Introduction
The hypoxic environment is a phenomenon specific to
cancer pathophysiology, in which quickly growing solid tumors
result in poor development of angiogenic vessels, thus leading to
an insufficient supply of oxygen (1,2).
Hypoxic responses are highly controlled by hypoxia-inducible
factors (HIFs), which are transcription factors with critical roles
in the development and progression of various tumors, including
colorectal cancer (CRC) (3–5). The
prototypic member of the HIF family is HIF-1, which is a
heterodimer consisting of an oxygen concentration-dependent α
subunit and a constitutively expressed β subunit (6,7). In
normal oxygen concentration, HIF-1α is rapidly degraded by prolyl
hydroxylases (PHDs) at proline residues within the oxygen-dependent
degradation domain. This degradation mediates interaction with PHDs
and the von Hippel-Lindau (pVHL) tumor suppressor protein,
eventually leading to HIF-1α degradation through a VHL-dependent
ubiquitin-proteasome pathway (8).
On the contrary, under hypoxic condition, the
O2-dependent PHDs are inhibited, thereby inhibiting the
interaction between HIF-1α and pVHL. Therefore, HIF-1α
ubiquitination/degradation is suppressed, resulting in increased
protein expression of HIF-1α (9).
One of the mechanisms by which HIF-1 promotes cancer
progression is by inducing the epithelial-mesenchymal transition
(EMT). EMT is a phenotypic change in which epithelial cells
transform to spindle-like motile cells that express mesenchymal
markers (10,11). Epithelial cells lose their
cell-cell adhesion properties and cell polarity and acquire
invasive properties, which permit access to the vessel (12). Through this process, tumor cells
experience migration and invasion, leading to cancer progression
and metastasis (13). Among the
HIFs, HIF-1α in particular has been studied to promote EMT in
several types of cancers by modulating EMT-associated genes,
including TWIST, snail, and slug (14–16).
Since hypoxia-induced EMT plays a key role in metastasis, EMT is
believed to be a promising target for developing new and effective
anticancer therapies.
Parthenolide (PT) can be isolated from extracts of
Mexican Indian medicinal plants that show anti-inflammatory
properties. Moreover, PT is currently used clinically to treat
migraines (17,18). The biological activities of PT are
mediated by suppression of nuclear factor-κB (NF-κB) signaling,
which entails inhibition of IκB kinase, alteration of NF-κB binding
activity and modification of the p65 protein (17,19–21).
We previously used a colitis-associated colorectal cancer (CAC)
model to demonstrate that PT inhibits NF-κB activation, ultimately
preventing CAC development and carcinogenesis (22).
NF-κB is a central molecule involved in inflammation
that regulates the expression of various target genes that promote
cell proliferation, modulate immune and inflammatory responses, and
play a role in the pathogenesis of various diseases, including
cancer (23,24). Both HIF-1α and NF-κB are involved
in cancer progression and have been implicated in the tumor
responses to hypoxia (25).
Moreover, positive correlations between HIF-1α and NF-κB have been
shown in various cancer cells including lung cancer cells, CRC
cells, osteosarcoma and gastric cancer cells (26–29).
However, the mechanism of HIF-1α and NF-κB activation under hypoxia
still remains unclear. Interestingly, several studies have reported
that activation of NF-κB is closely involved in the progression of
EMT by regulating EMT-specific genes in cancer cells (30–32).
Therefore, we hypothesized that NF-κB inhibition may regulate both
HIF-1α signaling and hypoxia-induced EMT progression.
We designed this study to test our hypothesis that
PT significantly inhibits hypoxic response and hypoxia mediated EMT
through blockade of NF-κB activation, thus resulting in suppression
of tumor growth, angiogenesis and invasion in CRC. We tested this
hypothesis using both in vitro and in vivo
models.
Materials and methods
Chemicals and reagenst
Parthenolide was from Calbiochem (San Diego, CA,
USA) and it was dissolved in dimethylsulfoxide (DMSO; Sigma, St.
Louis, MO, USA). Growth factor-reduced Matrigel was purchased from
BD Biosciences (San Diego, CA, USA). Anti-HIF-1α, anti-VHL,
anti-p65, anti-VEGF, anti-hexokinase II, anti-GLUT1, anti-COX2,
anti-PI3K, anti-TWIST, anti-Lamin B and anti-CA IX were purchased
from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA).
Anti-AKT, anti-phospho-AKT, anti-ERK1/2, anti-phospho-ERK1/2,
anti-MMP9, anti-E-cadherin, anti-β-catenin and anti-vimentin were
from Cell Signaling Technology (Danvers, MA, USA). Anti-MMP2,
anti-Slug and anti-Snail were from Abcam (Cambridge, UK).
Anti-actin was purchased from Sigma-Aldrich (St. Louis, MO,
USA).
Cell culture, treatments and observation
of morphological changes
HT-29, DLD-1 and HCT116 cells (American Type Culture
Collection ATCC, Rockville, MD, USA) were employed as
representative human CRC cells. The cells were cultured in
RPMI-1640 medium supplemented with 10% FBS, 100 U penicillin, and
100 U streptomycin. Human umbilical vein endothelium cells (HUVECs)
were also purchased from ATCC and cultured in endothelial cell
growth medium (EGM®-2 MV Bullet kit) (Lonza,
Walkersville, MD, USA) containing 2% fetal bovine serum (FBS).
HUVECs were used at 2–5 passages. Normoxia (general air condition,
21% O2) and hypoxia (1% O2, 5% CO2
and 94% N2) incubation was performed after PT treatment
for indicated times at 37°C.
The morphological alterations in the HT-29 cells
were observed using an inverted microscope. Images were captured
using an inverted microscope (Olympus IX71, USA).
Quantification of cell viability
The number of living cells was determined by
staining of the cells with the vital dye trypan blue. HUVECs and
CRC cells were plated at a density of 1×104 cells per
well in 24-well plates. Cells were treated with PT in normoxic or
hypoxic condition for 24 h. After the medium was removed, cells
were harvested with trypsin-EDTA and re-suspended in fresh medium.
The cells in medium was diluted with trypan blue, and counted
employing a hematocytometer.
Reverse transcription-PCR
Total RNA was isolated from cultured cells using
TRIzol (Invitrogen, Eugene, OR, USA) and cDNA was synthesized with
SuperScript II reverse-transcriptase (Invitrogen) according to the
manufacturer's protocol. The expression level of GAPDH gene was
used as a loading control. The following primer sequences were
used: HIF-1α, 5′-GCTGGCCCCAGCCGCTGGAG-3′ (forward) and
5′-GAGTGCAGGGTCAGCACTAC-3′ (reverse), generating 214-bp product;
VEGF, 5′-ACCCATGGCAGAAGGAG GAG-3′ (forward) and
5′-ACGCGAGTCTGTGTTTTTGC-3′ (reverse), generating 487-bp product.
After initial denaturation at 94°C for 5 min, PCR was performed for
30 cycles (30 sec at 94°C, 1 min at annealing temperature and 30
sec at 72°C) using Taq polymerase. Reaction products (10 μl)
were separated on 1.5–2% agarose gel, and stained with Redsafe™
(Intron, Daejeon, Korea). DNA band intensity was analyzed by
densitometry using NαBI imager and SET ROI software (Neogene
Science, Suwon, Korea).
Cytoplasmic and nuclear extract
preparation
Cells were harvested and resolved in a 400-μl
cytosolic lysis buffer [10 mM HEPES (pH 7.9), 10 mM KCl, 0.1 mM
EDTA, 1 mM dithiotreitol (DTT) and 1 mM
phenylmethylsulphonylfluoride (PMSF) and protease inhibitor
cocktail]. After reaction for 10 min on ice, 10% NP-40 was added
and strongly vortexed for 1 min. After centrifugation at 3,000 rpm
for 1 min, the supernatants were used as cytoplasmic extracts. The
resulting pellets were resolved in 100 μl of nuclear lysis
buffer [20 mM HEPES (pH 7.9), 420 mM NaCl, 1.5 mM MgCl2,
0.2 mM EDTA, 0.5 mM dithiotreitol, 0.5 mM
phenylmethylsulphonylfluoride and protease inhibitor cocktail] for
30 min on ice. After centrifugation at 14,000 rpm for 15 min, the
supernatants were used as nuclear extracts.
Electrophoretic mobility shift assay
(EMSA)
We performed EMSA for determination of NF-κB
activation using the biotin-labeled NF-κB probe:
5′-AGTTGAGGGGACTTTC CCAGGC-3′. Biotin-labeled NF-κB probe (2
μg of nuclear extracts) and binding buffer including
poly(dIdC) were incubated at 15°C for 30 min in a final volume of
10 μl. Specific binding was controlled by competition with a
cold NF-κB probe. The reaction mixture was separated by
electrophoresis on a non-denaturing polyacrylamide gel. After
electrophoresis at 120 V for 50 min, oligonucleotides were
transferred to a Biodyne B positively charged nylon membrane (Pall,
Basel, Switzerland) at 300 mA for 30 min by the wet transfer
method. The membrane was fixed for 1 h at 80°C, then blocked by
blocking buffer for 1 h then streptavidin-HRP was added in blocking
buffer. After washing, signals were detected by chemiluminescent
imaging according to the manufacturer's protocol (EMSA Gel Shift
kit; Panomics, Redwood City, CA, USA).
Western blotting and immunoprecipitation
analysis
The protein concentration in cell lysates was
determined using a Protein Quantification kit (Bio-Rad, TX, USA).
