Introduction
The PI3K/PTEN/AKT signaling pathway has
well-established roles in multiple cellular activities, including
cell proliferation, survival, metabolism, cytoskeleton
reorganization and membrane trafficking (1-3). The
abnormal activation of this signaling pathway leads to various
diseases such as diabetes, autoimmunity and cancer. About ten years
ago, our group, along with several other research groups,
simultaneously found that the somatic mutation frequency of the
PIK3CA gene in breast cancer is 20-30% (4-7). Our
research demonstrated that somatic mutation, rather than gain of
PIK3CA copy number, is one of the most frequent genetic
alterations contributing to human breast cancer progression
(7). In another study, we
comprehensively analyzed and compared the oncogenic properties of
nine different PIK3CA somatic mutations, which localized in
different domains of the PIK3CA gene and with different
frequencies in human breast cancer (8). The results of our study are
consistent with several other groups, using different research
systems, and strongly indicate that different PI3KCA mutants
exhibit different abilities in contributing to cell proliferation,
EGF independent growth, cell morphogenesis, transformation,
invasion and signaling (9-12). These findings collectively provide
fundamental biological evidence to support the critical role of the
PI3k/AkT signalling pathway in breast cancer progression. However,
to date, there is insufficient clinical data to support that PI3K
or AKT inhibitors can be powerful single agents for breast cancer
patients (13,14).
HER2 (ErbB2), a member of the HER family of tyrosine
kinase receptors (HER1-4), is a major driver of tumor growth in 20%
of breast cancers. Due to the well-studied nature of the
HER2 gene in breast cancer and the availability of the
monoclonal targeting antibody trastuzumab, targeting HER2 has been
the most successful targeted treatment for breast cancer patients
(15,16). However, targeting HER2 alone was
less effective for breast cancer patients with PIK3CA mutations in
clinical studies (17,18). In line with these observations,
several groups reported that HER2 amplification and mutation
of PIK3CA genes could be co-occurring in certain breast
cancer population (6,19-22).
However, the cooperative effect of these two genetic alterations in
comparison with either single genetic change on cell oncogenic
properties has not been well investigated.
In this study, we performed a genome-wide analysis
for amplification regions and corresponding genes that correlate to
mutant PIK3CA in 51 human breast cancer cell lines. We also
specifically examined the oncogenic properties driven by expressing
both mutant PIK3CA and HER2 and compare the effects
to cells with either genetic alteration alone. Additionally, we
tested the drug treatment response in cells with ectopic expression
of mutant PIK3CA and HER2 amplification. Finally, we
investigated the downstream target genes and cell signalling
pathways regulated by HER2, mutant PIK3CA and both of
these genetic alterations.
Materials and methods
Bioinformatics analysis for amplification
of regions that are correlated with mutant PIK3CA
A published database was used for bioinformatic
analysis. This database contains gene expression and copy number
information for 51 breast cancer cell lines (23) (http://caarraydb.nci.nih.gov/caarray/publicEx-perimentDetailAction.do?expId=1015897590151581
at http://cancer.lbl.gov/breastcancer/data.php). Among
these 51 cell lines, 13 cell lines contain PIK3CA mutations.
The other 38 are considered PIK3CA-wild type. We then used
the strategy as follows to identify amplified gene that could
synergize with mutant PIK3CA in breast cancer. a) Threshold
aCGH and gene expression data: copy number variation (CNV)
amplification based on a cut-off ≥0.2. Gene overexpression based on
a cut-off >143.767 (3-fold of the median of all samples). b) CNV
markers and genes with highly increased
amplification/overexpression frequency based on the following
criteria: i) frequency difference between cell line w/mutations and
w/o ≥0.25 or ii) Fisher exact test P-value of the difference
<0.05. c) Pairs of amplified CNV markers and overexpressed genes
which are close to each other (distance ≤2 Mb). d) Pairs of
amplified CNV markers and overexpressed genes which are close to
each other (distance ≤2 Mb) and positively correlated.
Cell culture
MCF 10A and HCC1954 cells were obtained from the
American Tissue culture collection. MCF10A cell lines expressing
LacZ (negative control), PI3KCA-H1047R, HER2, and
both PI3KCA-H1074R and HER2 genes were created in our
laboratory at the Barbara Ann karmanos cancer Institute (KCI).
Briefly, full-length PIK3CA-H1047R and HER2 were
subcloned into a pENTR vector and recombinated into the
pLenti-6/V5-DeST vector. The lentiviruses for the full-length genes
were generated using the pLenti-virus-expression system
(Invitrogen). The generated virus was used to infect targeted model
cells. Stable cells were generated after being selected with
blasticidin (10 μg/ml, Invivogen) and were grown at 37°C in
an incubator with 5% CO2Most of the cell lines used were
originally grown in standard SFIHE medium (serum-free medium with
insulin, hydrocortisone, and epidermal growth factor) made by Ham's
F12 media supplemented with 0.1% bovine serum albumin, 0.5
μg/ml fungizone, 5 μg/ml gentamycin, 5 mM
ethanolamine, 10 mM HEPES, 5 μg/ml transferrin, 10 μM
T3, 50 μM selenium, 1 μg/ml hydrocortisone, 10 ng/ml
EGF, and 5 μg/ml insulin. MCF10A cells expressing HER2 were
grown in SFHE medium (serum-free medium with hydrocortisone and
EGF). For experimentation, each individual cell line with its own
different overexpressed gene was first placed in SFIHE or SFHE
medium for a few days, and then placed in either SFIHE medium, SFIH
(lacking EGF or epidermal growth factor), SFHE (lacking insulin),
or SFH (lacking both EGF and insulin) medium in order to observe
cell growth and colony formation. For the invasion assay, cells
were first plated in each well insert with SFIH medium, and then
SFIHE with 5% FBS was placed outside in the well. Tests on
inhibition were done in SFIHE medium.
Cell proliferation/MTT assay
McF10A/HER2, McF10A/PIK3CA-h1047r,
McF10A/HER2/PIK3CA-H1047R and McF10A/LacZ were seeded in
triplicate at a density of 2×103 cells per well in
96-well plates on day 0. Cells were grown in SFIHE (growth medium
with insulin, hydrocortisone and EGF), SFIH (growth medium lacking
EGF) or SFHE (growth medium lacking insulin) or SFH (lacking both
EGF and insulin) medium. Cell proliferation was measured with an
MTT assay kit (Fisher Scientific, Pittsburgh, PA, USA), trypsinized
and counted with a particle counter (Beckman Coulter) with an
absorbance value of 540 nm on days 1, 3, 5, and 7 to make the
growth curve. Media was changed at each of these time points.
Colony formation
Each cell line was seeded in two wells of a 6-well
plate with a density of 2×103 cells per well for 1-2
weeks in SFIHE, SFIH, SFHE, or SFH culture mediums. As soon as
distinct clones were visible with a microscope and were almost
starting to merge, all plates were stained with crystal violet on
day 12 of growth for approximately 5 min and then exposed to water
gently. The numbers of individual clones were then counted and
recorded, and the plates were scanned for clear pictures of
clones.
Migration and invasion assay
A cell migration and cell invasion assay was
performed in BD control chamber and BioCoat Matrigel Invasion
chambers (BD Bioscience, Corning, NY, USA) according to the
manufacturer's instructions. Cells (5×104) were plated
in the upper chamber in 500 μl of SFIH growth medium without
FBS, and the lower chamber was filled with 750 μl of SFIHE
growth medium with 5% FBS. After 48-h incubation, the cells that
had invaded to the underside of the membrane were fixed and stained
with the HEMA 3 kit (Fisher Scientific). The migration and invasive
cells were then determined by counting the stained cells by
microscopy of ×100 and images were taken.
