Introduction
Approximately 7% of all non-small cell lung cancers
(NSCLCs) contain chromosomal rearrangements of anaplastic lymphoma
kinase (ALK), resulting in constitutively active ALK.
ALK rearranged NSCLCs are often highly sensitive to ALK
tyrosine kinase inhibitors (ALK TKIs) such as crizotinib. However,
the ALK TKI-sensitive ALK rearranged NSCLCs will eventually
develop targeted therapy resistance. Multiple different mechanisms
for ALK TKI resistance have been reported, such as secondary
mutations of ALK, or mutations in other somatic kinase
domains, and activation of alternative signaling pathways through
different receptor tyrosine kinases (RTKs), such as EGFR and HER2
(1–3).
Cancer stem-like cells (CSLCs) have often been
linked to tumor initiation and therapy resistance. They have shown
to be resistant both to the traditional chemo- and radiotherapies,
and to targeted therapies, causing tumor relapses (4–8).
Many signaling pathways have been described to be essential for
CSLCs and potential cancer therapy targets have been discovered
based on these pathways. Furthermore, some unspecific agents, like
salinomycin and metformin, have been shown to target CSLCs
(9,10). Currently, clinical utility of CSLC
targeting agents is still unknown. We have previously shown that
CSLCs can mediate therapy resistance in ALK translocated
NSCLC cell lines (H2228 and H3122) and that targeting both ALK and
CSLCs results in improved cell killing compared to either alone
(6).
ErbB/HER family consists of four members: EGFR,
HER2, HER3 and HER4. When activated, ErbB/HER family members form
either homo- or heterodimers, which can signal downstream to the
PI3K-AKT or Ras-Raf-MEK pathways. EGFR and HER2 receptors are
commonly altered in some cancers, like breast cancers and NSCLCs,
and act as cancer driving oncogenes. HER3 and HER4 have also shown
to be expressed in cancers, but their activating genetic
alterations are uncommon. Co-expression of different HER receptors
have been linked to a worse outcome/poor prognosis, especially EGFR
and HER2 co-expression (11–14).
Furthermore, HER family receptors, especially HER2, have been
linked to CSLCs. Overexpression of HER2 has been shown to increase
not only the amount of CSLC population in series of breast
carcinoma cell lines, but also the tumorigenity in NOD/SCID mice
(15–18). Since numerous clinically active
HER2 targeting agents, such as trastuzumab, pertuzumab and
lapatinib, are approved, characterization of CSLC dependency on
HER2 could lead to rapid clinical testing of the agents in the
context of CSLC targeting.
In the present study, we investigate the role of
HER2 for CSLCs in ALK translocated NSCLC cell lines. The
results suggest that HER2 plays an important role in the CSLC
phenotype.
Materials and methods
Cell lines, inhibitors and growth
factors
The cell lines used in the present study included
ALK translocated NSCLC lines H3122 and H2228, and their
counterparts modified to overexpress GFP or HER2
(H3122) or to knock down GFP or HER2 (H2228). The
original cell lines were a kind gift from Dr Pasi Jänne
(Dana-Farber Cancer Institute, Boston, MA, USA). The cell lines
were grown in RPMI-1640 medium (Thermo Fisher Scientific, Waltham,
MA, USA and Sigma-Aldrich, St. Louis, MO, USA) with 10% fetal
bovine serum (FBS) and 100 IU/ml penicillin and streptomycin
(Thermo Fisher Scientific). Cells were incubated at 37°C with 5%
CO2 in the atmosphere. Following inhibitors were used:
TAE684 (a gift from Dr Nathanael Gray, Dana-Farber Cancer
Institute), crizotinib, afatinib (LC Laboratories, Woburn, MA, USA)
and labatinib (Alexis Biochemicals, Lausen, Switzerland).
Neuregulin-1 (NRG1) and epidermal growth factor (EGF) were also
used (Thermo Fisher Scientific). Inhibitors were diluted in
dimethyl sulfoxide (DMSO) and stored at −20°C while growth factors
were diluted in sterile, distilled water and stored at −80°C.
Lentiviral knockdown and retroviral
overexpression
Lentiviral and retroviral vectors were used to
achieve overexpression or knockdown of GFP and HER2
in the H3122 and H2228 cell lines. HER2 shRNA vector was purchased
from Sigma-Aldrich. GFP shRNA vector and both retroviral vectors
used for overexpression were kind gifts from Dr Pasi Jänne
(Dana-Farber Cancer Institute). 293T cells were transfected with
lenti-/retroviral expression vectors and packaging plasmids using
FuGENE 6 reagent (Roche Diagnostics, Mannheim, Germany). Lentiviral
supernatants were collected 24 h and retroviral supernatants 48 h
after transfection. Both supernatants were filtered through 0.45
μm filter and applied to the target cells in the presence of
Polybrene (Sigma-Aldrich). After 48 h of infections, the target
cells were selected with puromycin (Sigma-Aldrich) for 72–96 h.
Western blot analysis
The cells were plated on 6-well plates, allowed to
attach for 1–2 days and treated with desired drugs for 5 h or 5
days, after which they were lysed with NP-40 lysis buffer (20 mM
Tris-HCl pH 8.0, 137 mM NaCl, 10% glycerol, 2 mM EDTA, 1 mM sodium
orthovanadate, 1% igepal CA-630, 10 μg/ml aprotinin and 10
μg/ml leupeptin). Protein concentrations were measured with
Bio-Rad Protein assay (Bio-Rad Laboratories, Hercules, CA, USA).
After equalizing the concentrations Laemmli buffer was added, the
samples were boiled and stored at −80°C.
Equal amounts of protein samples were separated on
SDS-PAGE and proteins were transferred to PVDF membranes, after
which the membranes were blocked with 5% BSA (1x PBS, 0.1%
Tween-20, 0.005% sodium azide) and incubated in primary antibodies
overnight at 4°C. Horseradish peroxidase (HRP)-linked secondary
antibody was used, the membranes were developed using
chemiluminescence and exposed to radiographic film. The
quantification of western blot images was performed with ImageJ
software, measuring the intensities (pixel percentages) for each
sample in the same membrane with equal, manually selected area.
