In response to various stresses such as DNA damage
or hypoxia, the tumor suppressor p53 can be activated to regulate
cell cycle, differentiation, apoptosis, senescence and autophagy
(1,2). Mutations of p53 in single allele may
lead to loss of the tumor suppressor functions, gain of oncogenic
functions, or exert dominant-negative effects which may disrupt the
normal functions of the wild-type allelic p53 (3). Under normal circumstances, p53 is
rapidly turned over by ubiquitinization through binding to MDM2.
Mutant p53 is usually much more stable than the wild-type p53 due
to the loss of the binding activity to MDM2, and is often
accumulated in tumor cells (4,5).
While the wild-type p53 predominantly functions as a transcription
factor, the mutant p53 also has the ability to transactivate
multiple genes involved in cell proliferation, apoptosis
inhibition, chemoresistance and matrix degradation (4).
On the other hand, oncogenic mutations of RAS are
detected in many cancer types including pancreatic, lung, ovarian
and colon cancers, which usually lead to chemo- and/or
radio-resistance of cancer cells (6,7). RAS
activates several downstream cascade branches including the
RAF/MEK/ERK, PI3K/AKT and RalGDS/Ral signal molecules critical for
cancer progression (8,9). Although p53 and RAS are individually
reported to contribute to cellular autophagy and EMT, how they
functionally interact with each other to cooperatively regulate the
downstream signaling cancer progression is unclear. In this mini
review, we will summarize the recent findings regarding the
functional interaction of mutant p53 and RAS in modulating cancer
progression through some key events of cell autophagy and EMT
(10–13).
Autophagy, an intracellular catabolic process in
response to stress and nutrient deprivation, plays multiple roles
during tumorigenesis and cancer therapy (14). To maintain metabolic homeostasis,
autophagy occurs to deliver excessive or unnecessary cytoplasmic
components as well as injured or aged organelles to the lysosomes
for degradation (15,16). As a homeostatic process, autophagy
has both tumor-promoting and tumor-suppressing properties depending
on cancer cell type and the tumorigenic context (17). The main regulators of autophagy
include the PI3K-Akt-mTOR pathway associated molecules, RAS and p53
(14). Several studies have shown
that the nuclear p53 stimulates cellular autophagy via the
transactivation of multiple target genes, while the cytoplasmic p53
inhibits autophagy in a transcription-independent manner, therefore
the subcellular localization of p53 may determine the outcome of
autophagy (18,19). On the other hand, RAS can modulate
autophagy via various signaling cascades in cancer cells,
conversely, autophagy also mediates and promotes the RAS-driven
cancer progression and invasion (20,21).
RAS renders mitochondrial health particularly reliance on autophagy
to the extent that RAS-driven cancer cells seem more
autophagy-dependent for survival to nutrient starvation than normal
cells. Thus, that RAS-driven cancers are susceptible to autophagy
inhibition therapy (22).
Both Ras and p53 are reported to interact with
several identical binding partners and signaling cascades during
the autophagy process, suggesting the possible interplay between
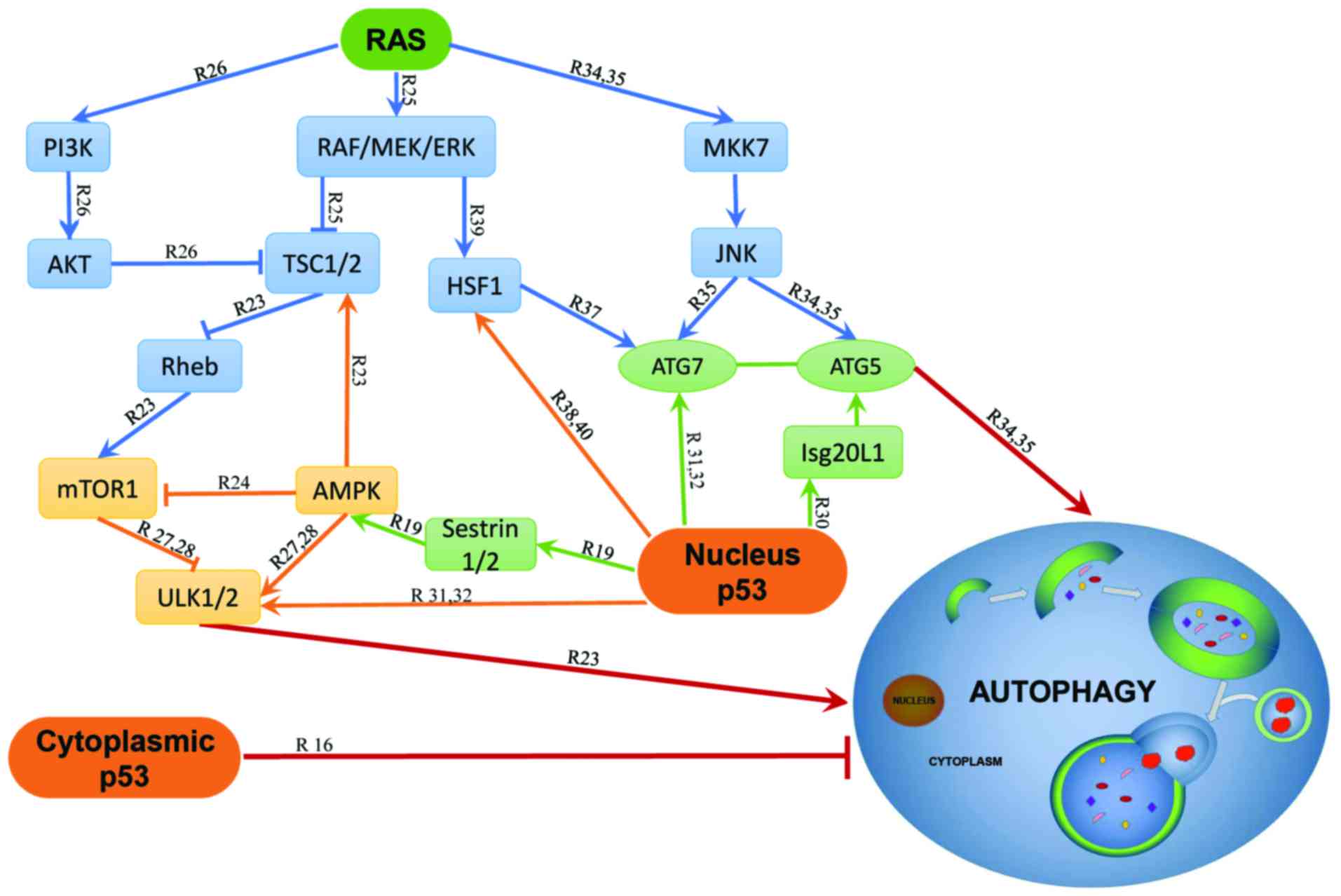
their corresponding pathways. In the nucleus, p53 activates
Sestrin1 (also known as p53-activated gene 26, PA26) and Sestrin2
(also known as hypoxia-inducible gene 95, Hi95), to induce
autophagy through the activation of adenosine
monophosphate-activated protein kinase (AMPK) (23). The activated AMPK inhibits mTOR1
activity by phosphorylating the mTORC1 binding factor Raptor or the
tumor suppressor tuberous sclerosis protein 1/2 (TSC1/2) complex
(24,25). Studies on metastatic pancreatic
ductal adenocarcinomas showed that two K-RAS activation pathways
RAF/MEK/ERK and PI3K/AKT also converge to the TSC1/2 (26). In addition, inhibition of mTORC1 or
activation of AMPK can activate the unc-51-like autophagy
activating kinase 1/2 (ULK1/2) to eventually initiate autophagy
(27,28).