Fifty micrograms of protein or 30 μg of nuclear extract
protein was loaded onto an SDS-polyacrylamide gel. After
transferring and blocking, the polyvinylidene difluoride (PVDF)
membrane was probed with various antibodies. The binding of
antibody was detected using enhanced ECL prime (GE Healthcare, NJ,
USA), captured and analyzed by the Las-3000 luminescent Image
Analyzer (Fuji Film, Tokyo, Japan).
For immunoprecipitation, 100 μg of cell
extracts was incubated overnight with 4 μg of anti-HIF-1α or
anti-mouse-IgG antibody. The resulting immunocomplex was
precipitated by adding 40 μl of protein A-agarose slurry at
4°C for 18 h. The complex was washed with lysis buffer, separated
by SDS-PAGE and transferred to a PVDF membrane. The membrane was
probed with an anti-VHL antibody and visualized using Las-3000
luminescent Image Analyzer.
Tube formation assay with HUVECs on
Matrigel
Two hundred microliter of Matrigel was added to a
24-well plate and solidified for 30 min at 37°C. The HUVECs
(1×104 cells per well) were seeded into each well of the
Matrigel-coated plate and then incubated in EGM-2 containing 2%
FBS. After overnight incubation, media were changed to fresh
endothelial basal media (EBM-2) in the absence and presence of PT.
Capillary-like tube formation was captured by an inverted
microscope (Olympus IX71) at ×40 magnification. The number of
polygons was counted and then tube formation was calculated as a
percentage by normalization with control cells under normoxic
condition.
In vitro migration and invasion
assays
Cell migration was evaluated by the wound-healing
assay. HT-29 cells (1×106) were seeded in 6-cm culture
plates and incubated at 37°C. The confluent cells were scratched
with a P200 pipette tip to generate a wound ~1 mm wide. After 48-h
incubation under normoxic or hypoxic condition, images were
captured at ×10 magnification, and the wound area was determined
using an inverted microscope (Olympus IX71). The ability of the
cells to close the wound, that is their motility, was evaluated by
determining the healed area.
Invasion assays were performed by using a 24-well
Transwell chamber (24-wells, 8 μm pore size with
poly-carbonate membrane; SPL, Pocheon, Korea) with growth
factor-reduced Matrigel. Briefly, 1×105 cells per well
were seeded into upper chamber with serum-free media. The low
chamber was filled with 800 μl medium containing 10% FBS as
a chemoattractant and allowed to invade for 6 h. Cells remaining
above the insert membrane were removed by suction. Then, the cells
on the lower side of the membrane were fixed in 3.8% formaldehyde
for 20 min and stained with 0.1% crystal violet solution. The
inserts were washed three times in 1X PBS and air-dried. The
numbers of invaded cells in five randomly selected fields were
counted using an inverted microscope (Olympus IX71) at ×10
magnification and analyzed statistically.
Gelatin zymography
Cells were treated with PT at 37°C for 24 h under
normoxic or hypoxic condition, and 100 μl of conditioned
media were collected. Conditioned media was concentrated by using a
speed vacuum concentrator (Thermo Scientific™, MA, USA). The
un-boiled samples were separated by 0.1% gelatin-8% SDS-PAGE
electrophoresis. The gels were washed with 2.5% Triton X-100 at
room temperature for 30 min and then incubated in reaction buffer
(10 mM CaCl2, 40 mM Tris-HCl and 0.01% NaN3,
pH 8.0) at 37°C overnight. Coomassie brilliant blue R-250 was then
used to stain the gel. Stained gel was located on light box and the
image was captured by digital camera.
Nude mouse tumor xenografts study
Six-week-old female athymic nude mice were purchased
from Orients Bio (Sungnam, Korea). Animal experiment protocol was
reviewed and approved by the Chonbuk National University Animal
Care and Use Committee (Approval no: CBU 2012-0035). The tumor was
established by injecting 6×106 CRC cells (HT-29) in 50
μl of Matrigel subcutaneously into right flanks. Mice were
randomized and assigned to control and treatment group and
intraperitoneally injected 3 times a week vehicle (DMSO) and 4
mg/kg PT, respectively. Tumor diameters were measured using a
caliper, and the volumes were also calculated using a formula:
volume = X × Y × Z × π/6. The experiment was terminated on day 27,
and the tumor xenografts were harvested for
immunohistochemistry.
Immunohistochemistry
Immunohistochemistry (IHC) was carried out in
paraffin-embedded 5-μm tissue sections. The sections were
immunostained with anti-carbonic anhydrase IX (CA IX), anti-p65,
anti-von Willebrand factor (VWF) and anti-vimentin antibody,
visualized by appropriate biotin-conjugated secondary antibodies
followed by immmunoperoxidase detection with the Vectastain ABC
Elite kit (Linaris, Germany) and diamino-benzidine (DAB) substrate
(Vector, UK). Five equal-sized fields were randomly chosen. Four
fields at ×10 magnification were selected and antibody-positive
cells were counted.
Statistical analysis
he data are presented as the mean ± standard
deviation (SD) of at least three independent experiments.
Representative blots are shown. All the data were entered into the
Microsoft Excel 5.0, and Graphpad Prism 5.0 was used to perform the
Student's t-tests (for differences between two groups) or the
analysis of the variance, where appropriate. P-values of <0.05
were considered statistically significant.
Results
Parthenolide downregulates HIF-1α protein
level in CRC cell lines
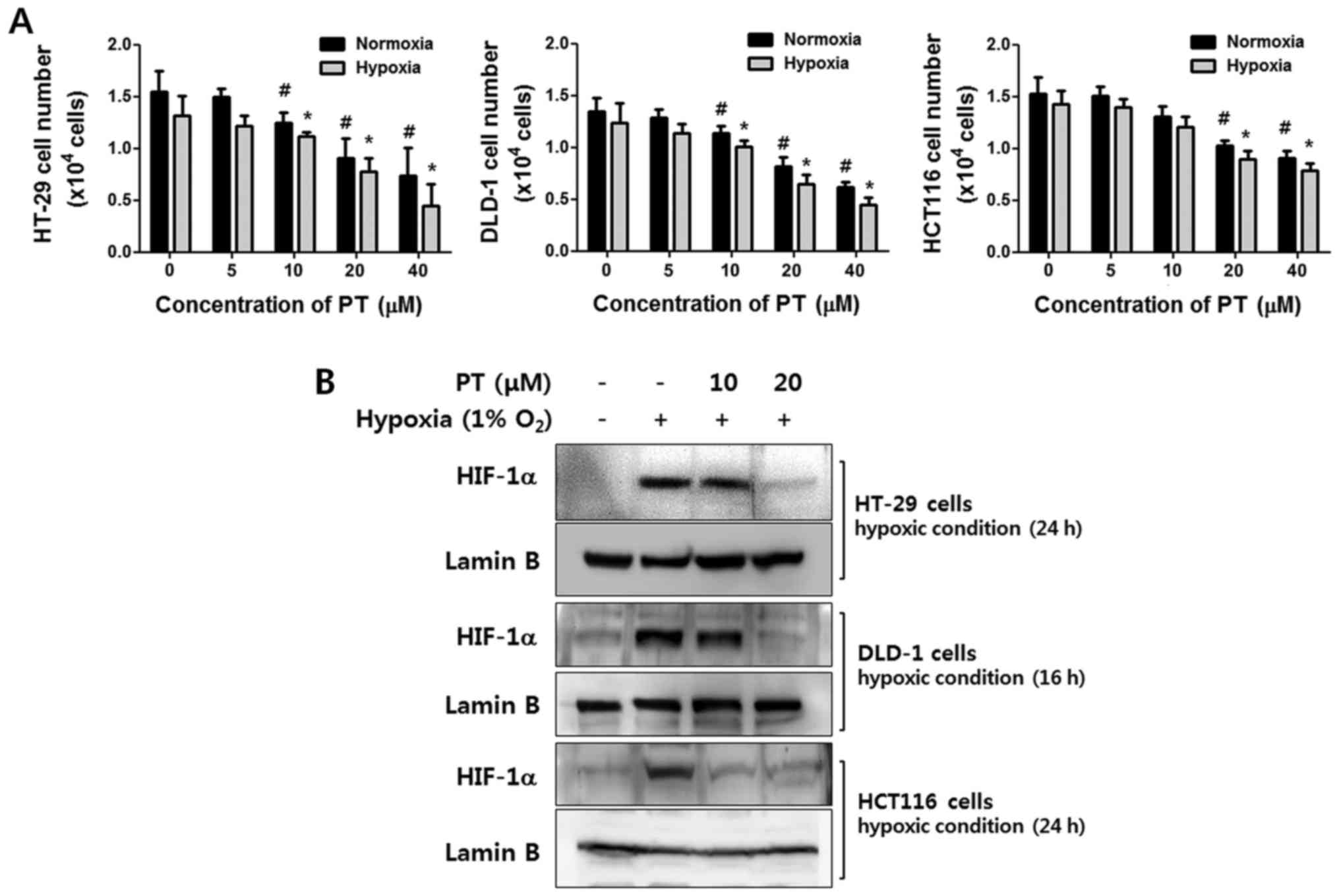
Human CRC cell lines, HT-29, DLD-1 and HCT116 cells
were treated with various concentrations of PT for 24 h in normoxic
or hypoxic conditions. As shown in Fig. 1A, PT exhibited a dose-dependent
anti-proliferative effect in both oxygen conditions in all CRC
cells. Especially, the viability of HT-29 cells was significantly
affected in dose-dependent manner (normoxic condition: vehicle
99.20±3.41%, 20 μM PT 87.94±2.77%, 40 μM PT
49.43±10.52%, hypoxic condition: vehicle 85.5.38%, 20 μM PT
55.89±8.17%, 40 μM PT 30.04±6.75%). These results indicate
that PT has inhibitory effect on growth regardless of oxygen
concentration.