Inhibition of cell proliferation or
colony formation by inhibitors
LY294002 [pan PI3K inhibitor (24,25)], and CP-724714 [selective HER2
inhibitor (26,27)] were diluted to the concentrations
of 3 and 10 μM and 0.1 and 1 μM in SFIHE medium,
respectively, according to previous data. SFIHE medium with either
concentration of the PI3K inhibitor, the HER2 inhibitor, or both
inhibitors at both concentrations were added to only the MCF10A
cell line expressing both the mutant PIK3CA-H1047R and the
HER2 gene in a 96-well plate for cell growth, and in 6-well
plates for colony formation with 2.5×104 cells/well.
Medium with fresh inhibitors was replaced every other day. For the
96-well plate, cells were trypsinized and counted with a particle
counter (Beckman coulter) with an absorbance value of 540 nm on the
9th day of growth. Clones in the 6-well plates were stained with
crystal violet on the 12th day of experimentation, and clones were
counted. The plates were also scanned for pictures of clones.
Affymetrix microarray
Gene expression levels for the four different cell
lines were measured with Human Gene 2.1 ST Array Strip at the
Genomics Core of University of Michigan. Microarray data reported
herein have been deposited at the NCBI Gene Expression Omnibus
(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE99512)
with the accession number GSE99512. Microarray analyses and pathway
analysis were performed by the Biostatistics core of the KCI. The
raw intensity probe-level values were background corrected, log2
transformed, quantile normalized, and summarized to probe set level
expression values. The 'rma' function from R package 'oligo', based
on robust multi-array average (RMA) method (28), was used for all above
pre-processions. Control type of probe sets, such as 'Background
probes', 'Poly-A controls' and 'Hybridization controls', were
excluded from further analyses. Probe sets with log2 expression
values less than 4 for any two cell lines compared were excluded
from the analyses of differential expression (DE) probe sets. The
k-mean clustering method was used for identifying DE probe sets
based on each of three comparisons PIK3CA vs. MCF10A_
Control, HER2 vs. MCF10A_Control, and
PIK3CA_HER2 vs. MCF10A_Control. The identified
up-/down-regulated probe sets were subjected to ingenuity pathway
analysis (IPA). IPA canonical pathway analysis identified the
enriched pathways that were most significant to the data set. The
significance of the association between the data set and the IPA
canonical pathway was measured by the Fisher's exact test. The −Log
(P-value) ≥3 was used as the cut-off for identifying the common or
unique pathways across three comparisons.
Results
Bioinformatics analysis reveals the
co-occurrence of mutant PIK3CA and HER2 amplification in breast
cancer cells
Six distinct chromosomal regions were identified
within chromosomes 3, 7, 12, 14, 16, and 17 as having increased
frequency of amplification in PIK3CA-mutant versus
PIK3CA-WT cell lines. The regions with the most
amplification include chromosome 7, 14, 16, and 17 (P<0.05,
Table I). In particular, the
ERBB2/HER2 region was one of the most correlated regions.
The percent difference of amplification of the HER2 region
between the cell lines with and without the PIK3CA mutation
was 37%, which indicated that there may be a synergistic
relationship between the PIK3CA mutation and HER2
gene amplification. In addition to the chromosome 17 region, which
contains HER2 and two other genes, multiple other
chromosomal regions were also found to be correlated with
PIK3CA mutation. Chromosome 7 region contains three genes.
Chromosome 12 region contains ten genes. Chromosome 14 region
contains seven genes and chromosome 16 contains 11 genes (Table II).
 | Table IChromosome regions show different CGH
frequency between cell lines with different status of PIK3CA. |
Table I
Chromosome regions show different CGH
frequency between cell lines with different status of PIK3CA.
| Chromosome | Difference in CGH
frequency (%) | Fisher
P-value |
|---|
| 3 | 33 | 0.07 |
| 7 | 39.40 | 0.04 |
| 12 | 30 | 0.09 |
| 14 | 36.60 | 0.04 |
| 16 | 46.10 | 0.002 |
| 17 | 38.40 | 0.04 |
| Table IIDifferential amplified chromosome
regions and genes. |
Table II
Differential amplified chromosome
regions and genes.
| Clone | Chrom | HUGO | Description |
|---|
| CTD-2172D17 | 3 | EIF4G1 | Eukaryotic
translation initiation factor 4 γ, 1 |
| CTD-2172D17 | 3 | ABCC5 | ATP-binding
cassette, sub-family C (CFTR/MRP), member 5 |
| CTD-2172D17 | 3 | EIF2B5 | Eukaryotic
translation initiation factor 2B, subunit 5 (ε, 82 kD) |
| CTC-329F6 | 7 | FTSJ2 | FtsJ homolog 2
(E. coli) |
| GS1-165K6 | 7 | AHR | Aryl hydrocarbon
receptor |
| GS1-165K6 | 7 | AGR2 | Anterior gradient 2
homolog (Xenepus laevis) |
| RP11-270J9 | 12 | U5-100K | prp28, U5 snRNP 100
kd protein |
| RP11-112M10 | 12 | GALNT6 | GalNAc-T6 |
| RP11-101H10 | 12 | TARBP2 | TAR (HIV) RNA
binding protein 2 |
| RP11-101H10 | 12 | MGC11308 | Hypothetical
protein MGC11308 |
| RP11-101H10 | 12 | AAAS | Achalasia,
adrenocortical insufficiency, alacrimia |
| RP11-132H4 | 12 | HOXC10 | Homeo box C10 |
| VYS12P2692 | 12 | SAS | Sarcoma amplified
sequence |
| RP11-234C19 | 12 | PXN | Paxillin |
| RP11-234C19 | 12 | MGC5139 | Hypothetical
protein MGC5139 |
| RP11-234C19 | 12 | GCN1L1 | GCN1 general
control of amino-acid synthesis 1-like 1 (yeast) |
| RP11-70F9 | 14 | THTP | Thiamine
triphosphate |
| RP11-189N14 | 14 | PCK2 | Phosphoenolpyruvate
carboxykinase 2 (mitochondrial) |
| RP11-189N14 | 14 | TM9SF1 | Transmembrane 9
superfamily member 1 |
| RP11-189N14 | 14 | FLJ23338 | Importin 4 |
| RP11-189N14 | 14 | LOC51016 | CGI-112
protein |
| RP11-189N14 | 14 | TINF2 | TERF1
(TRF1)-interacting nuclear factor 2 |
| RP11-111F22 | 14 | RNB6 | RNB6 |
| CTD-2271I17 | 16 | CES2 | Carboxylesterase 2
(intestine, liver) |
| CTD-2271I17 | 16 | ELMO3 | Engulfment and cell
motility 3 (ced-12 homolog, C. elegans) |
| RP11-354N7 | 16 | CDH1 | Cadherin 1, type 1,
E-cadherin (epithelial) |
| RP11-354N7 | 16 | VPS4A | Vacuolar protein
sorting factor 4A |
| RMC16P004 | 16 | LOC64146 | Peptide
deformylase-like protein |
| RP11-253O10 | 16 | RAP1 | TRF2-interacting
telomeric RAP1 protein |
| RP11-110L8 | 16 | MBTPS1 | Membrane-bound
transcription factor protease, site 1 |
| RP11-162I18 | 16 | USP10 | Ubiquitin specific
protease 10 |
| RP11-118F19 | 16 | LOC51659 | HSPC037
protein |
| RP11-122P17 | 16 | KIAA0182 | KIAA0182
protein |
| RMC16P026 | 16 | GALNS | Galactosamine
(N-acetyl)-6-sulfate sulfatase |
| RMC17P077 | 17 | GRB7 | Growth factor
receptor-bound protein 7 |
| RMC17P077 | 17 | ERBB2 | v-erb-b2
erythroblastic leukemia viral oncogene homolog 2 |
| RMC17P077 | 17 | MGC9753 | Hypothetical gene
MGC9753 |
Co-occuring HER2 and mutant PIK3CA-H1047R
synergistically enhance cell proliferation, colony formation, and
cell invasion in MCF10A cells
To specifically study the cooperative effect of
mutant PIK3CA and HER2 genes in breast cancer cell
growth, we chose to create MCF10A cell models expressing mutant
PIK3CA-h1047r (MCF10A/PIK3CA-H1047R), HER2
(MCF10A/HER2) or both mutant PIK3CA-H1047R and
HER2 genes (MCF10A/HER2/PIK3CA-H1047R). A MCF10A cell
line expressing LacZ was used as a control (MCF10A/LacZ) (Fig. 1A). A cell proliferation assay was
performed in SFIHE, SFIH, SFHE, and SFH media in order to address
proliferation under different growth stimulating conditions. In
SFIHE medium, all the cell models demonstrate similar proliferation
rate (Fig. 1B). However, in media
deprived of the growth factor EGF and insulin, the
MCF10A/HER2/PIK3CA-H1047R cells exhibit the greatest growth
advantage over MCF10A/PIK3CA-H1047R, MCF10A/HER2, and
MCF10A/LacZ (negative control) (Fig.
