Introduction
It has been >70 years since Waddington first
proposed the notion of 'epigenetics' (1), in which he defined it as 'the
relation between phenotypes and genotypes'. Although the full scope
of epigenetics is yet to be determined, its general meaning refers
to chemical modifications such as DNA methylation, histone
acetylation/methylation, protein glycosylation, and RNA methylation
that alter gene expression without changing nucleotide sequences.
To date, >100 RNA chemical modifications have been discovered
(2). Among them,
N6-methyladenosine (m6A) is the most
abundant modification on epitranscriptome in mRNAs, which is
introduced by the human methyltransferase complex composed of
methyltransferase-like 3 (METTL3), METTL14 and Wilms tumor
1-associated protein (WTAP) complex (3,4).
While m6A was identified in the 1970s (5,6),
even the precise distribution was seldom elucidated until recently.
With the advent of anti-m6A antibody and methylated RNA
immunoprecipitation sequencing (MeRIP-Seq) technology, there has
been significant progress in the field of m6A in recent
years. M6A site mapping reveals that m6A
peaks are typically located in the vicinity of the stop codon as
well as 5′-untranslated regions (UTRs) and m6A tends to
be found in the RR-A-CH (R = A/G, H = A, C, or U) consensus motif
(7-11). Previous studies reported that
m6A RNA methylation is a dynamic reversible phenomenon
(12,13) in which the METTL3-METTL14-WTAP
complex works as m6A writers; fat mass- and
obesity-associated protein (FTO) and AlkB homolog 5 (ALKBH5)
demethylate as m6A erasers (12,13);
and YTH domain family and heterogeneous nuclear ribonucleoprotein C
(HNRNPC) and its family proteins recognize m6A as
m6A readers (7,14). However, comprehensive understanding
of this dynamic regulatory system remains elusive. The presence of
m6A seems to affect mRNA fate and function in various
ways such as circadian clock (15), mRNA degradation (16,17),
translational alteration (10,18,19),
splicing effects (3,7,14,20),
microRNA maturation (21,22), cell reprogramming (11,23,24),
long non-coding RNA (lncRNA) inactivation, X-inactive specific
transcript (XIST) (25), and
RNA-mediated response to ultraviolet (UV)-induced DNA damage
(26). Using previously reported
MeRIP-Seq data, computational in silico analysis has built
an m6A-driven network of m6A-driven genes,
suggesting various roles of m6A (27).
Despite increased understanding about
m6A, direct or indirect target genes of METTL3 or
m6A remain unclear. Additionally, the precise role of
METTL3 in cancer cells is unknown. To evaluate the aforementioned
issues, we focused on the role of METTL3 in pancreatic cancer cells
in patients whose prognosis remains poor despite the recent
development of multidisciplinary therapies. We investigated the
role of METTL3 in vitro using human pancreatic cancer lines
and demonstrated the functional changes in METTL3 knockdown (KD)
cells.
Materials and methods
Cell lines and culture conditions
Human pancreatic adenocarcinoma cell lines, MIA
PaCa-2, were purchased from the the American Type Culture
Collection (ATCC; Manassas, VA, USA. The cells were maintained in
Dulbecco's modified Eagle's medium (DMEM) low glucose (Nacalai
Tesque, Inc., Kyoto, Japan) supplemented with 10% fetal bovine
serum (FBS; Thermo Fisher Scientific, Waltham, MA, USA) and 1%
penicillin/streptomycin (Life Technologies, Carlsbad, CA, USA) in
5% CO2 at 37°C.
Knockdown by short hairpin RNA
transfection
For lentiviral particle production, 293FT cells were
seeded 24 h before lentiviral infection and cotransfected with
pCMV-VSV-G-RSV-Rev and pCAG-HIVgp (provided by Dr Miyoshi from the
RIKEN BioResource, Tokyo, Japan) plasmid using Lipofectamine 3000
and P3000 Reagent (Thermo Fisher Scientific) as described in the
standard protocol of Lipofectamine 3000. Target sequences of METTL3
short hairpin RNA (shRNA) were 5′-CCAGTCATAAACCAGATGAAA-3′. The
collected viral soup was centrifuged at 12,000 x g for 5 min, and
the supernatant was filtered with 0.22-µm pores (Merck
Millipore, Billerica, MA, USA). Purified viral particles were
infected to target MIA PaCa-2 cells with polybrene (Sigma-Aldrich,
St. Louis, MO, USA). After a 24-h incubation, infected cells were
selected with 2 µg/ml of puromycin (InvivoGen, Inc., San
Diego, CA, USA) for 5 days.
RNA extraction and quantitative reverse
transcriptase polymerase chain reaction (qRT-PCR)
Total RNA was extracted from cultured cells using
TRIzol reagent (Thermo Fisher Scientific) and cDNA was synthesized
with ReverTra Ace (Toyobo Co., Ltd., Osaka, Japan). Quantitative
RT-PCR was performed with Thunderbird SYBR qPCR Mix (Toyobo) using
LightCycler 2.0 (Roche Molecular Systems, Inc., Pleasanton, CA,
USA). Furthermore, data were analyzed by the ΔΔCt method, in which
target genes were normalized to glyceraldehyde-3-phosphate
dehydrogenase (GAPDH). The following primers were used in the
present study: GAPDH-F, 5′-GCCCAATACGACCAAATCC-3′ and GAPDH-R,
5′-AGC CACATCGCTCAGACAC-3′; METLL3-F, 5′-CGTACTACA
GGATGATGGCTTTC-3′ and METTL3-R, 5′-TTTCATCTA CCCGTTCATACCC-3′.
Western blot analysis
Plated cells were carefully washed twice with
phosphate-buffered saline (PBS; Thermo Fisher Scientific). Next,
total protein was extracted with radioimmunoprecipitation assay
buffer (Pierce, Rockford, IL, USA) supplemented with 1% of Halt
Protease Inhibitor Cocktail (100X) and Halt Phosphatase Inhibitor
Cocktail (100X; Thermo Fisher Scientific). Isolated proteins were
electrophoresed on 4-20% Mini-PROTEAN TGX Precast protein gels
(Bio-Rad Laboratories, Hercules, CA, USA) and transferred to Blot 2
PVDF Mini Stack membranes (Thermo Fisher Scientific) using iBlot 2
Dry Blotting System (Life Technologies). The membrane was blocked
with 5% skim milk (Wako Pure Chemical Industries, Ltd., Osaka,
Japan) for 1 h at room temperature and reacted with appropriate
dilutions of primary antibody overnight at 4°C. The membrane was
washed with Tris-buffered saline-Tween (TBST) 6 times for 5 min
each, followed by incubation with HRP-linked, anti-rabbit
immunoglobulin G (GE Healthcare Biosciences, Little Chalfont, UK)
for 1 h at room temperature. The membrane was washed again with
TBST 6 times for 5 min each. Next, the secondary anti-bodies were
identified using detection reagent Clarity Western ECL substrate
(Bio-Rad Laboratories). Images were acquired with ImageQuant LAS
4000 (GE Healthcare Biosciences). The following primary antibodies
were used in this study: METTL3 (Proteintech Group Inc., Chicago,
IL, USA; rabbit, #15073-1-AP, 1:1,000) and β-actin (Cell Signaling
Technology, Danvers, MA, USA; rabbit, #4967, 1:10,000).
Proliferation assay
Cells were seeded in 96-well plates with 1,500
cells/well containing 100 µl of DMEM. After incubation for
48, 72 and 96 h, 10 µl of 25% glutaraldehyde (Wako Pure
Chemical Industries) was added to fixed cells, stained with 100
µl of 0.05% crystal violet (Sigma-Aldrich) with 20%
methanol, solubilized the stain in 150 µl of 50% ethanol
supplemented with 0.05 M NaH2PO4, and light
absorbance of the solution was measured at 595 nm on the EnSpire
micro-plate reader (Perkin-Elmer, Inc., Waltham, MA, USA).
