(5′-AGCGGGTAGGGGCGGGGCGGGGCGGGGGCGG-3′) oligonucleotide was
purchased from Sigma Genosys (Woodlands, TX, USA). Taq DNA
polymerase was purchased from Fermentas (Hanover, MD, USA).
Cell culture and media
The TT cell line was obtained from the American Type
Culture Collection (ATCC; Manassas, VA, USA) and maintained in
Dulbecco's modified Eagle's medium (DMEM)/F-12 medium (Cellgro,
Manassas, VA, USA) supplemented with 15% heat inactivated fetal
bovine serum (FBS). The TPC1 cells and another MTC derived cell
line, MZ-CRC-1 cells were provided by Dr Rebecca Schweppe
(University of Colorado, Denver, CO, USA). These cells were
maintained in RPMI-1640 medium supplemented with 9% FBS and
DMEM/F-12 medium supplemented with 15% FBS, respectively. The
isogenic cell line HEK293-RET, which carries the luciferase
reporter gene under the control of RET gene promoter was
generated as described in our previous study and grown in DMEM
medium supplemented with 9% FBS (23). The normal thyroid cell line,
Nthy-ori-3-1 was purchased from Sigma Genosys and cultured in
RPMI-1640 medium supplemented with 9% FBS. All the cell lines were
maintained in a humidified atmosphere containing 5% CO2
at 37°C. The stocks for all these cell lines were obtained from the
cell bank and utilized within 6 months. These cell lines were also
tested for mycoplasma contamination and were further authenticated
using STR profiling.
CD spectroscopy
The RET-WT oligonucleotide (5 µM) was allowed
to form the G-quadruplex structure in the presence of Tris-HCl
buffer (20 mM, pH 7.6) and 25 mM KCl by denaturing at 95°C for 5
min and slowly cooled to room temperature. The CD spectra were
recorded using a Jasco J-810 spectrophotometer (Jasco, Inc.,
Easton, MD, USA) using a quartz cell of 1 mm path length and
instrument scanning speed of 100 nm/min with a response time of 1
sec over a wavelength range of 230-330 nm as previously described
(23). The Tm was
determined by monitoring the molar ellipticity vs. temperature
profiles at 262 nm at increasing temperature from 20 to 90°C at a
gradient of 1°C/min.
Polymerase stop assay
The polymerase stop assay was performed on a DNA
template containing the G-quadruplex forming sequence, which is
present in the RET promoter region as previously described
(39). In brief, the template DNA
was annealed with a 5′-γ[32P] end-labelled primer (P28)
and purified on an 8% non-denaturing polyacrylamide gel. The
purified primer-DNA template was used in a primer extension assay
in the presence of Taq DNA polymerase.
Dimethyl sulfate (DMS) footprinting
The DMS footprinting was performed on the 5′FAM
labelled RET-WT oligonucle-otides as described in our previous
studies (23,24). In brief, the oligonucleotides were
allowed to form the G-quadruplex structure and were incubated in
the absence and presence of NSC311153 (5 equivalents) at room
temperature for 1 h. The samples were treated with DMS (0.2%) for 2
min and were resolved on an 8% non-denaturing polyacrylamide gel.
Each DNA band was recovered from the gel and treated with
piperidine (10%) after ethanol precipitation. The cleaved products
were resolved on a 16% denaturing polyacrylamide gel along with
purine and pyrimidine specific sequencing markers that were
generated according to the published procedure (40). The gel was dried and the
fluorescence was read on a Typhoon 8600 scanner (GE Healthcare Life
Sciences, Pittsburgh, PA, USA) for analysis.
Semi-quantitative RT-PCR analysis
Total RNA was extracted from the cells using the
RNeasy Mini QIAcube kit (Qiagen, Redwood City, CA, USA) according
to the manufacturer's protocol. The extracted RNA was subjected to
reverse-transcription using the oligo (dT)18 primer with QuantiTect
reverse-transcription kit (Qiagen) to generate single-stranded
cDNA. The primers used for RT-PCR were as follows: RET forward,
(5′-GCAGCATTGTTGGGGGACA-3′) and RET reverse,
(5′-CACCGGAAGAGGAGTAGCTG-3′); Rpl9 forward,
(5′-CTGAAGGGACGCACAGTTAT-3′) and Rpl9 reverse,
(5′-ACGGTAGCCAGTTCCTTTCT-3′). The PCR reactions involved an initial
denaturation at 95°C for 3 min followed by 33 and 23 cycles for RET
and Rpl9, respectively, at 95°C for 30 sec, 52°C for 30 sec and
72°C for 30 sec on a GeneAmp PCR system 9600 (Perkin-Elmer,
Waltham, MA, USA). The PCR products were analyzed on 1.5% agarose
gel electrophoresis.
Western blotting
The whole-cell protein extracts were prepared by
lysing the cells with 2% CHAPS lysis buffer in the presence of 10
mM Tris-HCl, pH 7.4, 0.15 M NaCl, 5 mM EDTA and Halt Protease
Inhibitor Cocktail (Thermo Fisher Scientific, Waltham, MA, USA).
The extracted proteins were resolved on a 4-12% polyacrylamide
SDS-PAGE, as previously described (23,24).
The primary antibodies used were as follows: anti-RET (#3220),
anti-RET/PTC1 (#14698), anti-cMYC (#5605), anti-p-mTOR (#2971) and
anti-mTOR (#2972) (dilution 1:1,000) were purchased from Cell
Signaling Technology (Beverly, MA, USA), anti-Bcl-2 (sc-7382),
anti-pERK (sc-7383), anti-ERK (sc-271270), anti-cyclin D1
(sc-20044) and anti-β-actin (sc-47778) (dilution 1:300) were
purchased from Santa Cruz Biotechnology. Anti-VEGF antibody was
purchased from GeneTex (Irvine, CA, USA; #GTX102643). Mouse and
rabbit IgG antibodies tagged with horseradish peroxidase (HRP)
(Bio-Rad Laboratories, Hercules, CA, USA) were used as secondary
antibodies (dilution 1:1,000). An enhanced chemiluminescence
substrate kit (#32106) purchased from Thermo Fischer Scientific was
used for detection.
Luciferase assay
The isogenic cell line HEK293-RET was exposed to
different concentrations of NSC311153 up to 24 h. Luciferase
expression level is determined using the ONE-Glo Luciferase Assay
system (Promega, Madison, WI, USA) following the manufacturer's
instruction.
Cell viability assay
Cells were plated at a concentration of 7,500
cells/well in a 96-well dish and incubated overnight, followed by
the treatment with NSC311153 at increasing concentrations up to 96
h. The cell viability was determined by using 0.33 mg/ml MTS dye in
the presence of phenazine methosulfate (PMS) (25 µM) as
previously described (41). The
absorbance was measured at 590 nm using a Synergy HT
multi-detection microplate reader (BioTek Instruments, Inc.,
Winooski, VT, USA).
Caspase-3 assay
TT cells were treated with different concentrations
of NSC311153 up to 48 h and caspase-3 activity was measured using
the ApoAlert Caspase Fluorescent Assay kit (Clontech Laboratories,
Inc., Mountain View, CA, USA) by following the manufacturer's
protocol.
In vivo studies
The in vivo antitumor efficacy of the
ellip-ticine derivative was evaluated using 8-10-week old male
severe combined immunodeficiency (SCID) mice, xenotrans-planted
with MTC derived TT cells. Animal experiments were conducted in
accordance with the Institutional Animal Care and Use Committee
(IACUC). The experiments were performed in the Experimental Mouse
Shared Resource (EMSR) Animal Facility Laboratory (University of
Arizona), which is accredited by the International Association for
Assessment and Accreditation of Laboratory Animal Care (AAALAC). In
brief, exponentially growing TT cells (1×107) were
subcutaneously injected into the flank of the mice and the tumor
growth was monitored every week by measuring the tumor diameters
using a vernier caliper. Tumor volume was calculated according to
the formula (b2×l)/2 where b and l are the shortest and
the longest diameters, respectively. Once the tumor volume reaches
100 mm3, the mice are randomly pair matched to vehicle
and treated group (n=8/group). The compound was dissolved in 90%
phosphate-buffered saline (PBS) and 10% DMSO and administered
intraperitoneally (i.p) at a single dose of 4 mg/kg for 5 days/week
up to 2 weeks. The antitumor efficacy was assessed based on the
percentage inhibition of tumor growth in treated vs. control group.
The toxicity of the compound was evaluated based on the loss of
average weight of mice. Tumor tissue from vehicle and treated group
were explanted at the end of last dosage to examine the effect of
the compound on the expression of proteins that contribute to tumor
growth.