1B). Epidermal growth factor (EGF) is an essential factor in
the acceleration of cell growth in cancer cells (29-31).
Insulin allows cells to use glucose for energy and maintains the
glucose level in blood and is tightly associated with cancer cell
growth and proliferation (32-34).
This finding suggests a cooperative effect of HER2 and
mutant PIK3CA that is insulin- and EGF-independent. A colony
formation assay showed similar synergistic effect of the two
genetic alterations on the promotion of colony formation (Fig. 1C and D).
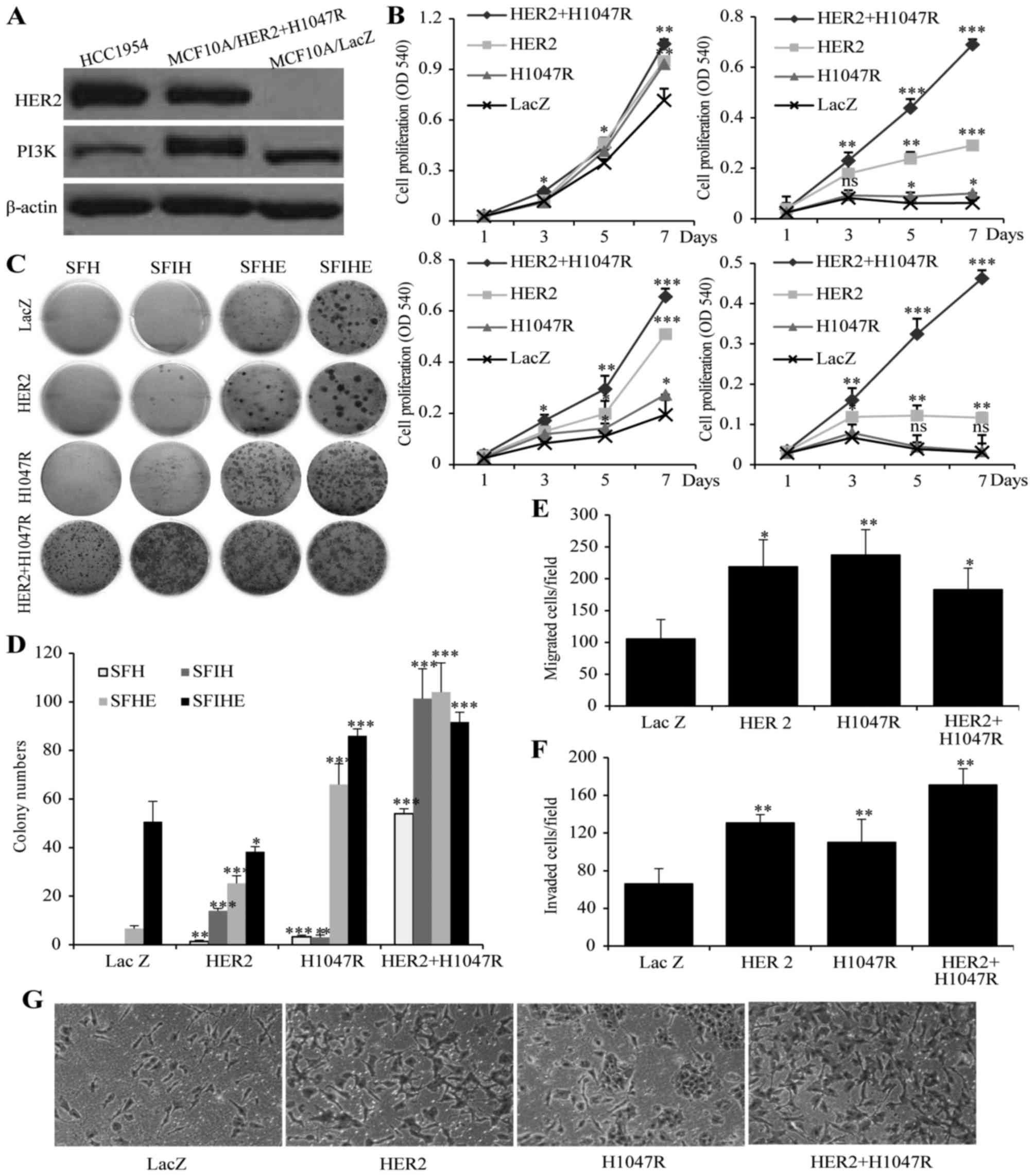 | Figure 1The effect of HER2 or/and
PIK3CA-H1047R on cell proliferation, invasiveness and colony
formation. (A) expression of PIK3CA and HER2 gene in
MCF10A/HER2/PIK3CA-H1047 cells and HCC1954 cells.
MCF10A/LacZ was used as negative control. (B) Cell proliferations
of MCF10A/HER2/PIK3CA-H1047R, MCF10A/HER2,
MCF10A/PIK3CA-H1047R and MCF10A/LacZ cells when cultured in
SFIHE (top left), SFIH (top right), SFHE (bottom left), and SFH
(bottom right) media. (c) Different capability in colony formation
of MCF10A/HER2/PIK3CA-H1047R, MCF10A/HER2,
MCF10A/PIK3CA-H1047R and MCF10A/LacZ cells when cultured in
SFH, SFHE, SFIH and SFIHE media. (D) Summary of the colony
formation assay under different cell culture conditions. Data are
mean ± SD. (e) MCF10A/HER2/PIK3CA-H1047R, MCF10A/HER2
and MCF10A/PIK3CA-H1047R exhibited higher migration ability
than MCF10A/Lac Z (negative control). (F) The effects of
MCF10A/HER2/PIK3CA-H1047R, MCF10A/HER2,
MCF10A/PIK3CA-H1047R and MCF10A/LacZ on cell invasion.
Columns mean of three independent experiments. Bars mean SD. The
MCF10A/LacZ was used as a control (100%). The invasion ability (%)
was measured by the invading cell number of each cell line
comparing to the invading cell number of MCF10A/LacZ cell line.
Student's t-test was used for statistical analysis.
