Introduction
Three signaling receptors are considered as
important markers for the grouping of human breast cancer types and
the subsequent planning of systemic therapy for breast cancer:
Estrogen receptor (ER), progesterone receptor (PR) and human
epidermal growth factor receptor 2 (HER2) (1). Triple-negative breast cancer (TNBC),
which comprises 15–20% of breast cancers (2–6), is
characterized by the immunohistochemical absence of both ER and PR,
as well as a lack of the amplification of HER2. Due to the absence
of these proteins, the treatment of TNBC with agents targeting
these proteins is inadequate. Although conventional adjuvant
chemotherapies employed, such as taxanes, anthracyclines,
capecitabines and/or platins show a desirable response with a
higher rate of pathological complete response (pCR) in initial
treatment (7); drug resistance to
these agents and distant metastasis commonly occur and are
associated with an overall poor prognosis (8–10).
The human epidermal growth factor receptor (HER)
family consists of 4 structurally related receptor tyrosine
kinases: HER1 [epidermal growth factor receptor (EGFR)], HER2,
HER3, and HER4 (11–13). Since EGFR is a well-established
oncogenic factor and therapeutic target in certain types of human
cancer, a number of humanized monoclonal antibodies and small
molecule kinase inhibitors have been approved by the US Food and
Drug Administration (FDA) for the treatment of several human
cancers (12,13). Unlike other types of hormone
therapy-sensitive breast cancer, the overexpression of EGFR is more
frequently found in TNBC (14–17)
and is closely associated with the aggressiveness and drug
resistance of malignant cancers, including breast cancer (11,13).
Up to 70–80% of metastatic breast cancers have been shown to
overexpress EGFR but not to overexpress HER2, the basis for
HER2-targeted therapy (18,19).
The activation of EGFR transmits signals of cell proliferation,
cell survival, drug resistance and metastasis via various signaling
cascades, including RAS-mitogen-activated protein kinase (MAPK),
phosphoinositide 3-kinase (PI3K)-AKT and SRC-signal transducer and
activator of transcription (STAT) pathways (20). Although numerous EGFR target drugs,
including protein kinase inhibitors (PKIs) and monoclonal
antibodies, are easily available, the majority of studies on the
therapeutic potential of EGFR targeted therapy have focused on lung
cancer, glioblastoma, colon cancer and head and neck cancers (21
and refs therein). In addition, despite the frequent overexpression
of EGFR in TNBC, the study of its potential role or therapeutic
benefits in TNBC remains still limited and is still under
evaluation (11,21,22).
Previously, we identified a potential therapeutic
benefit of blocking EGFR and PI3K with a gefitinib/PI-103
combination treatment in basal-like (BL) subtypes of TNBC in
vitro (23). However, a
mesenchymal stem-like (MSL) subtype of TNBC cells is relatively
resistant to this combination (23), probably due to the presence of
alternative intracellular signaling pathways that ensure cell
survival and proliferation even when EGFR is blocked (24–29).
We have also reported that the additional blocking of MET coupled
with EGFR inhibition effectively exerts anticancer effects in TNBC
cells of the MSL subtype with a decrease in p-/total RPS6 levels
(30). RPS6 is one of the
downstream target of mTOR kinase signaling. In addition, the
PI3K-AKT-mTOR signaling pathway, which is frequently deregulated in
TNBC, is indispensable for cancer cell survival and is associated
with drug resistance (31). In
this study, to identify PKIs that induce synthetic lethality in
combination with an EGFR inhibitor, we performed an MTT screening
in MDA-MB-231 cells with PKIs, including target agents against
PI3K, AKT, mTOR and SRC pathways in combination with gefitinib. As
a result, we identified and further characterized an AKT inhibitor,
MK-2206, as a synthetic lethal agent when used in combination with
gefitinib in TNBC cells of the MSL subtype and demonstrated that
the antitumor effect was associated with the downregulation of the
mammalian target of rapamycin complex 1
(mTORC1)/regulatory-associated protein of mTOR (RPTOR) signaling
coupled with a downregulation in the levels of ribosomal protein S6
(RPS6).
Materials and methods
Cell culture and reagents
The MDA-MB-231, HS578T and MDA-MB-468 cell lines
were purchased from the American Type Culture Collection (ATCC;
Manassas, VA, USA) and maintained in Dulbecco's modified Eagle's
medium (DMEM; HyClone, Logan, UT, USA) containing 10%
heat-inactivated fetal bovine serum (FBS; Corning, Manassas, VA,
USA) and 100 units/ml penicillin/streptomycin (Gibco, Grand Island,
NY, USA). Cell growth was monitored by trypan blue dye
(Sigma-Aldrich St. Louis, MO, USA) exclusion cell counting using
the Luna Automated Cell Counter (Logos Biosystems, Gyeonggi-do,
Korea). The PKIs used in these experiments were purchased from the
following sources: AZD0530, deforolimus, GDC-0941, GSK1059615,
IC-87114, KU-0063794, MK-2206, Perifosine, PIK-90, PIK-75, PI-103
TG100-115, TGX221 and WYE-354 were all obtained from SelleckChem
(Houston, TX, USA); (−)-Deguelin was purchased from Enzo Life
Sciences, Inc. (Farmingdale, NY, USA); BEZ235, dasatinib,
everolimus, FK506, pimecrolimus, staurosporine, temsirolimus and
ZSTK474 were all obtained from LC Laboratories (Woburn, MA, USA);
and LY294002, rapamycin and wortmannin were obtained from
Sigma-Aldrich. Each compound was dissolved in dimethyl sulfoxide
(DMSO) (Sigma-Aldrich) to create a stock solution and stored at
−20°C in small aliquots.
Screening of PKIs
Synthetic lethal screening was performed as
previously described (30). In
brief, the MDA-MB-231 cells, plated at 1,000 cells/well in 96-well
plates 24 h prior to testing, were treated with increasing
concentrations of gefitinib and PKIs in duplicate in a 6×5 matrix.
Cell viability was determined at 72 h after treatment by
3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay with 4 mg/ml MTT solution as previously described (25,33).
The synergism was determined by calculating the classification
index (CI) as previously described (30,32)
as follows: (viability of gefitinib) × (viability of
PKI)/(viability of the gefitinib and PKI combination).
Supra-additivity was defined as CI >1; additivity was defined as
CI=1; and subadditivity was defined as CI <1 (32,33).
The numbers of combination points with CI values >1.3 that were
generated from individual drug concentration points were summated
and further assigned as a quantitative index of synergism. The
average of CIs >1.3 was used as the qualitative index of
synergism.
Clonogenic cell survival assay
The cells were subcultured in 6-well plates at
appropriate densities: 1,000 cells/well for the HS578T cells and
2,000 cells/well for the MDA-MB-231 and MDA-MB-468 cells. At 1 day
after subculture, the cells were treated with 9 µM
gefitinib, 9 µM MK-2206, or a combination of these drugs for
24 h, and then washed and supplemented with fresh normal growth
media in the absence of the drugs. The cells were cultured for an
additional 10–14 days following treatment with replacement of
normal growth media twice per week. The colonies were stained as
previously described (30,34). After washing with distilled water
(DW), the colonies were imaged with a scanner. The relative amount
and number of colonies were quantified as follows: The crystal
violet stain for the staining of the colonies was dissolved in a
solubilizing buffer [1:1 mixture (v/v) of 0.1 M sodium phosphate
buffer (pH 4.5) and ethanol], and the absorbance of the dissolved
crystal violet was measured by using a 680 micro-plate reader
(Bio-Rad, Hercules, CA, USA).
Western blot analysis and antibodies
For PKI treatment, the HS578T and MDA-MB231 cells
were plated at 2×105/60-mm dish at 1 day prior to
treatment. The cells were then treated with gefitinib and MK-2206
for 24 or 2 h in the presence of normal serum-containing media. For
siRNA transfection, the HS578T and MDA-MB231 cells were plated at
5×104 cells/60-mm dish the day prior to transfection.
Following treatment with the drug or siRNA transfection, the cells
were lysed with Pierce™ RIPA buffer (Thermo Fisher Scientific,
Waltham, MA, USA) containing protease phosphatase inhibitor
cocktail (Thermo Fisher Scientific), and the protein concentration
was determined with a BCA protein assay kit (Thermo Fisher
Scientific). The following antibodies were used in this study: mTOR
(#4517), phosphorylated (p-)mTOR (S2448; #2971), extracellular
signal-regulated kinase (ERK)1/2 (#9102), p-ERK1/2 (T202/Y204;
#4370), SRC (#2108), p-SRC (Y416; #2101), eukaryotic translation
initiation factor 4E-binding protein 1 (4E-BP1; #9452), p-4E-BP1
(T37/46; #2855), RPS6 (#2217), p-RPS6 (#4856), proline-rich AKT
substrate of 40 kDa (PRAS40; #2691), p-PRAS40 (S235/236; #13175),
AKT (#9272), p-AKT (S473; #4060), glycogen synthase kinase (GSK)-3β
(#9315), p-GSK-3β (S9); #9315), RPTOR (#2280),
rapamycin-insensitive companion of mTOR (RICTOR) (#2140),
poly(ADP-ribose) polymerase-1 (PARP1; #9542), cleaved PARP (Asp214;
#5625), cleaved caspase-3 (Asp175; #9661) and peroxidase-conjugated
secondary antibodies (anti-rabbit IgG, #7074; anti-mouse IgG,
#7076) were all obtained from Cell Signaling Technology (Danvers,
MA, USA); X-linked inhibitor of apoptosis protein antibody (XIAP;
#610716) was from BD Sciences (San Jose, CA, USA); β-actin antibody
(A330-491A) was from Bethyl Laboratories, Inc. (Montgomery, TX,
USA); and β-tubulin antibody (T7816) was from Sigma-Aldrich.