Results
Interaction of ellipticine with RET
G-quadruplex
The molecular structure of ellipticine consists of
an aromatic pyridocarbazole ring, which allows the π-π stacking
interactions with the planar guanine residues present in the
terminal G-tetrad (Fig. 1)
(37). Not surprisingly, previous
studies have reported that ellipticine stabilizes different
G-quadruplex structures formed on the telomere region and on the
promoter region of c-Myc oncogene (37,38).
Hence, in the present study we investigated whether this compound
binds and stabilizes the RET G-quadruplex structure using CD
spectroscopic analysis. First, we examined whether ellipticine
alters the structural conformation of the RET G-quadruplex by
monitoring the CD spectra of RET G4 sequence (5 µM) in the
absence and presence of increasing concentrations of this compound.
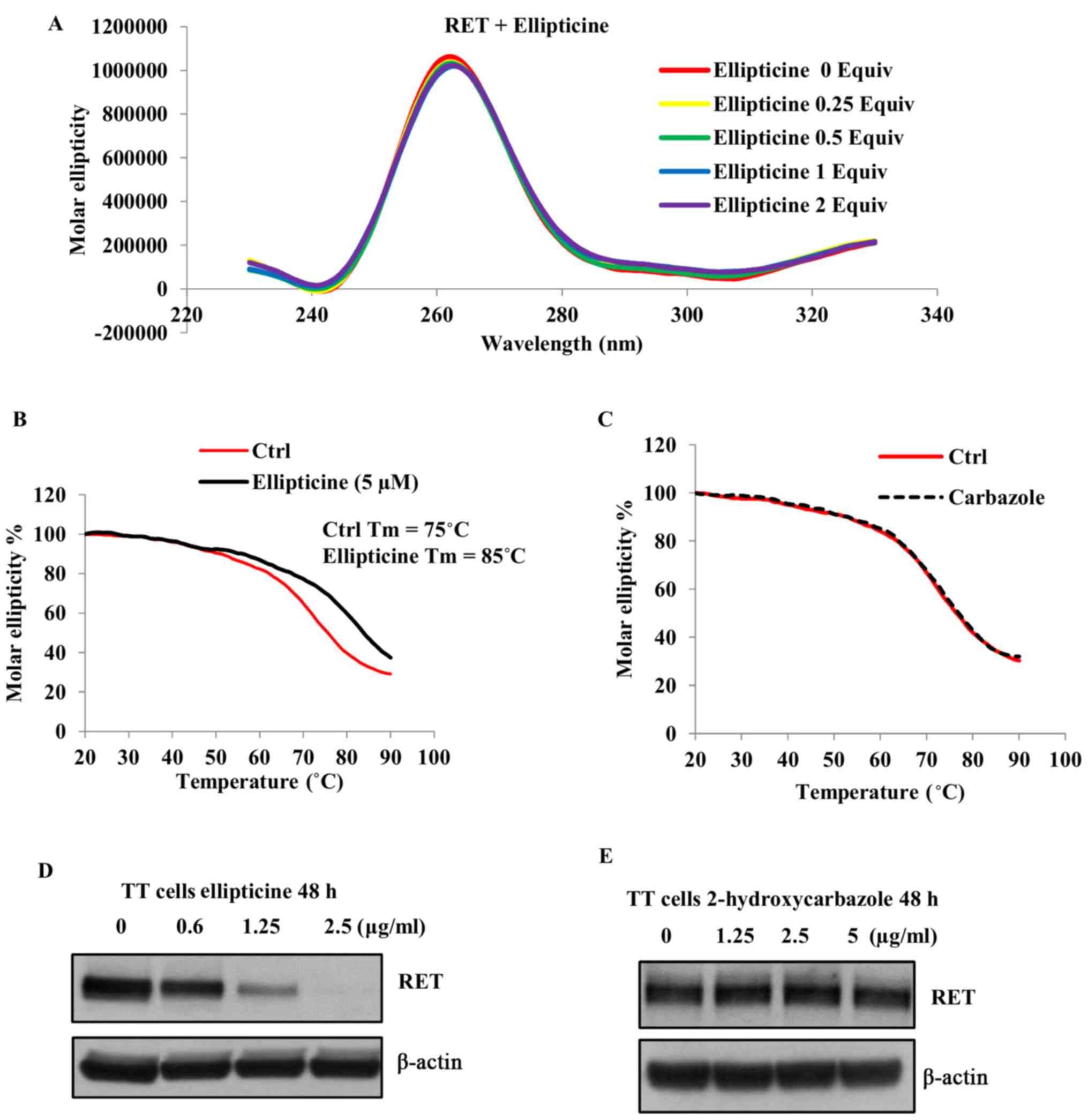
As shown in Fig. 2A, the positive
peak at 262 nm, which corresponds to a parallel G-quadruplex
structure (42), was not affected
in the presence of ellipticine, suggesting that the parallel
configuration of the RET G-quadruplex was not changed in the
presence of this molecule. Next, we examined the thermal stability
of the G-quadruplex structure by monitoring the CD melting curve in
the absence and presence of ellipticine (1 equivalent) at
increasing temperatures. As shown in Fig. 2B, the melting temperature
(Tm) of the RET G-quadruplex structure in the absence of
ellipticine was determined to be 75°C. Based on the melting curves,
the binding of ellipticine with the G-quadruplex structure
increases its melting temperature up to 85°C and the ∆Tm
was found to be 10°C (Fig. 2B). To
further understand the structural features required for the
interaction of ellipticine with the RET G-quadruplex structure, we
examined the binding of 2-hydroxycarbazole with this structure by
monitoring the CD melting profiles. As shown in Fig. 2C the Tm of the
G-quadruplex structure was not increased even in the presence of 5
equivalents of 2-hydroxycarbazole, suggesting that the presence of
pyridine ring in ellipticine allows additional π-π interactions
with the guanine residue. Overall these data imply that the
pyridocarbazole ring moiety is essential for the stabilization of
the RET G-quadruplex structure by ellipticine.
Effect of ellipticine on the RET
expression
In our previous studies we have clearly demonstrated
that the small-molecule mediated stabilization of the G-quadruplex
structure formed on the RET promoter region exerts
transcriptional inhibitory effect on this gene (23,24).
Hence, we determined whether ellipticine downregulates the
endogenous RET expression in the TT cell line in which the
RET gene transcription is regulated by the promoter region
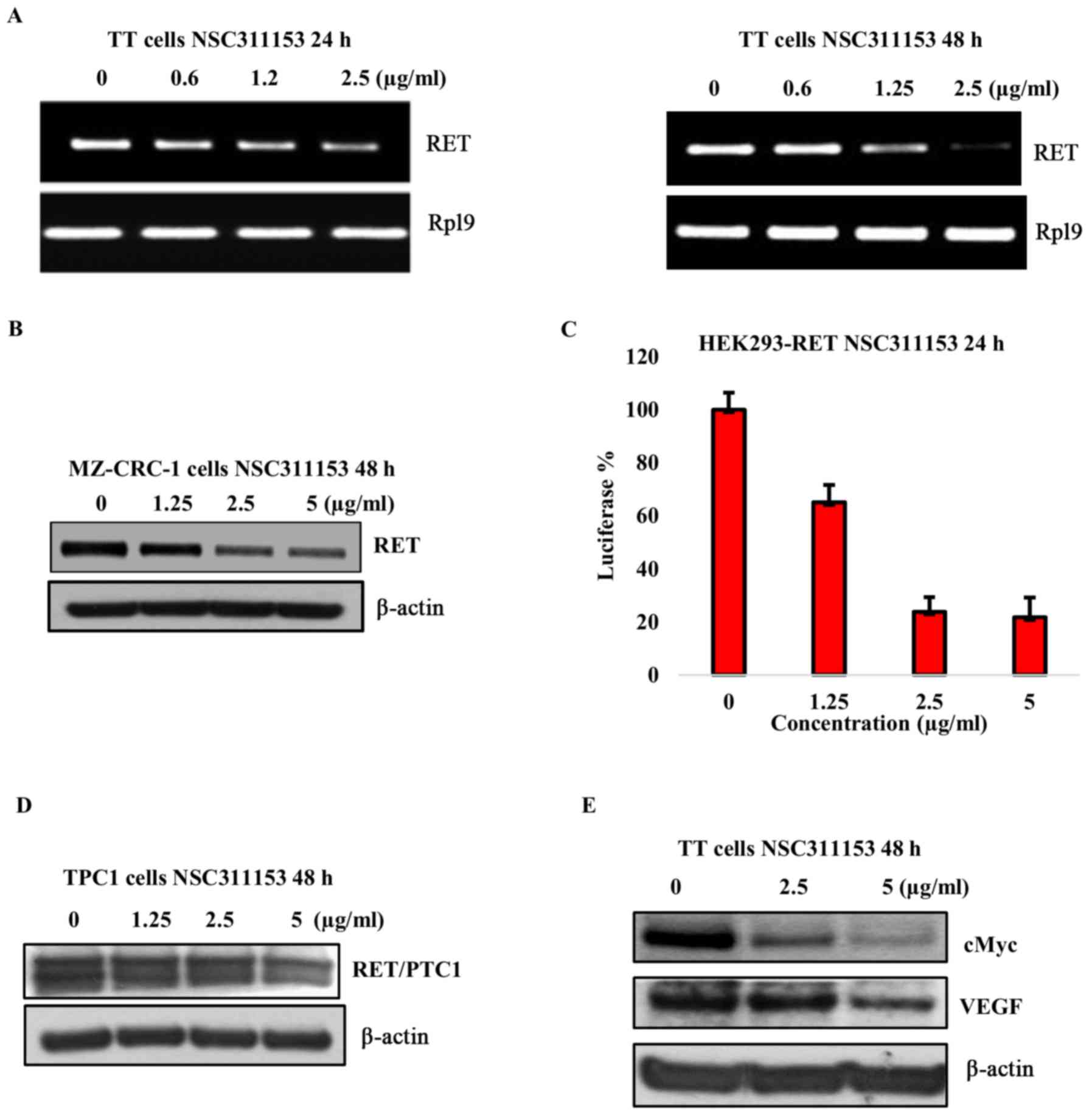
that contains the G-quadruplex forming sequence. As shown in
Fig. 2D, a concentration dependent
decrease in the RET protein expression was observed in the presence
of ellipticine following 48-h exposure. To further confirm that the
RET downregulation by ellipticine is mediated through the
stabilization of the G-quadruplex structure, we determined the
effect of 2-hydroxycarbazole on the RET expression in this cell
line. As shown in Fig. 2E the RET
protein expression was not decreased in the presence of this
compound even at high concentration (5 µg/ml). These data
are consistent with our previous studies that the stabilization of
the G-quadruplex structure formed within the RET promoter
region is responsible for silencing the expression of this
gene.