The autophagy related genes (ATG) have been
recognized to execute autophagy directly and the ATG proteins play
pivotal roles in the formation of the autophagosomes (29). p53 and RAS regulate autophagy
mostly relying on these ATG proteins. In vitro studies on
various cancer cell lines showed that overexpression of the p53
target gene Isg20L1 promotes autophagy that can be partially
rescued by ATG5 depletion (30).
The nucleus p53 induces autophagy through direct activation of
serial genes such as ULK1, ULK2 and ATG7 in multiple cell lines
such as MEFs, lung cancer cells and HCT116 cells (31,32),
indicating that the nucleus p53 induces autophagy at least
partially relying on ATG5/7. A recent study on ATG7-deletion
genetically engineered mouse models of K-RASG12D-driven
on small-cell lung cancer (NSCLC) showed that the functional status
of p53 determined the metabolic requirement for autophagy. During
tumor development, intact p53 with ATG7 deletion leads to the
premature p53 induction and blocks tumor proliferation, while p53
loss of function restored the proliferation and growth during ATG7
deletion (33). Finally, both
H-RASV12 and K-RASV12 can initiate autophagy
by upregulating ATG5 and ATG7 through the Rac1/mitogen-activated
kinase kinase 7 (MKK7)/c-Jun N-terminal kinase (JNK) signaling
pathways in normal fibroblasts and human breast epithelial cell
line MCF10A (34,35).
Activated RAS and mutant p53 may synergistically
regulate autophagy. In human pancreatic cancer cell lines CAPAN-2,
PANC-1 and Panc10.05, activated K-RAS and p53 loss of function
collaboratively upregulate Plac8 to facilitate
autophagosome-lysosome fusion (36). Both RAS and p53 signaling pathways
can regulate the heat shock transcription factor 1 (HSF1) that
stimulates autophagy through direct binding to the ATG7 promoter
and activating its expression during breast cancer progression
(37,38). The RAS/RAF/MEK/ERK signaling
pathway activates HSF1 through its phosphorylation at Ser326 in
human neurofibrosarcoma cell line MpNST while HSF1 and p53
interfere with each other during cancer development (39). The HSF1 signaling usually depends
on p53 mutation status and HSF1 is also required for the nuclear
localization of p53 in multiple cell lines (38,40).
Studies have also shown that autophagy plays a
cardinal role in response to hypoxia microenvironment of tumors.
For example, the hypoxia-inducible factor-1α (HIF-1α) can activate
autophagy and alter cancer metabolism (41). During anti-angiogenic therapy, some
cancer cells activated both AMPK and HIF-1α pathways to initiate
autophagy and thus survive under the hypoxic insult (42). A study
revealed that H-RAS can transform Rat1 fibroblasts through
upregulation of HIF-1α expression, but treatment with either MAPK
or PI3K inhibitors suppresses the HIF-1α level (43). HIF-1α also stabilizes p53 through
direct interaction with and inhibition of MDM2 (44,45).
Although it is known that HIF-1α interacts with both RAS and p53 in
hypoxia conditions, how these interactions induce autophagy is
still unclear. The detailed signaling pathways involving p53 and
RAS in autophagy are presented in Fig.
1.
Epithelial-mesenchymal transition (EMT) is a process
of certain cells switching from an epithelial to a mesenchymal
status (46). During EMT,
epithelial cells lose their characteristics as apical-basal
polarity and tight junction but gain the mesenchymal properties
such as reduced intercellular adhesion and increased motility
(47). EMT may play an important
role in the initiation and development of cancers and
chemoresistance of metastatic cancers (47–50).