Sphere formation assay
We performed sphere formation assay using the
limiting dilution method. Cultured cells were collected in
serum-free DMEM/F-12 with GlutaMAX, bFGF, EGF, B-27 and
N2 supplements (Thermo Fisher Scientific). Cell numbers
were diluted to 10 cells/ml, then 100-µl aliquots were
dispensed into each well in 96-Well Clear Round Bottom Ultra Low
Attachment Microplate (Costar culture plate; Corning Costar,
Corning, NY, USA). Sphere diameters were measured after 15 days of
incubation.
Chemosensitivity assay
Cells were plated in 96-well plates at a density of
900 cells/90 µl/well. After incubating for 24 h, 10
µl of fluorouracil (5-FU), cisplatin (CDDP), gemcitabine
(GEM) and PBS control were added. The final concentrations were
5-FU at 0, 10, 20, 40, 60, 80, 100 and 120 µM; CDDP at 0,
0.625, 1.25, 2.5, 5, 10, 20 and 50 µM; and GEM at 0, 8, 16,
32, 64 and 128 nM. After 3 to 5 days of incubation, crystal violet
staining was performed and absorbance at 595 nm was measured.
Radiosensitivity assay
Radiosensitivity assay was performed as previously
described (28). Cells were plated
into 6-cm dishes (N=4) at concentrations of 50, 100, 200, 400,
1,000, and 2,500 cells/dish and irradiated with 0, 2, 4, 6, 8 and
10 Gy, respectively, using Gammacell 40 Exactor (Nordion, Inc.,
Ottawa, ON, Canada) and incubated for 10 days. Cells were fixed
with 25% glutaraldehyde and stained with crystal violet. Colonies
with >50 cells were counted under the microscope.
Chemoradiosensitivity assay
Cells were seeded into four 96-well plates,
respectively, in the same way as the chemosensitivity assay. In 2
of 4 plates, 10 µl of drug was added to a final
concentration of 20 µM of CDDP or 16 nM of GEM after 24-h
incubation. Just after drug administration, a 4-Gy dose was
irradiated using the Gammacell 40 Exactor. After 5 days of
incubation, all plates were fixed and stained with crystal violet
and absorbance at 595 nm was measured.
Annexin V assays
GEM was added (final concentration 16 nM) after
4×104 cells were seeded in 10-cm dishes and incubated
for 24 h. PBS was used as a control. After incubation for 48 h,
apoptosis assay was performed using the Annexin V-FITC apoptosis
detection kit (Nacalai Tesque) in accordance with the instructional
protocols. Fluorescence intensity of Annexin V-FITC and PI were
measured after gating viable cells on FSC vs. SSC dot plots using
the BD FACSCanto II system (BD Biosciences, Franklin Lakes, NJ,
USA).
Human apoptosis proteome profiler
array
The relative expression levels of 35 human
apoptosis-related proteins were detected using the Proteome
Profiler Array Human Apoptosis Array kit (R&D Systems,
Minneapolis, MN, USA). Cells on a 10-cm dish were rinsed with PBS 3
times and solubilized with lysis buffer supplemented with 1% of
Halt Protease Inhibitor Cocktail (Thermo Fisher Scientific).
Experiments were performed in accordance with the manufacturer's
instructions.
cDNA expression study
TRIzol reagents were used to extract total RNA from
cultured cells, and RNA quality was checked (RNA concentration
>0.5 µg/µl and OD260/280, 1.8-2.0). These samples
were outsourced for DNA microarray analysis using 3D-Gene (Toray
Industries, Inc., Tokyo, Japan).
Data set comparison and Gene Ontology
enrichment analysis
Differentially expressed genes whose fold changes
were >2 or <1/2 in METTL3 KD cells underwent Gene Ontology
(GO) functional enrichment analysis using the Database for
Annotation, Visualization and Integrated Discovery (DAVID) v6.8
(29).
Protein-protein interaction network
construction
The functional interactions among differentially
expressed genes were conducted with the online Search Tool for the
Retrieval of Interacting Genes/Proteins (STRING, available at
https://string-db.org/) database (confidence
score ≥0.700) (30). Network edges
were visualized using a popular open source software Cytoscape
version 3.5.1 (available at http://www.cytoscape.org/) (31); subsequently, modules (highly
inter-connected regions) in a network were identified using the
Cytoscape plug-in MCODE (32),
version 1.4.2 (available at http://apps.cytoscape.org/apps/mcode), with parameters
of degree cut-off = 2, node score cut-off = 2, k-core = 2, and max
depth = 100. Next, genes in each module were analyzed with DAVID to
identify rich GO terms.
Statistical analysis
All data obtained were analyzed by the JMP Pro
12.2.0 software (SAS Institute, Inc., Cary, NC, USA) and described
as mean ± standard error (SE). Unless stated otherwise, Student's
t-test was used for statistical analysis for experimental results
and the differences were considered significant.
Results
shRNA-mediated KD of METTL3
We established METTL3 KD cell lines using shRNA.
Stable KD of METTL3 in RNA and protein expression was confirmed
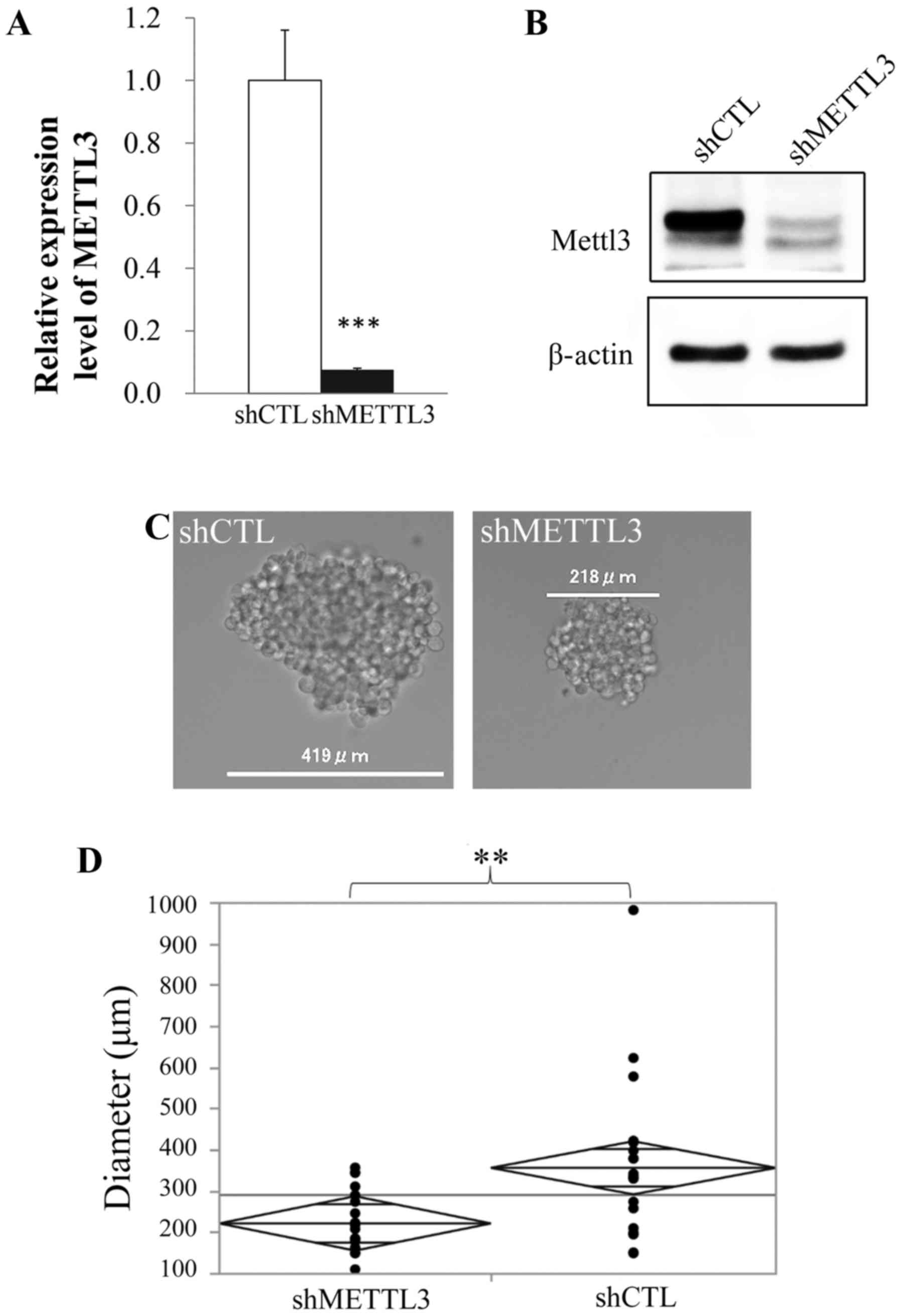
with qRT-PCR and western blot analysis, respectively (Fig. 1A and B). Although there was no
difference in morphology and proliferation rate between control and
METTL3 KD cells (data not shown), METTL3 KD cells in sphere
formation assay showed significantly lower ability than control
cells to form spheres (Fig. 1C and
D).