Identification of NSC311153 as RET
G-quadruplex binding compound
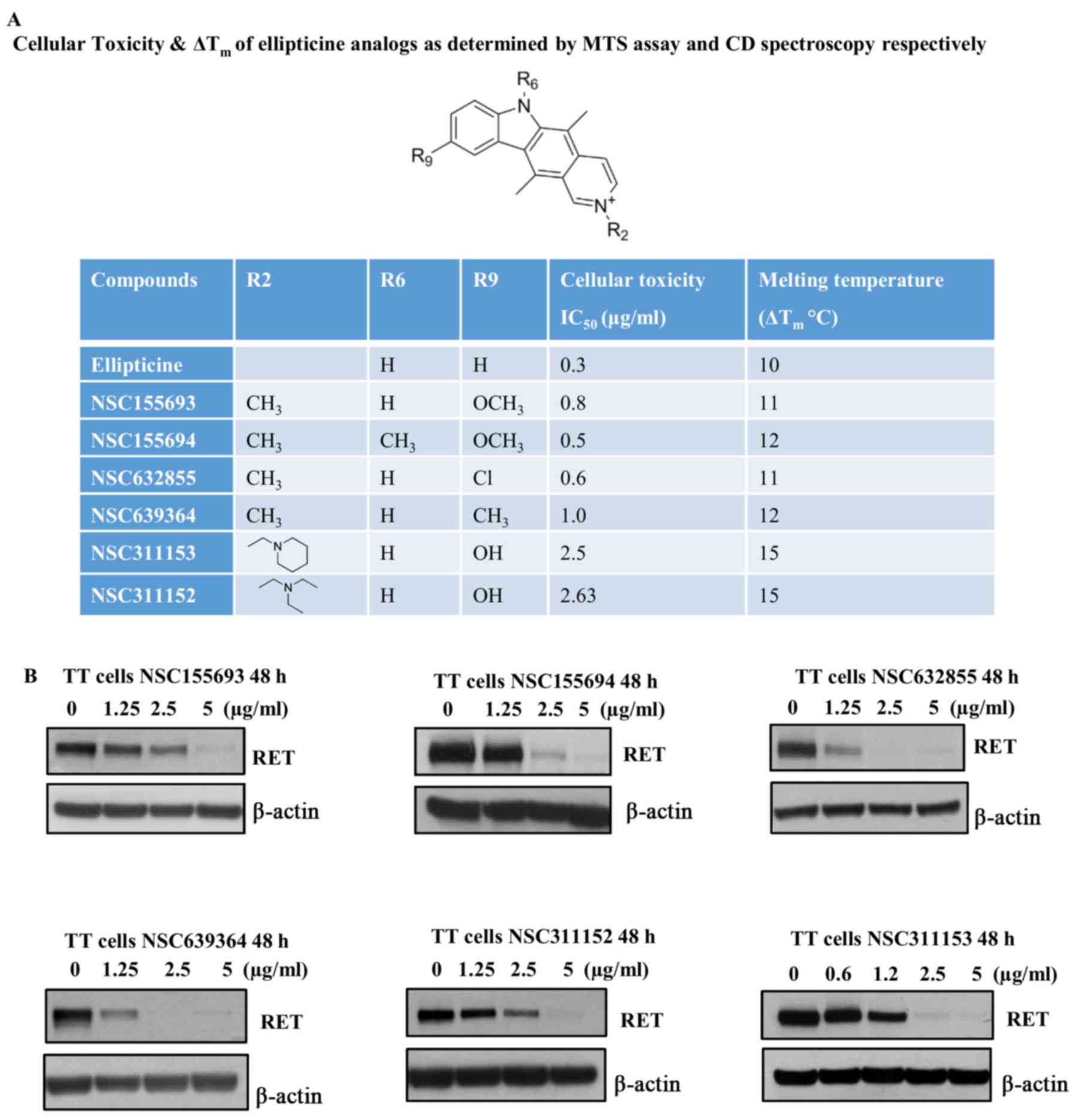
Although we identified ellipticine as a potent
RET transcriptional inhibitor, this compound was withdrawn
from this study due to its adverse cytotoxic effects in TT cells,
which was evaluated by MTS assay. The IC50 of
ellipticine was calculated to be 0.3 µg/ml after 96-h
treatment of TT cells in the presence of increasing concentration
of this compound (Fig. 3A). Hence,
we attempted to identify other structural analogs of ellipticine
from the NCI/DPT open chemicals repository that could suppress the
RET gene transcription at non-toxic concentrations without
affecting their ability to stabilize the RET G-quadruplex structure
(Figs. 1 and 3A). Based on the CD melting curves the
∆Tm was calculated individually for the ellipticine
analogs and the IC50 values for these compounds were
also determined by MTS assay (Fig.
3A). The ellipticine derivatives, which have different
substituents such as methoxy, chloride and methyl groups at
position C-9 of the pyridocarbazole ring stabilizes the RET
G-quadruplex structure by increasing the Tm >10°C
(Fig. 3A). These compounds also
possess RET inhibitory effects in TT cells, which is comparable to
their parent molecule, ellipticine (Fig. 3B). However, the structural
modifications in these compounds did not improve the cytotoxicity
in TT cells as compared to ellipticine (Fig. 3A). Notably, one of the ellip-ticine
analogs, NSC311153, which carries a hydroxyl group and a
2-piperidin-1-ylethy moiety at positions C-9 and N-2, respectively
showed an increased binding ability with the RET G-quadruplex
structure with an estimated ∆Tm as 15°C (Fig. 3A). The IC50 value of
this compound was also estimated to be 2.5 µg/ml after 96 h
of exposure, which is significantly higher than that of ellipticine
(Fig. 3A). Based on these data, we
selected NSC311153 as a lead compound to further proceed with the
in vitro studies.