Transfection of siRNA
For the siRNA-mediated knockdown of target gene
expression, AccuTarget™ premade siRNA was purchased from Bioneer
(Seoul, Korea) for the following target genes and control siRNA:
Human RPTOR (gene ID: 57521; #1079283), human RICTOR (gene ID:
253260; #1129356) and control siRNA (SN-1003). The transfection of
siRNA was carried out using Lipofectamine 2000 (Invitrogen,
Waltham, MA, USA) as previously described (30). In brief, the HS578T
(1×105 cells/well) or MDA-MB-231 (5×104
cells/well) cells in 6-well plates were transfected with 100 pmoles
of siRNA mixed and pre-incubated with Lipofectamine 2000 in
serum-free DMEM media. Following 4 h of incubation, the
transfection media was supplemented with an equal volume of DMEM
containing 20% FBS and 200 units/ml penicillin/streptomycin to
restore the normal growth condition, and the cells were further
incubated for 3 days. During the incubation, 9 µM gefitinib
or DMSO (vehicle) were added 1 day after the media supplement or
the last 2 h of the 3rd day of the incubation period for the cell
proliferation assay and western blot analyses.
Cell counting-based cell viability assay
and cell proliferation assay
The HS578T and MDA-MB231 cells were plated at
1×105 cells in a 60-mm dish at 1 day prior to treatment.
At the indicated time of siRNA transfection or drug treatment (0,
1, 2, 3 and 4 days), cells were detached with trypsin/EDTA (Gibco)
and suspended with 0.5 ml of DMEM containing 10% FBS. Cell
proliferation or number of viable cells were determined by the
trypan blue exclusion method using the Luna Automated Cell Counter
(Logos Biosystems) immediately after staining with trypan blue dye
(Sigma-Aldrich) to avoid cell death caused by prolonged incubation
(34).
Cell cycle analysis
The MDA-MB231 cells were plated at 1×105
cells in a 60-mm dish at 1 day prior to treatment. Following
treatment with the drugs, both attached and floating cells were
harvested and fixed with 70% ethanol at −20°C for >4 h. The
nuclei were then stained with propidium iodide (Sigma-Aldrich). The
chromosomal DNA content was measured using a FACSCalibur flow
cytometer (BD Sciences, Franklin Lakes, NJ, USA) according to the
manufacturer's instructions. The data were analyzed using CellQuest
Pro (BD Sciences) and the ModFit LT program (Verity Software House,
Topsham, ME, USA).
Statistical analysis
Triplicate experiments were repeated 3 times. Most
of the data are presented as the means ± standard deviation (SD).
One-way analysis of variance (ANOVA) with a post hoc Tukey's honest
significant difference (HSD) test was used to analyze the
significance of the differences between groups. Conventionally,
values of P<0.05 and P<0.01 were considered to indicate
statistically significant and highly statistically significant
differences, respectively.
Results
Identification of a set of small molecule
PKIs with synergistic effects in combination with gefitinib
In our previous studies, the combination of an EGFR
inhibitor (EGFRi) and PI3K/AKT inhibitor or MET inhibitors was
found to decrease the viability and proliferation of TNBC cells of
the BL subtype or MSL subtype, respectively (23,30).
In this study, to further identify PKIs that are effective in the
presence of an EGFR inhibitor in TNBC cells of the MSL subtype, a
set of known PKIs (Table I) was
screened with gefitinib in the MDA-MB231 cell line, and inhibitors
of the PI3K/AKT and mTOR signaling pathways were identified
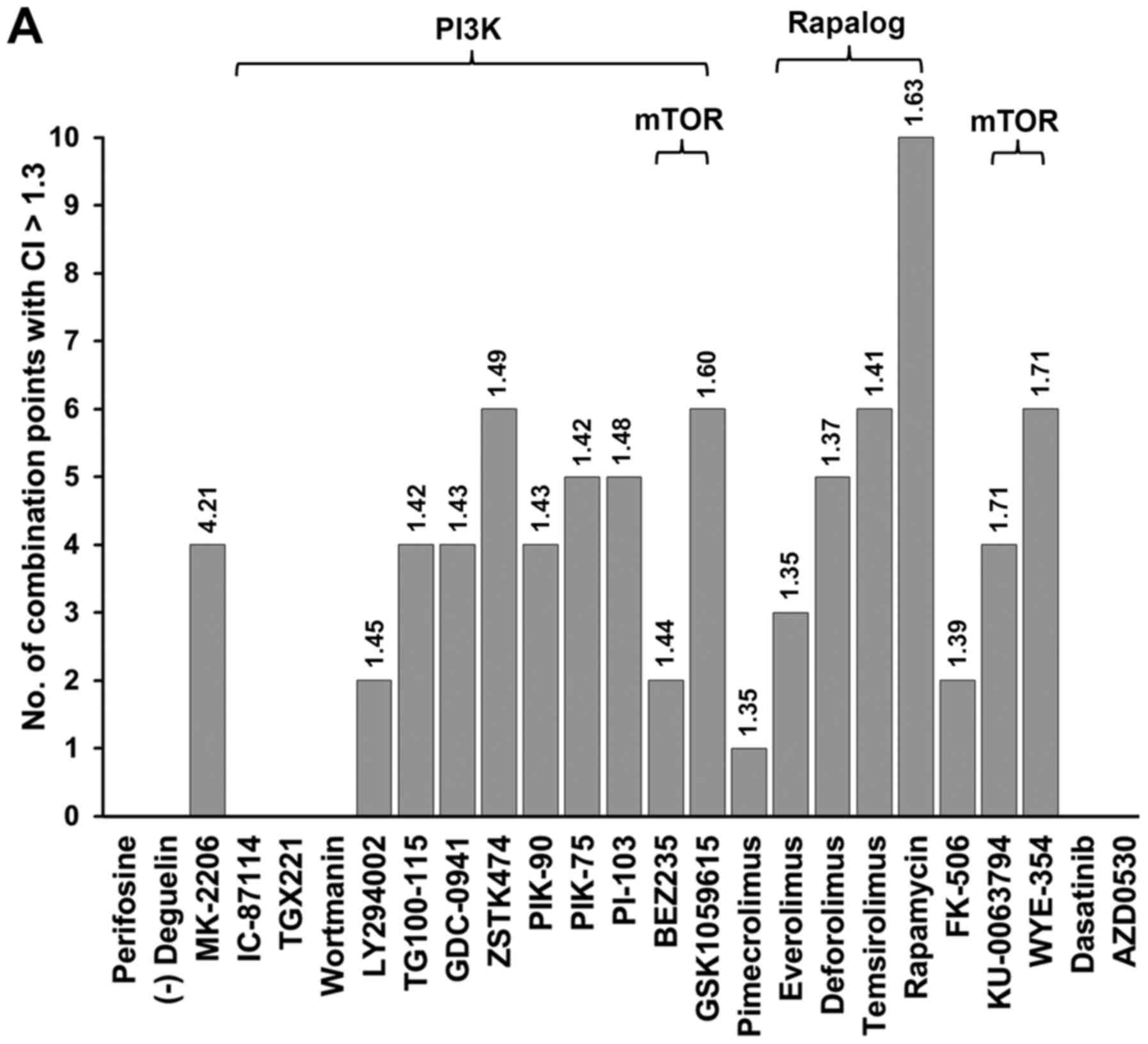
(Fig. 1A). For example, GSK1059615
(a PI3K/mTOR inhibitor), MK-2206 (an AKT inhibitor) and rapamycin
(an mTOR inhibitor) exerted synergistic effects with gefitinib at
various concentrations (Fig. 1A and
B). Although MK-2206/gefitinib treatment exhibited fewer points
of synergy than that of treatment with rapamycin, it exerted the
highest average CI, reflecting strong synergy. The majority of the
mTOR pathway inhibitors exerted a synergistic effect at 4–10 drug
combination points (Fig. 1A); the
overall lethal effects of rapamycin or other mTOR inhibitors in
combination with gefitinib were not as potent as those of MK-2206
in terms of the average CI value (Fig.