***P<0.001, **0.01>P>0.001;
*0.05>P>0.01; ns, not significant. (g)
representative images showing invading cells in Matrigel invasion
chambers for four cell models. Original magnification, ×100. |
We further investigated the effect of HER2,
the mutant PIK3CA-H1047R, or both genetic alterations on the
migration and invasion using a chamber assay. In the migration
assay, all the oncogene expressing cells showed higher migration
efficiency than control cell MCF10A/LacZ. MCF10A/HER2 and
MCF10A/PIK3CA-H1047R showed a similar migration cell
numbers, while MCF10A/HER2/PIK3CA-H1047R did not demonstrate
higher migration capability than either oncogene alone (Fig. 1E). In the invasion assay, all the
oncogene-expressing cells showed higher invasive efficiency than
control cell MCF10A/LacZ. Specifically,
MCF10A/HER2/PIK3CA-H1047R demonstrated a stronger invasion
capability than MCF10A/HER2 and MCF10A/PIK3CA-H1047R
cells (Fig. 1F and G).
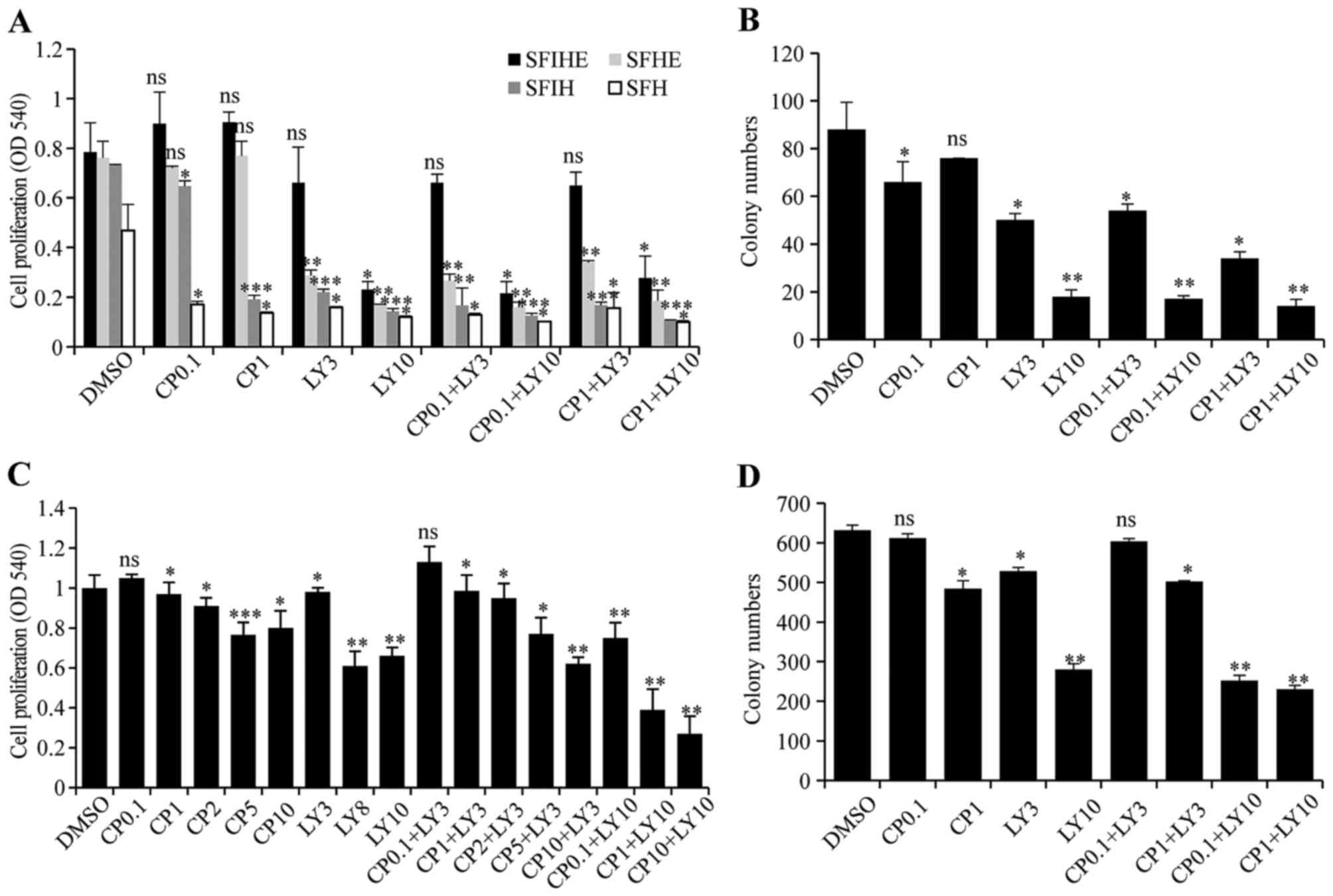
Pharmacological inhibition of HER2 and
PIK3CA-H1047R in MCF10A/HER2/PIK3CA-H1047R and HCC1954 cells
To test whether the cell model
(MCF10A/HER2/PIK3CA-H1047R) with both genetic alterations
can be synergistically inhibited by simultaneous targeting of both
genes, LY294002, a pan-PI3K inhibitor, was used for targeting
PIK3CA-H1047R and CP-724714, a selective HER2 inhibitor, was used
for targeting active HER2. The results showed that the cells
treated with CP-724714 had little to no change in cellular
proliferation. Low dose LY294002 (3 μM) alone also had a
minimal efficacy as an inhibitor of cell growth. However, cells
exposed to a combination of both inhibitors with 10 μM
LY294002 had a drastic decrease in cell proliferation. The combined
inhibition effects were more significant in SFIH, SFHE, and SFH
medium than in SFIHE medium (Fig. 2A
and B).
To validate the results obtained from the
established MCF10A/HER2/PIK3CA-H1047R cell model, we
repeated drug treatment using the HCC1954 cell line, which contains
both endogenous PIK3CA mutation and HER2
amplification. The treatment with different doses of CP724714 (0.1,
1, 2, 5, and 10 μM) marginally changed cell growth. The
treatment of HCC1954 cells with LY294002 only significantly
inhibited cell proliferation at high doses (8 and 10 μM).
The combination of these two drugs evinced a dose-dependent
inhibition in cell proliferation. With a low dose of LY294002 (3
μM) treatment, increasing the dose of CP724714 from 0.1 to
10 μM resulted in a cell proliferation inhibition effect
similar to the high dose of LY294002 (10 μM). However, when
a high dose of LY294002 (10 μM) was used, further treatment
with CP724714 at 0.1, 1, 10 μM obtained an even more
significant reduction in cell growth (Fig. 2C). A similar result was obtained in
colony formation assay when HCC1954 cells were treated with
different combinations (Fig.
2D).