Following antibodies were used: ALDH1 (BD
Transduction Laboratories, Franklin Lakes, NJ, USA), Akt,
phospho-Akt (S473), CD44, cleaved PARP, EGFR, phospho-EGFR (Y1068),
ERK1/2, phospho-ERK1/2 (T202/Y204), HER2, phospho-HER2
(Y1221/1222), HER3, phospho-HER3 (Y1289), HER4, phospho-HER4
(Y1284), anti-mouse/rabbit HRP-linked secondary antibody (Cell
Signaling Technology, Danvers, MA, USA) and β-actin
(Sigma-Aldrich). All antibodies were diluted in 5% BSA, and used at
1:1,000, 1:20,000 (β-actin), or 1:3,000 (secondary antibodies)
dilutions.
Colony formation assay
Cells (600–1,000) were plated on 24-well plates with
duplicates, allowed to attach for 1–2 days and treated with drugs.
After 7 days, the drugs were withdrawn and the cells were allowed
to recover and form colonies for several weeks. After clear
differences were observed in the growth of colonies, the cells were
fixed with ice-cold methanol and dyed with crystal violet
stain.
Tumor sphere formation assay
The cells were treated with or without ALK TKI for 5
days, after which the cells were plated on 6-well ultra-low
attachment plates (Corning Inc., Corning, NY, USA). A total of
5,000 (H3122) or 7,000 (H2228) cells were seeded on each well with
or without further ALK TKI treatment. The cells were cultured in
Dulbecco's modified Eagle's medium (DMEM)/F-12 media with 20 ng/ml
EGF, 20 ng/ml bFGF, 1% B27 supplement and 100 IU/ml penicillin and
streptomycin (Thermo Fisher Scientific). The spheres were allowed
to grow for 15 days, after which images were taken and the sphere
numbers were counted. Magnification (x10) in phase contrast
microscope was used when imaging the largest spheres from each
well.
Results
HER2 alters expression of CSLC
markers
We initially accessed whether two EML4-ALK
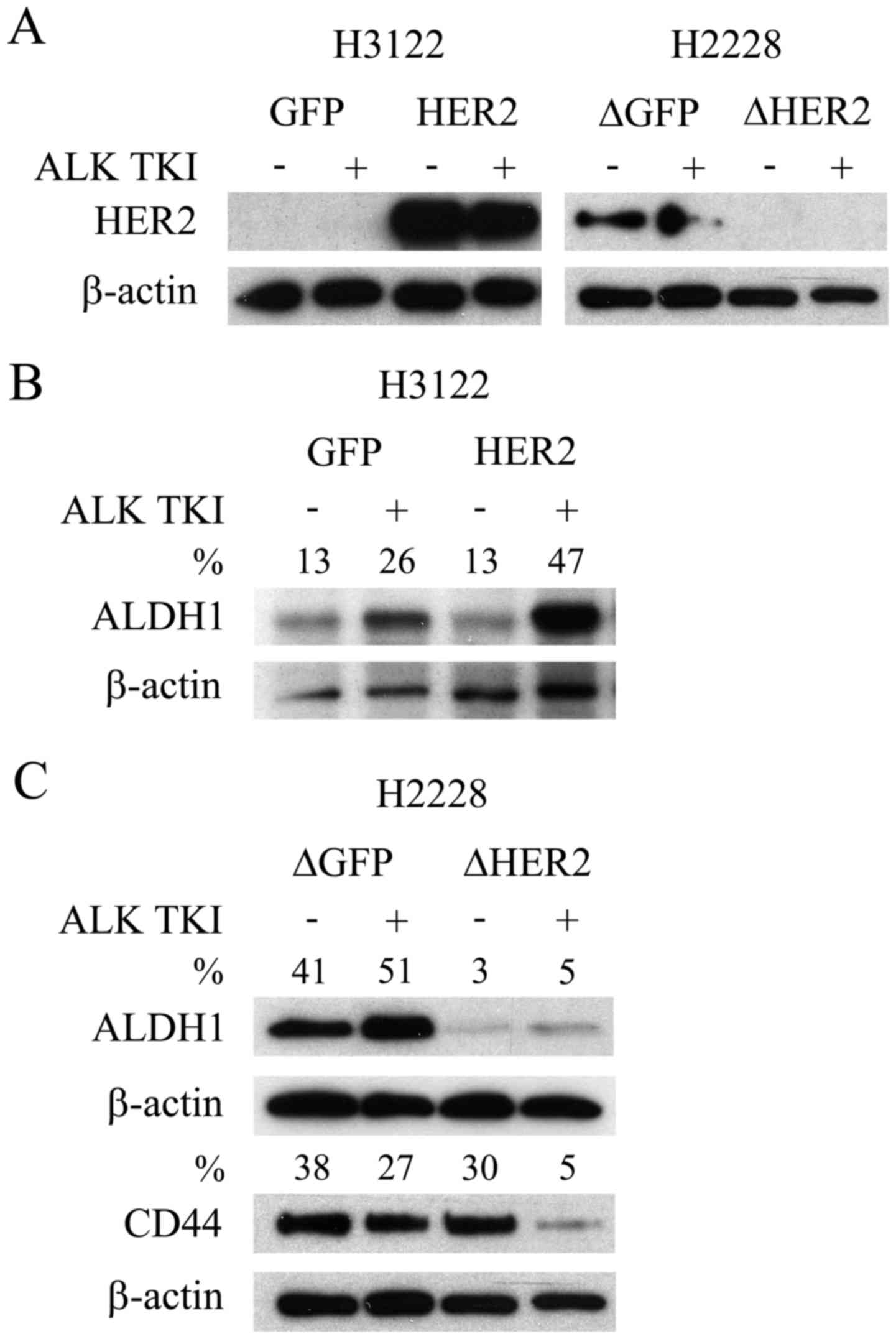
translocated NSCLC cell lines, H3122 and H2228, basally expressed
HER2. In the H3122 cells, which are very sensitive to ALK
inhibition, basal HER2 expression was undetectable (Fig. 1A). Conversely, the H2228 cells,
modestly sensitive to ALK inhibition, showed basal expression of
HER2 (Fig. 1A). The overexpression
of HER2 with a retroviral expression vector was successful in H3122
cells with a marked increase in the expression level of the protein
(Fig. 1A). Furthermore, knockdown
of HER2 with lentiviral shRNA vector induced a complete or
near complete downregulation of the protein expression in the H2228
line (Fig. 1A).
We next accessed whether overexpression or knockdown
of HER2 resulted in changes to CSLCs. We have previously
shown that CSLCs, indicated by specific markers ALDH1 (H3122) and
CD44/ALDH1 (H2228), can mediate the ALK TKI resistance (6). As in our previous studies, ALDH1
expression increased in the H3122 cells in response to ALK
inhibition (Fig. 1B), suggesting
CSLC mediated resistance. Overexpression of HER2 in H3122 did not
change the basal ALDH1 expression, but when the cells were
challenged with ALK TKI, this resulted in more pronounced
expression of CSLC marker (Fig.