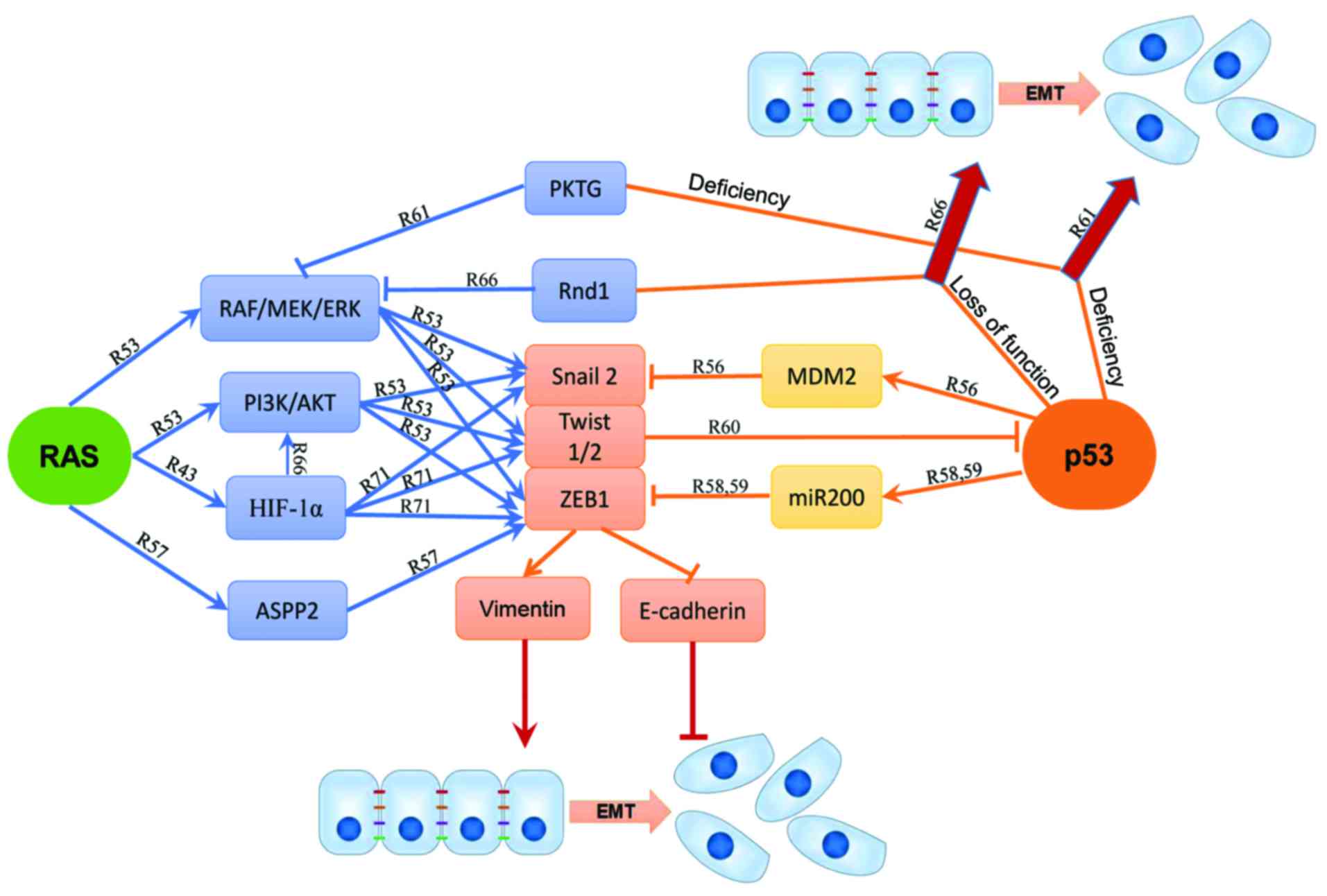
It is well established that oncogenic RAS promotes
EMT in collaboration with other pathways including p53 (51,52).
p53 inhibits the RAS-mediated EMT and EMT-associated stemness of
human mammary epithelial cells via the RAS/RAF/MEK/ERK and the
RAS/PI3K/AKT pathways. Moreover, inhibition of the RAS/RAF/MEK/ERK
pathway upregulates E-cadherin and β-catenin expression (53). Both RAS/PI3K/AKT and
RAS/RAF/MEK/ERK pathways stimulate EMT through the activation of
Snail2 (also known as Slug) expression and the reduction of
E-cadherin in multiple cell lines including colorectal carcinoma
cells HCT-116, HKe-3 and HKh-2, rat parotid gland epithelial cell
Pa4, and endometrial cancer cell lines Ishikawa and Hec251
(51,54,55).
In non-small cell lung cancer, mutation of p53 is associated with
high expression of Slug and low expression of E-cadherin, leading
to poor prognosis of patients (56). The study suggested that wt p53 can
bind to MDM2 and Slug simultaneously to form a p53-MDM2-Slug
complex, which then facilitates MDM2-mediated Slug degradation
(56). The
H-RASV12-induced EMT can be inhibited by ASPP2 without
p53 binding (57). In mouse
primary kidney epithelial cells, ASPP2 represses ZEB1 expression by
forming ASPP2-β-catenin-E-cadherin ternary complex at cell-cell
junctions to negatively regulate the WNT signaling (57). Although ASPP2 suppresses ZEB1
without regard to the p53 mutation status, in hepatocellular
carcinoma cell lines and immortal normal mammary epithelial cells,
p53 represses ZEB1 and ZEB2 expression through the transcriptional
activation of the miRNA-200 family members (58,59).
In murine and human cancer cells, Twist1 and Twist2 may also
cooperate with H-RASV12 to overcome premature senescence
of mouse embryonic fibroblasts through inhibition of the p53
pathway and promotion of EMT by suppressing E-cadherin and
stimulating vimentin expression (60).
Concurrent mutations of RAS and p53 have been found
to play a critical role in EMT and tumor metastasis via multiple
pathways. The Raf kinase trapping to Golgi (RKTG), the negative
regulator of the RAS/RAF/MEK/ERK pathway, may also collaborate with
p53 to regulate EMT (61).
Concomitant knockdown of p53 and RKTG in mice contribute to skin
cancer development and epidermal EMT. Studies of A431 and HepG2
cells suggested that loss of p53 and PKTG at the same time reduced
E-cadherin but increased vimentin to promote EMT (61). Furthermore, the AKT activator IGF-1
induces EMT with p53 silencing while the AKT inhibitor VIII blocks
the E-cadherin/β-catenin complex formation induced by p53 and RKTG,
implicating that the RAS/PI3K/AKT cascades enhance EMT function
likely through inhibition of the p53 function (61). On the other hand, miR-200 blocks
EMT and metastasis in syngeneic mice with metastatic lung
adenocarcinoma carrying both K-RASG12D and p53R172HΔG
mutations (62). Several studies
have shown that loss of p53 can enhance the RAS signaling induced
EMT. p53 may act as a checkpoint controller to inhibit EMT while
loss of p53 allows other signal cascades such as RAS activation to
induce EMT (61,63–65).
Activation of K-RASV12 and loss of p53 may cooperate to
induce EMT and cell motility by triggering the RhoA activity
(10). In metastatic mouse models,
depletion of the Rho-GTPase Rnd1 inhibits the RAS/RAF/MEK/ERK
pathway to promote EMT in collaboration with the loss of p53
(66).