To investigate whether PT regulates HIF-1α
expression in CRC cells under hypoxic conditions, we confirmed the
level of HIF-1α by western blot analysis after treatment with
various concentrations of PT. In response to hypoxia, HIF-1α
expression was dramatically increased in all cell lines.
Interestingly, under same conditions, PT significantly reduced
HIF-1α expression in a concentration-dependent manner. This
downregulation was specific to HIF-1α, as the protein level of
lamin B remained unchanged (Fig.
1B).
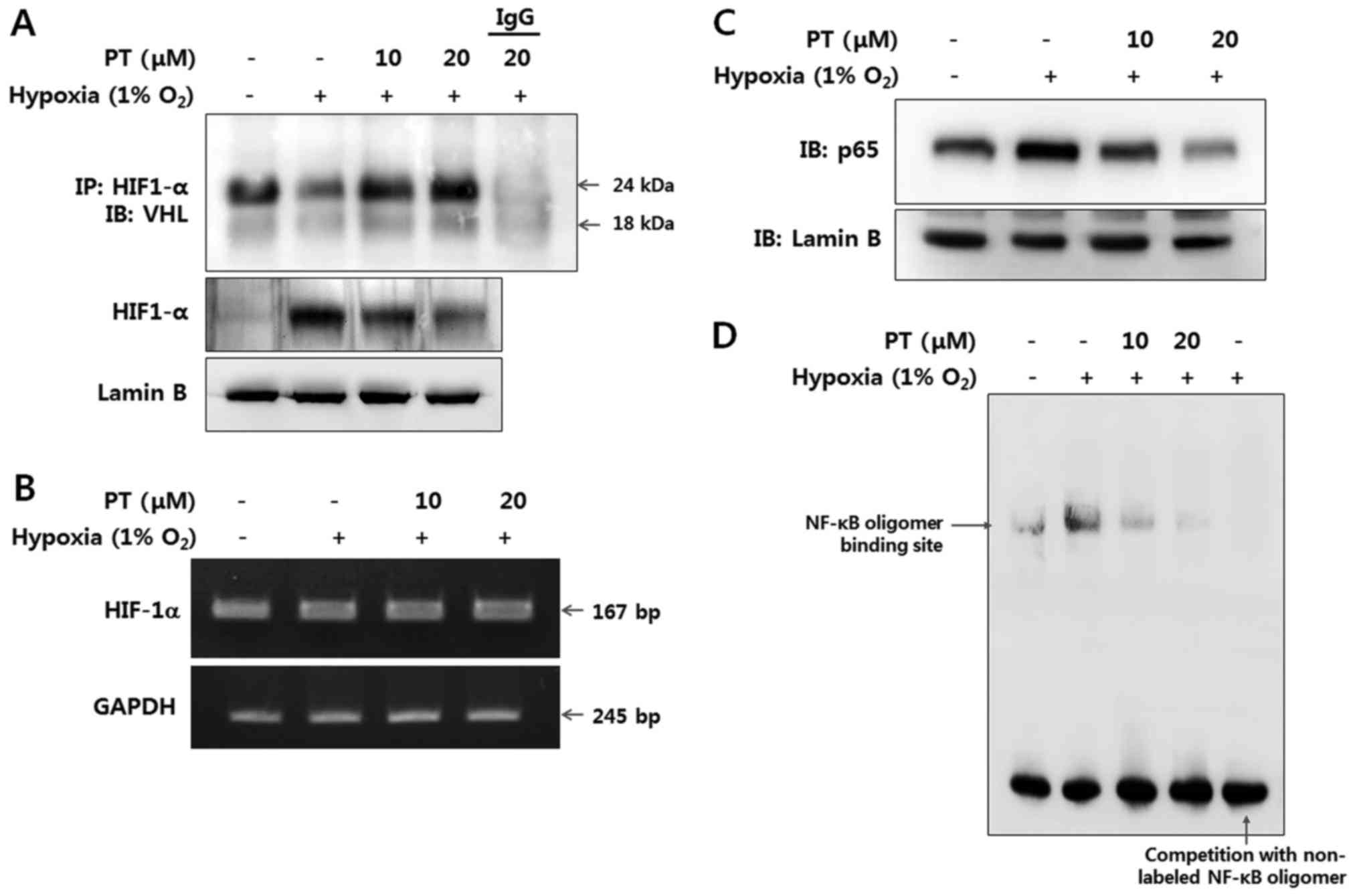
HIF-1α regulation by PT is correlated
with NF-κB activation
To investigate whether PT affects the interaction of
HIF-1α and VHL under hypoxia, we performed immunoprecipitation with
an anti-HIF-1α antibody. Interaction VHL and HIF-1α was decreased
under hypoxic conditions, while treatment with 20 μM of PT
dramatically increased VHL binding to HIF-1α, indicating increased
post-translational modification of HIF-1α (Fig. 2A).
Then, we analyzed the expression of HIF-1α at both
the transcriptional and translational levels for better
understanding of the molecular mechanisms by which PT inhibits
HIF-1α signaling in the CRC cells. When cultured in hypoxic
condition HIF-1α protein level was elevated, while HIF-1α mRNA
level was hardly affected. Cells treated with PT and cultured in
identical hypoxic conditions exhibited markedly reduced levels of
HIF-1α protein but similar levels of HIF-1α mRNA, indicating that
PT regulates HIF-1α on the post-transcriptional level (Fig. 2A and B).
Next, we investigated relation between HIF-1α and
hypoxia-induced NF-κB activation in CRC cells using PT treatment.
Since NF-κB activation results in nuclear translocation of the p65
subunit, we examined protein level of the p65 in nuclear extract by
western blotting. As shown in Fig.
2C, the expression level of p65 was dramatically elevated in
hypoxic condition without any NF-κB stimuli, whereas treatment with
PT resulted in a decreased level of nuclear p65, indicating that PT
prevents hypoxia-mediated nuclear translocation of p65 (Fig. 2C). Moreover, we observed the effect
of PT on the DNA-binding activities of NF-κB in normoxic and
hypoxic condition by EMSA. DNA-binding activity of NF-κB was
significantly increased in hypoxia, whereas its DNA-binding
activity was almost completely blocked by PT at the maximum
concentration tested (Fig. 2D).
These results demonstrate that NF-κB activation is correlated to
HIF-1α activation in CRC cells. Moreover, PT inhibits NF-κB and
downregulates hypoxia-induced NF-κB activation.
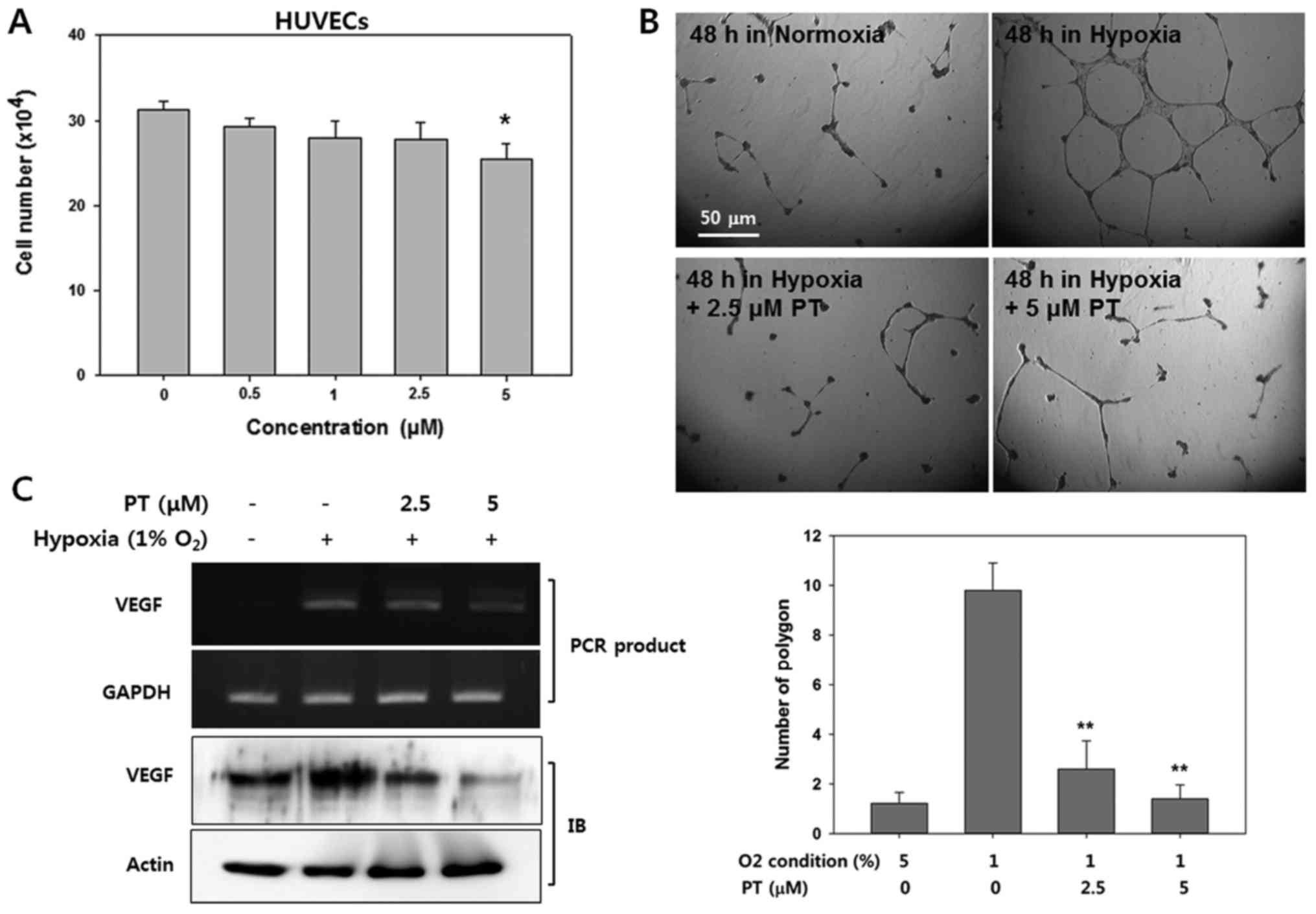
Parthenolide inhibits hypoxia-induced
angiogenesis
Vascular endothelial growth factor (VEGF) is a
downstream target whose expression is significantly induced by
HIF-1α (33). Thus, we next
determined whether PT exerts anti-angiogeneic effect due to its
inhibitory effect on HIF-1α expression. As an in vitro model
of angiogenesis, we used HUVECs in culture, and first we evaluated
viability of HUVECs in various concentration of PT. Treatment with
PT at concentrations ≤2.5 μM did not affect cell viability.