1B). Knockdown of HER2 in the H2228 cells resulted in
basal downregulation of ALDH1, but unaltered expression of CD44
(Fig. 1C), a marker previously
linked most strongly to CSLCs in this cell line (6). Moreover, marked CD44 downregulation
was only seen in HER2 knockdown H2228 cells when they were
challenged with ALK TKI (Fig.
1C).
Role of HER2 in cytotoxic response to ALK
TKI
The HER2 overexpressing H3122 cells or the
HER2 knockdown H2228 cells were next exposed to ALK TKIs to
see whether their colony formation and apoptotic responses to ALK
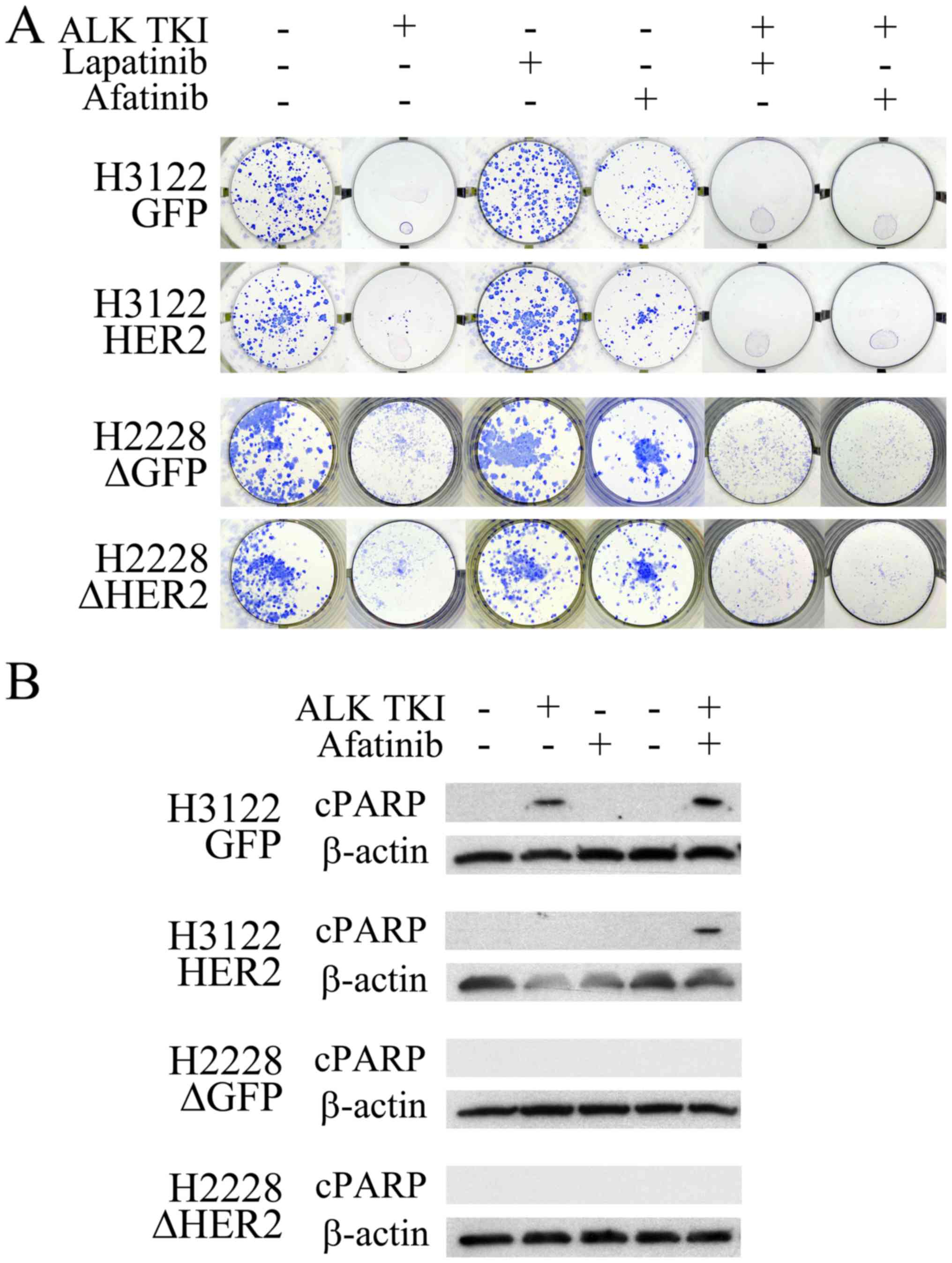
inhibition was altered. In the colony formation assay, major
cytotoxicity response to ALK inhibition remained unaffected by the
HER2 alterations (Fig. 2A).
However, the ALK inhibitor treatment in the H3122 cells with HER2
overexpression resulted in increased number of surviving colonies
compared to controls (Fig. 2A).
Analogously, the ALK inhibitor treated H2228 cells with HER2
knockdown had a modestly decreased number of surviving colonies
(Fig. 2A). When the H3122 and
H2228 cells were treated with a single HER2 specific inhibitor
lapatinib, no change in cell survival was seen (Fig. 2A). The pan-HER (EGFR, HER2 and
HER4) inhibitor afatinib, however, decreased the number of
surviving colonies modestly in both tested cell lines (Fig. 2A). There was no difference in
single-agent afatinib responses between the HER2 altered and
control cells (Fig. 2A).
Combination of ALK TKI with either lapatinib or afatinib resulted
in total inhibition of colony formation in HER2 overexpressing
H3122 cells (Fig. 2A). In H2228
cells, combined inhibition lead to a more pronounced colony
inhibition in the HER2 knockdown cells, afatinib being more
potent than lapatinib in this setting (Fig. 2A).
Next, we wanted to assess, whether HER2 alteration
would affect apoptotic response in the cell lines using western
blot analysis for the apoptotic marker cleaved PARP. In H3122
control cells, cleaved PARP was detected in cells treated with ALK
TKI and its combination with afatinib (Fig. 2B). In the H3122 cells
overexpressing HER2, cleaved PARP was only detected following a
combined treatment with ALK TKI and afatinib (Fig. 2B). In H2228 cells, analogously to
previous study (3), no cleaved
PARP signal was seen in control cells, and the HER2
knockdown did not have an effect on this (Fig. 2B).