Hypoxia-induced EMT, in particularly, is well-known
in several cancers such as breast, ovarian, hepatocellular
carcinomas and oesophageal squamous cell cancer (67–70).
HIF-1α targets several EMT transcriptional factors including Snail,
Slug, Twist and ZEB in hypoxia conditions (71). In response to hypoxia stress,
HIF-1α can activate PI3K/AKT to promote EMT and to enhance the
tumor cell metastatic potential (67). As mentioned above, p53 and RAS may
have an intimate crosstalk with HIF-1α, indicating the interactive
potentials among the three molecules during EMT or MET.
Since the first step of tumor metastasis is
characterized by the increased motility and invasiveness, it has
been implicated that EMT plays a cordial role in promoting
metastasis, although the role of EMT for invasion and metastasis
remains contested (72). Mutant
p53 and oncogenic RAS promote EMT while the upregulation of wt p53
suppresses RAS-induced EMT phenotypes. p53 may interact with the
RAS signaling to inhibit or promote EMT process via multiple
pathways depending on the p53 status and RAS activation level. The
detailed signaling pathways involving p53 and RAS in EMT are
depicted in Fig. 2.
Autophagy and EMT are two key processes during
cancer progression and linked in a close relationship with each
other according to recent studies. The interactions between
autophagy and EMT is complicated. Just like its dual role in
cancer, autophagy also has two-tier functions on EMT according to
the cellular type and the stage of tumor progression (73). Several studies showed the
controversial effect of autophagy on EMT. Autophagy inhibition
promotes EMT while autophagy activation reverses EMT mainly by
regulating several mesenchymal markers. A recent study on gastric
cancer cells indicated that autophagy deficiency increases the
expression of mesenchymal markers such as N-cadherin, vimentin and
Snail mainly through the ROS-NF-κB-HIF-1α pathway (74). Another research on human skin
squamous cell carcinoma and melanoma described that autophagy
deficiency facilitated EMT by stabilizing the pivotal mesenchymal
marker TWIST1 (75). Autophagy
stimulation downregulated two key regulators Slug and Snail in
glioblastoma cells while inhibition of ATG5 and ATG7 led to
overexpression of Slug and Snail (76). Studies on breast and colon cancers
described that the death effector domain-containing DNA-binding
protein (DEDD) negatively regulated EMT by activating autophagy and
then inducing the autophagy-mediated lysosomal degradation of Snail
and Twist (77). Considering its
special role in supporting cell viability during cancer progression
and migration, autophagy also has a positive effect on EMT. Li and
his colleagues (78) found that
the inhibition of autophagy by silencing ATG3 or ATG7 also
suppressed EMT and TGF-β/Smad3 signaling in hepatocellular
carcinoma cells HepG2 and BEL 7402. While starvation-induced
autophagy can promote EMT through the TGF-β/Smad3
signaling-dependent manner.
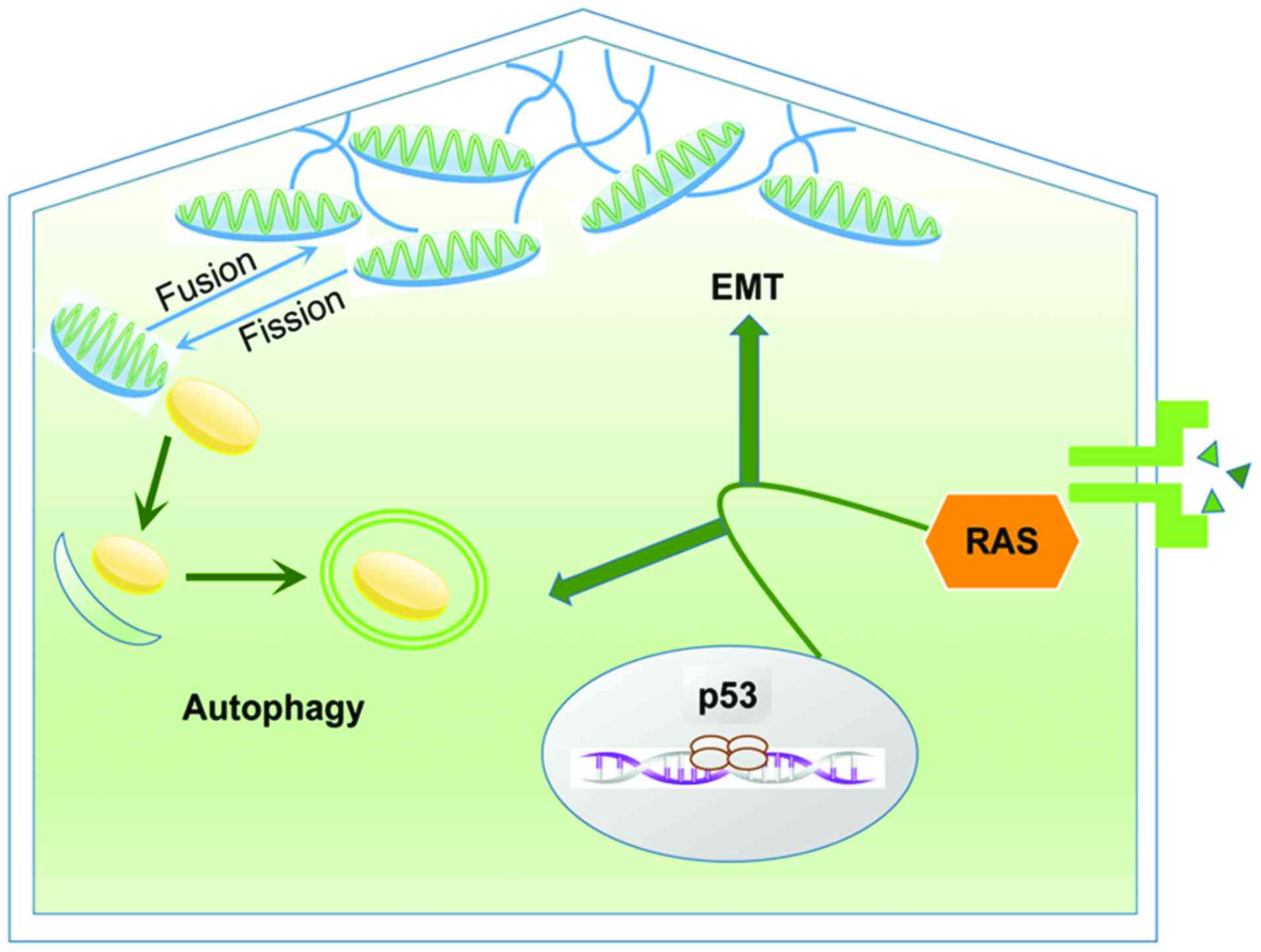
The correlation between autophagy and EMT is largely
based on the close relationship between cytoskeleton and
mitochondria and their pivotal function in modulating the two
processes. Cytoskeleton structures are essential to facilitate cell
movement and cytoskeleton remodeling is indispensable to accomplish
the process of EMT (79,80). While mitochondria are responsible
for ATP production and play fundamental roles in maintaining
cellular metabolic homeostasis (81). Mitochondria are dynamic organelles
that experience fusion and fission continuously (82). Fissile mitochondria are degraded
through autophagy to be reused as source of energy and thus
completed the recycling of metabolites (14,81).