Common and distinct downstream genes and
signaling pathways controlled by HER2 and/or mutant PIK3CA
The study of the biological function of three
different cell models showed that the
MCF10A/HER2/PIK3CA-H1047R cells harbor unexpected
characteristics in comparison to MCF10A/HER2 and
MCF10A/PIK3CA-H1047R including EGF and insulin-independent
growth, stronger invasion capability, and sensitive only to double
inhibition. These results suggest that the co-occuring oncogenic
HER2 and mutant PIK3CA could lead to activation of
novel cell signaling pathways which drive unique oncogenic
properties in cancer cells. To investigate the underlying mechanism
of the functional differences between cells with mutant
PIK3CA, HER2 or both mutant PIK3CA and
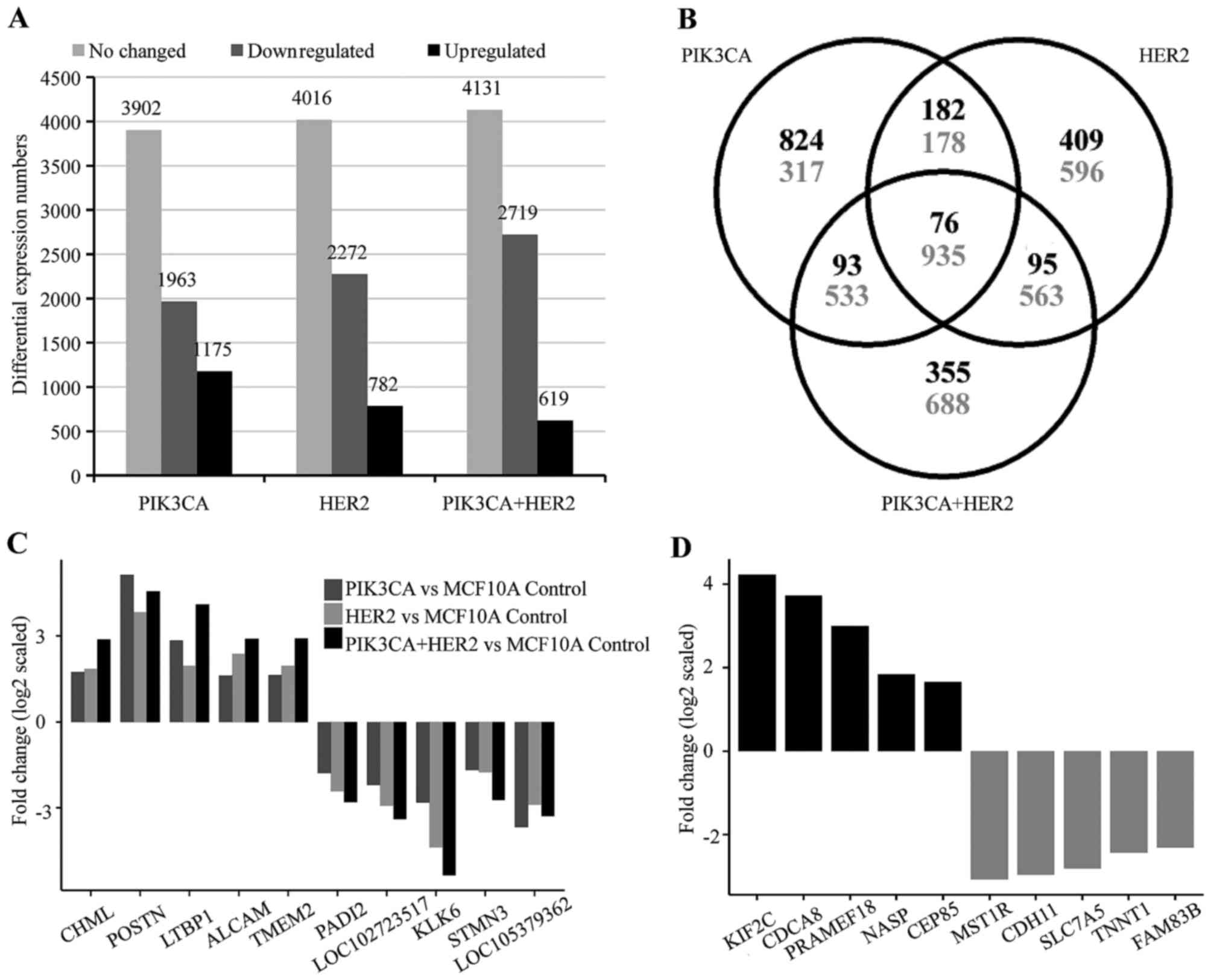
HER2, we performed an Affymetrix microarray analysis. After
normalization with MCF10A/LacZ control cells,
MCF10A/PIK3CA-H1047R cells showed 1,175 upregulated, 1,963
downregulated and 3,902 no changed genes; MCF10A/HER2 cells
showed 762 upregulated, 2,271 downregulated and 4,016 no changed
genes. The MCF10A/mutant PIK3CA/-HER2 showed 619
upregulated, 2,719 downregulated and 4,131 no changed genes
(Fig. 3A). Although there are 76
upregulated and 935 downregulated common genes shared in all three
cell models, each cell model was shown to contain hundreds of
unique genes (Fig. 3B–D).
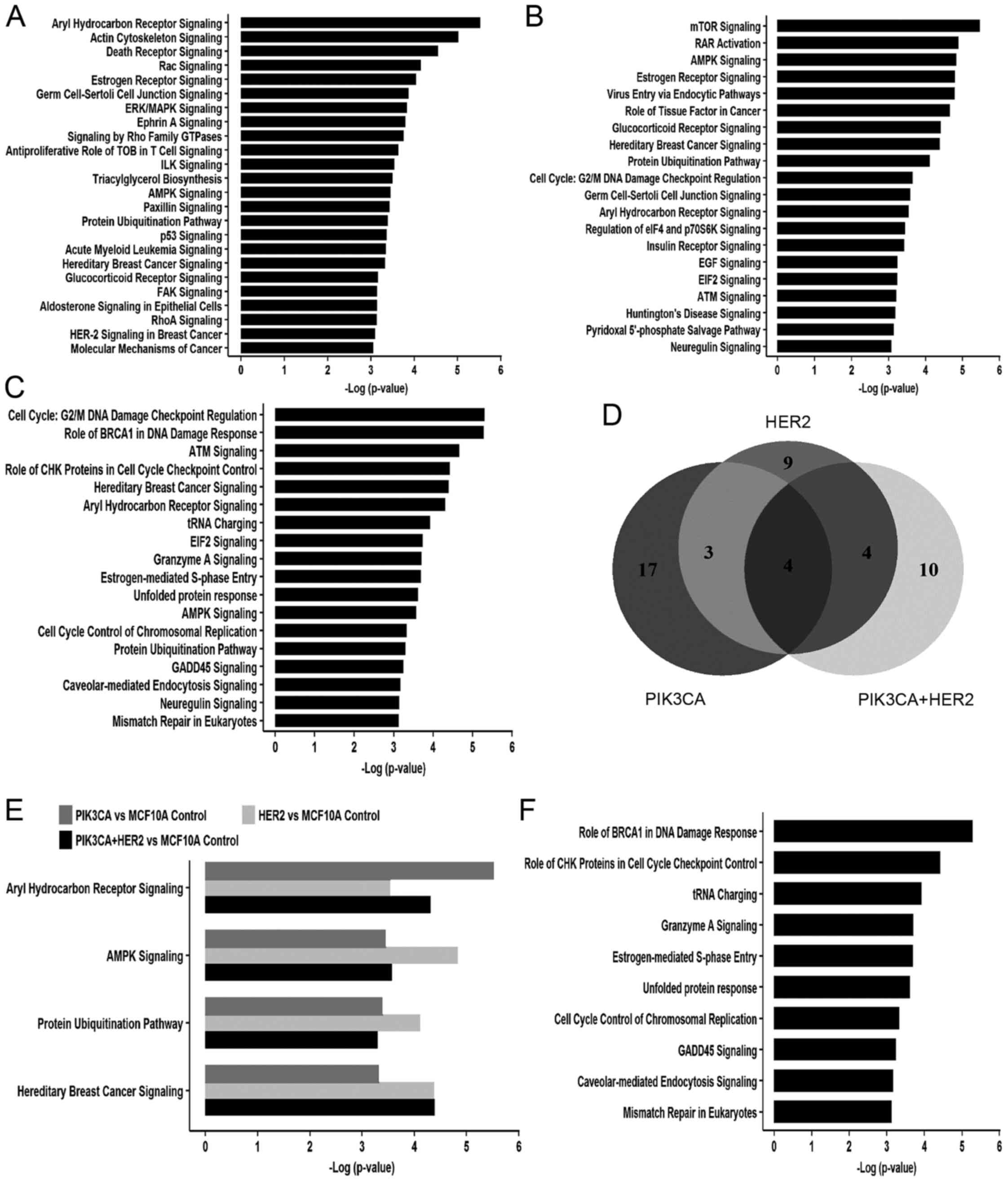
Further ingenuity pathway analysis showed that 24,
20 and 18 cell signaling pathways were enriched in
MCF10A/PIK3CA-H1047R, MCF10A/HER2 and
MCF10A/HER2/PIK3CA-H1047R, respectively, with a
cut-off of -log (P-value) ≥3 (Fig.