METTL3 depletion has crucial effects on
chemo- and radiosensitivity
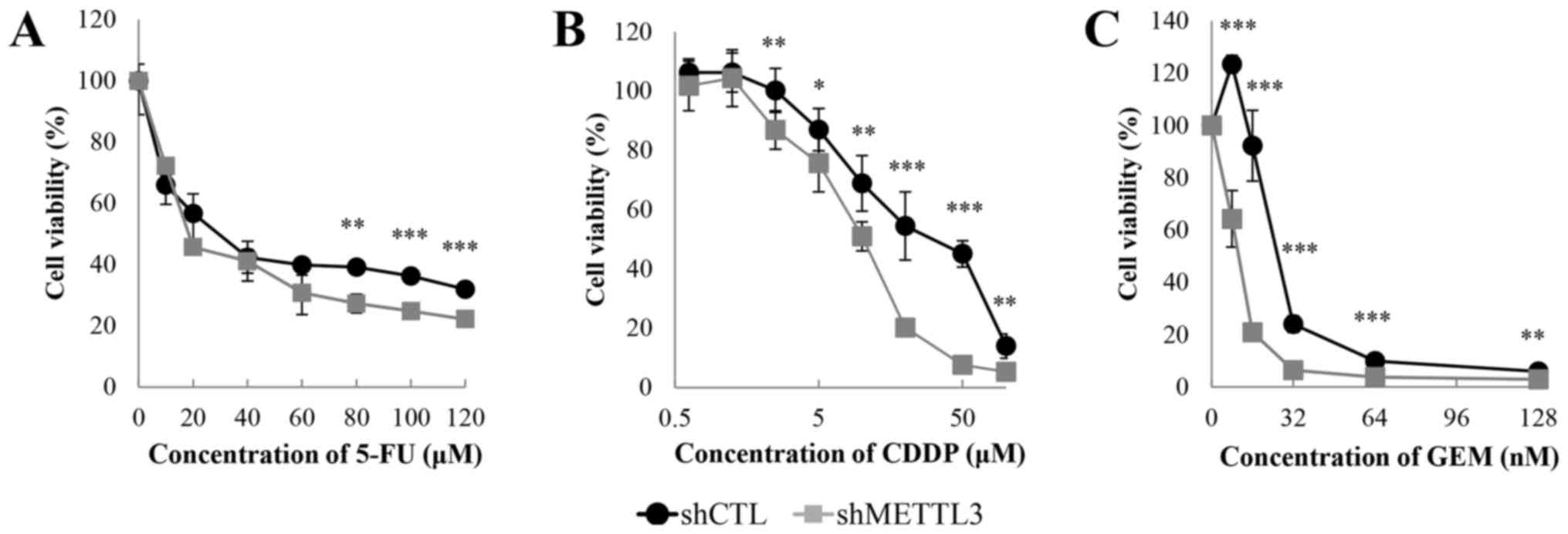
We performed crystal violet assay under
administration of 5-FU, CDDP and GEM to test whether METTL3 KD
alters sensitivity to anticancer agents. METTL3 depletion enhanced
the sensitivity to each drug (Fig.
2). The IC50 values of 5-FU, CDDP and GEM were
28.0/18.4 µM (shCTL/shMETTL3), 33.3/10.3 µM and
25.9/10.6 nM, respectively. There was an obvious change in the drug
sensitivity, especially in the presence of GEM. We also studied
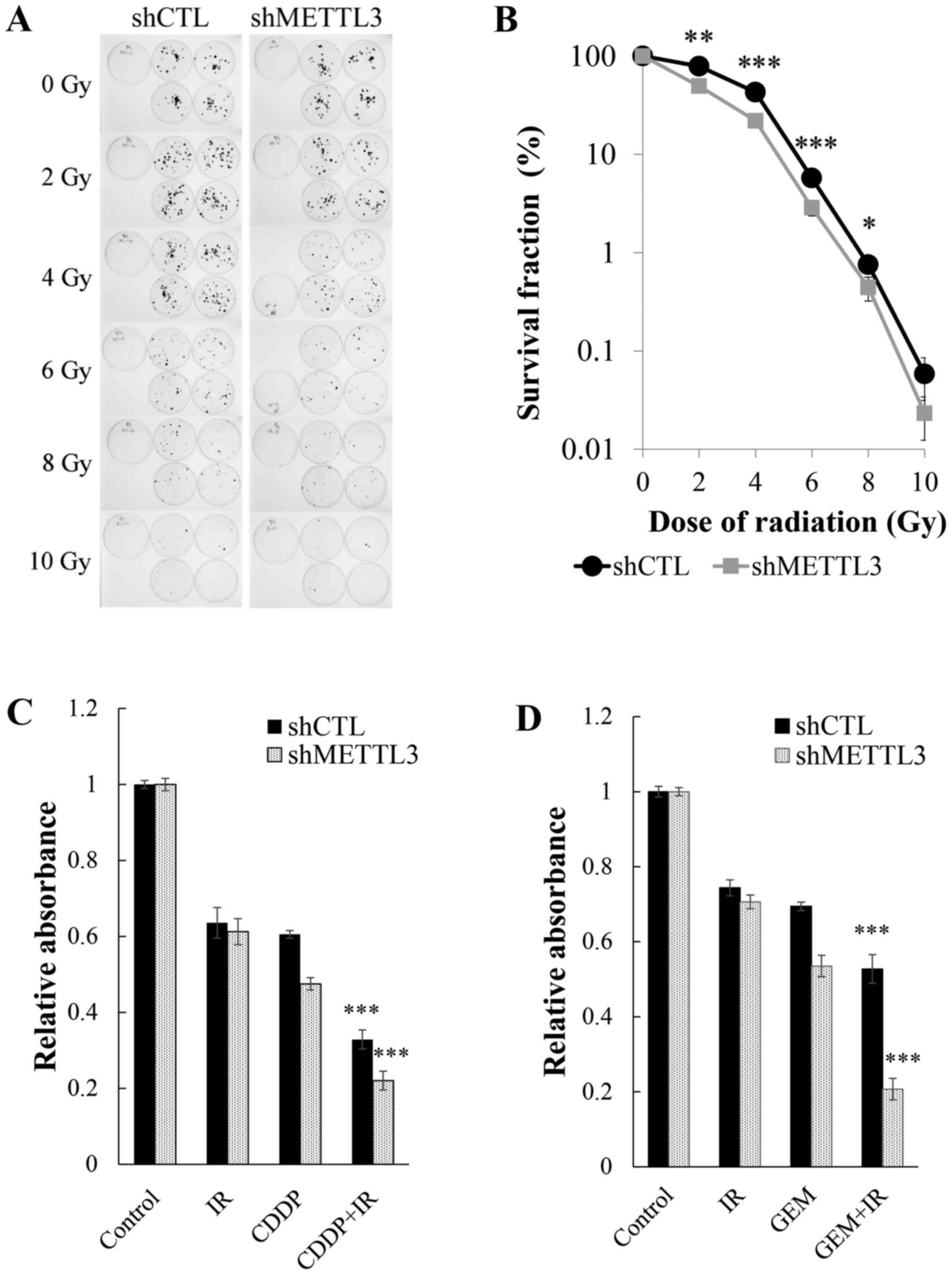
alterations of radiosensitivity derived from METTL3 KD. Clonogenic
assay (Fig. 3A) revealed that
METTL3 depletion resulted in enhancement of radiosensitivity with
2-8 Gy irradiation (Fig. 3B).