Mitochondria are reticular organelles characterized as high
plasticity to move across the cells through the cytoskeleton
(81). Amassing of mitochondria
below the cell membrane is essential to provide an abundance of ATP
to upgrade the formation of lamellipodia and filopodia, and then
assuring the cellular motility during EMT (83,84).
Thus, the close relationship between mitochondria
and cytoskeleton is correlated with both EMT and autophagy. During
cancer progression, mitochondrial dynamics provide ATP for
cytoskeleton remodeling to promote EMT while autophagy regulates
mitochondrial dynamics by eliminating the damaged mitochondria. The
relationship between autophagy and EMT in cancer is presented in
Fig. 3.
Inactivation of tumor suppressor genes and
activation of oncogenes may collaborate to induce cell malignant
transformation. RAS and p53 have been found most frequently mutated
in majority of human cancers. Early studies revealed that the
activated H-RASV12 cooperates with mutant p53 to induce
tumor progression (85–87). Recent studies report that mutant
p53 cooperates with activated RAS to stimulate highly invasive and
metastatic tumors with poor prognosis (88–92).
Since RAS and p53 pathways function as pivotal regulators in both
cancer cells and tumor microenvironment (93), the associated genes including
HIF-1α, HDAC, EHF and VGLL and their functions may be thoroughly
examined in autophagy and EMT. Retention of wt p53 can facilitate
the sensitivity to chemotherapy in some tumor types and inhibition
of the RAS downstream signaling factor AKT also represses survival,
invasiveness and drug resistance of cancer cells (94–96).
A variety of molecules and existing therapeutic agents targeting
the RAS and p53 pathways are currently in clinical trials (97,98).
Thus, identification of novel molecules or signaling cascades
involved with p53 or RAS mutations may greatly contribute to
precision medicine toward cancer treatment and prevention.
The present review was supported by grants from the
National Natural Science Foundation of China (nos. 81572553 and
81372797 to G.Y.).
|
1
|
Stępiński D: Nucleolus-derived mediators
in oncogenic stress response and activation of p53-dependent
pathways. Histochem Cell Biol. 146:119–139. 2016. View Article : Google Scholar
|
|
2
|
Merino D and Malkin D: p53 and hereditary
cancer. Subcell Biochem. 85:1–16. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Muller PA and Vousden KH: p53 mutations in
cancer. Nat Cell Biol. 15:2–8. 2013. View
Article : Google Scholar
|
|
4
|
Freed-Pastor WA and Prives C: Mutant p53:
One name, many proteins. Genes Dev. 26:1268–1286. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Silva JL, De Moura Gallo CV, Costa DC and
Rangel LP: Prion-like aggregation of mutant p53 in cancer. Trends
Biochem Sci. 39:260–267. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fang B: RAS signaling and anti-RAS
therapy: Lessons learned from genetically engineered mouse models,
human cancer cells, and patient-related studies. Acta Biochim
Biophys Sin (Shanghai). 48:27–38. 2016.
|
|
7
|
Kimmelman AC: Metabolic dependencies in
RAS-driven cancers. Clin Cancer Res. 21:1828–1834. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Stites EC and Ravichandran KS: A systems
perspective of ras signaling in cancer. Clin Cancer Res. 15(5):
1510–1513. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vandal G, Geiling B and Dankort D: Ras
effector mutant expression suggest a negative regulator inhibits
lung tumor formation. PLoS One. 9:e847452014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xia M and Land H: Tumor suppressor p53
restricts Ras stimulation of RhoA and cancer cell motility. Nat
Struct Mol Biol. 14:215–223. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Meylan E, Dooley AL, Feldser DM, Shen L,
Turk E, Ouyang C and Jacks T: Requirement for NF-kappaB signalling
in a mouse model of lung adenocarcinoma. Nature. 462:104–107. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Boiko AD, Porteous S, Razorenova OV,
Krivokrysenko VI, Williams BR and Gudkov AV: A systematic search
for downstream mediators of tumor suppressor function of p53
reveals a major role of BTG2 in suppression of Ras-induced
transformation. Genes Dev. 20:236–252. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Song H, Hollstein M and Xu Y: p53
gain-of-function cancer mutants induce genetic instability by
inactivating ATM. Nat Cell Biol. 9:573–580. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lorin S, Hamaï A, Mehrpour M and Codogno
P: Autophagy regulation and its role in cancer. Semin Cancer Biol.
23:361–379. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kumar A, Singh UK and Chaudhary A:
Targeting autophagy to overcome drug resistance in cancer therapy.
Future Med Chem. 7:1535–1542. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
White E and DiPaola RS: The double-edged
sword of autophagy modulation in cancer. Clin Cancer Res.
15:5308–53016. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tang J, Di J, Cao H, Bai J and Zheng J:
p53-mediated autophagic regulation: A prospective strategy for
cancer therapy. Cancer Lett. 363:101–107. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tasdemir E, Maiuri MC, Galluzzi L, Vitale
I, Djavaheri-Mergny M, D'Amelio M, Criollo A, Morselli E, Zhu C,
Harper F, et al: Regulation of autophagy by cytoplasmic p53. Nat
Cell Biol. 10:676–687. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schmukler E, Kloog Y and Pinkas-Kramarski
R: Ras and autophagy in cancer development and therapy. Oncotarget.