4A–C). Under this condition, four common cell signaling were
identified in all three cell models, including aryl hydrocarbon
receptor (AhR) signaling, AMP-activated protein kinase (AMPK)
signaling, protein ubiquitination pathways and hereditary breast
cancer signaling (Fig. 4D and E,
and Table III). Additionally,
25% (7 out of 24) signaling pathways in cells expressing
PIK3CA-H1047R overlapped with 35% (7 out of 20) signaling
pathways in HER2 overexpression cells (Fig. 4D). There is 55% (10 of 18)
signaling pathways enriched only in cells with both
PIK3CA-H1047R and HER2 and do not overlap with cells
expressing either mutant PIK3CA or HER2 alone
(Fig. 4F). Importantly, 50% (5 out
of 10) of these cell signaling pathways are related to DNA
replication stress, including role of BRCA1 in DNA damage response,
role of CHK1 in cell cycle checkpoint control, estrogen-mediated
S-phase entry, cell cycle control of chromosomal replication and
mismatch repair in eukaryotes (Fig.
4F and Table IV).
| Table IIIFour common pathways in HER2, mutant
PIK3CA and mutant PIK3CA+HER2 overexpression models. |
Table III
Four common pathways in HER2, mutant
PIK3CA and mutant PIK3CA+HER2 overexpression models.
| Ingenuity canonical
pathways | Common genes |
|---|
| Aryl hydrocarbon
receptor signaling | RELA, NFIX, MYC,
CTSD, ALDH7A1, TP53, MED1, NQO1, BAX, CYP1B1, CDKN1A, ALDH3B1,
NRIP1, HSPB1 |
| AMPK signaling | SMARCD2, MAPK13,
ELAVL1, CRTC2, TSC2, PIK3R2, AKT1S1, CPT1A, GRB2, STK11, PPP2R1A,
CDKN1A |
| Protein
ubiquitination pathway | USP5,
HSPA1A/HSPA1B, SACS, BAG1, USP36, USP19, PSMD3, SKP2, USP31,
PSMD11, BAP1, HSPB1, USP21, PSMB3, HLA-A |
| Hereditary breast
cancer signaling | SMARCD2, POLR2C,
PIK3R2, TP53, GRB2, POLR2E, CDKN1A, SMARCC1, HLTF, RFC3 |
| Table IVTop 10 signaling pathways enriched
only in mutant PIK3CA and HER2 co-occuring model. |
Table IV
Top 10 signaling pathways enriched
only in mutant PIK3CA and HER2 co-occuring model.
| Ingenuity canonical
pathways | Genes |
|---|
| Role of BRCA1 in
DNA damage response | ARID1A, FANCF,
SMARGD2, RBL1, CHEK1, RB1, FANCD2, SMARCB1, RFC2, BRCA1, BRIP1,
BLM, TP53, E2F4, PLK1, SMARCD1, RFC5, FANCL, NBN, MSH2, RFG4,
CDKN1A, BRCA2, SMARCC1, HLTF, FANCA, RFC3 |
| Role of CHK
proteins in cell cycle checkpoint control | TP53, GDG25G, E2F4,
PLK1, RFG5, GDK1, GHEK1, NBN, PPP2R1A, PGNA, RFG4, GDKN1A, HUS1,
ATMIN, RFG2, GLSPN, TLK2, BRGA1, GDK2, RFG3 |
| tRNA charging | GARS, MARS2, LARS2,
MARS, QARS, FARSA, WARS, YARS, FARS2, VARS2, AARS, VARS, SARS,
SARS2, IARS |
| Granzyme A
signaling | HIST1H1B, SET,
HIST1H1G, NME1, CREBBP, HMGB2, H1F0, APEX1, EP300 |
| Estrogen-mediated
S-phase entry | MYG, RB1, GGNA2,
E2F4, GGNE2, GDKN1A, RBL1, GGND1, GDK1, GDK2, SKP2 |
| Unfolded protein
response | SCAP, MAP2K7, ERN1,
INSIG1, HSPH1, HSPA1A/HSPA1B, HSPA9, GEBPD, GANX, OS9, MAP3K5,
GEBPB, GEBPG, MBTPS2, SEL1L, MBTPS1, PPP1R15A, AMFR |
| Cell cycle control
of chromosomal replication | MGM5, MGM3, MGM6,
PGNA, PRIM1, POLA1, GDG6, GDG7, ORG6, TOP2A, MGM4, DBF4, GDK2,
ORG1 |
| GADD45
signaling | TP53, PGNA, CCNE2,
CDKN1A, BRGA1, CCND1, GDK1, GDK2, CCNB1 |
| Caveolar-mediated
endocytosis signaling | B2M, FLNB, HLA-A,
ITGA2, GOPE, ABL1, ITGA6, ITGA10, ITGB8, GD55, ITGA3, FLOT2, AGTA2,
HLA-G, ITGB4, ITGB6, AGTG1, PTRF, DNM2, ITGB5, EGFR |
| Mismatch repair in
eukaryotes | PGNA, MSH2, RFG4,
RFG2, FEN1, RFG5, EXO1, RFG3 |
Discussion
Our current study represents the first large scale
genome-wide screening of gene amplifications that correlate with
the presence of activating PIK3CA mutations in a panel of 51
human breast cancer cell lines. Our results indicate that in the
human genome there are multiple other amplification regions besides
the HER2 region localized at different chromosomes that
correlate with the PIK3CA mutation in human breast cancer.
Further study of these novel regions could identify more novel
therapeutic targets in the future for breast cancer treatment.
Using the cell models established, we showed that
the co-occurrence of PIK3CA mutation and HER2 gene
amplification greatly enhances proliferation. In the MTT assay, the
MCF10A/HER2/PIK3CA-H1047R cell model exhibited
highest proliferation in all four media, and a synergistic effect
was evident in SFH (medium lacking insulin and EGF) and in SFIH
(medium lacking EGF). In the colony formation assay, the
MCF10A/HER2/PIK3CA-H1047R cell model had the most
colonies than any other cell model in all media. We also determined
that the cooperative action of HER2 and PIK3CA-H1047R
genes is EGF and insulin-signaling pathway independently, because
even in conditions lacking insulin and EGF, the growth of cells
with both genetic alterations is similar to that in regular
conditions (SFIHE medium) suggesting a strong self-sufficient
capability (Growth factor independent proliferation). Consistent
with previous studies (35,36),
the treatment with two inhibitors is more potent in inhibiting cell
proliferation and colony formation than using either inhibitor
alone in cells with both HER2 and PIK3CA
mutation.