1).
 | Table IList of protein kinase inhibitors
used in this study. |
Table I
List of protein kinase inhibitors
used in this study.
| Inhibitor | Other name | Known targets
[IC50 value (nM)] | (Refs.) |
|---|
| AZD0530 | Saracatinib | SRC (2.7), LCK
(<4), YES (4), EGFR (L861Q) (4), LYN (5), EGFR (L858R) (5), FYN
(10), FGR (10), BLK (11), ABL (30), EGFR (66), KIT (200) | (60) |
| BEZ235 | NVP-BEZ235 | PI3Kα (4/2), PI3Kβ
(75), PI3Kδ (7), PI3Kγ (5), mTOR (20.7/2), DNAPK (5), ATM (7), ATR
(21) | (61,62) |
| Dasatinib | BMS354825,
Sprycel | BCR-ABL (0.8), SRC
(0.5), LCK (0.4), YES (0.5), BLK (8), KIT (5), PDGFRβ (28), p38
(100), EGFR (180), EPHB2 (4.4), TXK (<0.3) | (63,64) |
| Deforolimus | Ridaforolimus
AP23573 MK-8669 | mTOR (0.2) | (65) |
| (−) Deguelin | | AKT signaling,
Hsp90 | (66) |
| Everolimus | SDZ-RAD, Certican,
RAD001 | mTOR (0.63) | (67) |
| FK-506 | Tacrolimus,
Fujimycin, Prograf | FKBP12 | (68) |
| GDC-0941 | | PI3Kα (3), PI3Kβ
(33), PI3Kδ (3), PI3Kγ (75), PI3KC2β (670), mTOR (580) | (69) |
| GSK1059615 | | PK3Kα (0.4), PI3Kβ
(0.6), PI3Kγ (5), PI3Kδ (2), mTOR (12) | (70) |
| IC-87114 | D-030 | PI3Kδ (130) | (71) |
| KU-0063794 | | mTOR1 (10), mTOR2
(10) | (72) |
| LY294002 | | PI3K (1400), PI3Kα
(500) PI3Kδ (570), PI3Kβ (970) | (73) |
| MK-2206 | | AKT1 (5), AKT2
(12), AKT3 (65) | (74) |
| Perifosine | KRX-0401 | AKT translocation
inhibitor | (75) |
| PI-103 | | DNAPK (2), PI3Kα
(8), PI3Kβ (88), PI3Kδ (48), PI3Kγ (150), mTORC1 (20), mTORC2
(83) | (71) |
| PIK-75 | | DNAPK (2), PI3Kα
(5.8), PI3Kγ (76) | (71) |
| PIK-90 | | PI3KC2a (47),
PI3KC2b (64), DNAPK (13), PI3Kα (11), PI3Kβ (350), PI3Kδ (58),
PI3Kγ (18) | (71) |
| Pimecrolimus | Elidel,
SDZ-ASM-981 | FKBP12 | (76) |
| Rapamycin | RAPA,
Sirolimus | mTOR (0.1) | (77) |
| Temsirolimus | CCI779,
Torisel | mTOR (1.76) | (78) |
| TG100–115 | | PI3Kγ (83), PI3Kσ
(235) | (79) |
| TGX221 | | PI3Kβ (10), PI3Kδ
(65) | (79) |
| Wortmannin | KY12420 | PI3K (0.3), AKT1
(35), AKT2 (30), AKT3 (60), PKCζ (24), p38α (6), p38β (2) | (80) |
| WYE-354 | | mTOR (5) | (81) |
| ZSTK474 | | PI3Kα, PI3Kβ (17),
PI3Kγ (53), PI3Kδ (6) | (82) |
Gefitinib/MK-2206 combination decreases
the viability and proliferation of TNBC cells
Among these PKIs, an allosteric AKT inhibitor,
MK-2206, was further determined to exert potent synergistic
anti-proliferative effects in 2 cell lines of the MSL subtype
(MDA-MB-231 and HS578T cells) and 1 cell line of the BL subtype
(MDA-MB-468 cells). As was expected, the MDA-MB-468 cells exhibited
greater sensitivity following combination treatment with
synergistic effect, as well as following treatment with gefitinib
or MK-2206 alone (Fig. 2A). Potent
synergistic effects were observed in the 2 cell lines of the MSL
subtype. In the MDA-MB-231 cells, gefitinib and MK-2206 alone had
little or no effect on cell viability; however, the combination of
these inhibitors exerted marked synergistic effects (Fig. 2A). The effects of treatment with
the gefitinib/MK-2206 combination were further assessed using a
long-term colony formation assay. Cells plated in 6-well plates
were treated as indicated for 24 h and further cultivated in normal
growth media for 14 days. All the TNBC cells tested were minimally
affected by treatment with gefitinib or MK-2206 alone. However, the
long-term survivals were significantly reduced by the combination
treatment (Fig. 2B). The effects
of the gefitinib/MK-2206 combination on cell proliferation were
also assessed by viable cell counting over time. As shown in
Fig. 2C, the gefitinib/MK-2206
combination markedly suppressed the proliferation of the HS578T and
MDA-MB-231 cells.
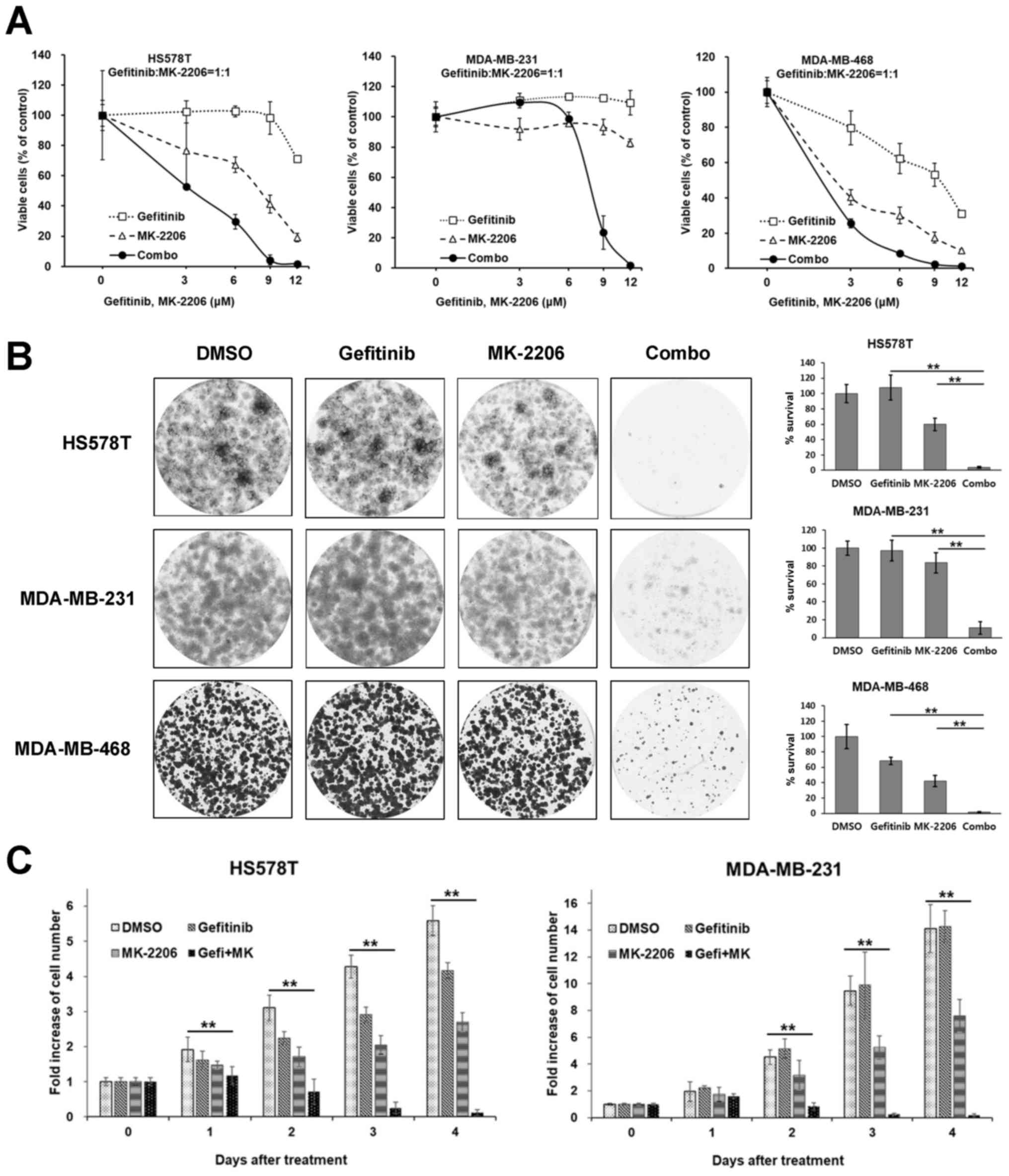 | Figure 2The gefitinib/MK-2206 combination
decreases the viability and proliferation of TNBC cells. (A) Cells
were treated with increasing concentrations of either gefitinib,
MK-2206 or a 1:1 molar ratio combination (Combo) of these drugs for
72 h, and the ratio of viable cells was measured by MTT assay. Data
are presented as the means ± SD from 3 independent experiments
performed in triplicate. (B) Cells were treated with 9 µM
gefitinib, 9 µM MK-2206 or a combination of these drugs for
24 h and further cultivated up to 10–14 days in normal growth
media. The colonies were stained with crystal violet as described
in the Materials and methods. Left panels, representative images
from 3 independent experiments performed in triplicate are shown;
right panels, the relative amounts of survived colonies were
determined as described in the Materials and methods.
**P<0.01, as obtained by the post hoc Tukey HSD test
after ANOVA. (C) The HS578T and MDA-MB-231 cells were treated with
9 µM gefitinib, 9 µM MK-2206, or a combination of
these drugs for the indicated number of days, and the number of
viable cells was determined by counting viable cells with trypan
blue dye staining as described in the Materials and methods.