However, at 5 μM PT, the viability of the HUVECs was
significantly decreased (81.212±6.91%, Fig. 3A).
Next, we examined whether PT affects hypoxia-induced
HUVEC tube formation on Matrigel. Endothelial cells can
spontaneously develop a capillary-like network on Matrigel, which
is an important feature of angiogenesis (34). Therefore, we evaluated whether PT
suppressed tube formation by counting the polygons (Fig. 3B). HUVECs did not form tubes under
normoxic conditions, whereas HUVECs obviously displayed
network-like structures after 24 h in hypoxic condition. However,
treatment with PT resulted in significantly reduced tube formation
under hypoxic condition. Followed by quantification of tube
formation by counting the number of polygons, hypoxic conditions
resulted in 10-fold more tubes that in normoxic conditions;
moreover, treatment with 2.5 μM PT decreased the polygon
number by ~70% compared with corresponding hypoxic control group.
This result indicates that the effect of PT on the tube formation
was not due to non-specific cytotoxic effects.
To elucidate the mechanism by which PT exerts its
anti-angiogenic activity, we determined the effect of PT on VEGF
expression in HUVECs (Fig. 3C).
Western blot analysis and reverse-transcriptase PCR revealed that
the treatment of the HUVECs with PT markedly decreased the levels
of VEGF protein and mRNA in a dose-dependent manner. These results
suggest that the anti-angiogenic effect of PT was mediated through
the decreased level of HIF-1α protein.
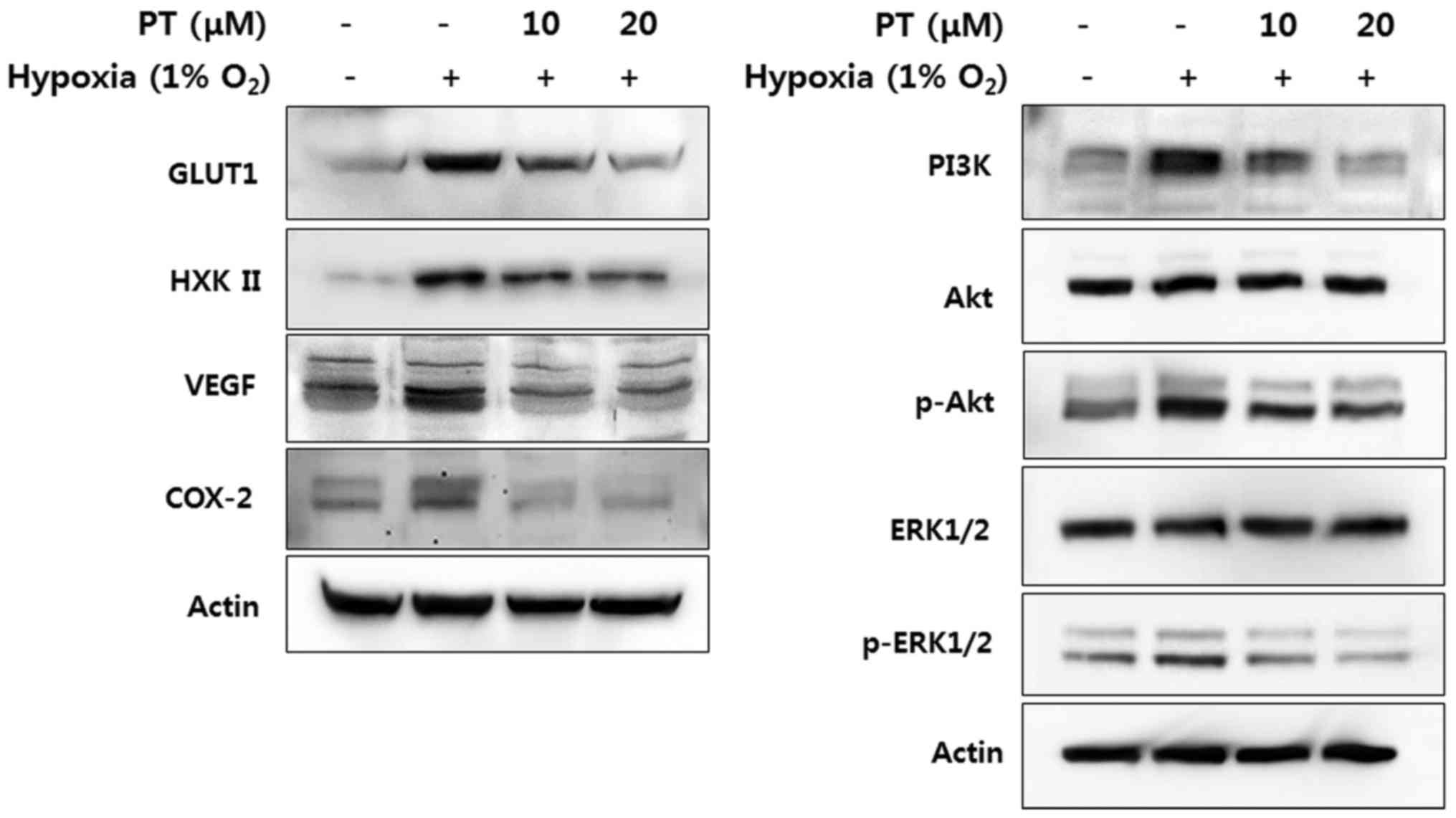
Parthenolide downregulates HIF-1α
dependent gene products
HIF-1α is ubiquitously expressed and binds hypoxia
response elements (HREs) on several hundred genes, thus
facilitating adaptation to hypoxia (35). To investigate whether treatment
with PT inhibits the level of target gene products involved in
energy metabolism, angiogenesis, survival and development, we
analyzed protein level of glucose transporter 1 (GLUT1) and
hexokinase II (HXKII), VEGF, phosphoinositide 3-kinase (PI3K)
phosphor-AKT, phosphor-ERK1/2 and cyclooxygenase-2 (COX-2) by
western blotting. All of target gene products are markedly
upregulated in response to hypoxia. We found that treatment with PT
dramatically inhibited level of these gene products in a
concentration-dependent manner (Fig.
4). Thus, PT can decrease glucose metabolism, angiogenesis,
survival and development modulated by hypoxia.