We demonstrated that AhR signaling, AMPK signaling,
protein ubiquitination pathway and hereditary breast cancer
signaling are four common signaling pathways in cells expressing
HER2, mutant PIK3CA and HER2 plus mutant
PIK3CA. The AhR is a ligand-activated transcription factor
that regulates a battery of genes in response to exposure to a
broad class of environmental poly aromatic hydrocarbons (PAH). AhR
is historically characterized for its role in mediating the
toxicity and adaptive responses to these chemicals. However,
mounting evidence has established a role for it in
ligand-independent physiological processes and pathological
conditions, including cancer. The AhR is overexpressed and
constitutively activated in advanced breast cancer cases and was
shown to drive the progression of breast cancer (37–39).
The AMPK is a central regulator of cellular metabolism and energy
homeostasis in mammalian tissues. The importance of such regulation
of fundamental process poses the AMPK signaling pathway in one of
the most attractive therapeutic targets in diabetes and cancer
(40–42). Ubiquitination is a protein
posttranslational modification reservedly regulated by a series of
ubiquitination-associated enzymes. The cellular functions of
ubiquitination span a wide spectrum that includes cell death, DNA
damage repair, autophagy, proteasomal degradation of proteins and
metabolism (43). Thus,
dysregulation ubiquitination has broad consequences that may lead
to aberration of tumor-promoting pathways and tumor-suppressing
pathways (44,45). Consistent with these published
results, the finding of our study highlights that these pathways
are basic and essential fundamental signaling pathways in
HER2 and PIK3CA driven breast cancer.
More importantly, we identified 10 signaling
pathways that were specifically enriched in the cells with both
mutant PIK3CA and HER2 overexpression. Among these
pathways, we found that 50% (5/10) are related to cell cycle
control and DNA damage response, including role of BRCA1 in DNA
damage response, role of CHK proteins in cell cycle checkpoint
control, estrogen-mediated S phase entry, cell cycle control of
chromosomal replication and mismatch repair in eukaryotes. Oncogene
expression has been shown to drive cell proliferation by
interfering with the regulatory pathways of cell cycle progression
control, which could further lead to replication stress, a term
that is characterized by DNA synthesis slow down and/or replication
fork stalling and is the primary cause of genome instability
(46-49). Our study for the first time showed
that co-occuring oncogenic HER2 and mutant PIK3CA
could induce replication stress in mammary epithelial cells. This
result highlights the acquisition of unique characteristics arising
from co-occurrence of two prominent oncogenes, which will lead to
us to re-think the tumor progression mechanism and open up new
possibilities for breast cancer diagnostics and treatment.
Abbreviations:
|
PI3k
|
phosphatidylinositol 3-kinase
|
|
ErbB2
|
v-erb-b2 erythroblastic leukemia viral
oncogene homolog
|
|
aCGH
|
array comparative genomic
hybridization
|
|
CNV
|
copy number variation
|
|
EGF
|
epidermal growth factor
|
|
SFIHE
|
serum-free medium with insulin,
hydrocortisone, and epidermal growth factor
|
|
SFHE
|
serum-free medium with hydrocortisone
and EGF
|
|
SFIH
|
serum-free medium with insulin and
hydrocortisone
|
|
SFH
|
serum-free medium with
hydrocortisone
|
|
AhR
|
aryl hydrocarbon receptor
|
|
IPA
|
ingenuity pathway analysis
|
|
PAH
|
poly aromatic hydrocarbons
|
|
AMPK
|
AMP-activated protein kinase
|
Acknowledgments
We would like to thank Ms. Elizabeth A. Katz of the
Karmanos Cancer Institute's Marketing and Communications Department
for editing our manuscript. This work was supported in part by the
NIH RO1 grant CA172480-01A1 (G.W.); MT program pilot grant (25RU21)
from the Karmanos Cancer Institute, and Grant Boost award from
Wayne State university (G.W.); the national natural Science
Foundation of china (grant no. 81572601, F.M.); and the natural
Science Foundation of Jiangsu Province (grant no. BK20151095)
(F.M.). The Biostatistics core and the Biorepository core of the
karmanos cancer Institute are supported by grant number
P30-CA022453-29.
References
|
1
|
Bader AG, Kang S, Zhao L and Vogt PK:
Oncogenic PI3K deregulates transcription and translation. Nat Rev
Cancer. 5:921–929. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cantley LC: The phosphoinositide 3-kinase
pathway. Science. 296:1655–1657. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-kinase AkT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bachman KE, Argani P, Samuels Y, Silliman
N, Ptak J, Szabo S, Konishi H, Karakas B, Blair Bg, Lin C, et al:
The PIK3CA gene is mutated with high frequency in human breast
cancers. Cancer Biol Ther. 3:772–775. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Campbell IG, Russell SE, Choong DY,
Montgomery KG, Ciavarella ML, Hooi CS, Cristiano BE, Pearson RB and
Phillips WA: Mutation of the PIK3CA gene in ovarian and breast
cancer. Cancer Res. 64:7678–7681. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Saal Lh, Holm K, Maurer M, Memeo L, Su T,
Wang X, YU JS, Malmström PO, Mansukhani M, Enoksson J, et al: IK3CA
mutations correlate with hormone receptors, node metastasis, and
ERBB2, and are mutually exclusive with PTEN loss in human breast
carcinoma. Cancer Res. 65:2554–2559. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wu G, Xing M, Mambo E, Huang X, Liu J, Guo
Z, Chatterjee A, Goldenberg D, Gollin SM, Sukumar S, et al: Somatic
mutation and gain of copy number of PIK3CA in human breast cancer.
Breast Cancer Res. 7:R609–R616. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang H, Liu G, Dziubinski M, Yang Z,
Ethier SP and Wu G: Comprehensive analysis of oncogenic effects of
PIK3CA mutations in human mammary epithelial cells. Breast Cancer
Res Treat. 112:217–227. 2008. View Article : Google Scholar
|
|
9
|
Isakoff SJ, Engelman JA, Irie HY, Luo J,
Brachmann SM, Pearline RV, Cantley LC and Brugge JS: Breast
cancer-associated PIK3CA mutations are oncogenic in mammary
epithelial cells. Cancer Res. 65:10992–11000. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kang S, Bader AG and Vogt PK:
Phosphatidylinositol 3-kinase mutations identified in human cancer
are oncogenic. Proc Natl Acad Sci USA. 102:802–807. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Samuels Y, Diaz LA Jr, Schmidt-kittler O,
Cummins JM, Delong L, Cheong I, Rago C, Huso DL, Lengauer C,
Kinzler KW, et al: Mutant PIK3CA promotes cell growth and invasion
of human cancer cells. Cancer Cell. 7:561–573. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhao JJ, Liu Z, Wang L, Shin E, Loda MF
and Roberts TM: The oncogenic properties of mutant p110alpha and
p110beta phosphatidylinositol 3-kinases in human mammary epithelial
cells. Proc Natl Acad Sci USA. 102:18443–18448. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Engelman JA: Targeting PI3K signalling in
cancer: Opportunities, challenges and limitations. Nat Rev Cancer.
9:550–562. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu P, Cheng H, Roberts TM and Zhao JJ:
Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev
Drug Discov. 8:627–644. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
De Laurentiis M, Cancello G, Zinno L,
Montagna E, Malorni L, Esposito A, Pennacchio R, Silvestro L,
Giuliano M, Giordano A, et al: Targeting HER2 as a therapeutic
strategy for breast cancer: A paradigmatic shift of drug
development in oncology. Ann Oncol. 16(Suppl 4): iv7–iv13. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li SG and Li L: Targeted therapy in
HER2-positive breast cancer. Biomed Rep. 1:499–505. 2013.
View Article : Google Scholar
|
|
17
|
Gajria D and Chandarlapaty S:
HER2-amplified breast cancer: Mechanisms of trastuzumab resistance
and novel targeted therapies. Expert Rev Anticancer Ther.