Validation of NSC311153 as RET G4
stabilizing agent
To further validate that NSC311153 stabilizes the
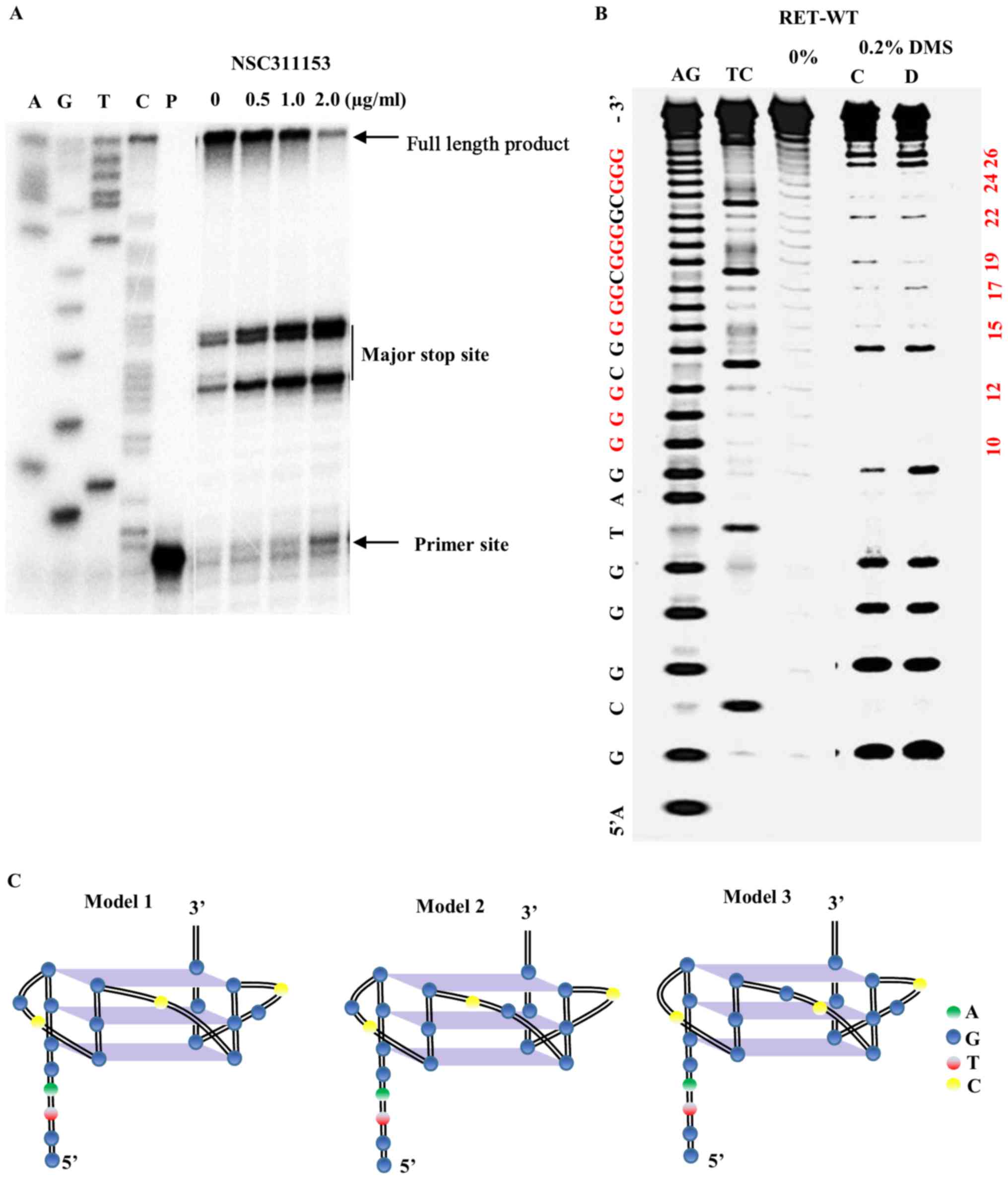
RET G-quadruplex structure formed on the promoter region of this
gene, a DNA polymerase stop assay was performed as previously
described (23). In this assay,
the ligand mediated stabilization of the G-quadruplex structure
that arises on the DNA template prevents the progression of the Taq
DNA polymerase during primer extension. As shown in Fig. 4A, in the presence of NSC311153 at
increasing concentrations, a dose-dependent increase in the amount
of arrested product was observed, indicating the potential
stabilization of the G-quadruplex structure by this compound.
Next, we investigated whether the interaction of
NSC311153 with the parallel RET G-quadruplex structure changes the
guanine residues that are involved in the G-tetrad formation using
dimethyl sulfate (DMS) footprinting. DMS footprinting is a
well-established technique to determine the guanine nucleotides
that are involved in the formation of G-quadruplex structures
(40,43). The N7 position of each of the
guanine residues that are involved in Hoogsteen base pairing to
form G-tetrads is inaccessible to methylation by DMS, which attacks
this position. As shown in Fig.
4B, the pattern of N7-guanine methylation produced by the RETG4
sequence in the presence of 100 mM K+ is consistent with
two parallel G-quadruplexes (lane C) in which either the guanines
(G19-G21) or (G20-G22) is involved in the G-tetrad formation
(Fig. 4C, models 1 and 2). As
shown in Fig. 4B, the binding of
NSC311153 (5 equivalents) changes the pattern of N7 guanine
methylation (lane D) in which the guanines (G14-G16) or (G15-G17)
is involved in the G-tetrad formation (Fig. 4C, models 1 and 3). The change in
the G-quadruplex structure in the presence of this compound clearly
suggest that NSC311153 binds with this secondary structure.
Effect of NSC311153 on the promoter
activity of RET gene
Since in our previous studies we reported that the
G-quadruplex structure formed on the promoter region of RET
gene acts as transcriptional silencer element, we next examined
whether the stabilization of RET G-quadruplex structure by
NSC311153 interferes with the transcriptional activation of this
gene in TT cell line. As shown in Fig.
5A, NSC311153 decreased the RET mRNA expression by
>50 and 90% at a non-toxic concentration of 2.5 µg/ml
following the exposure up to 24 and 48 h, respectively. We also
utilized the MZ-CRC-1 cell line, which harbors an M918T mutation in
the tyrosine kinase domain of the RET protein. Moreover, the
RET gene expression in this cell line is regulated by the
same promoter region as found in the TT cells, which harbors the
G-quadruplex forming motif (44).
As shown in Fig. 5B, a
dose-dependent decrease in the RET protein expression was observed
in the presence of NSC311153 after 48-h incubation.
To determine whether the decrease in RET mRNA
expression by NSC311153 is a direct effect of the promoter-specific
transcriptional inhibition of this gene, a bioluminescent reporter
gene assay was performed using an isogenic cell line HEK293-RET in
which the luciferase expression is under the control of the
RET promoter region as described in our previous study
(23). As shown in Fig. 5C, in the presence of NSC311153 a
dose-dependent decrease in the basal luciferase expression was
observed in the HEK293-RET cell line following 48-h incubation. To
further demonstrate that the mechanism of action of NSC311153 is
through stabilizing the G-quadruplex structure present on the
RET promoter region, we utilized PTC1 derived TPC1 cells as
a control. Chromosomal rearrangement between the RET kinase
domain coding region and the CCD6 gene results in a chimeric
RET/PTC1 expression, whose transcriptional activation is
controlled by the CCD6 promoter region that lacks the
G-quadruplex forming motif (45-49).
Notably, NSC311153 did not suppress the RET/PTC1 expression in this
cell line even after 48-h incubation (Fig. 5D). Overall, these data provide
clear evidence to support that NSC311153 intervenes in the
transcription mechanism of RET gene by targeting the
intracellular G-quadruplex structure formed on its promoter
region.