Role of HER2 in sphere formation
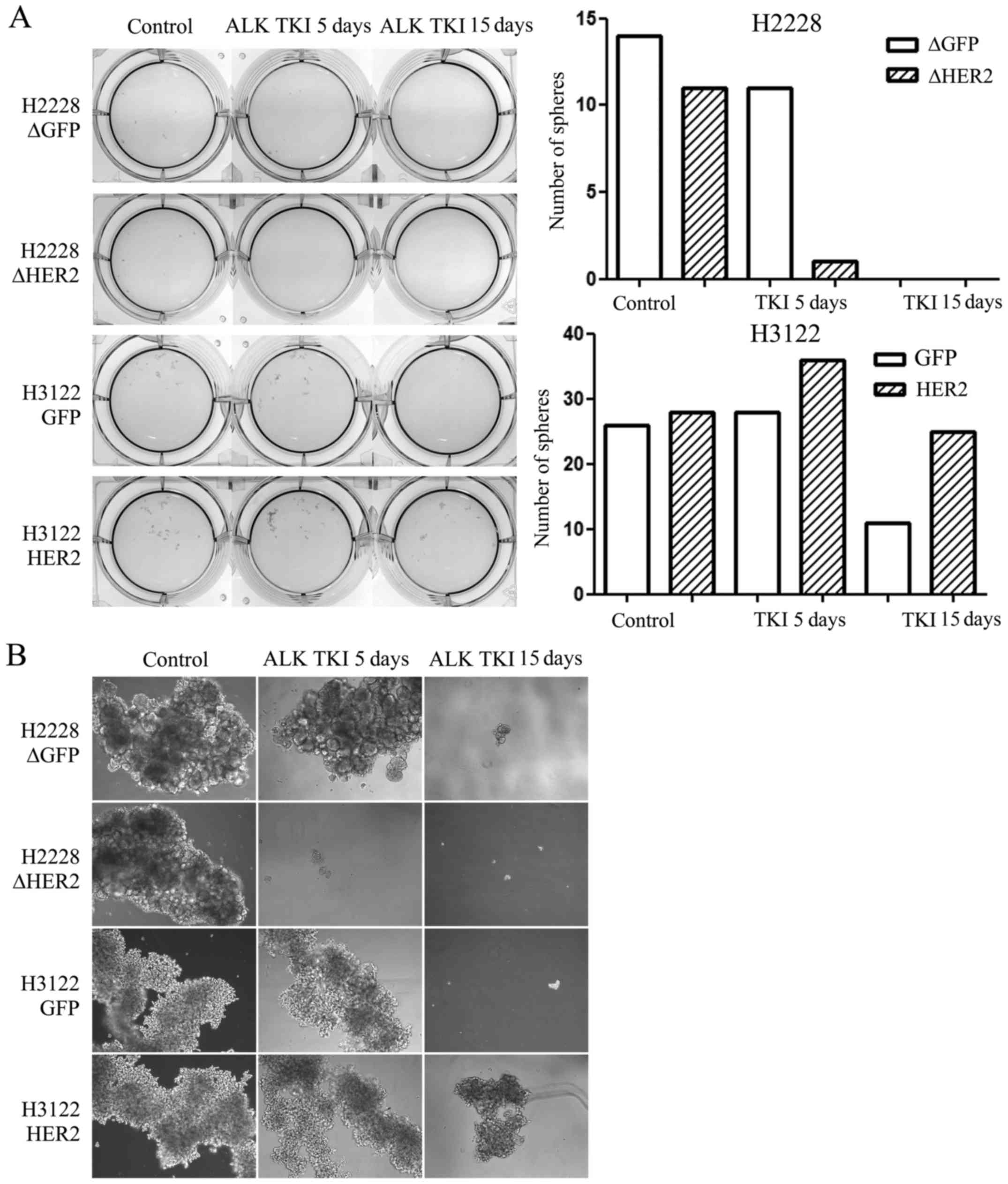
Sphere formation assay is one of the most used assay
to identify CSLCs in vitro and therefore, we assessed
whether HER2 alteration would modify the sphere formation of the
ALK translocated H3122 or H2228 cells. We exposed the
control and HER2 altered cells to ALK TKI in vitro for 5 or
15 days in stem cell enriching sphere formation environment with
special media and low-attaching culture plates. In untreated cells,
HER2 had no effect in the sphere formation capacity (Fig. 3). In H2228 cells treated with ALK
TKI, HER2 knockdown was able to inhibit sphere formation at
5 days compared to control cells while no surviving spheres where
seen at 15 days (Fig. 3). H3122
cells with HER2 overexpression were able to form spheres in the
presence of ALK TKI while this capacity was markedly reduced in
control cells treated with ALK TKI for 5 or 15 days (Fig. 3).
HER2 is essential to AKT and ERK1/2
downstream signaling in long-term exposure to ALK TKI
PI3K and MAPK pathways are one of the most important
downstream signaling pathways controlled by RTKs, such as ALK and
HER2 (3,19,20).