11:263–275. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rexer BN and Arteaga CL: Optimal targeting
of HER2-PI3K signaling in breast cancer: Mechanistic insights and
clinical implications. Cancer Res. 73:3817–3820. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Castaneda CA, Lopez-Ilasaca M, Pinto JA,
Chirinos-Arias M, Doimi F, Neciosup SP, Rojas KI, Vidaurre T, Balko
JM, Arteaga CL, et al: PIK3CA mutations in Peruvian patients with
HER2-amplified and triple negative non-metastatic breast cancers.
Hematol Oncol Stem Cell Ther. 7:142–148. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chakrabarty A, Rexer BN, Wang SE, Cook RS,
Engelman JA and Arteaga CL: H1047R phosphatidylinositol 3-kinase
mutant enhances HER2-mediated transformation by heregulin
production and activation of HER3. Oncogene. 29:5193–5203. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hanker AB, Pfefferle AD, Balko JM, Kuba
MG, Young CD, Sánchez V, Sutton CR, Cheng H, Perou CM, Zhao JJ, et
al: Mutant PIK3CA accelerates HER2-driven transgenic mammary tumors
and induces resistance to combinations of anti-HER2 therapies. Proc
Natl Acad Sci USA. 110:14372–14377. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kataoka Y, Mukohara T, Shimada H, Saijo N,
Hirai M and Minami H: Association between gain-of-function
mutations in PIK3CA and resistance to HER2-targeted agents in
HER2-amplified breast cancer cell lines. Ann Oncol. 21:255–262.
2010. View Article : Google Scholar
|
|
23
|
Neve RM, Chin K, Fridlyand J, Yeh J,
Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, et al: A
collection of breast cancer cell lines for the study of
functionally distinct cancer subtypes. Cancer Cell. 10:515–527.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Searl TJ and Silinsky EM: LY 294002
inhibits adenosine receptor activation by a mechanism independent
of effects on PI-3 kinase or casein kinase II. Purinergic Signal.
1:389–394. 2005. View Article : Google Scholar
|
|
25
|
Vlahos CJ, Matter WF, Hui KY and Brown RF:
A specific inhibitor of phosphatidylinositol 3-kinase,
2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J Biol
Chem. 269:5241–5248. 1994.PubMed/NCBI
|
|
26
|
Feng B, Xu JJ, Bi YA, Mireles R, Davidson
R, Duignan DB, Campbell S, Kostrubsky VE, Dunn MC, Smith AR, et al:
Role of hepatic transporters in the disposition and hepatotoxicity
of a HER2 tyrosine kinase inhibitor CP-724,714. Toxicol Sci.
108:492–500. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jani JP, Finn RS, Campbell M, Coleman KG,
Connell RD, Currier N, Emerson EO, Floyd E, Harriman S, Kath JC, et
al: Discovery and pharmacologic characterization of CP-724,714, a
selective ErbB2 tyrosine kinase inhibitor. Cancer Res.
67:9887–9893. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Irizarry RA, Hobbs B, Collin F,
Beazer-Barclay YD, Antonellis KJ, Scherf U and Speed TP:
exploration, normalization, and summaries of high density
oligonucleotide array probe level data. Biostatistics. 4:249–264.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim S, Han J, Lee SK, Koo M, Cho Dh, Bae
Sy, Choi My, Kim JS, Kim JH, Choe JH, et al: Smad7 acts as a
negative regulator of the epidermal growth factor (EGF) signaling
pathway in breast cancer cells. Cancer Lett. 314:147–154. 2012.
View Article : Google Scholar
|
|
30
|
Chen JX, XU LL, Wang XC, Qin HY and Wang
JL: Involvement of c-Src/STAT3 signal in EGF-induced proliferation
of rat spermatogonial stem cells. Mol cell Biochem. 358:67–73.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Guo Y, Fu P, Zhu H, Reed E, Remick SC,
Petros W, Mueller MD and YU JJ: correlations among ERCC1, XPB,
UBE2I, EGF, TAL2 and ILF3 revealed by gene signatures of
histological subtypes of patients with epithelial ovarian cancer.
Oncol Rep. 27:286–292. 2012.
|
|
32
|
Boyd DB: Insulin and cancer. Integr Cancer
Ther. 2:315–329. 2003. View Article : Google Scholar
|
|
33
|
Müssig K and Häring HU: Insulin signal
transduction in normal cells and its role in carcinogenesis. Exp
Clin Endocrinol Diabetes. 118:356–359. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pollak M: The insulin and insulin-like
growth factor receptor family in neoplasia: An update. Nat Rev
Cancer. 12:159–169. 2012.PubMed/NCBI
|
|
35
|
Rexer BN, Chanthaphaychith S, Dahlman K
and Arteaga CL: Direct inhibition of PI3K in combination with dual
HER2 inhibitors is required for optimal antitumor activity in
HER2+ breast cancer cells. Breast Cancer Res. 16:R92014.
View Article : Google Scholar
|
|
36
|
Lopez S, Cocco E, Black J, Bellone S,
Bonazzoli E, Predolini F, Ferrari F, Schwab CL, English DP, Ratner
E, et al: Dual HER2/IK3CA targeting overcomes single-agent acquired
resistance in HER2-amplified uterine serous carcinoma cell lines in
vitro and in vivo. Mol Cancer Ther. 14:2519–2526. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Feng S, Cao Z and Wang X: Role of aryl
hydrocarbon receptor in cancer. Biochim Biophys Acta. 1836:197–210.
2013.PubMed/NCBI
|
|
38
|
Murray IA, Patterson AD and Perdew GH:
Aryl hydrocarbon receptor ligands in cancer: Friend and foe. Nat
Rev Cancer. 14:801–814. 2014. View Article : Google Scholar
|
|
39
|
Powell JB, Goode GD and Eltom SE: The aryl
hydrocarbon receptor: A target for breast cancer therapy. J Cancer
Ther. 4:1177–1186. 2013. View Article : Google Scholar
|
|
40
|
Kuhajda FP: AMP-activated protein kinase
and human cancer: Cancer metabolism revisited. Int J Obes. 32(Suppl
4): S36–S41. 2008. View Article : Google Scholar
|
|
41
|
Faubert B, Vincent EE, Poffenberger MC and
Jones RG: The AMP-activated protein kinase (AMPK) and cancer: Many
faces of a metabolic regulator. Cancer Lett. 356:165–170. 2015.
View Article : Google Scholar
|
|
42
|
Jeon S-M and Hay N: The double-edged sword
of AMPK signaling in cancer and its therapeutic implications. Arch
Pharm Res. 38:346–357. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Popovic D, Vucic D and Dikic I:
Ubiquitination in disease pathogenesis and treatment. Nat Med.
20:1242–1253. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ohta T and Fukuda M: Ubiquitin and breast
cancer. Oncogene. 23:2079–2088. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu J, Shaik S, Dai X, Wu Q, Zhou X, Wang
Z and Wei W: Targeting the ubiquitin pathway for cancer treatment.
Biochim Biophys Acta. 1855:50–60. 2015.
|
|
46
|
Hills SA and Diffley JF: DNA replication
and oncogene-induced replicative stress. Curr Biol. 24:R435–R444.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mazouzi A, Velimezi G and Loizou JI: DNA
replication stress: causes, resolution and disease. Exp Cell Res.
329:85–93. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gaillard H, García-Muse T and Aguilera A:
replication stress and cancer. Nat Rev Cancer. 15:276–289. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Taylor EM and Lindsay HD: DNA replication
stress and cancer: cause or cure? Future Oncol. 12:221–237. 2016.
View Article : Google Scholar
|