Although no significant difference was observed with 10 Gy
irradiation by statistical analysis, there was a strong tendency
for shMETTL3 cells to be susceptible to irradiation, suggesting
that METTL3 has important roles in the acquisition of resistance to
anticancer drugs and irradiation. We next examined how toxic
effects were enhanced in METTL3 KD cells when treated concurrently
with drugs and irradiation at a dose of 4 Gy. The multifactor ANOVA
showed a very significant effect of drug (GEM or CDDP) or radiation
(P<0.0001) and significant synergistic effects of interaction
(P≤0.001) in control and METTL3 KD cells (Fig. 3C and D).
Low expression of METTL3 enhances
apoptotic response by GEM
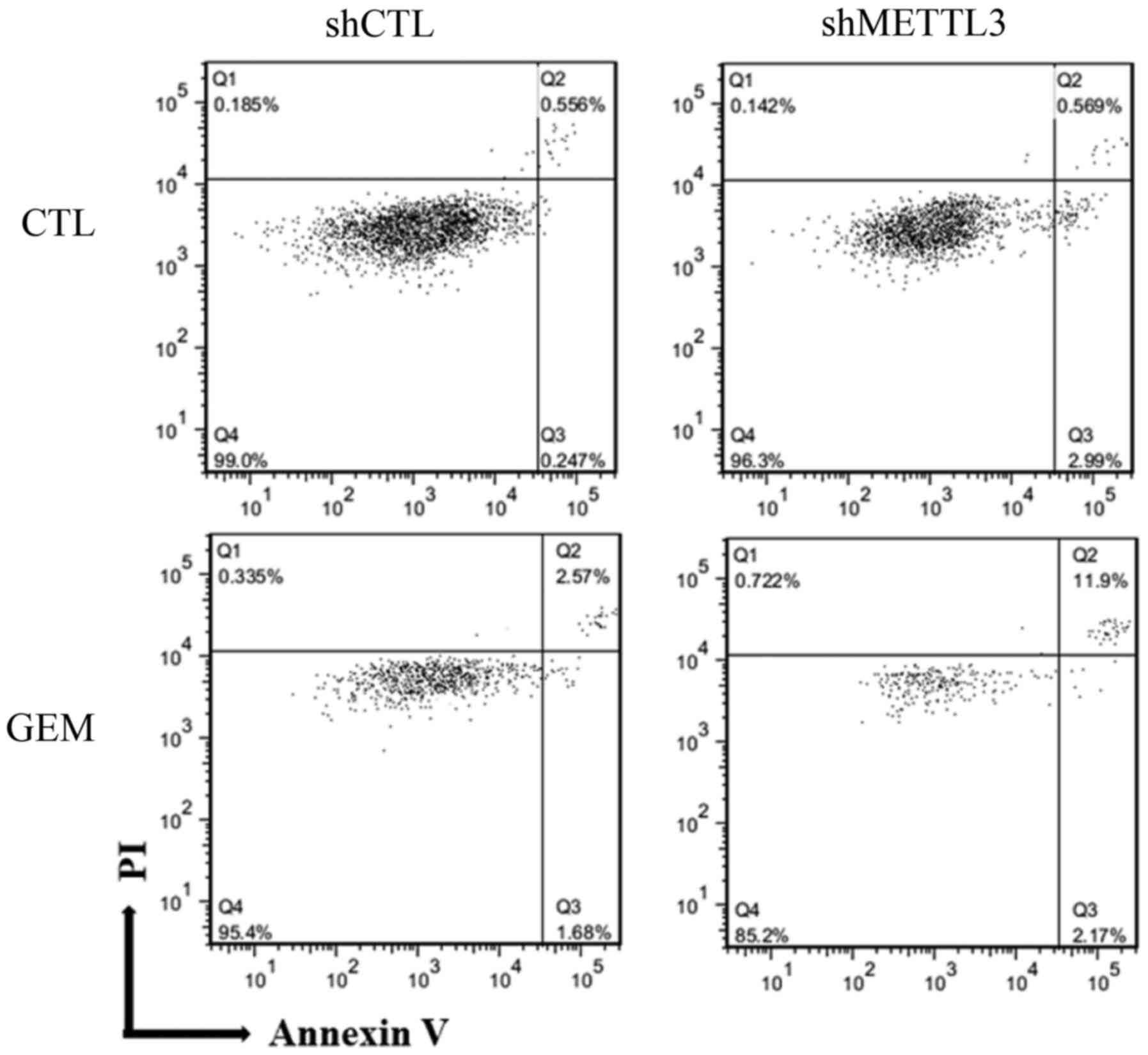
The aforementioned data suggested that METTL3 is
involved in apoptosis; therefore, Annexin V assay was conducted 48
h after treatment with GEM (Fig.
4). Compared with control cells, the percentage of late
apoptotic cells was increased from 2.6 to 12% in shMETTL3 cells
treated with GEM. Conversely, the percentage of live cells was
decreased from 95 to 85% in shMETTL3 cells treated with GEM,
suggesting that any apoptosis-related proteins were influenced by
METTL3 depletion. To confirm this, we screened 35 apoptosis-related
proteins using a human apoptosis proteome profiler array. No
significant alteration was confirmed between shCTL and shMETTL3
cells (data not shown), implying that unknown other METTL3 target
genes could be regulating apoptosis and
chemo-/radiosensitivities.