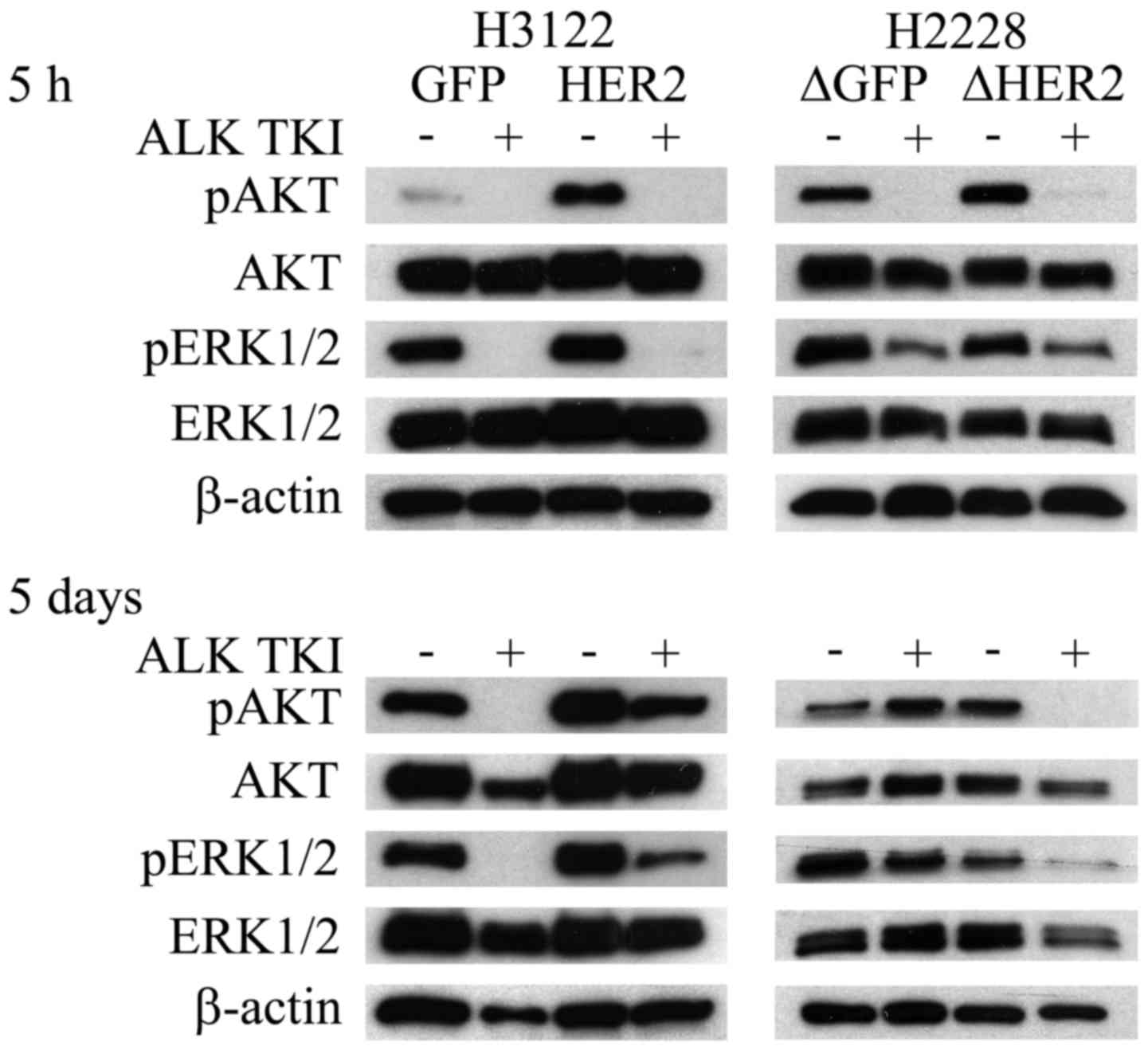
To examine the effects of HER2 overexpression and knockdown to
initial and long-term downstream signaling, phosphorylation of AKT
and ERK1/2 proteins was investigated at 5 h and 5 days after
initiation of ALK inhibition. The overexpression of HER2 in H3122
cells did not change the AKT or ERK1/2 responses to short-term (5
h) ALK TKI treatment (Fig. 4).
Long-term (5 days) treatment of HER2 overexpressing H3122 cells
with ALK TKIs, however, resulted in reformed downstream RTK
signaling with major upregulation of both phosphorylated AKT and
ERK1/2 (Fig. 4). In H2228 cells,
initial response (5 h) to ALK TKIs was similar in the HER2
knockdown cells (Fig. 4).
Similarly to H3122 cells, HER2 altered H2228 cells showed different
long-term (5 days) ALK TKI responses in downstream signaling
(Fig. 4). In control H2228 cells,
initial downregulation of AKT and ERK1/2 phosphorylation was
completely recovered at 5 days while the downregulation remained in
the HER2 knockdown cells (Fig.
4).
HER2 orchestrates ErbB-family
signaling
Next, we wanted to study whether HER2
overexpression or knockdown had any effects on other
ErbB/HER-family members and their activation, since HER2 forms not
only homodimers but also heterodimers with all the other members of
the protein family (21). In the
H3122 control cells, only EGFR and HER3 were expressed without
evidence of their activation by absence of phosphorylated protein
counterparts (Fig. 5A). HER2
overexpression increased both expression and phosphorylation of
EGFR and HER4 proteins, and phosphorylation of HER3 (Fig. 5A). In H2228 cells, only EGFR and
HER2 were basally expressed and no phosphorylation of the proteins
was detected (Fig. 5B). Knockdown
of HER2 resulted in downregulated expression of HER3
(Fig. 5B).
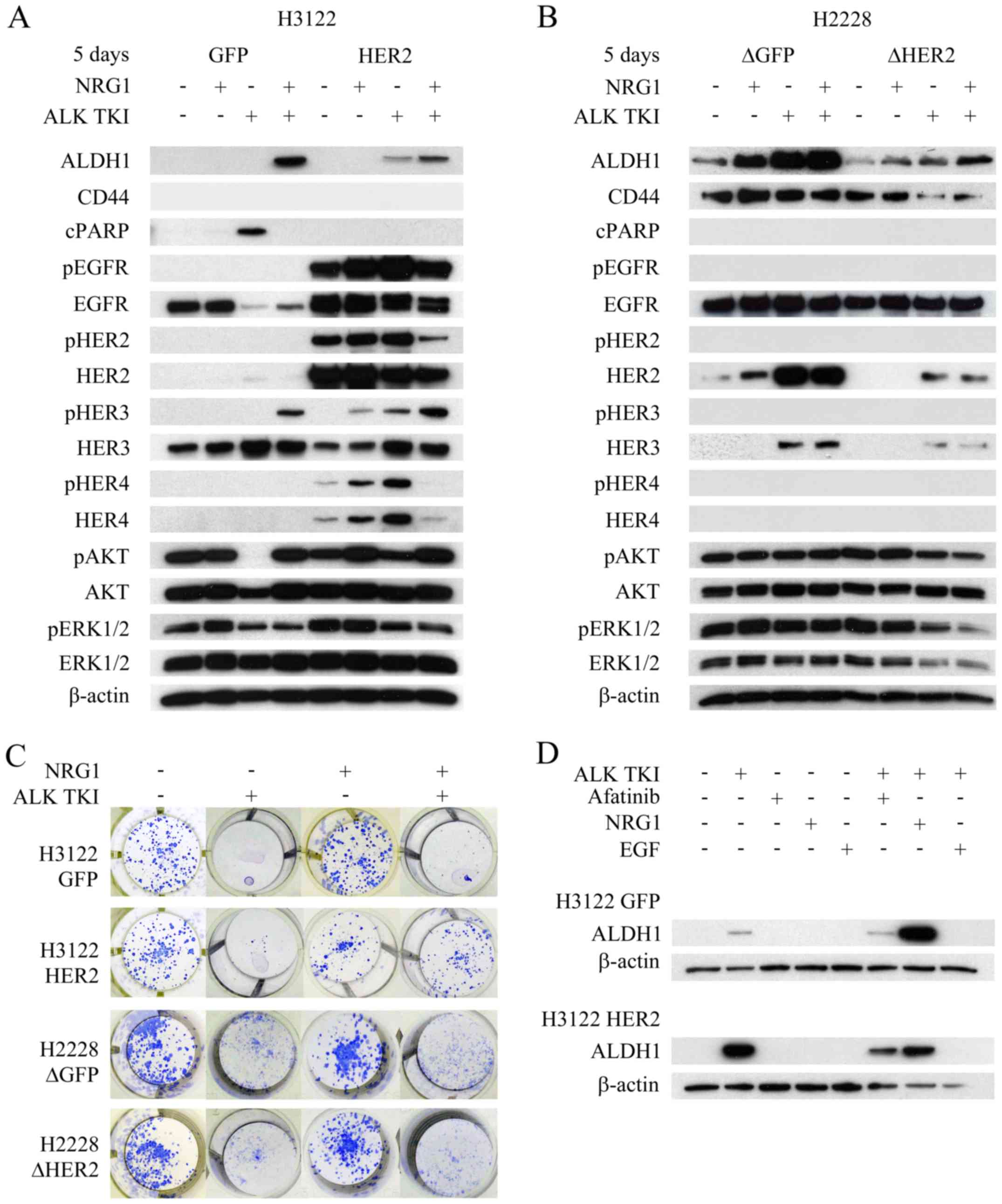
Since HER2 alteration resulted in changes to HER3
and/or HER4 in both cell lines, we accessed whether neuregulin-1
(NRG1), only known joint HER3 and HER4 ligand and linked to tumor
initiating cells (22), would
modify behavior of the cells. In colony formation assay, NRG1
itself did not alter the number of colonies (Fig. 5C). As previously described, H3122
cells with HER2 overexpression showed increased number of surviving
colonies in ALK TKI treated cells compared controls (Fig. 2A). When H3122 cells were treated
with NRG1 in combination with ALK TKI, there was marked difference
between the HER2 overexpressing and control cells (Fig. 5C). HER2 overexpressing cells showed
a marked increase in the number of surviving colonies while the
control cells showed only a minor increase (Fig. 5C). In H2228 cells, HER2
knockdown did not affect the NRG1 response (Fig. 5C).
The cells were further analyzed for cell signaling,
apoptotic response and/or CSLC marker expression after the NRG1
exposure. Especially in the H3122 line, NRG1 treatment altered all
the analyzed responses (Fig. 5A).
In H3122 cells, the most prominent effect of NRG1 was seen in HER3
phosphorylation (Fig. 5A). In
control cells, NRG1 was able to induce HER3 phosphorylation in the
ALK TKI treated cells, which was accompanied by upregulation of
phosphorylated AKT, inhibition of apoptosis (cleaved PARP) and
increased expression of CSLC markers (Fig. 5A). In HER2 overexpressing cells,
HER3 phosphorylation was already detectable in cells treated with
NRG1 alone and the most prominent expression was seen when it was