Parthenolide prevents hypoxia-induced
EMT
Next, we sought to determine if PT modulates
hypoxia-induced EMT. Epithelial and mesenchymal cells show distinct
phenotypes. Specifically, epithelial cells have apical-basal
polarity and form epithelial adherent junctions, whereas
mesenchymal cells lack cell polarity and exhibit a spindle-like
morphology (36). To assess the
effects of PT on hypoxia-induced EMT, we examined the morphological
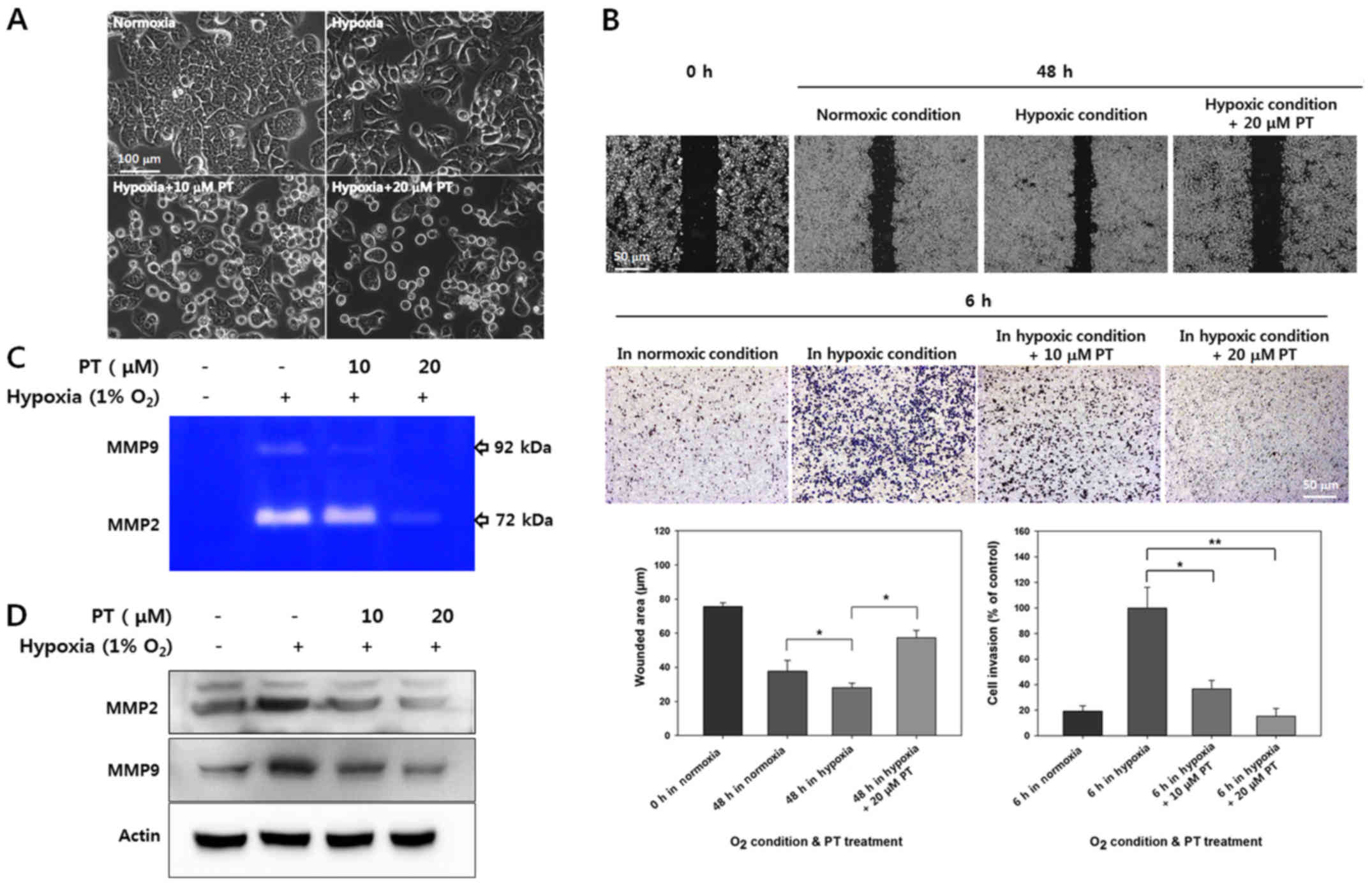
changes under hypoxia. As shown in Fig. 5A, HT-29 cells showed greater
isolation and a more spindle-like morphology under hypoxic
conditions than under normoxic conditions; these characteristics
are typically associated with EMT. However, these hypoxia-induced
morphological alteration was inhibited by PT treatment.
To verify that PT blocked hypoxia-induced EMT, cell
migration and invasion were evaluated in wound healing and invasion
assays using Matrigel coated Transwell chamber. As shown in
Fig. 5B, while hypoxia clearly
promoted cell migration and invasion, these effects were largely
counteracted by PT treatment. Quantification of the wounded area in
scratch tests revealed that hypoxia increased the migration
distance by 28.13±2.65 μm, whereas treatment with 20
μM PT significantly reduced by 57.32±4.29 μm.
Quantification of invading cells showed that 20 μM PT
significantly inhibited invasion by ~80% in HT-29 cells (Fig. 5B). These results demonstrate that
PT inhibits hypoxia-mediated cell migration and invasion.
MMPs have been identified as a powerful modulator of
the promoting invasion and metastasis, moreover HIF-1α promotes
MMPs in hypoxic condition in cancer cells (37). To determine whether PT regulates
hypoxia-mediated MMP activity, we analyzed MMP activity in culture
supernatants and quantified the protein levels of various MMPs in
cell extracts. As shown in Fig.
5C, the levels of hypoxia-mediated MMP2 and MMP9 activity were
decreased by PT in a dose-dependent manner. Similarly, PT inhibited
hypoxia-mediated induction of MMP2 and MMP9 protein expression
(Fig. 5D). Taken together, our
results indicate that PT inhibits hypoxia-induced EMT and reduces
the protein levels of MMP2 and MMP9.
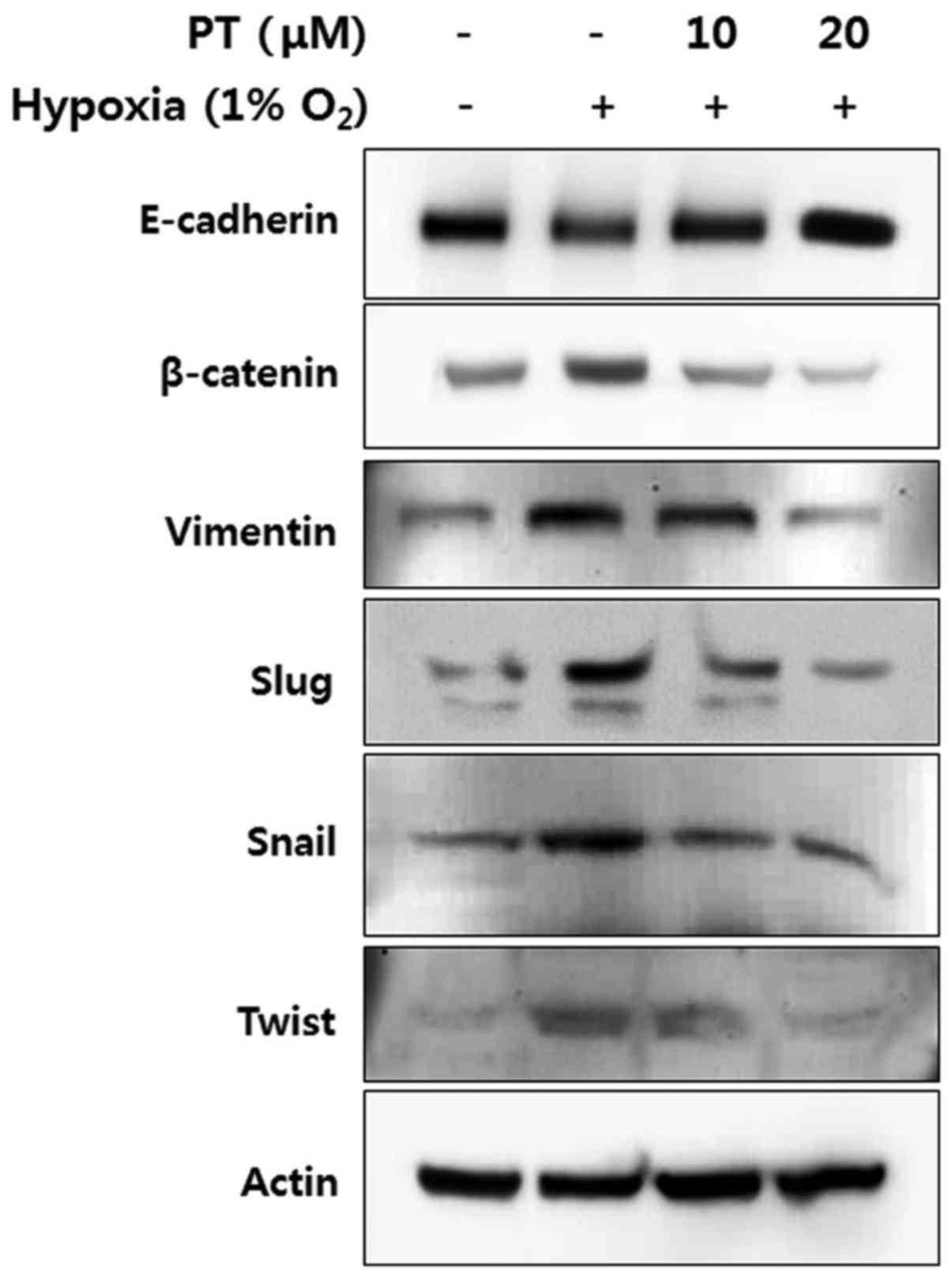
Parthenolide inhibits hypoxia-induced EMT
pathway
To elucidate mechanism by which PT inhibits
hypoxia-induced EMT, we examined the protein levels of
EMT-associated markers in CRC cells. First, the level of
E-cadherin, a representative epithelial marker, was analyzed by
western blotting. Hypoxia treatment decreased the level of
E-cadherin, whereas this level was recovered by PT treatment in a
dose-dependent manner (Fig. 6,
panel 1). In contrast, the expression levels of mesenchymal
markers, such as β-catenin, vimentin, Slug, Snail, and Twist were
increased under hypoxic conditions; moreover, PT significantly
decreased the expression of all mesenchymal markers in a
concentration-dependent manner (Fig.
6, panels 2-6). These results indicate that PT treatment
inhibits EMT through the upregulation of epithelial markers and the
downregulation of mesenchymal markers.