5:577–586. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lock R, Kenific CM, Leidal AM, Salas E and
Debnath J: Autophagy-dependent production of secreted factors
facilitates oncogenic RAS-driven invasion. Cancer Discov.
4:466–479. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
White E: Deconvoluting the
context-dependent role for autophagy in cancer. Nat Rev Cancer.
12:401–410. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Budanov AV: Stress-responsive sestrins
link p53 with redox regulation and mammalian target of rapamycin
signaling. Antioxid Redox Signal. 15:1679–1690. 2011. View Article : Google Scholar :
|
|
24
|
Huang J and Manning BD: The TSC1–TSC2
complex: A molecular switchboard controlling cell growth. Biochem
J. 412:179–190. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gwinn DM, Shackelford DB, Egan DF,
Mihaylova MM, Mery A, Vasquez DS, Turk BE and Shaw RJ: AMPK
phosphorylation of raptor mediates a metabolic checkpoint. Mol
Cell. 30:214–226. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kong B, Wu W, Cheng T, Schlitter AM, Qian
C, Bruns P, Jian Z, Jäger C, Regel I, Raulefs S, et al: A subset of
metastatic pancreatic ductal adenocarcinomas depends quantitatively
on oncogenic Kras/Mek/Erk-induced hyperactive mTOR signalling. Gut.
65:647–657. 2016. View Article : Google Scholar
|
|
27
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mizushima N, Yoshimori T and Ohsumi Y: The
role of Atg proteins in autophagosome formation. Annu Rev Cell Dev
Biol. 27:107–132. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Eby KG, Rosenbluth JM, Mays DJ, Marshall
CB, Barton CE, Sinha S, Johnson KN, Tang L and Pietenpol JA:
ISG20L1 is a p53 family target gene that modulates genotoxic
stress-induced autophagy. Mol Cancer. 9:952010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kenzelmann Broz D, Spano Mello S, Bieging
KT, Jiang D, Dusek RL, Brady CA, Sidow A and Attardi LD: Global
genomic profiling reveals an extensive p53-regulated autophagy
program contributing to key p53 responses. Genes Dev. 27:1016–1031.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gao W, Shen Z, Shang L and Wang X:
Upregulation of human autophagy-initiation kinase ULK1 by tumor
suppressor p53 contributes to DNA-damage-induced cell death. Cell
Death Differ. 18:1598–1607. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Guo JY, Karsli-Uzunbas G, Mathew R, Aisner
SC, Kamphorst JJ, Strohecker AM, Chen G, Price S, Lu W, Teng X, et
al: Autophagy suppresses progression of K-ras-induced lung tumors
to oncocytomas and maintains lipid homeostasis. Genes Dev.
27:1447–1461. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kim MJ, Woo SJ, Yoon CH, Lee JS, An S,
Choi YH, Hwang SG, Yoon G and Lee SJ: Involvement of autophagy in
oncogenic K-Ras-induced malignant cell transformation. J Biol Chem.
286:12924–12932. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kinsey C, Balakrishnan V, O'Dell MR, Huang
JL, Newman L, Whitney-Miller CL, Hezel AF and Land H: Plac8 links
oncogenic mutations to regulation of autophagy and is critical to
pancreatic cancer progression. Cell Rep. 7:1143–1155. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Desai S, Liu Z, Yao J, Patel N, Chen J, Wu
Y, Ahn EE, Fodstad O and Tan M: Heat shock factor 1 (HSF1) controls
chemoresistance and autophagy through transcriptional regulation of
autophagy-related protein 7 (ATG7). J Biol Chem. 288:9165–9176.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Vydra N, Toma A and Widlak W: Pleiotropic
role of HSF1 in neoplastic transformation. Curr Cancer Drug
Targets. 14:144–155. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dai C, Santagata S, Tang Z, Shi J, Cao J,
Kwon H, Bronson RT, Whitesell L and Lindquist S: Loss of tumor
suppressor NF1 activates HSF1 to promote carcinogenesis. J Clin
Invest. 122:3742–3754. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li Q and Martinez JD: P53 is transported
into the nucleus via an Hsf1-dependent nuclear localization
mechanism. Mol Carcinog. 50:143–152. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hu YL, Jahangiri A, De Lay M and Aghi MK:
Hypoxia-induced tumor cell autophagy mediates resistance to
anti-angiogenic therapy. Autophagy. 8:979–981. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chen C, Pore N, Behrooz A, Ismail-Beigi F
and Maity A: Regulation of glut1 mRNA by hypoxia-inducible
factor-1. Interaction between H-ras and hypoxia. J Biol Chem.
276:9519–9525. 2001. View Article : Google Scholar
|
|
44
|
Nieminen AL, Qanungo S, Schneider EA,
Jiang BH and Agani FH: Mdm2 and HIF-1alpha interaction in tumor
cells during hypoxia. J Cell Physiol. 204:364–369. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Robertson ED, Semenchenko K and Wasylyk B:
Crosstalk between Mdm2, p53 and HIF1-α: Distinct responses to
oxygen stress and implications for tumour hypoxia. Subcell Biochem.
85:199–214. 2014. View Article : Google Scholar
|
|
46
|
Hay ED: An overview of
epithelio-mesenchymal transformation. Acta Anat (Basel). 154:8–20.
1995. View Article : Google Scholar
|
|
47
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Puisieux A, Brabletz T and Caramel J:
Oncogenic roles of EMT-inducing transcription factors. Nat Cell
Biol. 16:488–494. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Fischer KR, Durrans A, Lee S, Sheng J, Li
F, Wong ST, Choi H, El Rayes T, Ryu S, Troeger J, et al:
Epithelial-to-mesenchymal transition is not required for lung
metastasis but contributes to chemoresistance. Nature. 527:472–476.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zheng X, Carstens JL, Kim J, Scheible M,
Kaye J, Sugimoto H, Wu CC, LeBleu VS and Kalluri R:
Epithelial-to-mesenchymal transition is dispensable for metastasis
but induces chemoresistance in pancreatic cancer. Nature.