Microarray data and GO analysis reveals
potent target genes of METTL3
Given that METTL3 functions as m6A
writers (12,13), we are interested in the global
effect on networks of mRNA expression and protein-protein
interaction (PPI). A total of 735 Ensembl ID-coded differentially
expressed genes were identified, of which 104 were upregulated and
631 were downregulated. To clarify the functional and pathway
enrichment of differentially expressed genes, GO analysis was
applied using DAVID and the top 5 GO terms and Reactome pathways of
upregulated and downregulated differentially expressed genes were
selected according to P-value (Tables
I and II). The upregulated
genes were associated primarily with immune or interferon
reactions, indicating effects by shRNA transfection. Conversely,
the downregulated differentially expressed genes were associated
mainly with regulation of mitogen-activated protein kinase (MAPK)
cascades and cellular component organization. We next focused on
PPI networks of downregulated differentially expressed genes in
shMETTL3 cells. The PPI analysis by STRING provided 209 nodes
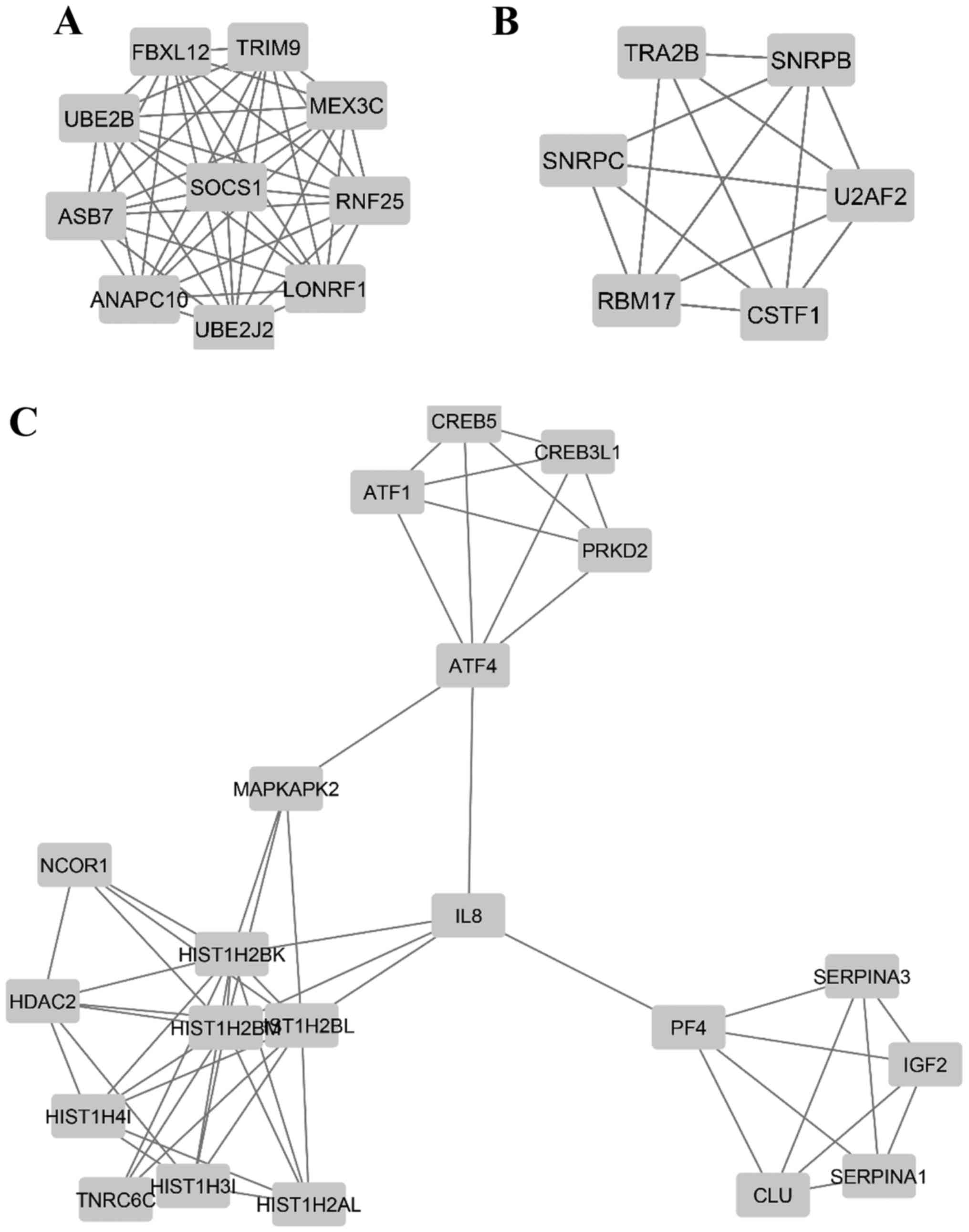
(genes) and 362 edges (interactions) in the network (Fig. 5), and additional MCODE analysis
identified 3 modules that were associated with a
ubiquitin-dependent process (including UBE2B, SOCS1, MEX3C, TRIM9,
RNF25, LONRF1 and UBE2J2), RNA splicing (including RBM17, U2AF2,
SNRPC, SNRPB and CSTF1), and regulation of cellular process
(including HDAC2, IGF2, MAPKAPK2 and CLU), respectively (Fig. 6 and Table III).
 | Table ITop five GO terms and Reactome
pathways of upregulated DEGs. |
Table I
Top five GO terms and Reactome
pathways of upregulated DEGs.
| Category | Term | Count | P-value | FDR |
|---|
| GOTERM_BP_FAT | GO:0071357~cellular
response to type I interferon | 6 | 2.80E-05 | 0.0488 |
| GOTERM_BP_FAT | GO:0060337~type I
interferon signaling pathway | 6 | 2.80E-05 | 0.0488 |
| GOTERM_BP_FAT | GO:0034340~response
to type I interferon | 6 | 3.62E-05 | 0.0630 |
| GOTERM_BP_FAT |
GO:0032020~ISG15-protein conjugation | 3 | 3.38E-04 | 0.5862 |
| GOTERM_BP_FAT | GO:0006955~immune
response | 18 | 9.54E-04 | 1.647 |
|
REACTOME_PATHWAY |
R-HSA-909733~Interferon α/β signaling | 6 | 3.41E-05 | 0.0417 |
|
REACTOME_PATHWAY |
R-HSA-168928~DDX58/IFIH1-mediated
induction of interferon-α/β | 3 | 3.99E-03 | 4.783 |
|
REACTOME_PATHWAY |
R-HSA-936440~Negative regulators of
DDX58/IFIH1 signaling | 3 | 1.55E-02 | 17.417 |
|
REACTOME_PATHWAY | R-HSA-1169408~ISG15
antiviral mechanism | 3 | 6.32E-02 | 55.089 |
|
REACTOME_PATHWAY |
R-HSA-1236977~Endosomal/vacuolar
pathway | 2 | 6.54E-02 | 56.377 |
| Table IITop five GO terms and Reactome
pathways of downregulated DEGs. |
Table II
Top five GO terms and Reactome
pathways of downregulated DEGs.
| Category | Term | Count | P-value | FDR |
|---|
| GOTERM_BP_FAT |
GO:0043408~regulation of MAPK cascade | 36 | 6.94.E-04 | 1.319 |
| GOTERM_BP_FAT |
GO:0051128~regulation of cellular
component organization | 93 | 9.23.E-04 | 1.749 |
| GOTERM_BP_FAT |
GO:0007010~cytoskeleton organization | 52 | 1.38.E-03 | 2.602 |
| GOTERM_BP_FAT |
GO:0000226~microtubule cytoskeleton
organization | 25 | 1.63.E-03 | 3.065 |
| GOTERM_BP_FAT | GO:0002821~positive
regulation of adaptive immune response | 9 | 2.01.E-03 | 3.769 |
|
REACTOME_PATHWAY |
R-HSA-2428928~IRS-related events triggered
by IGF1R | 3 | 1.21.E-02 | 16.127 |
|
REACTOME_PATHWAY |
R-HSA-112412~SOS-mediated signaling | 3 | 1.67.E-02 | 21.4931 |
|
REACTOME_PATHWAY |
R-HSA-450302~activated TAK1 mediates p38
MAPK activation | 4 | 2.31.E-02 | 28.560 |
|
REACTOME_PATHWAY |
R-HSA-171007~p38MAPK events | 3 | 5.51.E-02 | 55.772 |
|
REACTOME_PATHWAY |
R-HSA-418359~Reduction of cytosolic
Ca++ levels | 3 | 6.30.E-02 | 60.840 |
| Table IIITop three GO terms of modules in PPI
of downregulated DEGs. |
Table III
Top three GO terms of modules in PPI
of downregulated DEGs.