Parthenolide inhibits CRC progression and
angiogenesis by affecting the NF-κB/HIF-1α/EMT signaling
pathway
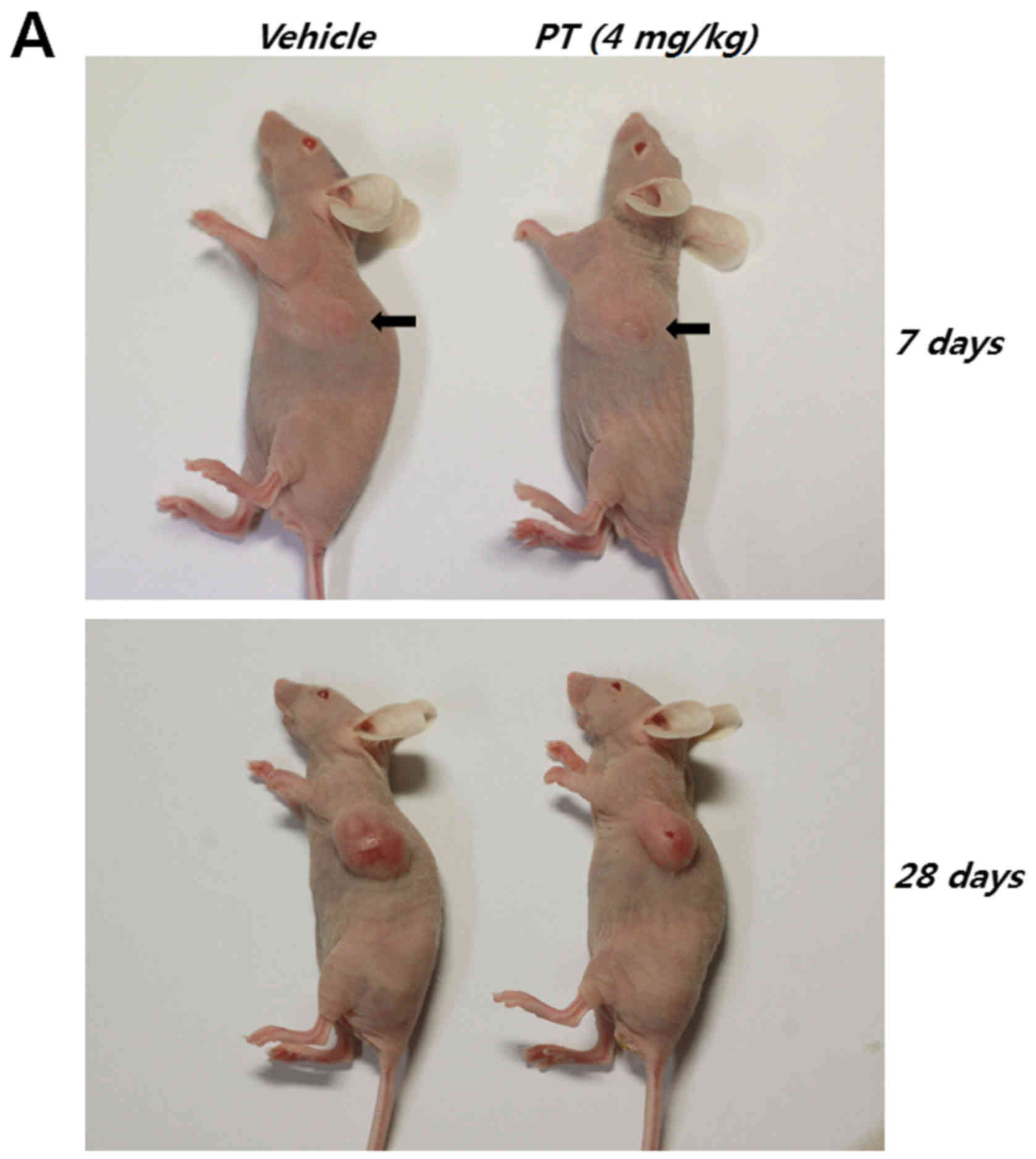
Finally, human CRC cells were inoculated
subcutaneously into athymic nude mice to examine whether PT affects
tumorigenicity and cancer progression (Table I and Fig. 7A). At ~1 week after the start of
treatment, the mean tumor volumes of control and PT-treated mice
were 24.852±11.581 and 23.386±4.986 mm3, respectively.
However, after 4 weeks, the mean tumor volume of PT-treated mice
was significantly lower than that of control mice (control mice:
592.181±132.425 mm3, PT-treated mice: 330.018±52.096
mm3).
 | Table IThe effect of PT on tumor growth in
mice bearing the HT-29 tumor. |
Table I
The effect of PT on tumor growth in
mice bearing the HT-29 tumor.
| Group | n | Tumor volume
(mm3)
|
|---|
| 7 days | 15 days | 27 days |
|---|
| Vehicle | 6 | 24.852±11.581 | 181.172±59.177 |
592.181±132.425 |
| PT (4 mg/kg) | 6 | 23.386±4.986 | 118.48±28.375 |
330.018±52.096a |
To verify the in vitro effects of PT, we
examined the expression of CA IX, p65, VWF and vimentin in
paraffin-embedded CRC tissue from xenograft models by IHC. First,
CA IX is a novel member of the carbonic anhydrase family and the
overexpression of CA IX in cancer tissues is strongly regulated by
hypoxia, through the HIF-1 mediated transcription (38). Therefore, CA IX is a commonly used
hypoxia marker. As shown in Fig.
7B (panel 1), positive staining of CA IX was observed in
control tissue. However, treatment with PT revealed a significant
reduction in the number of CA IX-positive cells (Fig. 7B, panel 1; p<0.05). Next, cells
positively stained for NF-κB subunit p65 were detected in tissues
from control group; however, the number of p65 positively stained
cells was significantly reduced by PT treatment, indicating that PT
inhibited NF-κB activation and NF-κB is implicated in HIF-1α
expression (Fig. 7B, panel 2).
Next, VWF, a well-established marker for angiogenesis, was
examined. This staining revealed that the blood vessel networks
were reduced in the tumor tissue samples from PT-treated mice
compared with the networks in control mice, suggesting that PT
inhibits hypoxia-induced angiogenesis in vivo (Fig. 7B, panel 3; p<0.05). To further
evaluate the effects of PT on EMT, the expression patterns of
vimentin were investigated. The number of vimentin-positive cells
was significantly decreased in the PT-treated mice compared with
the control mice (Fig. 7B, panel
4). Collectively, our IHC results indicate that PT suppresses CRC
progression and angiogenesis through the NF-κB/HIF-1α/EMT pathway,
a finding consistent with our in vitro experiments.
Discussion
In the pro-metastatic environment, hypoxia exerts
its effects predominantly by stabilizing HIF-1α. Many clinical and
preclinical studies have demonstrated that hypoxia and HIF-1α are
promising anticancer drug targets (4). Therefore, searching pharmacologically
active compound from natural sources to suppress cancer progression
and metastasis by modulating the hypoxic tumor microenvironment is
possible to be an attractive approach to anticancer drug discovery.
In the present study, we have identified for the first time that
the PT, isolated from medicinal plants, effectively inhibits cancer
progression and angiogenesis by inducing HIF-1α degradation and
suppressing hypoxia-induced EMT of CRC cells.
PT exerts a wide spectrum of anticancer activities,
ranging from preventing cancer development to enhancing anticancer
drug effect in combined therapies. It is well known that the main
mechanism of PT in cancer treatment is based on NF-κB signaling
pathway through many published studies. In a previous study, we
demonstrated that PT inhibits phosphorylation of IκB-α and NF-κB
activation, resulting in initiation of apoptosis, suppression of
CRC tumor growth and colitisassociated cancer development (22,39).
While the mechanism linking PT to NF-κB activation has been
extensively studied, the effects of PT on HIF-1α expression and
hypoxia-induced EMT has not been previously investigated. In this
study, we demonstrate that PT suppresses hypoxia-induced HIF-1α
upregulation and also inhibits EMT by preventing NF-κB activation.
These findings support the utility of PT for inhibiting HIF-1α
mediated CRC progression and angiogenesis, thus implying that PT
could be used to treat metastatic CRC.
HIF-1α induction has been demonstrated to be
dependent upon NF-κB transcriptional activity and this activity is
well correlated with HIF-1α protein level. Moreover, many of the
stimuli that induce HIF-1 in normoxic conditions are known to
activate a number of other transcription factors such as NF-κB. In
2003, Jung et al reported that IL-1β upregulates functional
HIF-1α protein through NF-κB signaling pathway, resulted in
upregulation of VEGF, a potent angiogenic factor required for tumor
growth and metastasis (27). Van
Uden et al have presented that TNFα-induced NF-κB activation
increases the expression level of HIF-1α and activity, leading to
transactivation of target genes under normoxic condition (28). Furthermore, Bonello et al
identified that H2O2 upregulates HIF-1α
transcription by activating NF-κB, thus linking these important
pathways in a common mechanism induced by ROS (40). Conversely, exposure to hypoxia has
been shown to activate NF-κB signaling. In 2006, it was reported
that hypoxia releases repression of NF-κB activity through
decreased PHD-dependent hydroxylation of IKK-β (41). Koong et al also reported
that hypoxia activates NF-κB through the phosphorylation of IκB-α
on tyrosine residues (42).
Interestingly, several studies have identified a high degree of
crosstalk between the NF-κB and HIF signaling pathways. Rius et
al demonstrated that NF-κB activation upregulates HIF-1α on the
transcriptional level and also that basal NF-κB is a prerequisite
for constitutive HIF-1α expression (43). In addition, Belaiba et al
reported that the human HIF-1α promoter includes a NF-κB binding
site at a site -197/-188 bp upstream of the transcriptional start
site, mutation of which leads to a loss of hypoxic HIF-1α
upregulation (44). Here, we
demonstrated that a low oxygen concentration resulted in NF-κB
activation and that the HIF-1α signaling pathway was suppressed by
NF-κB inhibition. Thus, our findings are consistent with the
presence of NF-κB-HIF crosstalk.
Inhibition of HRE activation under hypoxic condition
can be explained by either a reduction in the expression level of
HIF-1α or the interference of HIF-1α binding to these elements.
Here, we showed that PT inhibited HIF-1α accumulation under hypoxic
condition in a dose-dependent manner. Thus, the expression levels
of hypoxia-related elements (including VEGF) are likely decreased
due to the reduction in HIF-1α expression. Since VEGF plays a
central role in angiogenesis, antibodies and soluble proteins that
block VEGF and VEGF receptors have been investigated for
anti-angiogenic therapies. Moreover, a number of studies have
indicated that VEGF regulation depends on NF-κB activation. Growth
factors for angiogenesis are upregulated by NF-κB signaling, and
COX2 is known to promote angiogenesis. Moreover, COX2 expression
can be induced by NF-κB activation (45,46).