527:525–530. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wang Y, Ngo VN, Marani M, Yang Y, Wright
G, Staudt LM and Downward J: Critical role for transcriptional
repressor Snail2 in transformation by oncogenic RAS in colorectal
carcinoma cells. Oncogene. 29:4658–4670. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Gonzalez DM and Medici D: Signaling
mechanisms of the epithelial-mesenchymal transition. Sci Signal.
7:re82014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zhang J, Lei Y, Gao X, Liang Q, Li L, Feng
J, Hou P, Han L, Zhang Y, Huang B, et al: p53 Attenuates the
oncogenic Ras-induced epithelial-mesenchymal transition in human
mammary epithelial cells. Biochem Biophys Res Commun. 434:606–613.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wang Z, Wade P, Mandell KJ, Akyildiz A,
Parkos CA, Mrsny RJ and Nusrat A: Raf 1 represses expression of the
tight junction protein occludin via activation of the zinc-finger
transcription factor slug. Oncogene. 26:1222–1230. 2007. View Article : Google Scholar
|
|
55
|
Saegusa M, Hashimura M, Kuwata T and
Okayasu I: Requirement of the Akt/beta-catenin pathway for uterine
carcinosarcoma genesis, modulating E-cadherin expression through
the transactivation of slug. Am J Pathol. 174:2107–2115. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wang SP, Wang WL, Chang YL, Wu CT, Chao
YC, Kao SH, Yuan A, Lin CW, Yang SC, Chan WK, et al: p53 controls
cancer cell invasion by inducing the MDM2-mediated degradation of
Slug. Nat Cell Biol. 11:694–704. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wang Y, Bu F, Royer C, Serres S, Larkin
JR, Soto MS, Sibson NR, Salter V, Fritzsche F, Turnquist C, et al:
ASPP2 controls epithelial plasticity and inhibits metastasis
through β-catenin-dependent regulation of ZEB1. Nat Cell Biol.
16:1092–1104. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Kim T, Veronese A, Pichiorri F, Lee TJ,
Jeon YJ, Volinia S, Pineau P, Marchio A, Palatini J, Suh SS, et al:
p53 regulates epithelial-mesenchymal transition through microRNAs
targeting ZEB1 and ZEB2. J Exp Med. 208:875–883. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Chang CJ, Chao CH, Xia W, Yang JY, Xiong
Y, Li CW, Yu WH, Rehman SK, Hsu JL, Lee HH, et al: p53 regulates
epithelial-mesenchymal transition and stem cell properties through
modulating miRNAs. Nat Cell Biol. 13:317–323. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Ansieau S, Bastid J, Doreau A, Morel AP,
Bouchet BP, Thomas C, Fauvet F, Puisieux I, Doglioni C, Piccinin S,
et al: Induction of EMT by twist proteins as a collateral effect of
tumor-promoting inactivation of premature senescence. Cancer Cell.
14:79–89. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Jiang Y, Xie X, Li Z, Wang Z, Zhang Y,
Ling ZQ, Pan Y, Wang Z and Chen Y: Functional cooperation of RKTG
with p53 in tumorigenesis and epithelial-mesenchymal transition.
Cancer Res. 71:2959–2968. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Gibbons DL, Lin W, Creighton CJ, Rizvi ZH,
Gregory PA, Goodall GJ, Thilaganathan N, Du L, Zhang Y,
Pertsemlidis A, et al: Contextual extracellular cues promote tumor
cell EMT and metastasis by regulating miR-200 family expression.
Genes Dev. 23:2140–2151. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Roger L, Jullien L, Gire V and Roux P:
Gain of oncogenic function of p53 mutants regulates E-cadherin
expression uncoupled from cell invasion in colon cancer cells. J
Cell Sci. 123:1295–1305. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Ohashi S, Natsuizaka M, Wong GS,
Michaylira CZ, Grugan KD, Stairs DB, Kalabis J, Vega ME, Kalman RA,
Nakagawa M, et al: Epidermal growth factor receptor and mutant p53
expand an esophageal cellular subpopulation capable of
epithelial-to-mesenchymal transition through ZEB transcription
factors. Cancer Res. 70:4174–4184. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Jiang Z, Deng T, Jones R, Li H,
Herschkowitz JI, Liu JC, Weigman VJ, Tsao MS, Lane TF, Perou CM, et
al: Rb deletion in mouse mammary progenitors induces luminal-B or
basal-like/EMT tumor subtypes depending on p53 status. J Clin
Invest. 120:3296–3309. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Okada T, Sinha S, Esposito I, Schiavon G,
López-Lago MA, Su W, Pratilas CA, Abele C, Hernandez JM, Ohara M,
et al: The Rho GTPase Rnd1 suppresses mammary tumorigenesis and EMT
by restraining Ras-MAPK signalling. Nat Cell Biol. 17:81–94. 2015.
View Article : Google Scholar
|
|
67
|
Gao T, Li JZ, Lu Y, Zhang CY, Li Q, Mao J
and Li LH: The mechanism between epithelial mesenchymal transition
in breast cancer and hypoxia microenvironment. Biomed Pharmacother.
80:393–405. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Imai T, Horiuchi A, Wang C, Oka K, Ohira
S, Nikaido T and Konishi I: Hypoxia attenuates the expression of
E-cadherin via up-regulation of SNAIL in ovarian carcinoma cells.
Am J Pathol. 163:1437–1447. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Zhang L, Huang G, Li X, Zhang Y, Jiang Y,
Shen J, Liu J, Wang Q, Zhu J, Feng X, et al: Hypoxia induces
epithelial-mesenchymal transition via activation of SNAI1 by
hypoxia-inducible factor-1α in hepatocellular carcinoma. BMC
Cancer. 13:1082013. View Article : Google Scholar
|
|
70
|
Cui Y, Li YY, Li J, Zhang HY, Wang F, Bai
X and Li SS: STAT3 regulates hypoxia-induced epithelial mesenchymal
transition in oesophageal squamous cell cancer. Oncol Rep.
36:108–116. 2016.PubMed/NCBI
|
|
71
|
Tsai YP and Wu KJ: Hypoxia-regulated
target genes implicated in tumor metastasis. J Biomed Sci.