Integrated data are presented as the means ± SD from 3 independent
experiments performed in triplicate. **P<0.01, as
obtained by the post hoc Tukey HSD test after ANOVA. |
The gefitinib/MK-2206 combination reduces
the protein level of RPS6 in TNBC cells of the MSL subtype
To determine the intracellular signaling pathway
responsible for the effects of the gefitinib/MK-2206 combination, a
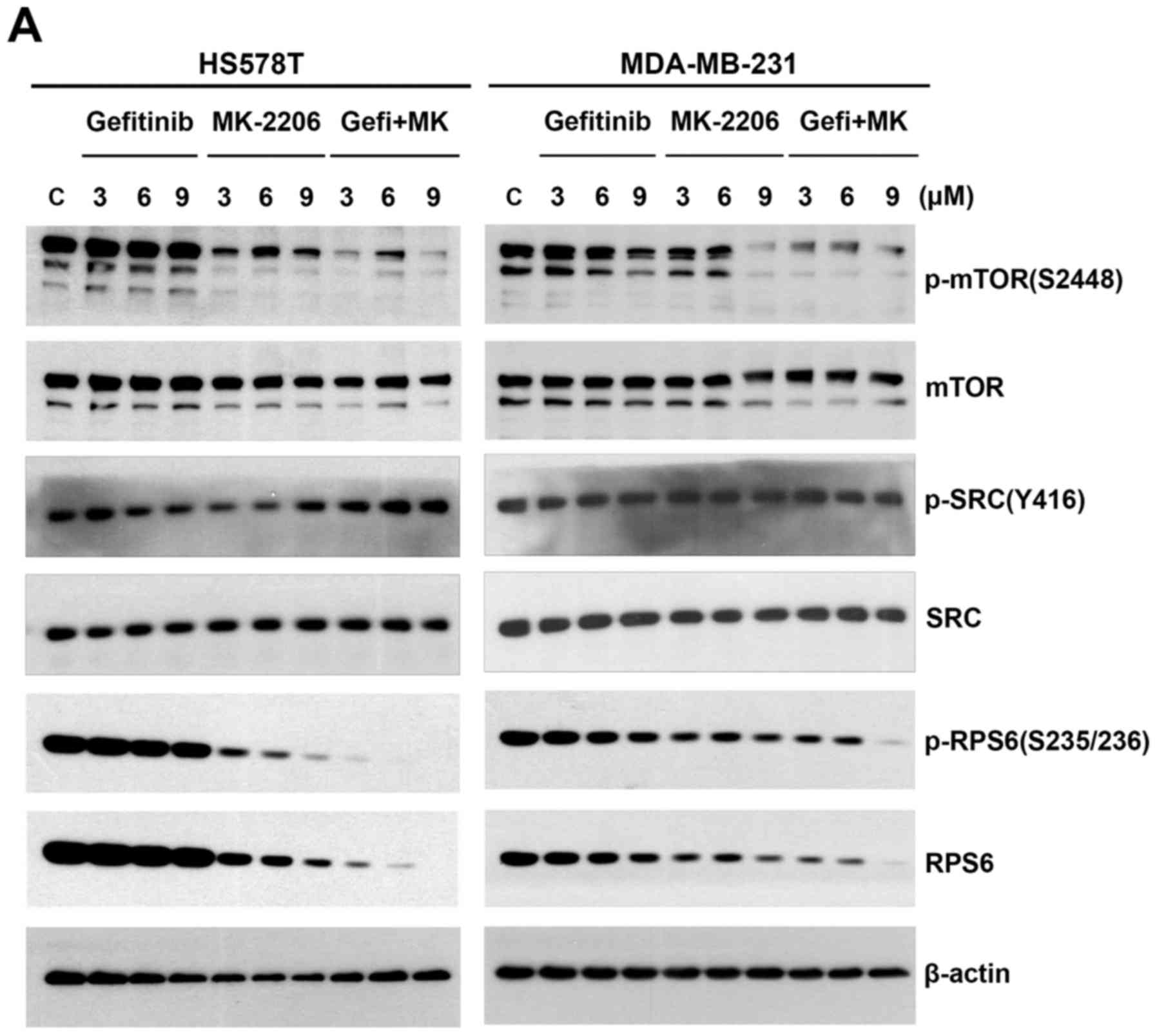
series of western blot analyses were carried out. First, the cells
were treated with increasing concentrations of gefitinib and
MK-2206 in an equimolar ratio for 24 h, and the levels of p-mTOR
(S2448), p-SRC (Y416) and p-RPS6 (S235/236) were examined. Of note,
both the phosphorylated and total RPS6 levels were decreased in a
concentration-dependent manner both in the cells treated with
MK-2206 alone and in those that were treated with the
gefitinib/MK-2206 combination (Fig.
3A). However, the decrease in the phosphorylated and total RPS6
levels was more prominent with the combination treatment.
Previously, we found a similar synergistic decrease in the RPS6
levels following combination treatment with gefitinib and SU11274,
a MET inhibitor, in the same TNBC cell line (30). As the overexpression of EGFR in
breast cancer is often accompanied by the co-overexpression of SRC,
providing synergistic tumorigenic effects between EGFR and SRC
(35), TNBC cells of the MSL type
exhibit greater sensitivity to dasatinib, an SRC and ABL kinase
inhibitor (5,36). Therefore, in this study, we also
examined the changes in SRC expression following treatment with
gefitinib/MK-2206. The level of p-SRC (Y416) (37) was not affected under these
conditions (Fig. 3A). However, the
level of p-mTOR (S2448) (38) was
more markedly decreased by the gefitinib/MK-2206 combination than
with treatment with either agent alone (Fig. 3A).
The effect of the gefitinib/MK-2206 combination was
further analyzed with a fixed concentration of both PKIs. Unlike
p-ERK1/2 (T202/Y204), which exhibited no apparent change in the
MDA-MB-231 cells and even increased in the HS578T cells, the
expression of p-AKT (S473) and p-PRAS40 (T246) (39), a downstream substrate of AKT,
disappeared in the cells treated with MK-2206 and the
gefitinib/MK-2206 combination (Fig.
3B). In addition, the level of p-mTOR was decreased by the
combination treatment. The phosphorylation of 4E-BP1, which is
known to be mediated by mTORC1 (39,40),
at the threonine 37 and 46 residues, was apparently diminished by
gefitinib/MK-2206 combination treatment.
To identify the immediate intracellular signaling
changes that occur prior to the onset of autonomous feedback
regulation, we further assessed the immediate-early phosphorylation
status of the AKT and mTOR signaling molecules. As shown in
Fig. 3C, the level of p-ERK1/2
(T202/Y204) was not affected, even in the HS578T and MDA-MB-231
cells treated with the gefitinib/MK-2206 combination, suggesting
that ERK1/2 may be uncoupled from EGFR in these cells. By contrast,
p-AKT (S473) expression disappeared both in the cells treated with
MK-2206 alone and in those treated with the gefitinib/MK-2206
combination (Fig. 3C).
Accordingly, the phosphorylation of AKT substrates such as PRAS40
(T246) and GSK-3β (S9) was reduced in MK-2206-treated cells and
further decreased in gefitinib/MK-2206 combination-treated cells
within 2 h. However, the phosphorylation status of mTOR (S2448)
differed between the HS578T and MDA-MB-231 cells. The level of
p-mTOR was synergistically decreased in the HS578T cells, whereas
the change in p-mTOR expression in the MDA-MB-231 cells was not as
evident. Under these conditions, the level of p-4E-BP1 (T37) was
synergistically decreased by the gefitinib/MK-2206 combination
treatment. Of note, the levels of phosphorylated and total RPS6
were decreased as early as 2 h after treatment in both cell lines,
and the gefitinib/MK-2206 combination further decreased the levels
of phosphorylated/total RPS6 in both the HS578T and MDA-MB231
cells. A similar early decrease in p-RPS6 and RPS6 expression
within 2 h was also previously observed with MET
inhibitor/gefitinib combination treatment in the same TNBC cell
lines (30).