combined to ALK TKI (Fig. 5A).
Furthermore, NRG1 and ALK TKI combination induced the strongest
expression of CSLC markers (Fig.
5A). In H2228 cells, NRG1 induced increase in ALDH1 expression,
most notably in control cells (Fig.
5B). Only minor changes were seen in the CD44 expression, some
rescue of ALK TKI induced downregulation of the marker detected in
the HER2 knockdown cells by NRG1 (Fig. 5B). We also assessed the effects of
EGF, another HER ligand, mainly activating EGFR, on CSLC marker
expression in the H3122 cells with HER2 alterations. However, EGF
was unable to stimulate CSLC marker expression alone or in
combination with ALK TKI (Fig.
5D).
Discussion
CSLCs have been linked to chemo-, radio- and
targeted therapy resistance (4,7,8).
Molecular mechanisms behind CSLC phenotype are largely unknown, but
some signaling pathways such as wnt/β-catenin, TGF-β and HER2
pathways have been linked to it (23–25).
Understanding molecular mechanisms of CSLCs would enable more
efficient cancer treatment with combinatory approaches. Early phase
clinical trials are testing some agents suggested to target CSLCs
but no clear evidence of their effectiveness have been
presented.
ALK translocated NSCLC represents a subgroup
of disease in which patients are highly sensitive to ALK
inhibitors, such as crizotinib. As with other targeted agents,
acquired resistance to ALK inhibitors develops ~10–12 months after
therapy initiation. Molecular mechanisms of ALK inhibitor
resistance includes secondary mutations in tyrosine kinase domain
of ALK, activation of by-pass signaling mechanisms and CSLC
phenotype (3,6,26).
Many previous reports have linked the ErbB-pathway activation
mediated by-pass signaling mechanisms to ALK TKI resistance
(3,6,27–29).
The present study assessed whether ErbB-signaling affects the CSLC
acquired in ALK translocated NSCLC models. We used two model
cell lines, which we have previously shown to be either sensitive
or modestly sensitive to ALK inhibition and CSLC phenotype to be
related to therapy resistance.
Of the ErbB-pathways, HER2 has been linked most
strongly to CSLC phenotype (17).
HER2 targeting antibody trastuzumab is approved only for the
treatment of HER2 amplified breast cancer. However,
reassessment of studies of HER2 amplification have
identified that some patients without amplification can benefit
from adjuvant trastuzumab therapy (30,31).
It has been speculated that benefit without HER2
amplification could relate to CSLC targeting activity of
trastuzumab. Our results showed that HER2 expression correlated
with CSLC markers and sphere formation in ALK translocated
models. More precisely, HER2 overexpression resulted more
pronounced stem-like cell marker in response to ALK TKI while
knockdown of the gene inhibited TKI induced stem-like cell
phenotype. These results further highlight the importance of HER2
in CSLC. Our results showed not only the correlation between
stem-like cell marker expression and HER2 but also pointed towards
functionality of HER2 to stem-like cells assessed by colony or
sphere formation. Genomic or pharmacologic alteration of HER2
modifies colony and sphere formation ability of ALK
translocated models, HER2 correlating with increased capability.
Since numerous agents targeting ErbB-signaling components are
available in clinic, it would be interesting to test them in
context of CSLC targeting as a combinatory approach.
