Introduction
Breast cancer is one of the most prevalent cancer
types among females worldwide. In addition to the sharp increase in
the rate of morbidity, the average age of morbidity has gradually
decreased (1). Triple-negative
breast cancer (TNBC) is a clinical phenotype characterized by the
lack of three proteins: Estrogen receptor, progesterone receptor
and human epidermal growth factor receptor 2 (HER2). TNBC tends to
be more aggressive and has a higher mortality rate compared with
the other breast cancer subtypes (2). As a result of the deficiency of
certain receptors, hormone therapy and anti-HER2 targeted therapy
are ineffective. Treatment primarily relies on chemotherapy
(3). However, treatment failure
has become a growing trend, accompanied by tumor relapse and
chemotherapeutic resistance (4).
Consequently, there is an urgent requirement to ascertain the
precise molecular mechanisms of TNBC and seek novel strategies for
its treatment.
Apoptosis-stimulating p53-binding protein 2 (ASPP2),
also known as 53BP2L, is the long form of the two splicing variants
encoded by the tumor protein p53 binding protein 2 gene through
alternative splicing (5). ASPP2
was originally identified as an activator of the p53 family of
proteins that specifically enhanced their transcriptional
activities toward pro-apoptosis genes (but not genes in association
with cell-cycle arrest) by binding to them (6,7).
However, emerging evidence has suggested that ASPP2 is associated
with a series of p53-independent biological pathways, rather than
simply inducing apoptosis dependent on p53 (8). One study indicated that ASPP2 could
promote Ras-induced senescence through the direct interaction of
its N-terminus with Ras-GTP (9).
Furthermore, it serves as a pivotal regulator of cell polarity and
the autophagy process (10,11).
ASPP2 has also been confirmed to bind and co-localize with PAR3,
thereby inhibiting tumor metastasis as a molecular switch of
epithelial-mesenchymal transition (EMT), and the reduction of ASPP2
results in the poor survival and prognosis of patients with
hepatocellular carcinoma and breast cancer (12). In the majority of human cancer
types, including hepatocellular carcinoma (13), pancreatic cancer and cervical
cancer (14,15), ASPP2 is considered to be a tumor
suppressor, usually with low expression. Notably, widespread p53
mutations take place in TNBC (16), which may limit the role of ASPP2
with regard to p53-dependent pro-apoptosis, since it acts only on
wild type-p53 genes rather than mutant-p53 genes (17,18).
The mechanisms and functions of ASPP2 in the presence of p53
mutations are much less well known, particularly in TNBC, in which
p53 mutation frequently occurs.
In the present study, the function and associated
mechanisms of ASPP2 in TNBC were investigated. The aims of the
present study were to examine the relative expression of ASPP2 in
breast cancer samples and cell lines, to investigate its functional
roles in cell proliferation, migration and invasion using specific
small interfering RNA (siRNA), and to investigate the possibility
of its target signaling pathways as potential molecular targets for
therapeutic agents.
Materials and methods
Patients and samples
The breast tumor tissues and paired normal adjacent
tissues were collected from patients who underwent surgical
resection at the Department of Breast and Thyroid Surgery of the
Shanghai Tenth People’s Hospital (Shanghai, China) between December
2016 and February 2017. The patients were women between the ages of
32 and 71 years, with a mean age of 53 years. None of the patients
had received any chemotherapy (19) or radiotherapy prior to surgery.
Patients with distant metastases or a history of a previous or
concomitant malignancy were excluded. The samples were immediately
snap-frozen in liquid nitrogen. Tumor and normal tissues were
histologically confirmed by more than one experienced pathologist
according to the World Health Organization guidelines (19), using hematoxylin and eosin
staining. All specimens were embedded in 10% formalin solution for
12–24 h at room temperature and cut into 5-μm thick
sections. The sections were stained with hematoxylin for 3–10 min
and with eosin for 60 sec at room temperature, and then observed
under a light microscope at ×40 magnification. The specimen
collection and use was approved by the Institutional Ethics
Committees of Tongji University (Shanghai, China). All patients
provided written informed consent. The data of the patients are not
shown.
Cell culture and reagent
Human breast cancer cell lines, MDA-MB-231,
HCC-1937, MCF-7, BT-549 and MDA-MB-468, and the human mammary
epithelial cell line, MCF-10A, were purchased from the Chinese
Academy of Sciences (Shanghai, China). The BT-549 cells were
cultured in RPMI-1640 medium (Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) supplemented with 100 U/ml penicillin, 100
μg/ml streptomycin (Enpromise, Hangzhou, China) and 10%
fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.).
MCF-10A cells were cultured in mammary epithelial basal medium
(Cambrex Corporation, East Rutherford, NJ, USA). The remaining
cells were grown in Dulbecco’s modified Eagle’s medium (DMEM)
supplemented with 10% FBS (both Gibco; Thermo Fisher Scientific,
Inc.), penicillin (100 U/ml) and streptomycin (100 μg/ml)
(both from Enpromise). All cells were incubated at 37°C in a
humidified chamber containing 5% CO2.