The gefitinib/MK-2206 combination
decreases XIAP expression without affecting the levels of apoptotic
markers
As the gefitinib/MK-2206 combination effectively
inhibited cell proliferation and clonogenicity, we analyzed cell
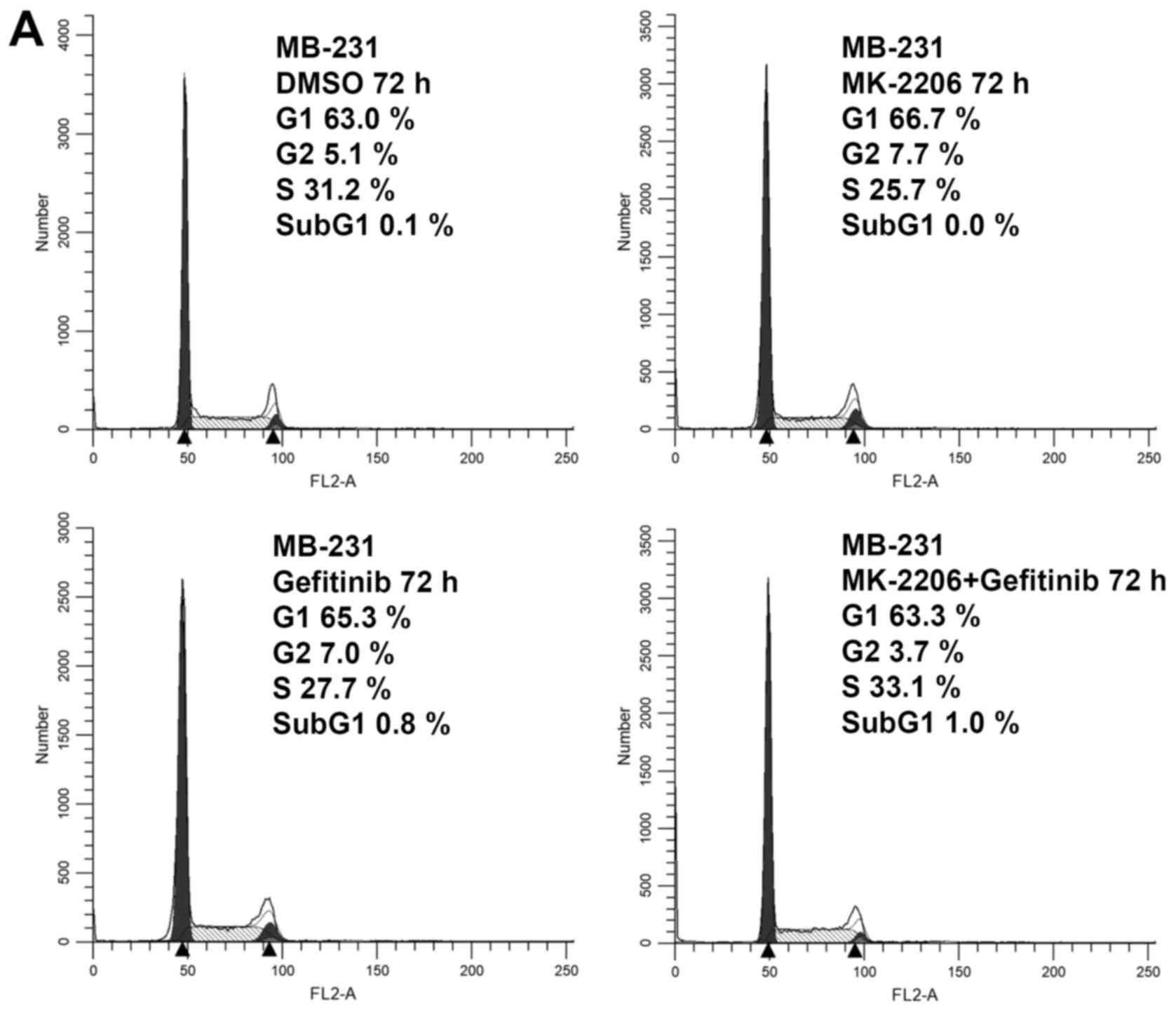
cycle progression following treatment with the drug. The MDA-MB-231
cells were treated as indicated for 72 h and were then subjected to
flow cytometric analysis. As shown in Fig. 4A, no apparent cell cycle arrest or
accumulation of apoptotic cells was observed in the MDA-MB-231
cells. We also used western blot analysis to examine PARP cleavage
and XIAP expression following treatment of the HS578T and
MDA-MB-231 cells with the drugs for 24 or 72 h. As shown in
Fig. 4B, the expression of
full-length PARP was decreased by treatment with the drugs in both
cell lines. However, the cleaved product of PARP was not observed.
The level of XIAP was decreased in the cells treated with the
gefitinib/MK-2206 combination for 72 h. Additional western blot
analyses were performed using antibodies for the cleaved forms of
caspase-3 and PARP, and staurosporine-treated samples were
simulatnously tested as positive controls. Staurosporine induced
the cleavage of both caspase-3 and PARP in the TNBC cells tested.
On the contrary, no evident cleavage of capase-3 and PARP was
observed in the TNBC cells treated with gefitinib, MK-2206 or their
combination (Fig. 4C).
Gefitinib with RPTOR knockdown decreases
the level of p-/total RPS6 and the viability of TNBC cells of the
MSL subtype
In addition to MK-2206, several mTOR pathway
inhibitors and a subset of PI3K/AKT pathway inhibitors consistently
exhibited synergism with gefitinib in the initial PKI screening
(Fig. 1). Based on these findings,
we speculated that the resistance to EGFR inhibition and the
effects of the gefitinib/MK-2206 combination in TNBC cells of the
MSL subtype are to be attributed to the mTOR pathway. mTOR is a
protein kinase in two distinct protein complexes, mTORC1 and
mTORC2. Therefore, to further elucidate which mTOR pathway
contributes to the resistance to EGFR inhibition, the mTORC1 and
mTORC2 pathways were selectively blocked by the siRNA-based
knockdown (KD) of RPTOR or RICTOR, respectively, in both the
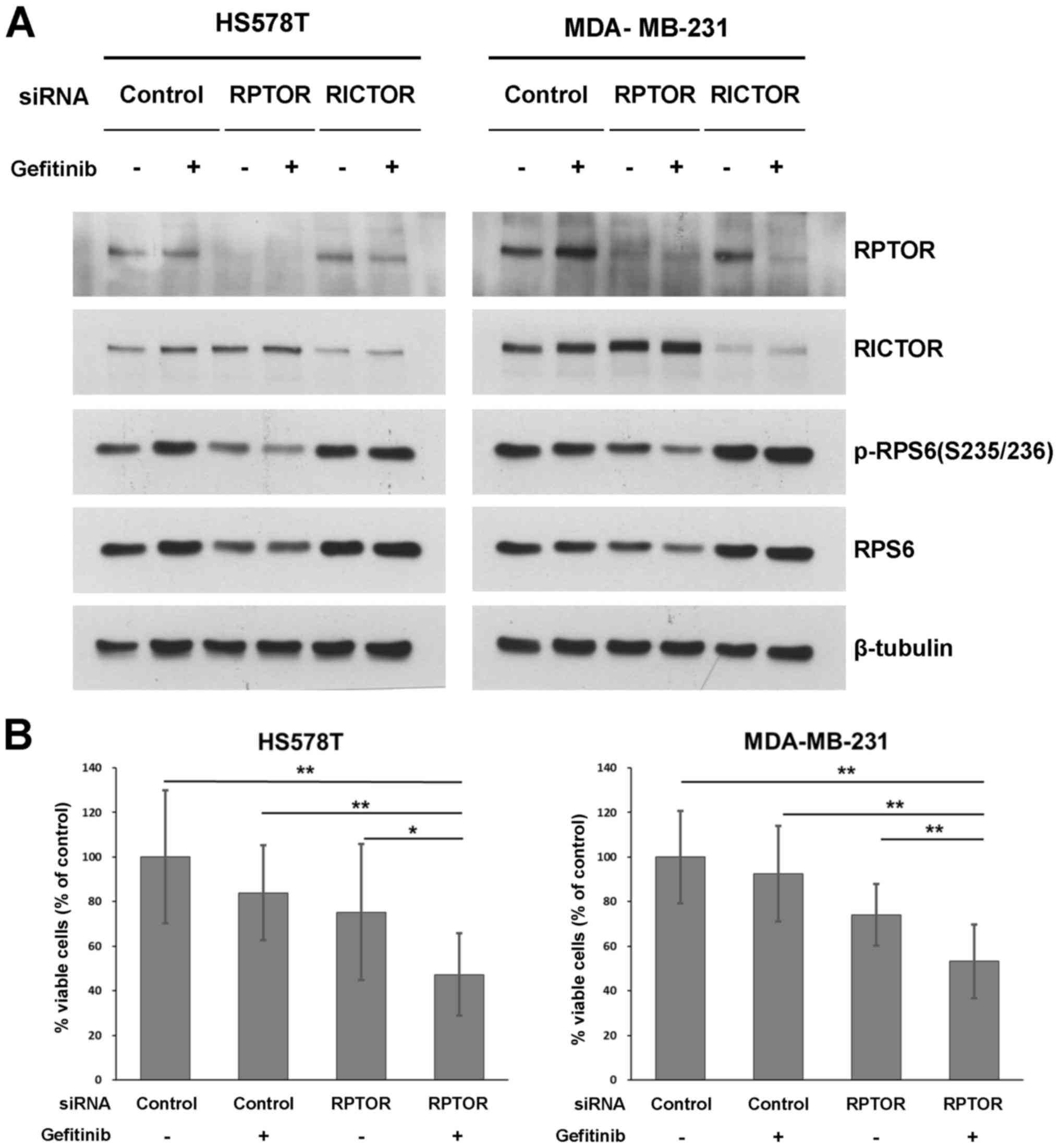
MDA-MB231 and HS578T cell lines. RPTOR KD decreased the level of
phosphorylated/total RPS6, and the effect was potentiated by the
addition of gefitinib in both cell lines (Fig. 5A). However, RICTOR KD with
gefitinib treatment did not affect the level of RPS6 protein. As
RPTOR KD potentiated the de-phosphorylation of RPS6, we examined
the effects of the gefitinib/RPTOR KD combination on cell
proliferation. As shown in Fig.
5B, the percentage cell viability was significantly decreased
to approximately 50% inhibited by the combination of RPTOR KD and
gefitinib treatment. To determine the synergistic inhibitory
effects on cell proliferation, the CI values (33) of each group after RPTOR KD and/or
gefitinib treatment were calculated. The CI values from 5
independent cell proliferation experiments indicated that this
combination was supra-additive, with mean CI values of 1.38±0.31
and 1.34±0.25 in the HS578T and MDA-MB-231 cells, respectively.
Discussion
Due to the lack of appropriate therapies and a poor
prognosis of patients with TNBC, the identification of a
'druggable' target is an unmet requirement for the development of
an effective TNBC therapy. As numerous types of cancer, including
breast cancer have limited driver oncogene activation or tumor
suppressor gene mutations (41),
proper control of the key surrogate/redundant pathway crosstalk
involved in EGFR resistance can provide appropriate therapeutic
strategies against the particular type of breast cancer with EGFR
activation/overexpression (22–24,26–28).
Among the TNBC subgroups classified according to Lehmann et
al, the BL and MSL subtype exhibits activated EGFR signaling
(5). In addition to the activation
of the epithelial-mesenchymal transition, the MSL-type TNBC
exhibits relatively greater resistance to EGFR inhibitors than the
BL subtype, regardless of the over-expression status of EGFR
(23,30).