Our experimentation showed that initial cytotoxic
response or downstream receptor signaling (occurring in hours) to
ALK TKIs was not changed by HER2 alterations. Notably, there was a
marked change in the number of surviving colonies and downstream
receptor signaling after long-term exposure (days) to ALK TKIs
according HER2 status. More precisely, HER2 overexpression was able
to markedly reactivate the AKT and ERK signaling after long-term
exposure to ALK TKIs compared to controls. Analogously, HER2
knockdown resulted in less recovery of AKT and ERK signaling after
long ALK TKI treatment. More pronounced effects of HER2 alterations
were seen in AKT rather than in ERK signaling. Many previous
studies have linked AKT-mTOR signaling to CSLC phenotype and
targeting this pathway has been shown to inhibit CSLCs (24,32–34).
In ALK translocated cancers, AKT and ERK signaling is mainly
driven by ALK and signaling recovery after long exposures to ALK
inhibition is generally unknown. The present study suggests the
importance of HER2 in this signaling recovery.
ErbB-family members can form homo- or heterodimers,
which signal downstream of AKT and ERK with variable preference
(19,21). In HER2 amplified breast
cancer, HER2-HER3 heterodimer is thought to be the most important
signaling component, which preferentially signals though AKT-mTOR
(35,36). HER3/HER4 ligand NRG1 mainly
promotes AKT-mTOR signaling and interestingly, has previously been
linked to tumor initiating cells/CSLCs (22). This study suggests that HER2
orchestrates all other ErbB-family members. Expression of CSLC
marker ALDH1 followed most closely HER3. NRG1 and long ALK TKI
treatment promoted CSLC marker expression and HER3, which was
accompanied by increased colony formation. This suggests that
HER2/HER3 heterodimer could play an important role in CSLCs of
ALK translocated lung cancers.
This study investigated the role of HER2 in CSLCs
using ALK translocated lung cancer as a model. The results
of the study suggest that HER2 has an important role in CSLC
phenotype in vitro mainly orchestrated by HER2/HER3
heterodimers.
Glossary
Abbreviations
Abbreviations:
|
ALDH1
|
aldehyde dehydrogenase 1
|
|
ALK
|
anaplastic lymphoma kinase
|
|
CSLC
|
cancer stem-like cell
|
|
EGF
|
epidermal growth factor
|
|
ErbB
|
erythroblastic leukemia viral oncogene
homolog
|
|
HER
|
human epidermal growth factor
receptor
|
|
NRG1
|
neuregulin-1
|
|
NSCLC
|
non-small cell lung cancer
|
|
RTK
|
receptor tyrosine kinase
|
|
TKI
|
tyrosine kinase inhibitor
|
Acknowledgments
The present study was supported by the Cancer
Foundation of Finland.
References
|
1
|
Doebele RC, Pilling AB, Aisner DL,
Kutateladze TG, Le AT, Weickhardt AJ, Kondo KL, Linderman DJ,
Heasley LE, Franklin WA, et al: Mechanisms of resistance to
crizotinib in patients with ALK gene rearranged non-small cell lung
cancer. Clin Cancer Res. 18:1472–1482. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Soda M, Choi YL, Enomoto M, Takada S,
Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K,
Hatanaka H, et al: Identification of the transforming EML4-ALK
fusion gene in non-small-cell lung cancer. Nature. 448:561–566.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Koivunen JP, Mermel C, Zejnullahu K,
Murphy C, Lifshits E, Holmes AJ, Choi HG, Kim J, Chiang D, Thomas
R, et al: EML4-ALK fusion gene and efficacy of an ALK kinase
inhibitor in lung cancer. Clin Cancer Res. 14:4275–4283. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q,
Hjelmeland AB, Dewhirst MW, Bigner DD and Rich JN: Glioma stem
cells promote radioresistance by preferential activation of the DNA
damage response. Nature. 444:756–760. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
De Cola A, Volpe S, Budani MC, Ferracin M,
Lattanzio R, Turdo A, D'Agostino D, Capone E, Stassi G, Todaro M,
et al: miR-205-5p-mediated downregulation of ErbB/HER receptors in
breast cancer stem cells results in targeted therapy resistance.
Cell Death Dis. 6:e18232015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jokinen E, Laurila N, Koivunen P and
Koivunen JP: Combining targeted drugs to overcome and prevent
resistance of solid cancers with some stem-like cell features.
Oncotarget. 5:9295–9307. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shien K, Toyooka S, Yamamoto H, Soh J,
Jida M, Thu KL, Hashida S, Maki Y, Ichihara E, Asano H, et al:
Acquired resistance to EGFR inhibitors is associated with a
manifestation of stem cell-like properties in cancer cells. Cancer
Res. 73:3051–3061. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li X, Lewis MT, Huang J, Gutierrez C,
Osborne CK, Wu MF, Hilsenbeck SG, Pavlick A, Zhang X, Chamness GC,
et al: Intrinsic resistance of tumorigenic breast cancer cells to
chemotherapy. J Natl Cancer Inst. 100:672–679. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Takebe N, Miele L, Harris PJ, Jeong W,
Bando H, Kahn M, Yang SX and Ivy SP: Targeting Notch, Hedgehog, and
Wnt pathways in cancer stem cells: Clinical update. Nat Rev Clin
Oncol. 12:445–464. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gupta PB, Onder TT, Jiang G, Tao K,
Kuperwasser C, Weinberg RA and Lander ES: Identification of
selective inhibitors of cancer stem cells by high-throughput
screening. Cell. 138:645–659. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Abd El-Rehim DM, Pinder SE, Paish CE, Bell
JA, Rampaul RS, Blamey RW, Robertson JF, Nicholson RI and Ellis IO:
Expression and co-expression of the members of the epidermal growth
factor receptor (EGFR) family in invasive breast carcinoma. Br J
Cancer. 91:1532–1542. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lynch TJ, Bell DW, Sordella R,
Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat
SM, Supko JG, Haluska FG, et al: Activating mutations in the
epidermal growth factor receptor underlying responsiveness of
non-small-cell lung cancer to gefitinib. N Engl J Med.
350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Slamon DJ, Leyland-Jones B, Shak S, Fuchs
H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M,
et al: Use of chemotherapy plus a monoclonal antibody against HER2
for metastatic breast cancer that overexpresses HER2. N Engl J Med.