19:1022012. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Tsai JH and Yang J: Epithelial-mesenchymal
plasticity in carcinoma metastasis. Genes Dev. 27:2192–2206. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Gugnoni M, Sancisi V, Manzotti G, Gandolfi
G and Ciarrocchi A: Autophagy and epithelial-mesenchymal
transition: An intricate interplay in cancer. Cell Death Dis.
7:e25202016. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Qin W, Li C, Zheng W, Guo Q, Zhang Y, Kang
M, Zhang B, Yang B, Li B, Yang H, et al: Inhibition of autophagy
promotes metastasis and glycolysis by inducing ROS in gastric
cancer cells. Oncotarget. 6:39839–39854. 2015.PubMed/NCBI
|
|
75
|
Qiang L and He YY: Autophagy deficiency
stabilizes TWIST1 to promote epithelial-mesenchymal transition.
Autophagy. 10:1864–1865. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Catalano M, D'Alessandro G, Lepore F,
Corazzari M, Caldarola S, Valacca C, Faienza F, Esposito V,
Limatola C, Cecconi F, et al: Autophagy induction impairs migration
and invasion by reversing EMT in glioblastoma cells. Mol Oncol.
9:1612–1625. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Lv Q, Hua F and Hu ZW: DEDD, a novel tumor
repressor, reverses epithelial-mesenchymal transition by activating
selective autophagy. Autophagy. 8:1675–1676. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Li J, Yang B, Zhou Q, Wu Y, Shang D, Guo
Y, Song Z, Zheng Q and Xiong J: Autophagy promotes hepatocellular
carcinoma cell invasion through activation of
epithelial-mesenchymal transition. Carcinogenesis. 34:1343–1351.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Wei SC and Yang J: Forcing through tumor
metastasis: The interplay between tissue rigidity and
epithelial-mesenchymal transition. Trends Cell Biol. 26:111–120.
2016. View Article : Google Scholar :
|
|
80
|
Tojkander S, Gateva G and Lappalainen P:
Actin stress fibers-assembly, dynamics and biological roles. J Cell
Sci. 125:1855–1864. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Ni HM, Williams JA and Ding WX:
Mitochondrial dynamics and mitochondrial quality control. Redox
Biol. 4:6–13. 2015. View Article : Google Scholar :
|
|
82
|
Youle RJ and van der Bliek AM:
Mitochondrial fission, fusion, and stress. Science. 337:1062–1065.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Zhao J, Zhang J, Yu M, Xie Y, Huang Y,
Wolff DW, Abel PW and Tu Y: Mitochondrial dynamics regulates
migration and invasion of breast cancer cells. Oncogene.
32:4814–4824. 2013. View Article : Google Scholar
|
|
84
|
Ketschek A and Gallo G: Nerve growth
factor induces axonal filopodia through localized microdomains of
phosphoinositide 3-kinase activity that drive the formation of
cytoskeletal precursors to filopodia. J Neurosci. 30:12185–12197.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Parada LF, Land H, Weinberg RA, Wolf D and
Rotter V: Cooperation between gene encoding p53 tumour antigen and
ras in cellular transformation. Nature. 312:649–651. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Jenkins JR, Rudge K and Currie GA:
Cellular immortalization by a cDNA clone encoding the
transformation-associated phosphoprotein p53. Nature. 312:651–654.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Eliyahu D, Raz A, Gruss P, Givol D and
Oren M: Participation of p53 cellular tumour antigen in
transformation of normal embryonic cells. Nature. 312:646–649.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
DuPage M, Dooley AL and Jacks T:
Conditional mouse lung cancer models using adenoviral or lentiviral
delivery of Cre recombinase. Nat Protoc. 4:1064–1072. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Hingorani SR, Wang L, Multani AS, Combs C,
Deramaudt TB, Hruban RH, Rustgi AK, Chang S and Tuveson DA:
Trp53R172H and KrasG12D cooperate to promote
chromosomal instability and widely metastatic pancreatic ductal
adenocarcinoma in mice. Cancer Cell. 7:469–483. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Tsumura H, Yoshida T, Saito H,
Imanaka-Yoshida K and Suzuki N: Cooperation of oncogenic K-ras and
p53 deficiency in pleomorphic rhabdomyosarcoma development in adult
mice. Oncogene. 25:7673–7679. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Zheng S, El-Naggar AK, Kim ES, Kurie JM
and Lozano G: A genetic mouse model for metastatic lung cancer with
gender differences in survival. Oncogene. 26:6896–6904. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Muñoz DM, Tung T, Agnihotri S, Singh S,
Guha A, Zadeh G and Hawkins C: Loss of p53 cooperates with K-ras
activation to induce glioma formation in a region-independent
manner. Glia. 61:1862–1872. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Solomon H, Brosh R, Buganim Y and Rotter
V: Inactivation of the p53 tumor suppressor gene and activation of
the Ras oncogene: Cooperative events in tumorigenesis. Discov Med.
9:448–454. 2010.PubMed/NCBI
|
|
94
|
Jackson JG and Lozano G: The mutant p53
mouse as a preclinical model. Oncogene. 32:4325–4330. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Bertheau P, Turpin E, Rickman DS, Espié M,
de Reyniès A, Feugeas JP, Plassa LF, Soliman H, Varna M, de
Roquancourt A, et al: Exquisite sensitivity of TP53 mutant and
basal breast cancers to a dose-dense epirubicin-cyclophosphamide
regimen. PLoS Med. 4:e902007. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Cassinelli G, Zuco V, Gatti L, Lanzi C,
Zaffaroni N, Colombo D and Perego P: Targeting the Akt kinase to
modulate survival, invasiveness and drug resistance of cancer
cells. Curr Med Chem. 20:1923–1945. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Gurpinar E and Vousden KH: Hitting
cancers' weak spots: Vulnerabilities imposed by p53 mutation.
Trends Cell Biol. 25:486–495. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Bournet B, Buscail C, Muscari F, Cordelier
P and Buscail L: Targeting KRAS for diagnosis, prognosis, and
treatment of pancreatic cancer: Hopes and realities. Eur J Cancer.
54:75–83. 2016. View Article : Google Scholar : PubMed/NCBI
|