In this study, we screened a series of known PKIs in
the presence of gefitinib in typical EGFR inhibitor-resistant TNBC
cells of the MSL subtype (MDA-MB-231 cells) and identified that
MK-2206, an AKT inhibitor, exerts a potent lethal effect with
gefitinib combination treatment. In addition, a group of inhibitors
against mTOR kinase, including rapamycin, temsirolimus, WYE-354 and
GSK1059615 also synergistically inhibited cell viability with
gefitinib at multiple points of combination than those of MK2206.
However, to investigate resistance to gefitinib in TNBC cells of
the MSL-subtype, the investigation of the effects of the
MK-2206/gefitinib combination, which exhibited potent lethal
synergistic effects (average CI value, 4.21; Fig. 1A), would be more feasible to
dissect the downstream signaling cascade to unveil the mechanism of
resistance to EGFR inhibitors. MK-2206 treatment alone, which
effectively blocked AKT activation and phosphorylation of its
downstream substrates, had minimal effect in TNBC cells, indicating
that EGFR signaling is indispensable for TNBC cell viability. In
addition, gefitinib alone had little effect on the TNBC cells of
the MSL subtype tested, whereas the gefitinib/MK-2206 combination
was synthetic lethal. The synergistic effects of the
gefitinib/MK-2206 combination were also demonstrated by a long-term
clonogenic assay following a 24-h treatment with this combination.
These results suggest that the AKT-mTOR pathway may contribute to
resistance to EGFR inhibitors in MSL TNBC cells.
The decreased phosphorylation of both mTOR and
4E-BP1 (Thr 37/46) suggested that the nodal signaling pathways of
the MK-2206/gefitinib combination converge on mTOR signaling. As a
downstream signal transducer of the PI3K/AKT pathway, mTORC1,
composed of mTOR, RPTOR, PRAS40, mammalian lethal with SEC13
protein 8 (mLST8) and DEP-domain-containing mTOR-interacting
protein (DEPTOR), mediates protein synthesis for cell proliferation
and cell cycle progression by promoting mRNA translocation as a
serine-threonine kinase (42,43).
A closely related, but structurally heterologous complex, mTORC2
[composed of mTOR, RICTOR, GβL, mammalian stress-activated protein
kinase interacting protein-1 (mSIN1), Protor 1/2 and DEPTOR]
mediates AKT activation and cytoskeletal actin remodeling (43,44).
mTOR pathway activation is more frequent in TNBC than non-TNBC and
is associated with a poor prognosis in early-stage TNBC (45,46).
In addition, the activated PI3K mutation and loss of PTEN function,
along with the recent identification of mTOR activation in TNBC,
rationalizes the use of mTOR-targeted therapy (31,47).
The safety and efficacy of an oral mTOR inhibitor, everolimus, was
previously evaluated in phase II clinical studies of patients with
metastatic or recurrent breast cancer (48). In a recent report of phase II
clinical trials, the use of everolimus in combination with
paclitaxel and cisplatin neoadjuvant therapy in TNBC patients
revealed more adverse effects without a clinically significant
improvement (49). Other
independent phase II clinical trials of everolimus combined with
conventional chemotherapeutics, such as paclitaxel with
anthracyclines also revealed no significant benefit in TNBC
patients (50). To date, several
phase I/II clinical trials targeting EGFR and mTOR pathways in an
advanced setting of breast cancers, including TNBC have been
carried out or are still undergoing: Erlotinib and everolimus
treatment in metastatic breast cancer (NCT00574366, completed);
lapatinib and everolimus treatment in TNBC (NCT01272141,
terminated); letrozole, lapatinib and everolimus treatment in
advanced endocrine resistant breast cancer (NCT01499160,
terminated); temsirolimus, cisplatin and erlotinib treatment in
TNBC (NCT00998036, completed); lapatinib and erlotinib treatment in
HER2-positive metastatic breast cancer (NCT01283789, active); and
everolimus, lapatinib and capecitabine treatment in HER2-positive
breast cancer with CNS metastasis (NCT01783756, active) (31,51).
While the effectiveness of the combination of EGFR inhibitors and
rapalogs with the appearance of apoptotic cells has been reported
in some TNBC cell lines, the mechanisms responsible for these
synergistic effects have not been reported (52). The phosphorylation of eukaryotic
translation initiation factor 4B (eIF4B) in TNBC is a possible
fragile point in EGFRi/rapalog synergy (53). As a component of mTORC1, RPTOR is a
stoichiometric adaptor molecule for mTOR kinase (54). In the present study, the
suppression of mTORC1 via siRNA-mediated RPTOR KD with gefitinib
decreased the level of both phosphorylated and total RPS6 protein
and inhibited cell viability. These results suggest that mTORC1
provides, at least partially, resistance to EGFR inhibitors in TNBC
cells of the MSL subtype, and targeting mTORC1 is a potential
alternative therapeutic strategy to overcome resistance to EGFR
inhibitors in a subset of TNBCs. Previously, we identified RPS6
dephosphorylation/degradation as a readout pathway of the
gefitinib/MET inhibitor combination treatment, and RPS6 KD
eventually reduced TNBC cell proliferation (30). A high level of phosphorylated RPS6
is closely associated with active mTOR and is a poor prognostic
marker of various cancers including non-small cell lung cancer
(NSCLC), hepatocellular carcinoma, sarcoma and pancreatic
neuroendocrine tumors (55–58).
Importantly, the inhibition of mTOR reduces the level of p-RPS6 in
renal carcinoma cells (59).
In this study, we demonstrated the necessity for
mTORC1-targeted therapy in EGFRi-resistant TNBC cells and that the
dephosphorylation/degradation of RPS6 is closely associated with
the mTORC1 signaling pathway along with the survival of a subgroup
of TNBC cells. Although we demonstrated an association between
resistance to gefitinib and mTORC1 activation, other possible
crosstalk mechanisms need to be explored to overcome resistance to
EGFR inhibitors and for the successful intervention and control of
TNBC. In addition, whether the regulation of RPS6 protein is under
the control of a cancer-specific growth signaling pathway remains
unclear. Further investigations are warranted in order to dissect
the RPS6 de-phosphorylation and/or the degradation pathway and to
identify the cancer-specific altered homeostasis of the RPS6
pathway to further develop therapeutic targets in malignant
cancers, including TNBCs.
Acknowledgments
This study was supported by a grant (NRF-2015R1D1A1A
01057893 to Y.-S.S.) funded by the National Research Foundation of
Korea.
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Mohamed A, Krajewski K, Cakar B and Ma CX:
Targeted therapy for breast cancer. Am J Pathol. 183:1096–1112.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Brenton JD, Carey LA, Ahmed AA and Caldas
C: Molecular classification and molecular forecasting of breast
cancer: Ready for clinical application? J Clin Oncol. 23:7350–7360.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Morris GJ, Naidu S, Topham AK, Guiles F,
Xu Y, McCue P, Schwartz GF, Park PK, Rosenberg AL, Brill K, et al:
Differences in breast carcinoma characteristics in newly diagnosed
African-American and Caucasian patients: A single-institution
compilation compared with the National Cancer Institute's
Surveillance, Epidemiology, and End Results database. Cancer.
110:876–884. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Podo F, Buydens LM, Degani H, Hilhorst R,
Klipp E, Gribbestad IS, Van Huffel S, van Laarhoven HW, Luts J,
Monleon D, et al FEMME Consortium: Triple-negative breast cancer:
Present challenges and new perspectives. Mol Oncol. 4:209–229.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lehmann BD, Bauer JA, Chen X, Sanders ME,
Chakravarthy AB, Shyr Y and Pietenpol JA: Identification of human
triple-negative breast cancer subtypes and preclinical models for
selection of targeted therapies. J Clin Invest. 121:2750–2767.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lehmann BD and Pietenpol JA:
Identification and use of biomarkers in treatment strategies for
triple-negative breast cancer subtypes. J Pathol. 232:142–150.
2014. View Article : Google Scholar :
|
|
7
|
Liedtke C, Mazouni C, Hess KR, André F,
Tordai A, Mejia JA, Symmans WF, Gonzalez-Angulo AM, Hennessy B,
Green M, et al: Response to neoadjuvant therapy and long-term
survival in patients with triple-negative breast cancer. J Clin
Oncol. 26:1275–1281. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kassam F, Enright K, Dent R, Dranitsaris
G, Myers J, Flynn C, Fralick M, Kumar R and Clemons M: Survival
outcomes for patients with metastatic triple-negative breast
cancer: Implications for clinical practice and trial design. Clin
Breast Cancer. 9:29–33. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Costa R, Shah AN, Santa-Maria CA, Cruz MR,
Mahalingam D, Carneiro BA, Chae YK, Cristofanilli M, Gradishar WJ
and Giles FJ: Targeting Epidermal Growth Factor Receptor in triple
negative breast cancer: New discoveries and practical insights for
drug development. Cancer Treat Rev. 53:111–119. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dent R, Trudeau M, Pritchard KI, Hanna WM,
Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P and Narod SA:
Triple-negative breast cancer: Clinical features and patterns of
recurrence. Clin Cancer Res. 13:4429–4434. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Eccles SA: The epidermal growth factor
receptor/Erb-B/HER family in normal and malignant breast biology.
Int J Dev Biol. 55:685–696. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wheeler DL, Dunn EF and Harari PM:
Understanding resistance to EGFR inhibitors-impact on future
treatment strategies. Nat Rev Clin Oncol. 7:493–507. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yarden Y and Pines G: The ERBB network: At
last, cancer therapy meets systems biology. Nat Rev Cancer.