| Category | Term | Count | P-value | FDR |
|---|
| Hub nodes 1 | | | | |
| GOTERM_BP_FAT | GO:0016567~protein
ubiquitination | 9 | 1.74E-10 | 2.53E-07 |
| GOTERM_BP_FAT | GO:0032446~protein
modification by small protein conjugation | 9 | 5.40E-10 | 7.84E-07 |
| GOTERM_BP_FAT | GO:0070647~protein
modification by small protein conjugation or removal | 9 | 1.81E-09 | 2.62E-06 |
| Hub nodes 2 | | | | |
| GOTERM_BP_FAT | GO:0000377~RNA
splicing, via transesterification reactions with bulged adenosine
as nucleophile | 5 | 1.02E-07 | 1.20E-04 |
| GOTERM_BP_FAT | GO:0000398~mRNA
splicing, via spliceosome | 5 | 1.02E-07 | 1.20E-04 |
| GOTERM_BP_FAT Hub
nods 3 | GO:0000375~RNA
splicing, via transesterification reactions | 5 | 1.08E-07 | 1.26E-04 |
| GOTERM_BP_FAT | GO:0010557~positive
regulation of macromolecule biosynthetic process | 10 | 8.61E-07 | 1.38.E-03 |
| GOTERM_BP_FAT | GO:0031328~positive
regulation of cellular biosynthetic process | 10 | 1.70E-06 | 2.72.E-03 |
| GOTERM_BP_FAT | GO:0009891~positive
regulation of biosynthetic process | 10 | 1.97E-06 | 3.16.E-03 |
Discussion
In the present study, we demonstrated that METTL3
was a key protein in pancreatic cancer therapy and addressed
potential targets of METTL3. METTL3 KD MIA PaCa2 cells showed lower
self-renewal abilities in sphere formation assay (Fig. 1C and D) and enhanced apoptotic
reactions to GEM (Fig. 4),
although no significant changes were seen in morphology,
proliferation rate and apoptotic reaction without drug treatment
(Fig. 4). In a previous study,
shRNA-mediated METTL3 KD A549 cells showed a lower proliferation
rate and increased apoptosis (10). Another study showed high levels of
apoptosis in small interfering RNA (siRNA)-mediated METTL3 KD HepG2
(7). For undifferentiated cells,
shRNA-mediated METTL3 KD mouse embryonic stem cells (mESCs) led to
a morphological change of colonies and decreased proliferation rate
(17), whereas another study
reported enhanced proliferation rate in CRISPR/Cas9-mediated METTL3
knockout (KO) mESCs (11). These
different phenotypes could be explained by different roles of
METTL3 in cell types as well as which and how many RNAs are
affected by m6A.
While investigating whether METTL3 KD affected
chemo- or radiotherapy, we found that anticancer agents, 5-FU, CDDP
and GEM, as well as radiation treatment significantly suppressed
cell proliferation in METTL3 KD cells (Figs. 2 and 3B). Moreover, combination use of chemo-
and radiotherapy led to significant synergistic effects in both
cells (Fig. 3C and D). While
treatment of radiation alone in chemoradiosensitivity assay was not
significantly effective (Fig. 3C and
D), METTL3 KD cell showed elevated sensitivity to irradiation
in colony formation assay (Fig.
3B). These contrasting results could be explained by the
difference of endpoints in each assay. Irradiated cells containing
lethal DNA damage survived only a short time; however, they were
unable to form colonies for a few weeks. A recent study revealed
that m6A was induced rapidly at the sites of DNA damage
after ultraviolet irradiation (UVC), and METTL3 KO cells showed
delayed repair of UVC-induced DNA damage and higher sensitivity to
UVC compared to controls (26),
suggesting the importance of METTL3 in the recovery from
UVC-induced DNA damage. It is interesting to note that, in that
study, γ-irradiation did not induce m6A within 1 min.
Therefore, our results provided the possibility of another unknown
mechanism by which METTL3 regulates radiation-induced DNA damage.
Today, GEM- and 5-FU-based chemotherapy as well as concur-rent
chemoradiation therapy are key treatments in advanced pancreatic
cancer (33). Taken together, our
findings indicate that METTL3 is a potent target for pancreatic
cancer treatment.
From cDNA microarray data, several processes and
cascades emerged as potential targets of METTL3: MAPK cascades,
ubiquitin-dependent process, RNA splicing and regulation of the
cellular process (Fig. 6 and
Tables II and III). MAPK cascades are involved in
various essential cellular processes such as proliferation,
differentiation, stress response, DNA repair, apoptosis and
survival (34,35). Histone deacetylase 2 (HDAC2) plays
a crucial role in DNA double-strand break repair and cancer
malignancy (36,37). Insulin-like growth factor 2 (IGF-2)
and IGF signaling are important for cancer development and
progression (38). MAP
kinase-activated protein kinase 2 (MAPKAPK2), a member of the
Ser/Thr protein kinase family, is shown to participate in
inflammatory response, gene transcription, and cell cycle
regulation (39,40). Clusterin (CLU) is a key protein in
stress response and cell survival. Accumulating data revealed that
CLU is associated with resistance to chemotherapy and radiotherapy,
through various pathways including inhibition of BAX (bcl-2-like
protein 4), activation of ERK1/2 and phosphoinositide 3-kinase
(PI3K)-Akt signaling pathway (41,42).
Collectively, our finding raises the possibility that METTL3
modulates MAPK cascade and cellular processes, resulting in
resistance to chemo- and radiotherapy.
This study also showed that ubiquitin-dependent
process is a possible target of METTL3 (Fig. 6A and Table III). Ubiquitination requires 3
steps, activation, conjugation, and ligation, and is performed
using ubiquitin-activating enzymes (E1s), ubiquitin-conjugating
enzymes (E2s) and ubiquitin ligases (E3s), respectively. According
to recent reports, these proteins play an important role in cancer
malignancy. Ubiquitin-conjugating enzyme E2B (UBE2B, also called
Rad6B), which works as a DNA repair protein, is associated with
chemoresistance and cell stemness in ovarian cancer cells (43). Suppressor of cytokine signaling 1
(SOCS1) is generally known to provide a negative feedback of
cytokine signaling though the JAK/STAT3 pathway and suppress
insulin signaling by ubiquitin-mediated degradation of insulin
receptor substrate (IRS) (44).
Notably, SOCS1 is reported as an oncogene and a tumor suppressor in
cancer (45). Mex-3 RNA-binding
family member C (MEX-3C), a family of RNA-binding E3
ubiquitin-protein ligase, is suggested to be involved in mRNA decay
(46). Recently, MEX-3C was
suggested as a new chromosomal instability (CIN) suppressor in
CIN+ colorectal cancer (47), possibly contributing to chromosome
segregation errors and DNA damage. Taken together, our data
indicate that METTL3 alters ubiquitination-related protein
expression, leading to genomic instability and insufficient DNA
damage repair.