Cellular effects mediated by NSC311153
due to RET down-regulation
The oncogenic RET activation is mainly involved in
mediating the cell proliferation and survival and hence the effect
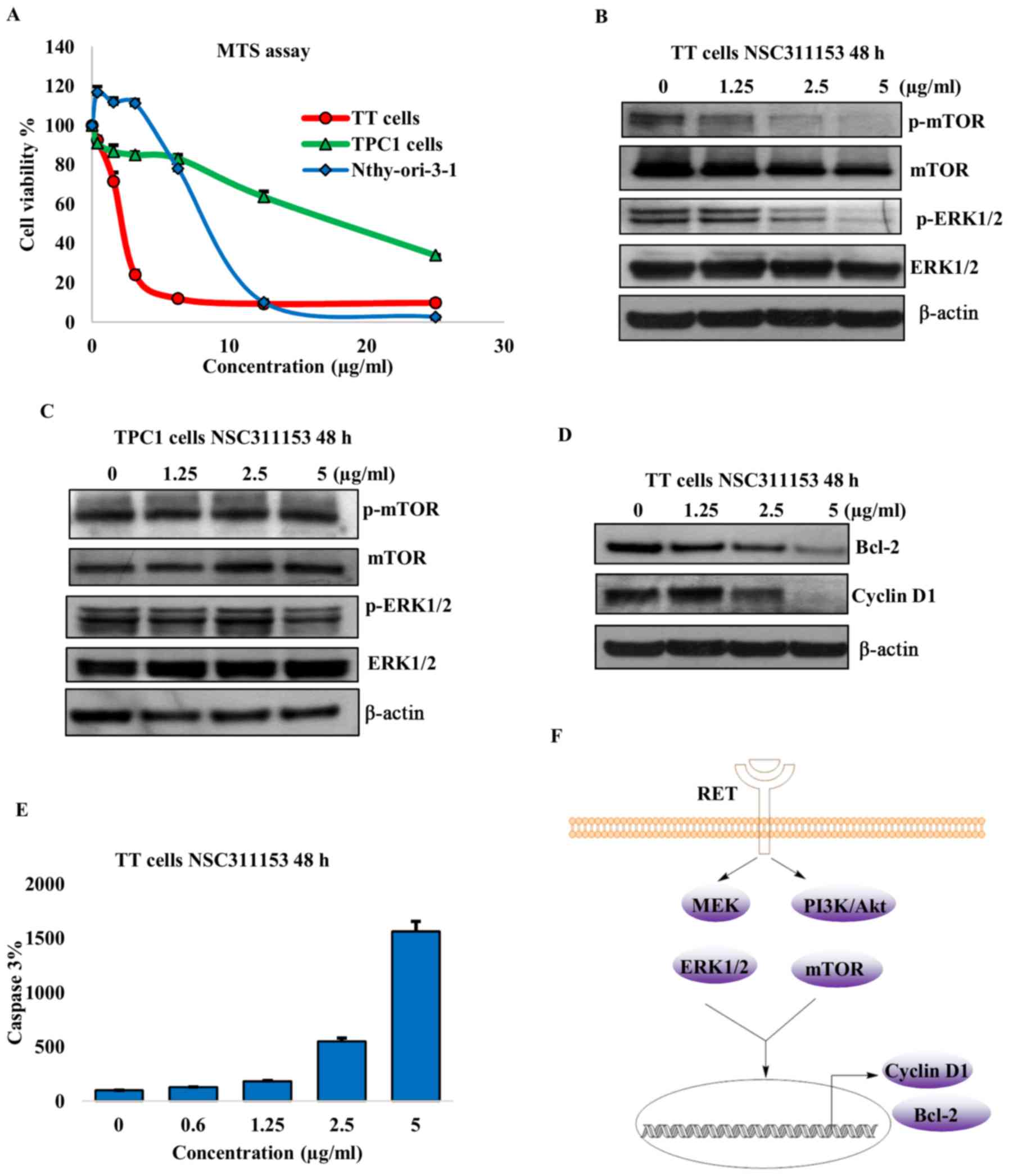
of NSC311153 on the viability of TT cell line was determined using
the MTS assay. As shown in Fig. 6A
the proliferation of the TT cells decreased with increasing
concentrations of NSC311153 and the IC50 was found to be
~2.5 µg/ml after 96-h exposure. The TPC1 cell line in which
the RET/PTC1 expression was not inhibited by NSC311153 was less
sensitive to this compound and the IC50 was 10-fold more
than the TT cells (Fig. 6A). This
clearly shows that the inhibition of cell growth by NSC311153 is
mediated through the downregulation of the RET gene. To
further determine whether the anti-proliferative effect of
NSC311153 is cancer cell-specific, we included a normal thyroid
cell line, Nthy-ori-3-1 in the present study. Based on the MTS
data, the IC50 of NSC311153 in the Nthy-ori-3-1 cell
line was estimated to be 10 µg/ml (Fig. 6A), which is 4-fold higher than that
of TT cells, suggesting that this compound is selectively sensitive
to the mutant RET driven thyroid cancer.
To further characterize the mechanism through which
NSC311153 suppresses the TT cell proliferation, we investigated
whether this compound inhibits the RET mediated downstream signal
transduction pathways. Previous studies have revealed that RET
activates the Raf/MEK and PI3K/Akt downstream signaling pathways,
which in turn phosphorylate and activate ERK1/2 and mammalian
target of rapamycin (mTOR) proteins, respectively (10-12).
As shown in Fig. 6B, the
phosphorylation status of ERK1/2 and mTOR were decreased in a
dose-dependent manner following the exposure of TT cells in the
presence of NSC311153 up to 48 h. However, in TPC1 cells the
phosphorylation levels of these proteins were not altered by
NSC311153 (Fig. 6C). These data
clearly suggest that the inhibitory effect of NSC311153 on this
pathway is a consequence of RET downregulation.
The Raf/MEK/ERK and PI3K/Akt/mTOR pathways are known
to promote cancer cell survival through the activation of cyclin D1
and Bcl-2, which are involved in enhancing cell-cycle progression
and inhibiting the apoptosis mechanism, respectively (50-52).
As shown in Fig. 6D, NSC311153
also decreased the expression of cyclin D1 and Bcl-2 in TT cells in
a concentration-dependent manner. Furthermore, the down-regulation
of Bcl-2 by NSC311153 is further accompanied by the increase in the
caspase-3 activity, which is also a known indicator of apoptosis
(Fig. 6E).
In vivo antitumor activity of ellipticine
derivative
In the final step of the present study, we evaluated
the in vivo effect of the ellipticine derivative on the
tumor growth of MTC xenografts through subcutaneous injection of
the TT cells into SCID mice. Although NSC311153 showed potential
anti-proliferative effects in TT cells in vitro, the poor
solubility of this compound hindered its in vivo antitumor
examination. However, a previous study reported that a water
soluble ellipticine analog, NSC311152 (datelliptium) was well
tolerated in different cancer patients in a phase-I clinical trial
(53). As shown in Fig. 1, the structure of NSC311152 is very
similar to NSC311153 with the presence of a
diethylaminoethyl-moiety at position N-2 that improves the
solubility of this compound compared to that of NSC311153. Based on
our preliminary data, the inhibitory effect of NSC311152 on RET
expression and its IC50 value is the same as that of
NSC311153, suggesting that the slight structural modification does
not alter the efficacy of this compound. Hence, we decided to
pursue with NSC311152 to test the in vivo antitumor
activity. Dosing regimen and treatment schedules were determined
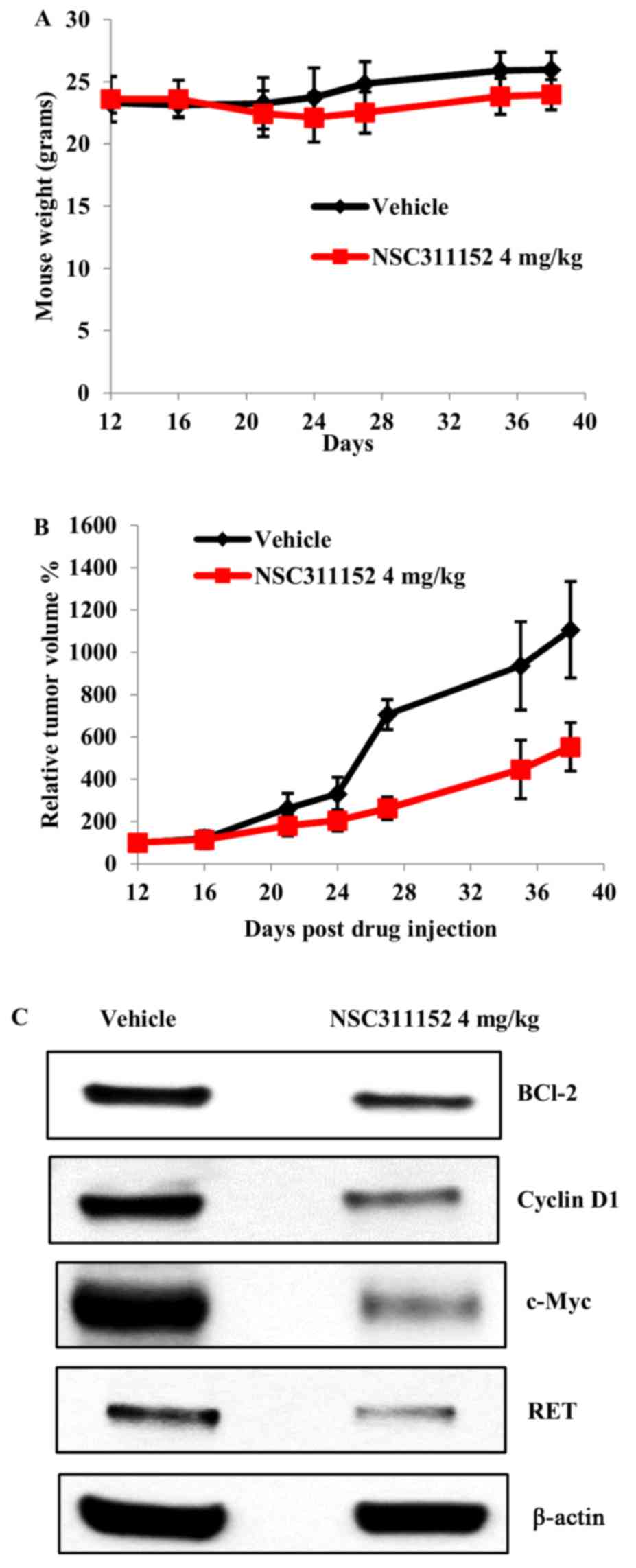
based on our preliminary dose optimization studies. Upon systemic
administration via i.p, NSC311152 (4 mg/kg) was well tolerated
without any significant decrease in the average body weight
(Fig. 7A). As shown in Fig. 7B, ~60% inhibition of the tumor
growth was observed in mice treated with NSC311152 compared to the
vehicle treated group. To further validate whether the inhibition
of tumor growth by NSC311152 in vivo is mediated through
target specific effect, we determined the RET expression in tumor
tissues of vehicle- and NSC311152-treated mice using western
blotting. As shown in Fig. 7C, the
RET protein expression was decreased in the presence of NSC311152
compared to that of vehicle. Furthermore, NSC311152 reduced the
expression of other proteins such as c-Myc, Bcl-2 and cyclin D1
that contribute to cell proliferation, which is in accordance with
the in vitro studies. Overall, these data suggest that
NSC311152 possesses potent in vivo antitumor activity
through RET downregulation.