We previously reported that PT inhibits angiogenesis by
downregulating VEGF/VEGFRs in CRC cells and HUVECs (47). Similarly, here we showed that PT
blocks hypoxia-induced VEGF expression and tube formation in
HUVECs. We also showed that PT reduces microvessel formation and
tumor growth in vivo. Taken together, these results indicate
that PT-mediated inhibition of HIF-1α/NF-κB signaling effectively
suppresses hypoxia-mediated angiogenesis in CRC.
We also confirmed protein level of HIF-1α-dependent
genes to examine the mechanisms underlying adaptation to hypoxic
conditions. Since the oxidative phosphorylation pathway in
mitochondria is impaired in hypoxia, several glycolytic enzymes are
induced to maintain the supply of ATP required for cell growth and
survival (48). Based on these
findings, PT-mediated inhibition of GLUT1 and HXK II expression
probably prevents adaptation to hypoxia by decreasing the rate of
glycolysis. The involvement of the PI3K/Akt pathways in
hypoxia-mediated regulation of HIF-1 remains controversial.
However, growing evidence has indicated that the PI3K/Akt pathway
is affected by hypoxic conditions. Under hypoxic condition, Akt is
targeted by PI3K in the inner leaflet of the plasma membrane.
Subsequently, Akt is phosphorylated by a phosphoinositide-dependent
protein kinase, thereby regulating cancer development and survival
(49,50). Moreover, Huang et al
reported that decreased nuclear accumulation of HIF-1α resulted in
reduced hypoxia-induced Akt phosphorylation upon treatment with
folic acid (51). Numerous studies
have also demonstrated that MEK-1/p42/p44 MAPK pathway is involved
in hypoxia. Hur et al have also reported that the MAPK
inhibitor PD98059 decreases the transactivation ability of HIF-1α
thereby reducing hypoxia-induced transcription of target gene and a
hypoxia-responsive reporter gene (52). In the present study, we reported
that PT-mediated downregulation of HIF-1α significantly decreases
hypoxia-induced survival and development in vitro, in
addition to tumor growth in vivo, by inhibiting the PI3K/Akt
and MAPK pathways.
HIF-1α modulates important steps of the metastatic
processes, especially EMT, which is one of the most crucial events
resulting in cell migration and invasion in early stage of tumor
metastasis (53). Under hypoxic
condition, the tumor microenvironment generates major
EMT-triggering pathways such as transforming growth factor (TGF)-β,
Notch and NF-κB signaling pathways (54). Inflammatory cytokines (including
TNF-α, IL-1 and IL-6) have been reported to be critical to SNAIL
stability and EMT induction through the activation of NF-κB
signaling (55–57). Furthermore, Cheng et al
identified that activation of the HIF-1α and NF-κB loop is
mechanically linked with EMT phenotype of pancreatic cancer cells
under hypoxic condition showing downregulation of hypoxia-induced
EMT by p65 siRNA (58). In the
present study, hypoxia-induced EMT was characterized by
morphological changes, cell migration and invasion, MMPs enzyme
activity, suppression of epithelial marker (E-cadherin) and
enhanced expression of mesenchymal markers (β-catenin, vimentin,
Slug, Snail and Twist). We also showed that suppression of HIF-1α
by NF-κB inhibitor led to changes of the cell morphology to
epithelial phenotype and decreased hypoxiainduced migration and
invasion, and regulation of EMT markers. In addition, we confirmed
that PT treatment inhibits NF-κB activation, thereby reducing its
binding to CA IX (a representative hypoxia marker) and vimentin in
CRC tissue samples obtained from an in vivo xenograft model.
Thus, our data indicate that hypoxia-mediated EMT is intimately
connected to NF-κB activation. Moreover, our findings implicate
NF-κB as a principal mediator of HIF-1α-induced EMT in CRC under
hypoxic conditions.
Further studies using metastatic animal model will
be needed to determine the precise mechanisms by which PT potently
prevents metastasis of CRC. However, these data provide strong
evidence that this mechanism involves effective inhibition of the
HIF-1α signaling pathway and hypoxia-induced EMT in CRC.
Acknowledgments
This study was supported by Basic Science Research
Program through the National Research Foundation of Korea (NRF)
funded by the Ministry of Education (NRF-2014R1A6A3A01057354) and
by Fund of Biomedical Research Institute, Chonbuk National
University Hospital.
References
|
1
|
Rajaganeshan R, Prasad R, Guillou PJ,
Poston G, Scott N and Jayne DG: The role of hypoxia in recurrence
following resection of Dukes' B colorectal cancer. Int J Colorectal
Dis. 23:1049–1055. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lunt SJ, Chaudary N and Hill RP: The tumor
microenvironment and metastatic disease. Clin Exp Metastasis.
26:19–34. 2009. View Article : Google Scholar
|
|
3
|
Guillemin K and Krasnow MA: The hypoxic
response: Huffing and HIFing. Cell. 89:9–12. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Semenza GL: Targeting HIF-1 for cancer
therapy. Nat Rev Cancer. 3:721–732. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Poon E, Harris AL and Ashcroft M:
Targeting the hypoxia-inducible factor (HIF) pathway in cancer.
Expert Rev Mol Med. 11:e262009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schofield CJ and Ratcliffe PJ: Oxygen
sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 5:343–354.
2004. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Semenza GL: Regulation of mammalian
O2 homeostasis by hypoxia-inducible factor 1. Annu Rev
Cell Dev Biol. 15:551–578. 1999. View Article : Google Scholar
|
|
8
|
Maxwell PH, Wiesener MS, Chang GW,
Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER and
Ratcliffe PJ: The tumour suppressor protein VHL targets
hypoxia-inducible factors for oxygen-dependent proteolysis. Nature.
399:271–275. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Semenza GL: HIF-1, O(2), and the 3 PHDs:
How animal cells signal hypoxia to the nucleus. Cell. 107:1–3.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang J and Weinberg RA:
Epithelial-mesenchymal transition: At the crossroads of development
and tumor metastasis. Dev Cell. 14:818–829. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hur K, Toiyama Y, Takahashi M, Balaguer F,
Nagasaka T, Koike J, Hemmi H, Koi M, Boland CR and Goel A:
MicroRNA-200c modulates epithelial-to-mesenchymal transition (EMT)
in human colorectal cancer metastasis. Gut. 62:1315–1326. 2013.
View Article : Google Scholar :
|
|
12
|
Turley EA, Veiseh M, Radisky DC and
Bissell MJ: Mechanisms of disease: Epithelial-mesenchymal
transition - does cellular plasticity fuel neoplastic progression?
Nat Clin Pract Oncol. 5:280–290. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bates RC and Mercurio AM: The
epithelial-mesenchymal transition (EMT) and colorectal cancer
progression. Cancer Biol Ther. 4:365–370. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hill RP, Marie-Egyptienne DT and Hedley
DW: Cancer stem cells, hypoxia and metastasis. Semin Radiat Oncol.
19:106–111. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang MH, Wu MZ, Chiou SH, Chen PM, Chang
SY, Liu CJ, Teng SC and Wu KJ: Direct regulation of TWIST by
HIF-1alpha promotes metastasis. Nat Cell Biol. 10:295–305. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhou G, Dada LA, Wu M, Kelly A, Trejo H,
Zhou Q, Varga J and Sznajder JI: Hypoxia-induced alveolar
epithelial-mesenchymal transition requires mitochondrial ROS and
hypoxia-inducible factor 1. Am J Physiol Lung Cell Mol Physiol.
297:L1120–L1130. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bork PM, Schmitz ML, Kuhnt M, Escher C and
Heinrich M: Sesquiterpene lactone containing Mexican Indian
medicinal plants and pure sesquiterpene lactones as potent
inhibitors of transcription factor NF-kappaB. FEBS Lett. 402:85–90.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Murphy JJ, Heptinstall S and Mitchell JR:
Randomised double-blind placebo-controlled trial of feverfew in
migraine prevention. Lancet. 2:189–192. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hehner SP, Heinrich M, Bork PM, Vogt M,
Ratter F, Lehmann V, Schulze-Osthoff K, Dröge W and Schmitz ML:
Sesquiterpene lactones specifically inhibit activation of NF-kappa
B by preventing the degradation of I kappa B-alpha and I kappa
B-beta. J Biol Chem. 273:1288–1297. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Oka D, Nishimura K, Shiba M, Nakai Y, Arai
Y, Nakayama M, Takayama H, Inoue H, Okuyama A and Nonomura N:
Sesquiterpene lactone parthenolide suppresses tumor growth in a
xenograft model of renal cell carcinoma by inhibiting the
activation of NF-kappaB. Int J Cancer. 120:2576–2581. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kishida Y, Yoshikawa H and Myoui A:
Parthenolide, a natural inhibitor of nuclear factor-kappaB,
inhibits lung colonization of murine osteosarcoma cells. Clin
Cancer Res. 13:59–67. 2001. View Article : Google Scholar
|
|
22
|
Kim SL, Liu YC, Seo SY, Kim SH, Kim IH,
Lee SO, Lee ST, Kim DG and Kim SW: Parthenolide induces apoptosis
in colitis-associated colon cancer, inhibiting NF-κB signaling.