Furthermore, we found that the RNA splicing process
was a potent target process of METTL3 (Fig. 6B and Table III). Alternative pre-mRNA
splicing in which intronic sequences are removed stepwise is an
essential process in producing encoded proteins. Aberrations of
splicing by altered spliceosome proteins lead to gain-, loss- and
opposite-of-function mutations, contributing to alteration of tumor
characteristics (48,49). A recent report suggested that
splicing factor U2AF 65 kDa subunit (U2AF2) is required for
efficient DNA repair (50). In
addition, while its mechanism is still unknown, RNA-binding motif
protein 17 (RBM17, also called SPF45) induces resistance to various
anticancer drugs (51). Small
nuclear ribonucleoprotein-associated proteins B and B′ (SNRPB) are
suggested to be involved in RNA processing and DNA repair in
glioblastoma (52). Notably, SNRPB
is associated with crucial genes including RTK, PI3K, MAPK, RAS,
AKT, RB and p53, which are involved in essential cellular processes
(52). Collectively, our findings
indicate that METTL3 alters splicing regulator expression,
resulting in unexpected splicing events including insufficient DNA
repair.
We note several limitations to this study. First,
our in vitro experiments and results are based on a single
cell line; however, additional confirming studies are necessary in
other cell lines. Second, possible targets of METTL3 are derived
from microarray data mining. Currently, microarray technology is a
powerful tool to screen gene-expression profiling. However, actual
protein expression levels are not confirmed. In addition, genes
with a <2-fold change in expression are excluded from GO
analysis and PPI analysis, which may lead to inaccurate
conclusions.
In summary, the present study demonstrates that
METTL3 is associated with therapeutic resistance and is a potential
therapeutic target of pancreatic cancer. Additionally, our findings
suggest several critical pathways, including MAPK cascades,
ubiquitin-dependent process, RNA splicing and regulation of
cellular process, as possible targets of METTL3. Alteration of
these processes triggers various aberrant biological behaviors that
could lead to cancer progression and therapeutic resistance.
Further studies on the interactions between METTL3 and these
processes are critical for understanding the functional mechanisms
of METTL3.
Acknowledgments
The authors thank our laboratory members for
fruitful discussions, N. Nishida, K. Otani and K. Tamari for
helpful suggestions, and M. Ozaki and Y. Noguchi for excellent
technical support. Flow cytometric analysis was performed with
equipment in the Center for Medical Research and Education,
Graduate School of Medicine, Osaka University, Japan. This study
received financial support from grants-in-aid for Scientific
Research and P-DIRECT and P-CREATE Grants from the Ministry of
Education, Culture, Sports, Science, and Technology, MEXT (MK, YD,
MM, HI, and KO); Kobayashi Foundation for Cancer Research (HI);
Kobayashi International Scholarship Foundation (MK, HI); and a
grant-in-aid from the Ministry of Health, Labor, and Welfare (MK,
YD, MM, HI, and KO). Institutional endowments were received from
Taiho Pharmaceutical Co., Ltd. (Tokyo, Japan), Evidence Based
Medical Research Center INC. (Osaka, Japan), UNITECH Co., Ltd.
(Chiba, Japan), IDEA Consultants, Inc. (Tokyo, Japan), and
Kinshukai Medical Corporation (Osaka, Japan). These funders had no
role in the main experimental equipment, supply expenses, study
design, data collection and analysis, decision to publish, or
preparation of the present study.
References
|
1
|
Waddington CH: The epigenotype. Endeavour.
1:18–20. 1942.
|
|
2
|
Cantara WA, Crain PF, Rozenski J,
McCloskey JA, Harris KA, Zhang X, Vendeix FA, Fabris D and Agris
PF: The RNA Modification Database, RNAMDB: 2011 update. Nucleic
Acids Res. 39(Database): D195–D201. 2011. View Article : Google Scholar :
|
|
3
|
Ping XL, Sun BF, Wang L, Xiao W, Yang X,
Wang WJ, Adhikari S, Shi Y, Lv Y, Chen YS, et al: Mammalian WTAP is
a regulatory subunit of the RNA N6-methyladenosine
methyltrans-ferase. Cell Res. 24:177–189. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang
L, Jia G, Yu M, Lu Z, Deng X, et al: A METTL3-METTL14 complex
mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem
Biol. 10:93–95. 2014. View Article : Google Scholar :
|
|
5
|
Perry RP and Kelley DE: Existence of
methylated messenger RNA in mouse L cells. Cell. 1:37–42. 1974.
View Article : Google Scholar
|
|
6
|
Desrosiers R, Friderici K and Rottman F:
Identification of methylated nucleosides in messenger RNA from
Novikoff hepatoma cells. Proc Natl Acad Sci USA. 71:3971–3975.
1974. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dominissini D, Moshitch-Moshkovitz S,
Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K,
Jacob-Hirsch J, Amariglio N, Kupiec M, et al: Topology of the human
and mouse m6A RNA methylomes revealed by
m6A-seq. Nature. 485:201–206. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Meyer KD, Saletore Y, Zumbo P, Elemento O,
Mason CE and Jaffrey SR: Comprehensive analysis of mRNA methylation
reveals enrichment in 3′ UTRs and near stop codons. Cell.
149:1635–1646. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han
D, Fu Y, Parisien M, Dai Q, Jia G, et al:
N6-methyladenosine-dependent regulation of messenger RNA
stability. Nature. 505:117–120. 2014. View Article : Google Scholar
|
|
10
|
Lin S, Choe J, Du P, Triboulet R and
Gregory RI: The m6A Methyltransferase METTL3 promotes
translation in human cancer cells. Mol Cell. 62:335–345. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Batista PJ, Molinie B, Wang J, Qu K, Zhang
J, Li L, Bouley DM, Lujan E, Haddad B, Daneshvar K, et al:
m6A RNA modification controls cell fate transition in
mammalian embryonic stem cells. Cell Stem Cell. 15:707–719. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang
Y, Yi C, Lindahl T, Pan T, Yang YG, et al:
N6-methyladenosine in nuclear RNA is a major substrate
of the obesity-associated FTO. Nat Chem Biol. 7:885–887. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zheng G, Dahl JA, Niu Y, Fedorcsak P,
Huang CM, Li CJ, Vågbø CB, Shi Y, Wang WL, Song SH, et al: ALKBH5
is a mammalian RNA demethylase that impacts RNA metabolism and
mouse fertility. Mol Cell. 49:18–29. 2013. View Article : Google Scholar :
|
|
14
|
Liu N, Dai Q, Zheng G, He C, Parisien M
and Pan T: N6-methyladenosine-dependent RNA structural
switches regulate RNA-protein interactions. Nature. 518:560–564.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fustin JM, Doi M, Yamaguchi Y, Hida H,
Nishimura S, Yoshida M, Isagawa T, Morioka MS, Kakeya H, Manabe I,
et al: RNA-methylation-dependent RNA processing controls the speed
of the circadian clock. Cell. 155:793–806. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Du H, Zhao Y, He J, Zhang Y, Xi H, Liu M,
Ma J and Wu L: YTHDF2 destabilizes m6A-containing RNA
through direct recruitment of the CCR4-NOT deadenylase complex. Nat
Commun. 7:126262016. View Article : Google Scholar
|
|
17
|
Wang Y, Li Y, Toth JI, Petroski MD, Zhang
Z and Zhao JC: N6-methyladenosine modification
destabilizes developmental regulators in embryonic stem cells. Nat
Cell Biol. 16:191–198. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Meyer KD, Patil DP, Zhou J, Zinoviev A,
Skabkin MA, Elemento O, Pestova TV, Qian SB and Jaffrey SR: 5′ UTR
m6A promotes cap-independent translation. Cell.