Discussion
RET was previously described as a regulator of
several intracellular signaling events that contribute to the
well-recognized biological processes such as cell survival,
proliferation, migration and invasion (54). Hence, it is easy to understand why
the constitutive activation of this protein due to point mutations
in the functional domains is mainly involved in the progression of
MTC. The clinical responses to standard chemotherapy and radiation
therapy in patients with MTC have been shown to be less effective
thereby representing MTC as a promising disease for the field of
targeted drug therapy (55-57).
Therapeutic approaches that target the RET kinase activity using
small-molecule kinase inhibitors such as vandetanib and
cabozantinib have proved to be clinically valuable in MTC (58,59).
However, these kinase inhibitors possess potential inhibitory
effects on other tyrosine-kinase receptors like VEGFR, EGFR, MET
and hence the development of a specific inhibitor for RET still
remains challenging (60,61). Moreover, a previous study
demonstrated that the RET kinase carrying a substitution mutation
of V804 to a bulky hydrophobic leucine or methionine amino-acids at
the gatekeeper region in ATP binding pocket confers resistance to
the kinase inhibitors (62).
Hence, these gatekeeper mutations will likely emerge as one of the
obstacles in the long-term use of vandetanib and cabozan-tinib in
the treatment of patients with advanced MTC.
Another possible approach for the treatment of RET
associated MTC involves the use of small interfering RNAs (siRNAs)
to silence the RET gene expression. In a previous
investigation the transfection of TT cell line with RET siRNA
exerted anti-proliferative effects on this cell line and also
inhibited the growth of tumor xenografts in vivo (63). However, the development of siRNAs
as drug-like molecules possesses several pitfalls that mainly
include difficulty in delivering these molecules into target cells
and their extracellular instability (64,65).
The siRNAs are large and negatively charged molecules that greatly
affect their permeability into plasma membrane and prevent
intracellular accumulation at their site of action. Furthermore,
siRNAs are highly susceptible to degradation by many extracellular
enzymes that undermine the clinical implications of these molecules
(65). To overcome these
challenges, this study mainly focuses in targeting the
transcription of the RET proto-oncogene using small
molecules.
In our previous studies we have clearly demonstrated
that the polypurine/polypyrimidine tract within the RET gene
promoter region has a propensity to undergo strand separation that
leads to conformational transition between duplex DNA and
G-quadruplex structures (28,29).
Moreover, we have also shown that the stabilization of these
structures using small molecules emerged as potential
transcriptional repressors of RET gene (23,24).
The present study is based on a previous report, which revealed the
interaction of a putative anticancer agent, ellipticine with the
G-quadruplex structure formed by the human telomeric sequence
(37). However, due to the adverse
cytotoxic effects of ellipticine, we investigated the interaction
of other ellipticine analogs with the RET G-quadruplex structure
using in vitro biochemical assays. Notably, we identified an
ellipticine derivative, NSC311153, which has a
2-piperidin-1-ylethyl moiety at position N-2 as a potent stabilizer
of the RET G-quadruplex structure. The structure activity
relationship (SAR) analysis clearly revealed that the presence of
1-ethylpiperidine at the N-2 position improves the binding of
NSC311153 with the RET G-quadruplex and also significantly
decreases the cellular toxicity compared to its parent molecule,
ellipticine. The stabilization of the G-quadruplex structure by
NSC311153 also exerted inhibitory effects on the RET promoter
activity, which was confirmed using bioluminescent reporter assay
in which the luciferase gene expression is driven by the RET
promoter region. This compound further inhibited the RET mRNA and
protein expression in TT cells, which harbor a MEN2A-type mutation.
The transcriptional inhibitory effect of NSC31153 on other
oncogenes like VEGF and c-MYC that also harbor the
G-quadruplex forming sequences on their promoter regions was also
investigated (66,67). As shown in Fig. 5E, the c-MYC expression showed a
dose-dependent decrease in the presence of NSC311153, which is
consistent with a previous study (37). Moreover, the VEGF expression was
also partially inhibited by this compound suggesting that the
G-quadruplex structure could be a potential intra-cellular target
for NSC311153 (Fig. 5E).
In the present study, we also addressed that
NSC311153 inhibited the proliferation of TT cells through the
down-regulation of RET expression. The oncogenic RET activation
promotes cell growth and survival by transducing a cascade of
intracellular signaling pathways. In a previous study by Drosten
et al (10) the RET
associated downstream signaling pathways that are required for
tumor maintenance and progression were well characterized in the TT
cell line. In their study, they used adenoviral vector expressing
the dominant negative truncated RET protein, which lacks the entire
intracellular tyrosine kinase domain to disrupt the phosphorylation
and activation of RET protein in TT cells. This resulted in the
downregulation of Raf/MEK/ERK and PI3K/Akt/mTOR pathways suggesting
that these two pathways are mainly involved in RET mediated
transformation (10). Consistent
with this study, we also observed that the suppression of
RET expression by NSC311153 inhibited the phosphorylation of
ERK and mTOR and further decreased the expression of cyclin D1 and
Bcl-2 that are tightly regulated by Raf/MEK/ERK and PI3K/Akt/mTOR
pathways (Fig. 6F). Notably, we
also observed that the normal thyroid cells, Nthy-ori-3-1 showed
significant resistance to NSC311153 with an IC50 of 10
µg/ml suggesting that this compound is more selective to
mutant RET driven thyroid cancer.
To validate the drug-target selectivity, we utilized
TPC1 cell line in this study, which is more robust and direct in
demonstrating the RET G4-targeted activity of NSC311153. The
chromosomal rearrangement between the RET tyrosine kinase domain
coding region with the 5′ terminal region of the coiled-coil domain
containing gene 6 (CCD6) at chromosome 10q11.2 is a common
genetic alteration identified in TPC1 cell line (46). This results in a chimeric fusion
protein RET/PTC1, which is capable of ligand independent
homodimerization due to the dimerization domain present in the
CCD6 gene thereby resulting in the constitutive activation
of this protein (47). Due to
chromosomal inversion the transcription of the RET/PTC1 gene
is regulated by the CCD6 gene promoter region in TPC1 cells,
which does not have the GC box region and thus is unable to form
the G-quadruplex structure (49).
In the present study, the RET/PTC1 expression was not decreased in
the presence of NSC311153, suggesting that the presence of
G-quadruplex structure on the promoter region of RET gene is
essential to mediate the inhibitory effect of this compound. Based
on the MTS data, the IC50 of this compound in TPC1 cell
line was 10-fold higher than that in TT cells, which clearly
suggest that the anti-proliferative effect of NSC311153 is
specifically mediated through RET downregulation.
In the present study, we also reported the in
vivo antitumor activity of a water soluble NSC311153 analog,
NSC311152 (datelliptium) in MTC xenograft mouse models. A phase I
clinical study has been previously carried out using NSC311152 in
patients with metastatic breast, ovarian, gastric and colorectal
cancers (53). Based on that
study, the maximum tolerated dose in humans was determined to be 9
mg/kg with minimal side-effects such as nausea, mild diarrhea, dry
mouth and fatigue (53). Since
NSC311152 has been investigated in clinical trial, repurposing this
compound as a potent anticancer agent for MTC therapy has better
advantages in terms of safety and other pharmacokinetic parameters
compared to NSC311153. Moreover, the structural modification in
NSC311152 did not affect the potency of this molecule in
downregulating the RET expression in vitro. Hence, in this
study we attempted to examine the in vivo effects of
NSC31112.