12:553–563. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Reis-Filho JS and Tutt AN: Triple negative
tumours: A critical review. Histopathology. 52:108–118. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livasy CA, Karaca G, Nanda R, Tretiakova
MS, Olopade OI, Moore DT and Perou CM: Phenotypic evaluation of the
basal-like subtype of invasive breast carcinoma. Mod Pathol.
19:264–271. 2006. View Article : Google Scholar
|
|
16
|
Nielsen TO, Hsu FD, Jensen K, Cheang M,
Karaca G, Hu Z, Hernandez-Boussard T, Livasy C, Cowan D, Dressler
L, et al: Immunohistochemical and clinical characterization of the
basal-like subtype of invasive breast carcinoma. Clin Cancer Res.
10:5367–5374. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nakai K, Hung MC and Yamaguchi H: A
perspective on anti-EGFR therapies targeting triple-negative breast
cancer. Am J Cancer Res. 6:1609–1623. 2016.PubMed/NCBI
|
|
18
|
Reis-Filho JS, Milanezi F, Carvalho S,
Simpson PT, Steele D, Savage K, Lambros MB, Pereira EM, Nesland JM,
Lakhani SR, et al: Metaplastic breast carcinomas exhibit EGFR, but
not HER2, gene amplification and overexpression:
Immunohistochemical and chromogenic in situ hybridization analysis.
Breast Cancer Res. 7:R1028–R1035. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Reis-Filho JS, Pinheiro C, Lambros MB,
Milanezi F, Carvalho S, Savage K, Simpson PT, Jones C, Swift S,
Mackay A, et al: EGFR amplification and lack of activating
mutations in metaplastic breast carcinomas. J Pathol. 209:445–453.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yarden Y and Sliwkowski MX: Untangling the
ErbB signalling network. Nat Rev Mol Cell Biol. 2:127–137. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Burness ML, Grushko TA and Olopade OI:
Epidermal growth factor receptor in triple-negative and basal-like
breast cancer: Promising clinical target or only a marker? Cancer
J. 16:23–32. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Alvarez RH, Valero V and Hortobagyi GN:
Emerging targeted therapies for breast cancer. J Clin Oncol.
28:3366–3379. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yi YW, Hong W, Kang HJ, Kim HJ, Zhao W,
Wang A, Seong YS and Bae I: Inhibition of the PI3K/AKT pathway
potentiates cytotoxicity of EGFR kinase inhibitors in
triple-negative breast cancer cells. J Cell Mol Med. 17:648–656.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jin Q and Esteva FJ: Cross-talk between
the ErbB/HER family and the type I insulin-like growth factor
receptor signaling pathway in breast cancer. J Mammary Gland Biol
Neoplasia. 13:485–498. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Karamouzis MV, Konstantinopoulos PA and
Papavassiliou AG: Targeting MET as a strategy to overcome
crosstalk-related resistance to EGFR inhibitors. Lancet Oncol.
10:709–717. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu P, Cheng H, Roberts TM and Zhao JJ:
Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev
Drug Discov. 8:627–644. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nahta R, Yu D, Hung MC, Hortobagyi GN and
Esteva FJ: Mechanisms of disease: Understanding resistance to
HER2-targeted therapy in human breast cancer. Nat Clin Pract Oncol.
3:269–280. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yamaguchi H, Chang SS, Hsu JL and Hung MC:
Signaling crosstalk in the resistance to HER family receptor
targeted therapy. Oncogene. 33:1073–1081. 2014. View Article : Google Scholar
|
|
29
|
Baselga J: Targeting tyrosine kinases in
cancer: The second wave. Science. 312:1175–1178. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yi YW, You K, Bae EJ, Kwak SJ, Seong YS
and Bae I: Dual inhibition of EGFR and MET induces synthetic
lethality in triple-negative breast cancer cells through
downregulation of ribosomal protein S6. Int J Oncol. 47:122–132.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Massihnia D, Galvano A, Fanale D, Perez A,
Castiglia M, Incorvaia L, Listì A, Rizzo S, Cicero G, Bazan V, et
al: Triple negative breast cancer: Shedding light onto the role of
pi3k/akt/mtor pathway. Oncotarget. 7:60712–60722. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Goldstein D, Bushmeyer SM, Witt PL, Jordan
VC and Borden EC: Effects of type I and II interferons on cultured
human breast cells: Interaction with estrogen receptors and with
tamoxifen. Cancer Res. 49:2698–2702. 1989.PubMed/NCBI
|
|
33
|
Duong HQ, You KS, Oh S, Kwak SJ and Seong
YS: Silencing of NRF2 reduces the expression of ALDH1A1 and ALDH3A1
and sensitizes to 5-FU in pancreatic cancer cells. Antioxidants.
6:62017. View Article : Google Scholar
|
|
34
|
Kim SI, Kim HJ, Lee HJ, Lee K, Hong D, Lim
H, Cho K, Jung N and Yi YW: Application of a non-hazardous vital
dye for cell counting with automated cell counters. Anal Biochem.
492:8–12. 2016. View Article : Google Scholar
|
|
35
|
Biscardi JS, Ishizawar RC, Silva CM and
Parsons SJ: Tyrosine kinase signalling in breast cancer: Epidermal
growth factor receptor and c-Src interactions in breast cancer.
Breast Cancer Res. 2:203–210. 2000. View
Article : Google Scholar
|
|
36
|
Finn RS, Dering J, Ginther C, Wilson CA,
Glaspy P, Tchekmedyian N and Slamon DJ: Dasatinib, an orally active
small molecule inhibitor of both the src and abl kinases,
selectively inhibits growth of basal-type/'triple-negative' breast
cancer cell lines growing in vitro. Breast Cancer Res Treat.
105:319–326. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Feder D and Bishop JM: Purification and
enzymatic characterization of pp60c-src from human platelets. J
Biol Chem. 265:8205–8211. 1990.PubMed/NCBI
|
|
38
|
Sekulić A, Hudson CC, Homme JL, Yin P,
Otterness DM, Karnitz LM and Abraham RT: A direct linkage between
the phosphoinositide 3-kinase-AKT signaling pathway and the
mammalian target of rapamycin in mitogen-stimulated and transformed
cells. Cancer Res. 60:3504–3513. 2000.
|
|
39
|
Kovacina KS, Park GY, Bae SS, Guzzetta AW,
Schaefer E, Birnbaum MJ and Roth RA: Identification of a
proline-rich Akt substrate as a 14-3-3 binding partner. J Biol
Chem. 278:10189–10194. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gingras AC, Gygi SP, Raught B, Polakiewicz
RD, Abraham RT, Hoekstra MF, Aebersold R and Sonenberg N:
Regulation of 4E-BP1 phosphorylation: A novel two-step mechanism.
Genes Dev. 13:1422–1437. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Vogelstein B, Papadopoulos N, Velculescu
VE, Zhou S, Diaz LA Jr and Kinzler KW: Cancer genome landscapes.
Science. 339:1546–1558. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wullschleger S, Loewith R and Hall MN: TOR
signaling in growth and metabolism. Cell. 124:471–484. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Laplante M and Sabatini DM: mTOR signaling
at a glance. J Cell Sci. 122:3589–3594. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sabatini DM: mTOR and cancer: Insights
into a complex relationship. Nat Rev Cancer. 6:729–734. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ueng SH, Chen SC, Chang YS, Hsueh S, Lin
YC, Chien HP, Lo YF, Shen SC and Hsueh C: Phosphorylated mTOR
expression correlates with poor outcome in early-stage triple
negative breast carcinomas. Int J Clin Exp Pathol. 5:806–813.
2012.PubMed/NCBI
|
|
46
|
Walsh S, Flanagan L, Quinn C, Evoy D,
McDermott EW, Pierce A and Duffy MJ: mTOR in breast cancer:
Differential expression in triple-negative and non-triple-negative
tumors. Breast. 21:178–182. 2012. View Article : Google Scholar
|
|
47
|
Montero JC, Esparís-Ogando A, Re-Louhau
MF, Seoane S, Abad M, Calero R, Ocaña A and Pandiella A: Active
kinase profiling, genetic and pharmacological data define mTOR as
an important common target in triple-negative breast cancer.
Oncogene. 33:148–156. 2014. View Article : Google Scholar
|
|
48
|
Ellard SL, Clemons M, Gelmon KA, Norris B,
Kennecke H, Chia S, Pritchard K, Eisen A, Vandenberg T, Taylor M,
et al: Randomized phase II study comparing two schedules of
everolimus in patients with recurrent/metastatic breast cancer:
NCIC Clinical Trials Group IND.163. J Clin Oncol. 27:4536–4541.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Jovanović B, Mayer IA, Mayer EL, Abramson
VG, Bardia A, Sanders ME, Kuba MG, Estrada MV, Beeler JS, Shaver
TM, et al: A Randomized phase II neoadjuvant study of cisplatin,
paclitaxel with or without everolimus in patients with stage II/III
triple-negative breast cancer (TNBC): Responses and long-term
outcome correlated with increased frequency of DNA damage response
gene mutations, TNBC subtype, AR status, and Ki67. Clin Cancer Res.
23:4035–4045. 2017. View Article : Google Scholar
|
|
50
|
Gonzalez-Angulo AM, Akcakanat A, Liu S,
Green MC, Murray JL, Chen H, Palla SL, Koenig KB, Brewster AM,
Valero V, et al: Open-label randomized clinical trial of standard
neoadjuvant chemotherapy with paclitaxel followed by FEC versus the
combination of paclitaxel and everolimus followed by FEC in women
with triple receptor-negative breast cancer. Ann Oncol.