Oncol Lett. 9:2135–2142. 2015.PubMed/NCBI
|
|
23
|
Luo JL, Kamata H and Karin M:
IKK/NF-kappaB signaling: Balancing life and death - a new approach
to cancer therapy. J Clin Invest. 115:2625–2632. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hayden MS and Ghosh S: Signaling to
NF-kappaB. Genes Dev. 18:2195–2224. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Royds JA, Dower SK, Qwarnstrom EE and
Lewis CE: Response of tumour cells to hypoxia: Role of p53 and
NFκB. Mol Pathol. 51:55–61. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kwon HC, Kim SH, Oh SY, Lee S, Kwon KA,
Lee JH, Choi HJ, Park KJ, Lee HS, Roh MS, et al:
Clinicopathological significance of nuclear factor-kappa B, HIF-1
alpha, and vascular endothelial growth factor expression in stage
III colorectal cancer. Cancer Sci. 101:1557–1561. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jung YJ, Isaacs JS, Lee S, Trepel J and
Neckers L: IL-1beta-mediated up-regulation of HIF-1alpha via an
NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between
inflammation and oncogenesis. FASEB J. 17:2115–2117.
2003.PubMed/NCBI
|
|
28
|
van Uden P, Kenneth NS and Rocha S:
Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. Biochem
J. 412:477–484. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nam SY, Ko YS, Jung J, Yoon J, Kim YH,
Choi YJ, Park JW, Chang MS, Kim WH and Lee BL: A hypoxia-dependent
upregulation of hypoxia-inducible factor-1 by nuclear factor-κB
promotes gastric tumour growth and angiogenesis. Br J Cancer.
104:166–174. 2011. View Article : Google Scholar
|
|
30
|
Wu Y and Zhou BP:
TNF-alpha/NF-kappaB/Snail pathway in cancer cell migration and
invasion. Br J Cancer. 102:639–644. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yu L, Mu Y, Sa N, Wang H and Xu W: Tumor
necrosis factor α induces epithelial-mesenchymal transition and
promotes metastasis via NF-κB signaling pathway-mediated TWIST
expression in hypopharyngeal cancer. Oncol Rep. 31:321–327. 2014.
View Article : Google Scholar
|
|
32
|
Cheng ZX, Sun B, Wang SJ, Gao Y, Zhang YM,
Zhou HX, Jia G, Wang YW, Kong R, Pan SH, et al: Nuclear
factor-κB-dependent epithelial to mesenchymal transition induced by
HIF-1α activation in pancreatic cancer cells under hypoxic
conditions. PLoS One. 6:e237522011. View Article : Google Scholar
|
|
33
|
Forsythe JA, Jiang BH, Iyer NV, Agani F,
Leung SW, Koos RD and Semenza GL: Activation of vascular
endothelial growth factor gene transcription by hypoxia-inducible
factor 1. Mol Cell Biol. 16:4604–4613. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tozer GM, Kanthou C and Baguley BC:
Disrupting tumour blood vessels. Nat Rev Cancer. 5:423–435. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xia X, Lemieux ME, Li W, Carroll JS, Brown
M, Liu XS and Kung AL: Integrative analysis of HIF binding and
transactivation reveals its role in maintaining histone methylation
homeostasis. Proc Natl Acad Sci USA. 106:4260–4265. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Thiery JP and Sleeman JP: Complex networks
orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell
Biol. 7:131–142. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhu S, Zhou Y, Wang L, Zhang J and Wu H,
Xiong J, Zhang J, Tian Y, Wang C and Wu H: Transcriptional
upregulation of MT2-MMP in response to hypoxia is promoted by
HIF-1α in cancer cells. Mol Carcinog. 50:770–780. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ambrosio MR, Di Serio C, Danza G, Rocca
BJ, Ginori A, Prudovsky I, Marchionni N, Del Vecchio MT and
Tarantini F: Carbonic anhydrase IX is a marker of hypoxia and
correlates with higher Gleason scores and ISUP grading in prostate
cancer. Diagn Pathol. 11:452016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kim SL, Trang KT, Kim SH, Kim IH, Lee SO,
Lee ST, Kim DG and Kim SW: Parthenolide suppresses tumor growth in
a xenograft model of colorectal cancer cells by inducing
mitochondrial dysfunction and apoptosis. Int J Oncol. 41:1547–1553.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bonello S, Zähringer C, BelAiba RS,
Djordjevic T, Hess J, Michiels C, Kietzmann T and Görlach A:
Reactive oxygen species activate the HIF-1alpha promoter via a
functional NFkappaB site. Arterioscler Thromb Vasc Biol.
27:755–761. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cummins EP, Berra E, Comerford KM,
Ginouves A, Fitzgerald KT, Seeballuck F, Godson C, Nielsen JE,
Moynagh P, Pouyssegur J, et al: Prolyl hydroxylase-1 negatively
regulates IkappaB kinase-beta, giving insight into hypoxia-induced
NFkappaB activity. Proc Natl Acad Sci USA. 103:18154–18159. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Koong AC, Chen EY and Giaccia AJ: Hypoxia
causes the activation of nuclear factor kappa B through the
phosphorylation of I kappa B alpha on tyrosine residues. Cancer
Res. 54:1425–1430. 1994.PubMed/NCBI
|
|
43
|
Rius J, Guma M, Schachtrup C, Akassoglou
K, Zinkernagel AS, Nizet V, Johnson RS, Haddad GG and Karin M:
NF-kappaB links innate immunity to the hypoxic response through
transcriptional regulation of HIF-1alpha. Nature. 453:807–811.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Belaiba RS, Bonello S, Zähringer C,
Schmidt S, Hess J, Kietzmann T and Görlach A: Hypoxia up-regulates
hypoxia-inducible factor-1alpha transcription by involving
phosphatidylinositol 3-kinase and nuclear factor kappaB in
pulmonary artery smooth muscle cells. Mol Biol Cell. 18:4691–4697.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Huang S, DeGuzman A, Bucana CD and Fidler
IJ: Nuclear factor-kappaB activity correlates with growth,
angiogenesis, and metastasis of human melanoma cells in nude mice.
Clin Cancer Res. 6:2573–2581. 2000.PubMed/NCBI
|
|
46
|
Tsujii M, Kawano S, Tsuji S, Sawaoka H,
Hori M and DuBois RN: Cyclooxygenase regulates angiogenesis induced
by colon cancer cells. Cell. 93:705–716. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kim SL, Lee ST, Trang KT, Kim SH, Kim IH,
Lee SO, Kim DG and Kim SW: Parthenolide exerts inhibitory effects
on angiogenesis through the downregulation of VEGF/VEGFRs in
colorectal cancer. Int J Mol Med. 33:1261–1267. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Semenza GL, Roth PH, Fang HM and Wang GL:
Transcriptional regulation of genes encoding glycolytic enzymes by
hypoxia-inducible factor 1. J Biol Chem. 269:23757–23763.
1994.PubMed/NCBI
|
|
49
|
Joshi S, Singh AR and Durden DL: MDM2
regulates hypoxic hypoxia-inducible factor 1α stability in an E3
ligase, proteasome, and PTEN-phosphatidylinositol
3-kinase-AKT-dependent manner. J Biol Chem. 289:22785–22797. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Mottet D, Dumont V, Deccache Y, Demazy C,
Ninane N, Raes M and Michiels C: Regulation of hypoxia-inducible
factor-1alpha protein level during hypoxic conditions by the
phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3beta
pathway in HepG2 cells. J Biol Chem. 278:31277–31285. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Huang X, He Z, Jiang X, Hou M, Tang Z,
Zhen X, Liang Y and Ma J: Folic acid represses hypoxia-induced
inflammation in THP-1 cells through inhibition of the
PI3K/Akt/HIF-1α pathway. PLoS One. 11:e01515532016. View Article : Google Scholar
|
|
52
|
Hur E, Chang KY, Lee E, Lee SK and Park H:
Mitogen-activated protein kinase kinase inhibitor PD98059 blocks
the transactivation but not the stabilization or DNA binding
ability of hypoxia-inducible factor-1alpha. Mol Pharmacol.
59:1216–1224. 2001.PubMed/NCBI
|
|
53
|
Cannito S, Novo E, Compagnone A, Valfrè di
Bonzo L, Busletta C, Zamara E, Paternostro C, Povero D, Bandino A,
Bozzo F, et al: Redox mechanisms switch on hypoxia-dependent
epithelial-mesenchymal transition in cancer cells. Carcinogenesis.
29:2267–2278. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Jiang J, Tang YL and Liang XH: EMT: A new
vision of hypoxia promoting cancer progression. Cancer Biol Ther.
11:714–723. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Chuang MJ, Sun KH, Tang SJ, Deng MW, Wu
YH, Sung JS, Cha TL and Sun GH: Tumor-derived tumor necrosis
factor-alpha promotes progression and epithelial-mesenchymal
transition in renal cell carcinoma cells. Cancer Sci. 99:905–913.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Sullivan NJ, Sasser AK, Axel AE, Vesuna F,
Raman V, Ramirez N, Oberyszyn TM and Hall BM: Interleukin-6 induces
an epithelial-mesenchymal transition phenotype in human breast
cancer cells. Oncogene. 28:2940–2947. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
John MA St, Dohadwala M, Luo J, Wang G,
Lee G, Shih H, Heinrich E, Krysan K, Walser T, Hazra S, et al:
Proinflammatory mediators upregulate snail in head and neck
squamous cell carcinoma. Clin Cancer Res. 15:6018–27. 2009.
View Article : Google Scholar
|
|
58
|
Cheng ZX, Wang DW, Liu T, Liu WX, Xia WB,
Xu J, Zhang YH, Qu YK, Guo LQ, Ding L, et al: Effects of the HIF-1α
and NF-κB loop on epithelial-mesenchymal transition and
chemoresistance induced by hypoxia in pancreatic cancer cells.
Oncol Rep. 31:1891–1898. 2014.PubMed/NCBI
|