Overall, the present study supports the notion that
the G-quadruplex mediated RET transcriptional inhibition may be a
valid therapeutic approach for the treatment of advanced and
metastatic MTC. The identified RET transcriptional inhibitors could
also be used in combination with other clinically available kinase
inhibitors to improve their therapeutic efficacy in MTC
patients.
Acknowledgments
We thank the U.S. NCI/DTP Open Chemical Repository
for providing the chemicals used in the present study.
References
|
1
|
Takahashi M and Cooper GM: ret
transforming gene encodes a fusion protein homologous to tyrosine
kinases. Mol Cell Biol. 7:1378–1385. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Patel A, Harker N, Moreira-Santos L,
Ferreira M, Alden K, Timmis J, Foster K, Garefalaki A, Pachnis P,
Andrews P, et al: Differential RET signaling pathways drive
development of the enteric lymphoid and nervous systems. Sci
Signal. 5:ra552012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Takahashi M, Buma Y, Iwamoto T, Inaguma Y,
Ikeda H and Hiai H: Cloning and expression of the ret
proto-oncogene encoding a tyrosine kinase with two potential
transmembrane domains. Oncogene. 3:571–578. 1988.PubMed/NCBI
|
|
4
|
Takahashi M, Buma Y and Hiai H: Isolation
of ret proto-oncogene cDNA with an amino-terminal signal sequence.
Oncogene. 4:805–806. 1989.PubMed/NCBI
|
|
5
|
Anders J, Kjar S and Ibáñez CF: Molecular
modeling of the extracellular domain of the RET receptor tyrosine
kinase reveals multiple cadherin-like domains and a calcium-binding
site. J Biol Chem. 276:35808–35817. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Durbec P, Marcos-Gutierrez CV, Kilkenny C,
Grigoriou M, Wartiowaara K, Suvanto P, Smith D, Ponder B,
Costantini F, Saarma M, et al: GDNF signalling through the Ret
receptor tyrosine kinase. Nature. 381:789–793. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Trupp M, Arenas E, Fainzilber M, Nilsson
AS, Sieber BA, Grigoriou M, Kilkenny C, Salazar-Grueso E, Pachnis
V, Arumäe U, et al: Functional receptor for GDNF encoded by the
c-ret proto-oncogene. Nature. 381:785–789. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cosma MP, Cardone M, Carlomagno F and
Colantuoni V: Mutations in the extracellular domain cause RET loss
of function by a dominant negative mechanism. Mol Cell Biol.
18:3321–3329. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Iwashita T, Asai N, Murakami H, Matsuyama
M and Takahashi M: Identification of tyrosine residues that are
essential for transforming activity of the ret proto-oncogene with
MEN2A or MEN2B mutation. Oncogene. 12:481–487. 1996.PubMed/NCBI
|
|
10
|
Drosten M, Hilken G, Böckmann M, Rödicker
F, Mise N, Cranston AN, Dahmen U, Ponder BA and Pützer BM: Role of
MEN2A-derived RET in maintenance and proliferation of medullary
thyroid carcinoma. J Natl Cancer Inst. 96:1231–1239. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pitt SC and Chen H: The
phosphatidylinositol 3-kinase/akt signaling pathway in medullary
thyroid cancer. Surgery. 144:721–724. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Melillo RM, Santoro M, Ong SH, Billaud M,
Fusco A, Hadari YR, Schlessinger J and Lax I: Docking protein FRS2
links the protein tyrosine kinase RET and its oncogenic forms with
the mitogen-activated protein kinase signaling cascade. Mol Cell
Biol. 21:4177–4187. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen Z, Qi X, Fei J, Yu X, Zhao Y, Zhao J,
Jin H, Wang J, Ying R and Zhang X: Multiple endocrine neoplasia
type 2A caused by a p.C618RRET proto-oncogene mutation in a Chinese
pedigree. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 31:348–351. 2014.In
Chinese. PubMed/NCBI
|
|
14
|
Lodish MB and Stratakis CA: RET oncogene
in MEN2, MEN2B, MTC and other forms of thyroid cancer. Expert Rev
Anticancer Ther. 8:625–632. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mulligan LM, Kwok JB, Healey CS, Elsdon
MJ, Eng C, Gardner E, Love DR, Mole SE, Moore JK, Papi L, et al:
Germ-line mutations of the RET proto-oncogene in multiple endocrine
neoplasia type 2A. Nature. 363:458–460. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Asai N, Iwashita T, Matsuyama M and
Takahashi M: Mechanism of activation of the ret proto-oncogene by
multiple endocrine neoplasia 2A mutations. Mol Cell Biol.
15:1613–1619. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Aboelnaga EM and Ahmed RA: Difference
between papillary and follicular thyroid carcinoma outcomes: An
experience from Egyptian institution. Cancer Biol Med. 12:53–59.
2015.PubMed/NCBI
|
|
18
|
Milan SA, Sosa JA and Roman SA: Current
management of medullary thyroid cancer. Minerva Chir. 65:27–37.
2010.PubMed/NCBI
|
|
19
|
Cabanillas ME, Hu MI, Jimenez C, Grubbs EG
and Cote GJ: Treating medullary thyroid cancer in the age of
targeted therapy. Int J Endocr Oncol. 1:203–216. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
de Groot JW, Links TP, Plukker JT, Lips CJ
and Hofstra RM: RET as a diagnostic and therapeutic target in
sporadic and hereditary endocrine tumors. Endocr Rev. 27:535–560.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Plaza-Menacho I, Burzynski GM, de Groot
JW, Eggen BJ and Hofstra RM: Current concepts in RET-related
genetics, signaling and therapeutics. Trends Genet. 22:627–636.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wells SA Jr and Santoro M: Targeting the
RET pathway in thyroid cancer. Clin Cancer Res. 15:7119–7123. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shin YJ, Kumarasamy V, Camacho D and Sun
D: Involvement of G-quadruplex structures in regulation of human
RET gene expression by small molecules in human medullary thyroid
carcinoma TT cells. Oncogene. 34:1292–1299. 2015. View Article : Google Scholar
|
|
24
|
Kumarasamy VM, Shin YJ, White J and Sun D:
Selective repression of RET proto-oncogene in medullary thyroid
carcinoma by a natural alkaloid berberine. BMC Cancer. 15:5992015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Andrew SD, Capes-Davis A, Delhanty PJ,
Marsh DJ, Mulligan LM and Robinson BG: Transcriptional repression
of the RET proto-oncogene by a mitogen activated protein
kinase-dependent signalling pathway. Gene. 298:9–19. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bachetti T, Borghini S, Ravazzolo R and
Ceccherini I: An in vitro approach to test the possible role of
candidate factors in the transcriptional regulation of the RET
proto-oncogene. Gene Expr. 12:137–149. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Andrew SD, Delhanty PJ, Mulligan LM and
Robinson BG: Sp1 and Sp3 transactivate the RET proto-oncogene
promoter. Gene. 256:283–291. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Guo K, Pourpak A, Beetz-Rogers K, Gokhale
V, Sun D and Hurley LH: Formation of pseudosymmetrical G-quadruplex
and i-motif structures in the proximal promoter region of the RET
oncogene. J Am Chem Soc. 129:10220–10228. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sun D, Guo K, Rusche JJ and Hurley LH:
Facilitation of a structural transition in the
polypurine/polypyrimidine tract within the proximal promoter region
of the human VEGF gene by the presence of potassium and
G-quadruplex-interactive agents. Nucleic Acids Res. 33:6070–6080.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sun D and Hurley LH: The importance of
negative superhelicity in inducing the formation of G-quadruplex
and i-motif structures in the c-Myc promoter: Implications for drug
targeting and control of gene expression. J Med Chem. 52:2863–2874.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Parkinson GN, Lee MP and Neidle S: Crystal
structure of parallel quadruplexes from human telomeric DNA.
Nature. 417:876–880. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Burge S, Parkinson GN, Hazel P, Todd AK
and Neidle S: Quadruplex DNA: Sequence, topology and structure.
Nucleic Acids Res. 34:5402–5415. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Paoletti C, Le Pecq JB, Dat-Xuong N, Juret
P, Garnier H, Amiel JL and Rouesse J: Antitumor activity,
pharmacology, and toxicity of ellipticines, ellipticinium, and
9-hydroxy derivatives: Preliminary clinical trials of
2-methyl-9-hydroxy ellipticinium (NSC 264-137). Recent Results
Cancer Res. 74:107–123. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rouëssé J, Spielmann M, Turpin F, Le
Chevalier T, Azab M and Mondésir JM: Phase II study of elliptinium
acetate salvage treatment of advanced breast cancer. Eur J Cancer.