344:783–792. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Suo Z, Risberg B, Kalsson MG, Willman K,
Tierens A, Skovlund E and Nesland JM: EGFR family expression in
breast carcinomas. c-erbB-2 and c-erbB-4 receptors have different
effects on survival. J Pathol. 196:17–25. 2002. View Article : Google Scholar
|
|
15
|
Ithimakin S, Day KC, Malik F, Zen Q,
Dawsey SJ, Bersano-Begey TF, Quraishi AA, Ignatoski KW, Daignault
S, Davis A, et al: HER2 drives luminal breast cancer stem cells in
the absence of HER2 amplification: Implications for efficacy of
adjuvant trastuzumab. Cancer Res. 73:1635–1646. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Clark PA, Iida M, Treisman DM, Kalluri H,
Ezhilan S, Zorniak M, Wheeler DL and Kuo JS: Activation of multiple
ERBB family receptors mediates glioblastoma cancer stem-like cell
resistance to EGFR-targeted inhibition. Neoplasia. 14:420–428.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Korkaya H, Paulson A, Iovino F and Wicha
MS: HER2 regulates the mammary stem/progenitor cell population
driving tumorigenesis and invasion. Oncogene. 27:6120–6130. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sette G, Salvati V, Mottolese M, Visca P,
Gallo E, Fecchi K, Pilozzi E, Duranti E, Policicchio E, Tartaglia
M, et al: Tyr1068-phosphorylated epidermal growth factor receptor
(EGFR) predicts cancer stem cell targeting by erlotinib in
preclinical models of wild-type EGFR lung cancer. Cell Death Dis.
6:e18502015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schulze WX, Deng L and Mann M:
Phosphotyrosine interactome of the ErbB-receptor kinase family. Mol
Syst Biol. 1:00082005. View Article : Google Scholar
|
|
20
|
Serra V, Scaltriti M, Prudkin L, Eichhorn
PJ, Ibrahim YH, Chandarlapaty S, Markman B, Rodriguez O, Guzman M,
Rodriguez S, et al: PI3K inhibition results in enhanced HER
signaling and acquired ERK dependency in HER2-overexpressing breast
cancer. Oncogene. 30:2547–2557. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hynes NE and Lane HA: ERBB receptors and
cancer: The complexity of targeted inhibitors. Nat Rev Cancer.
5:341–354. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hegde GV, de la Cruz CC, Chiu C, Alag N,
Schaefer G, Crocker L, Ross S, Goldenberg D, Merchant M, Tien J, et
al: Blocking NRG1 and other ligand-mediated Her4 signaling enhances
the magnitude and duration of the chemotherapeutic response of
non-small cell lung cancer. Sci Transl Med. 5:171ra182013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Malanchi I, Peinado H, Kassen D, Hussenet
T, Metzger D, Chambon P, Huber M, Hohl D, Cano A, Birchmeier W, et
al: Cutaneous cancer stem cell maintenance is dependent on
beta-catenin signalling. Nature. 452:650–653. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Korkaya H, Paulson A, Charafe-Jauffret E,
Ginestier C, Brown M, Dutcher J, Clouthier SG and Wicha MS:
Regulation of mammary stem/progenitor cells by
PTEN/Akt/beta-catenin signaling. PLoS Biol. 7:e10001212009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Anido J, Sáez-Borderías A, Gonzàlez-Juncà
A, Rodón L, Folch G, Carmona MA, Prieto-Sánchez RM, Barba I,
Martínez-Sáez E, Prudkin L, et al: TGF-β receptor inhibitors target
the CD44high/Id1high glioma-initiating cell
population in human glioblastoma. Cancer Cell. 18:655–668. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Katayama R, Shaw AT, Khan TM,
Mino-Kenudson M, Solomon BJ, Halmos B, Jessop NA, Wain JC, Yeo AT,
Benes C, et al: Mechanisms of acquired crizotinib resistance in
ALK-rearranged lung Cancers. Sci Transl Med. 4:120ra172012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Isozaki H, Ichihara E, Takigawa N, Ohashi
K, Ochi N, Yasugi M, Ninomiya T, Yamane H, Hotta K, Sakai K, et al:
Non-small cell lung cancer cells acquire resistance to the ALK
inhibitor alectinib by activating alternative receptor tyrosine
kinases. Cancer Res. 76:1506–1516. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sasaki T, Koivunen J, Ogino A, Yanagita M,
Nikiforow S, Zheng W, Lathan C, Marcoux JP, Du J, Okuda K, et al: A
novel ALK secondary mutation and EGFR signaling cause resistance to
ALK kinase inhibitors. Cancer Res. 71:6051–6060. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tanizaki J, Okamoto I, Okabe T, Sakai K,
Tanaka K, Hayashi H, Kaneda H, Takezawa K, Kuwata K, Yamaguchi H,
et al: Activation of HER family signaling as a mechanism of
acquired resistance to ALK inhibitors in EML4-ALK-positive
non-small cell lung cancer. Clin Cancer Res. 18:6219–6226. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Paik S, Kim C and Wolmark N: HER2 status
and benefit from adjuvant trastuzumab in breast cancer. N Engl J
Med. 358:1409–1411. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Perez EA, Reinholz MM, Hillman DW, Tenner
KS, Schroeder MJ, Davidson NE, Martino S, Sledge GW, Harris LN,
Gralow JR, et al: HER2 and chromosome 17 effect on patient outcome
in the N9831 adjuvant trastuzumab trial. J Clin Oncol.
28:4307–4315. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Todaro M, Gaggianesi M, Catalano V,
Benfante A, Iovino F, Biffoni M, Apuzzo T, Sperduti I, Volpe S,
Cocorullo G, et al: CD44v6 is a marker of constitutive and
reprogrammed cancer stem cells driving colon cancer metastasis.
Cell Stem Cell. 14:342–356. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xia P and Xu XY: PI3K/Akt/mTOR signaling
pathway in cancer stem cells: From basic research to clinical
application. Am J Cancer Res. 5:1602–1609. 2015.PubMed/NCBI
|
|
34
|
Zhu Y, Zhang X, Liu Y, Zhang S, Liu J, Ma
Y and Zhang J: Antitumor effect of the mTOR inhibitor everolimus in
combination with trastuzumab on human breast cancer stem cells in
vitro and in vivo. Tumour Biol. 33:1349–1362. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Holbro T, Beerli RR, Maurer F, Koziczak M,
Barbas CF III and Hynes NE: The ErbB2/ErbB3 heterodimer functions
as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor
cell proliferation. Proc Natl Acad Sci USA. 100:8933–8938. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Vaught DB, Stanford JC, Young C, Hicks DJ,
Wheeler F, Rinehart C, Sánchez V, Koland J, Muller WJ, Arteaga CL,
et al: HER3 is required for HER2-induced preneoplastic changes to
the breast epithelium and tumor formation. Cancer Res.
72:2672–2682. 2012. View Article : Google Scholar : PubMed/NCBI
|