25:1122–1127. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Oualla K, El-Zawahry HM, Arun B, Reuben
JM, Woodward WA, Gamal El-Din H, Lim B, Mellas N, Ueno NT and Fouad
TM: Novel therapeutic strategies in the treatment of
triple-negative breast cancer. Ther Adv Med Oncol. 9:493–511. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Liu T, Yacoub R, Taliaferro-Smith LD, Sun
SY, Graham TR, Dolan R, Lobo C, Tighiouart M, Yang L, Adams A, et
al: Combinatorial effects of lapatinib and rapamycin in
triple-negative breast cancer cells. Mol Cancer Ther. 10:1460–1469.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Madden JM, Mueller KL, Bollig-Fischer A,
Stemmer P, Mattingly RR and Boerner JL: Abrogating phosphorylation
of eIF4B is required for EGFR and mTOR inhibitor synergy in
triple-negative breast cancer. Breast Cancer Res Treat.
147:283–293. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kim DH, Sarbassov DD, Ali SM, King JE,
Latek RR, Erdjument-Bromage H, Tempst P and Sabatini DM: mTOR
interacts with raptor to form a nutrient-sensitive complex that
signals to the cell growth machinery. Cell. 110:163–175. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Chen B, Tan Z, Gao J, Wu W, Liu L, Jin W,
Cao Y, Zhao S, Zhang W, Qiu Z, et al: Hyperphosphorylation of
ribosomal protein S6 predicts unfavorable clinical survival in
non-small cell lung cancer. J Exp Clin Cancer Res. 34:1262015.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Iwenofu OH, Lackman RD, Staddon AP,
Goodwin DG, Haupt HM and Brooks JS: Phospho-S6 ribosomal protein: A
potential new predictive sarcoma marker for targeted mTOR therapy.
Mod Pathol. 21:231–237. 2008. View Article : Google Scholar
|
|
57
|
Komori Y, Yada K, Ohta M, Uchida H,
Iwashita Y, Fukuzawa K, Kashima K, Yokoyama S, Inomata M and Kitano
S: Mammalian target of rapamycin signaling activation patterns in
pancreatic neuroendocrine tumors. J Hepatobiliary Pancreat Sci.
21:288–295. 2014. View Article : Google Scholar
|
|
58
|
Masuda M, Chen WY, Miyanaga A, Nakamura Y,
Kawasaki K, Sakuma T, Ono M, Chen CL, Honda K and Yamada T:
Alternative mammalian target of rapamycin (mTOR) signal activation
in sorafenib-resistant hepatocellular carcinoma cells revealed by
array-based pathway profiling. Mol Cell Proteomics. 13:1429–1438.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Knoll M, Macher-Goeppinger S, Kopitz J,
Duensing S, Pahernik S, Hohenfellner M, Schirmacher P and Roth W:
The ribosomal protein S6 in renal cell carcinoma: Functional
relevance and potential as biomarker. Oncotarget. 7:418–432. 2016.
View Article : Google Scholar :
|
|
60
|
Green TP, Fennell M, Whittaker R, Curwen
J, Jacobs V, Allen J, Logie A, Hargreaves J, Hickinson DM,
Wilkinson RW, et al: Preclinical anticancer activity of the potent,
oral Src inhibitor AZD0530. Mol Oncol. 3:248–261. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Maira SM, Stauffer F, Brueggen J, Furet P,
Schnell CC, Brachmann S, Chène P, De Pover A, Schoemaker K, et al:
Identification and characterization of NVP-BEZ235, a new orally
available dual phosphatidylinositol 3-kinase/mammalian target of
rapamycin inhibitor with potent in vivo antitumor activity. Mol
Cancer Ther. 7:1851–1863. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Toledo LI, Murga M, Zur R, Soria R,
Rodriguez A, Martinez S, Oyarzabal J, Pastor J, Bischoff JR and
Fernandez-Capetillo O: A cell-based screen identifies ATR
inhibitors with synthetic lethal properties for cancer-associated
mutations. Nat Struct Mol Biol. 18:721–727. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
O'Hare T, Walters DK, Stoffregen EP, Jia
T, Manley PW, Mestan J, Cowan-Jacob SW, Lee FY, Heinrich MC,
Deininger MW, et al: In vitro activity of Bcr-Abl inhibitors AMN107
and BMS-354825 against clinically relevant imatinib-resistant Abl
kinase domain mutants. Cancer Res. 65:4500–4505. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Shah NP, Lee FY, Luo R, Jiang Y, Donker M
and Akin C: Dasatinib (BMS-354825) inhibits KITD816V, an
imatinib-resistant activating mutation that triggers neoplastic
growth in most patients with systemic mastocytosis. Blood.
108:286–291. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Rivera VM, Squillace RM, Miller D, Berk L,
Wardwell SD, Ning Y, Pollock R, Narasimhan NI, Iuliucci JD, Wang F,
et al: Ridaforolimus (AP23573; MK-8669), a potent mTOR inhibitor,
has broad antitumor activity and can be optimally administered
using intermittent dosing regimens. Mol Cancer Ther. 10:1059–1071.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Chun KH, Kosmeder JW II, Sun S, Pezzuto
JM, Lotan R, Hong WK and Lee HY: Effects of deguelin on the
phosphatidylinositol 3-kinase/Akt pathway and apoptosis in
premalignant human bronchial epithelial cells. J Natl Cancer Inst.
95:291–302. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Schuler W, Sedrani R, Cottens S, Häberlin
B, Schulz M, Schuurman HJ, Zenke G, Zerwes HG and Schreier MH: SDZ
RAD, a new rapamycin derivative: Pharmacological properties in
vitro and in vivo. Transplantation. 64:36–42. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Flanagan WM, Corthésy B, Bram RJ and
Crabtree GR: Nuclear association of a T-cell transcription factor
blocked by FK-506 and cyclosporin A. Nature. 352:803–807. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Folkes AJ, Ahmadi K, Alderton WK, Alix S,
Baker SJ, Box G, Chuckowree IS, Clarke PA, Depledge P, Eccles SA,
et al: The identification of
2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine
(GDC-0941) as a potent, selective, orally bioavailable inhibitor of
class I PI3 kinase for the treatment of cancer. J Med Chem.
51:5522–5532. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Carnero A: Novel inhibitors of the PI3K
family. Expert Opin Investig Drugs. 18:1265–1277. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Knight ZA, Gonzalez B, Feldman ME, Zunder
ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth
B, et al: A pharmacological map of the PI3-K family defines a role
for p110alpha in insulin signaling. Cell. 125:733–747. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
García-Martínez JM, Moran J, Clarke RG,
Gray A, Cosulich SC, Chresta CM and Alessi DR: Ku-0063794 is a
specific inhibitor of the mammalian target of rapamycin (mTOR).
Biochem J. 421:29–42. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Vlahos CJ, Matter WF, Hui KY and Brown RF:
A specific inhibitor of phosphatidylinositol 3-kinase,
2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J Biol
Chem. 269:5241–5248. 1994.PubMed/NCBI
|
|
74
|
Hirai H, Sootome H, Nakatsuru Y, Miyama K,
Taguchi S, Tsujioka K, Ueno Y, Hatch H, Majumder PK, Pan BS, et al:
MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy
by standard chemotherapeutic agents or molecular targeted drugs in
vitro and in vivo. Mol Cancer Ther. 9:1956–1967. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Gills JJ and Dennis PA: Perifosine: Update
on a novel Akt inhibitor. Curr Oncol Rep. 11:102–110. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Nghiem P, Pearson G and Langley RG:
Tacrolimus and pimecrolimus: From clever prokaryotes to inhibiting
calcineurin and treating atopic dermatitis. J Am Acad Dermatol.
46:228–241. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Edwards SR and Wandless TJ: The
rapamycin-binding domain of the protein kinase mammalian target of
rapamycin is a destabilizing domain. J Biol Chem. 282:13395–13401.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Shor B, Zhang WG, Toral-Barza L, Lucas J,
Abraham RT, Gibbons JJ and Yu K: A new pharmacologic action of
CCI-779 involves FKBP12-independent inhibition of mTOR kinase
activity and profound repression of global protein synthesis.
Cancer Res. 68:2934–2943. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Marone R, Cmiljanovic V, Giese B and
Wymann MP: Targeting phosphoinositide 3-kinase: Moving towards
therapy. Biochim Biophys Acta. 1784:159–185. 2008. View Article : Google Scholar
|
|
80
|
Somwar R, Niu W, Kim DY, Sweeney G,
Randhawa VK, Huang C, Ramlal T and Klip A: Differential effects of
phosphatidylinositol 3-kinase inhibition on intracellular signals
regulating GLUT4 translocation and glucose transport. J Biol Chem.
276:46079–46087. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Yu K, Toral-Barza L, Shi C, Zhang WG,
Lucas J, Shor B, Kim J, Verheijen J, Curran K, Malwitz DJ, et al:
Biochemical, cellular, and in vivo activity of novel
ATP-competitive and selective inhibitors of the mammalian target of
rapamycin. Cancer Res. 69:6232–6240. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Yaguchi S, Fukui Y, Koshimizu I, Yoshimi
H, Matsuno T, Gouda H, Hirono S, Yamazaki K and Yamori T: Antitumor
activity of ZSTK474, a new phosphatidylinositol 3-kinase inhibitor.
J Natl Cancer Inst. 98:545–556. 2006. View Article : Google Scholar : PubMed/NCBI
|