29A:856–859. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fossé P, René B, Charra M, Paoletti C and
Saucier JM: Stimulation of topoisomerase II-mediated DNA cleavage
by ellipticine derivatives: Structure-activity relationship. Mol
Pharmacol. 42:590–595. 1992.PubMed/NCBI
|
|
36
|
Barrett JF, Gootz TD, McGuirk PR, Farrell
CA and Sokolowski SA: Use of in vitro topoisomerase II assays for
studying quinolone antibacterial agents. Antimicrob Agents
Chemother. 33:1697–1703. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ghosh S, Kar A, Chowdhury S and Dasgupta
D: Ellipticine binds to a human telomere sequence: An additional
mode of action as a putative anticancer agent? Biochemistry.
52:4127–4137. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Brown RV, Danford FL, Gokhale V, Hurley LH
and Brooks TA: Demonstration that drug-targeted down-regulation of
MYC in non-Hodgkins lymphoma is directly mediated through the
promoter G-quadruplex. J Biol Chem. 286:41018–41027. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Han H, Hurley LH and Salazar M: A DNA
polymerase stop assay for G-quadruplex-interactive compounds.
Nucleic Acids Res. 27:537–542. 1999. View Article : Google Scholar
|
|
40
|
Sun D and Hurley LH: Biochemical
techniques for the characterization of G-quadruplex structures:
EMSA, DMS footprinting, and DNA polymerase stop assay. Methods Mol
Biol. 608:65–79. 2010. View Article : Google Scholar
|
|
41
|
Riss TL, Moravec RA, Niles AL, Duellman S,
Benink HA, Worzella TJ and Minor L: Cell Viability Assays. Assay
Guidance Manual. Sittampalam GS, Coussens NP, Nelson H, et al:
Bethesda (MD): 2004, View Article : Google Scholar
|
|
42
|
Gray DM, Gray CW, Mou TC and Wen JD: CD of
single-stranded, double-stranded, and G-quartet nucleic acids in
complexes with a single-stranded DNA-binding protein. Enantiomer.
7:49–58. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
González V, Guo K, Hurley L and Sun D:
Identification and characterization of nucleolin as a c-myc
G-quadruplex-binding protein. J Biol Chem. 284:23622–23635. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhu W, Hai T, Ye L and Cote GJ: Medullary
thyroid carcinoma cell lines contain a self-renewing
CD133+ population that is dependent on ret
proto-oncogene activity. J Clin Endocrinol Metab. 95:439–444. 2010.
View Article : Google Scholar
|
|
45
|
Grieco M, Santoro M, Berlingieri MT,
Melillo RM, Donghi R, Bongarzone I, Pierotti MA, Della Porta G,
Fusco A and Vecchio G: PTC is a novel rearranged form of the ret
proto-oncogene and is frequently detected in vivo in human thyroid
papillary carcinomas. Cell. 60:557–563. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Fusco A, Grieco M, Santoro M, Berlingieri
MT, Pilotti S, Pierotti MA, Della Porta G and Vecchio G: A new
oncogene in human thyroid papillary carcinomas and their
lymph-nodal metastases. Nature. 328:170–172. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Nikiforov YE: RET/PTC rearrangement in
thyroid tumors. Endocr Pathol Spring. 13:3–16. 2002. View Article : Google Scholar
|
|
48
|
Schweppe RE: Thyroid cancer cell line
misidentification: An update. J Clin Endocrinol Metab. 98:956–957.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tong Q, Li Y, Smanik PA, Fithian LJ, Xing
S, Mazzaferri EL and Jhiang SM: Characterization of the promoter
region and oligomerization domain of H4 (D10S170), a gene
frequently rearranged with the ret proto-oncogene. Oncogene.
10:1781–1787. 1995.PubMed/NCBI
|
|
50
|
Baldin V, Lukas J, Marcote MJ, Pagano M
and Draetta G: Cyclin D1 is a nuclear protein required for cell
cycle progression in G1. Genes Dev. 7:812–821. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Tsujimoto Y: Role of Bcl-2 family proteins
in apoptosis: Apoptosomes or mitochondria? Genes Cells. 3:697–707.
1998. View Article : Google Scholar
|
|
52
|
McCubrey JA, Steelman LS, Chappell WH,
Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M,
Tafuri A, et al: Roles of the Raf/MEK/ERK pathway in cell growth,
malignant transformation and drug resistance. Biochim Biophys Acta.
1773:1263–1284. 2007. View Article : Google Scholar
|
|
53
|
Khayat D, Borel C, Azab M, Paraisot D,
Malaurie E, Bouloux C and Weil M: Phase I study of Datelliptium
chloride, hydrochloride given by 24-h continuous intravenous
infusion. Cancer Chemother Pharmacol. 30:226–228. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Jain S: The many faces of RET dysfunction
in kidney. Organogenesis. 5:177–190. 2009. View Article : Google Scholar
|
|
55
|
Matuszczyk A, Petersenn S, Bockisch A,
Gorges R, Sheu SY, Veit P and Mann K: Chemotherapy with doxorubicin
in progressive medullary and thyroid carcinoma of the follicular
epithelium. Horm Metab Res. 40:210–213. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Terezakis SA and Lee NY: The role of
radiation therapy in the treatment of medullary thyroid cancer. J
Natl Compr Canc Netw. 8:532–540; quiz 541. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Ferrari SM, Fallahi P, Politti U,
Materazzi G, Baldini E, Ulisse S, Miccoli P and Antonelli A:
Molecular targeted therapies of aggressive thyroid cancer. Front
Endocrinol (Lausanne). 6:1762015.
|
|
58
|
Wells SA Jr, Robinson BG, Gagel RF, Dralle
H, Fagin JA, Santoro M, Baudin E, Elisei R, Jarzab B, Vasselli JR,
et al: Vandetanib in patients with locally advanced or metastatic
medullary thyroid cancer: A randomized, double-blind phase III
trial. J Clin Oncol. 30:134–141. 2012. View Article : Google Scholar
|
|
59
|
Elisei R, Schlumberger MJ, Müller SP,
Schöffski P, Brose MS, Shah MH, Licitra L, Jarzab B, Medvedev V,
Kreissl MC, et al: Cabozantinib in progressive medullary thyroid
cancer. J Clin Oncol. 31:3639–3646. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Chau NG and Haddad RI: Vandetanib for the
treatment of medullary thyroid cancer. Clin Cancer Res. 19:524–529.
2013. View Article : Google Scholar
|
|
61
|
Yakes FM, Chen J, Tan J, Yamaguchi K, Shi
Y, Yu P, Qian F, Chu F, Bentzien F, Cancilla B, et al: Cabozantinib
(XL184), a novel MET and VEGFR2 inhibitor, simultaneously
suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer
Ther. 10:2298–2308. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Carlomagno F, Guida T, Anaganti S, Vecchio
G, Fusco A, Ryan AJ, Billaud M and Santoro M: Disease associated
mutations at valine 804 in the RET receptor tyrosine kinase confer
resistance to selective kinase inhibitors. Oncogene. 23:6056–6063.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Koga K, Hattori Y, Komori M, Narishima R,
Yamasaki M, Hakoshima M, Fukui T and Maitani Y: Combination of RET
siRNA and irinotecan inhibited the growth of medullary thyroid
carcinoma TT cells and xenografts via apoptosis. Cancer Sci.
101:941–947. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Agrawal N, Dasaradhi PV, Mohmmed A,
Malhotra P, Bhatnagar RK and Mukherjee SK: RNA interference:
Biology, mechanism, and applications. Microbiol Mol Biol Rev.
67:657–685. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Gavrilov K and Saltzman WM: Therapeutic
siRNA: Principles, challenges, and strategies. Yale J Biol Med.
85:187–200. 2012.PubMed/NCBI
|
|
66
|
Sun D, Guo K and Shin YJ: Evidence of the
formation of G-quadruplex structures in the promoter region of the
human vascular endothelial growth factor gene. Nucleic Acids Res.
39:1256–1265. 2011. View Article : Google Scholar :
|
|
67
|
Siddiqui-Jain A, Grand CL, Bearss DJ and
Hurley LH: Direct evidence for a G-quadruplex in a promoter region
and its targeting with a small molecule to repress c-MYC
transcription. Proc Natl Acad Sci USA. 99:11593–11598. 2002.
View Article : Google Scholar : PubMed/NCBI
|