163:999–1010. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhou J, Wan J, Gao X, Zhang X, Jaffrey SR
and Qian SB: Dynamic m6A mRNA methylation directs
translational control of heat shock response. Nature. 526:591–594.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xiao W, Adhikari S, Dahal U, Chen YS, Hao
YJ, Sun BF, Sun HY, Li A, Ping XL, Lai WY, et al: Nuclear
m6A reader YTHDC1 regulates mRNA splicing. Mol Cell.
61:507–519. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Alarcón CR, Lee H, Goodarzi H, Halberg N
and Tavazoie SF: N6-methyladenosine marks primary
microRNAs for processing. Nature. 519:482–485. 2015. View Article : Google Scholar
|
|
22
|
Alarcón CR, Goodarzi H, Lee H, Liu X,
Tavazoie S and Tavazoie SF: HNRNPA2B1 is a mediator of
m6A-dependent nuclear RNA processing events. Cell.
162:1299–1308. 2015. View Article : Google Scholar
|
|
23
|
Chen T, Hao YJ, Zhang Y, Li MM, Wang M,
Han W, Wu Y, Lv Y, Hao J, Wang L, et al: m6A RNA
methylation is regulated by microRNAs and promotes reprogramming to
pluripotency. Cell Stem Cell. 16:289–301. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Geula S, Moshitch-Moshkovitz S,
Dominissini D, Mansour AA, Kol N, Salmon-Divon M, Hershkovitz V,
Peer E, Mor N, Manor YS, et al: Stem cells m6A mRNA
methylation facilitates resolution of naïve pluripotency toward
differentiation. Science. 347:1002–1006. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Patil DP, Chen CK, Pickering BF, Chow A,
Jackson C, Guttman M and Jaffrey SR: m6A RNA methylation
promotes XIST-mediated transcriptional repression. Nature.
537:369–373. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xiang Y, Laurent B, Hsu CH, Nachtergaele
S, Lu Z, Sheng W, Xu C, Chen H, Ouyang J, Wang S, et al: RNA
m6A methylation regulates the ultraviolet-induced DNA
damage response. Nature. 543:573–576. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang SY, Zhang SW, Liu L, Meng J and
Huang Y: m6A-Driver: Identifying context-specific mRNA
m6A methylation-driven gene interaction networks. PLOS
Comput Biol. 12:e10052872016. View Article : Google Scholar
|
|
28
|
Franken NA, Rodermond HM, Stap J, Haveman
J and van Bree C: Clonogenic assay of cells in vitro. Nat Protoc.
1:2315–2319. 2006. View Article : Google Scholar
|
|
29
|
Huang W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar
|
|
30
|
Szklarczyk D, Morris JH, Cook H, Kuhn M,
Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, et al:
The STRING database in 2017: Quality-controlled protein-protein
association networks, made broadly accessible. Nucleic Acids Res.
45(D1): D362–D368. 2017. View Article : Google Scholar
|
|
31
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bader GD and Hogue CW: An automated method
for finding molecular complexes in large protein interaction
networks. BMC Bioinformatics. 4:22003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tempero MA, Malafa MP, Al-Hawary M, Asbun
H, Bain A, Behrman SW, Benson AB III, Binder E, Cardin DB, Cha C,
et al: Pancreatic adenocarcinoma, version 2.2017, NCCN Clinical
Practice Guidelines in Oncology. J Natl Compr Cancer Netw.
15:1028–1061. 2017. View Article : Google Scholar
|
|
34
|
Dhillon AS, Hagan S, Rath O and Kolch W:
MAP kinase signalling pathways in cancer. Oncogene. 26:3279–3290.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Corre I, Paris F and Huot J: The p38
pathway, a major pleiotropic cascade that transduces stress and
metastatic signals in endothelial cells. Oncotarget. 8:55684–55714.
2017.PubMed/NCBI
|
|
36
|
Gong F and Miller KM: Mammalian DNA
repair: HATs and HDACs make their mark through histone acetylation.
Mutat Res. 750:23–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shan W, Jiang Y, Yu H, Huang Q, Liu L, Guo
X, Li L, Mi Q, Zhang K and Yang Z: HDAC2 overexpression correlates
with aggressive clinicopathological features and DNA-damage
response pathway of breast cancer. Am J Cancer Res. 7:1213–1226.
2017.PubMed/NCBI
|
|
38
|
Livingstone C: IGF2 and cancer. Endocr
Relat Cancer. 20:R321–R339. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cargnello M and Roux PP: Activation and
function of the MAPKs and their substrates, the MAPK-activated
protein kinases. Microbiol Mol Biol Rev. 75:50–83. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Moens U, Kostenko S and Sveinbjørnsson B:
The role of mitogen-activated protein kinase-activated protein
kinases (MAPKAPKs) in inflammation. Genes (Basel). 4:101–133. 2013.
View Article : Google Scholar
|
|
41
|
Koltai T: Clusterin: A key player in
cancer chemoresistance and its inhibition. Onco Targets Ther.
7:447–456. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Albany C and Hahn NM: Heat shock and other
apoptosis-related proteins as therapeutic targets in prostate
cancer. Asian J Androl. 16:359–363. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Somasagara RR, Spencer SM, Tripathi K,
Clark DW, Mani C, Madeira da Silva L, Scalici J, Kothayer H,
Westwell AD, Rocconi RP, et al: RAD6 promotes DNA repair and stem
cell signaling in ovarian cancer and is a promising therapeutic
target to prevent and treat acquired chemoresistance. Oncogene. Aug
14–2017.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Rui L, Yuan M, Frantz D, Shoelson S and
White MF: SOCS-1 and SOCS-3 block insulin signaling by
ubiquitin-mediated degradation of IRS1 and IRS2. J Biol Chem.
277:42394–42398. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Beaurivage C, Champagne A, Tobelaim WS,
Pomerleau V, Menendez A and Saucier C: SOCS1 in cancer: An oncogene
and a tumor suppressor. Cytokine. 82:87–94. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cano F, Rapiteanu R, Sebastiaan Winkler G
and Lehner PJ: A non-proteolytic role for ubiquitin in
deadenylation of MHC-I mRNA by the RNA-binding E3-ligase MEX-3C.
Nat Commun. 6:86702015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Burrell RA, McClelland SE, Endesfelder D,
Groth P, Weller MC, Shaikh N, Domingo E, Kanu N, Dewhurst SM,
Gronroos E, et al: Replication stress links structural and
numerical cancer chromosomal instability. Nature. 494:492–496.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Eblen ST: Regulation of chemoresistance
via alternative messenger RNA splicing. Biochem Pharmacol.
83:1063–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhang J and Manley JL: Misregulation of
pre-mRNA alternative splicing in cancer. Cancer Discov.
3:1228–1237. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Savage KI, Gorski JJ, Barros EM, Irwin GW,
Manti L, Powell AJ, Pellagatti A, Lukashchuk N, McCance DJ,
McCluggage WG, et al: Identification of a BRCA1-mRNA splicing
complex required for efficient DNA repair and maintenance of
genomic stability. Mol Cell. 54:445–459. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Perry WL III, Shepard RL, Sampath J, Yaden
B, Chin WW, Iversen PW, Jin S, Lesoon A, O'Brien KA, Peek VL, et
al: Human splicing factor SPF45 (RBM17) confers broad multidrug
resistance to anticancer drugs when overexpressed - a phenotype
partially reversed by selective estrogen receptor modulators.
Cancer Res. 65:6593–6600. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Correa BR, de Araujo PR, Qiao M, Burns SC,
Chen C, Schlegel R, Agarwal S, Galante PA and Penalva LO:
Functional genomics analyses of RNA-binding proteins reveal the
splicing regulator SNRPB as an oncogenic candidate in glioblastoma.
Genome Biol. 17:1252016. View Article : Google Scholar : PubMed/NCBI
|