ASPP2 siRNA and negative control siRNA (NC siRNA)
oligonucleotides were chemosynthesized by Sangon Biotech Co., Ltd.
(Shanghai, China). The sequence of the ASPP2 siRNA was
5′-GCCCAGUAGAAAUCCAGAATT-3′ (sense) and 5′-UUCUGGAUUUCUACUGGGCTT-3′
(antisense), while the sequence of the NC siRNA was
5′-UUCUCCGAACGUGUCACGUTT-3′ (sense) and 5′-ACGUGACACGUUCGGAGAATT-3′
(antisense).
Transfection assay
The MDA-MB-231, MCF-7 and HCC-1937 cells
(8×104/well) were cultured in a 6-well plate with serum
and antibiotic-free DMEM for transfection. When the confluence
reached 30–50%, transfection of ASPP2 siRNA and NC siRNA was
performed using the Lipofectamine® 2000 Transfection kit
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer′s protocols, at working concentrations. The
concentration of siRNAs used was 100 nmol/l, and the ratio of
mimics to Lipofectamine 2000 was 1.25:1.00 (volume). The medium was
replaced by DMEM with 10% FBS after 4–6 h of incubation. The cells
were used for future analysis after 48 h of transfection.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) assay
Total cellular RNA was extracted from the
transfected MDA-MB-231, MCF-7 or HCC-1937 cells using TRIzol
(Invitrogen; Thermo Fisher Scientific, Inc.) and stored at −80°C.
For ASPP2 detection, cDNA was generated by RT using the PrimeScript
RT-PCR kit (Takara Bio, Inc., Otsu, Japan) in accordance with the
manufacturer′s protocols. Conditions of the RT reaction were 37°C
for 15 min, then 85°C for 5 sec. RT-qPCR was performed using
SYBR-Green PCR master mix (Takara Bio, Inc.) on a 7900HT Fast
RT-PCR instrument (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The amplification procedure was as follows: 3 min at 95°C,
followed by 40 cycles at 95°C for 3 sec and 60°C for 30 sec. The
relative expression was evaluated following the relative
quantification 2−ΔΔCt method (20). Each sample was tested in
triplicate. The primers used in the RT-PCR were as follows: ASPP2
forward, 5′-CTGTGCAAA GAACCCGGCG-3′ and reverse,
5′-CAACTGGACGTTCAGAGCCACA-3′; and β-actin forward,
5′-CAGAGCCTCGCCTTTGCC-3′ and reverse, 5′-GTCGCCCACATAGGAATC-3′.
Western blot assay
The transfected MDA-MB-231, MCF-7 or HCC-1937 cells
were harvested and lysed in radioimmunoprecipitation assay lysis
buffer (80 μl/well; Beyotime Institute of Biotechnology,
Jiangsu, China) after 48–72 h of transfection. The protein
concentration was quantified with a bicinchoninic acid protein
assay kit (Beyotime Institute of Biotechnology). Next, equal
amounts of protein (30–50 μg) were separated by 8 or 10%
sodium dodecyl sulfate-polyacrylamide gel electrophoresis (Beyotime
Institute of Biotechnology), and then transferred to 0.45-μm
nitrocellulose membranes using the cold transfer buffer (3.03 g
Tris + 14.4 g glycine + 200 ml methanol + 800 ml deionized water).
Subsequent to blocking at room temperature for 1 h in 5% skimmed
milk diluted with phosphate-buffered saline plus Tween-20 (PBST),
the membranes were hybridized overnight at 4°C with specified
primary antibodies in PBST containing 5% skimmed milk.
Subsequently, the membranes were washed with PBST and incubated
with IRDye 680 donkey anti-mouse IgG-(H+L) (1:1,000 dilution; cat.
no. 926-68072) or goat anti-rabbit IRDye 800CW secondary antibody
(1:1,000 dilution; cat. no. 926-32211; LI-COR Biosciences, Lincoln,
NE, USA) for 1 h at a room temperature. Protein bands were detected
with an Odyssey Scanning system (LI-COR Biosciences).
Antibodies used were follows: Anti-ASPP2 (1:20,000
dilution; cat. no. ab181377; Abcam, Cambridge, UK), anti-β-actin
(1:2,000 dilution; cat. no. sc-47778; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA), anti-caspase-9 (1:1,000 dilution; cat. no.
ab202068; Abcam), anti-caspase-3 (1:1,000 dilution; cat. no. 9662;
Cell Signaling Technology, Inc., Danvers, MA, USA), anti-poly
(ADP-ribose) polymerase (PARP; 1:1,000 dilution; cat. no. ab191217;
Abcam), anti-Bax (1:1,000 dilution; cat. no. 2772), anti-E-cadherin
(1:750 dilution; cat. no. 3195) (both from Cell Signaling
Technology, Inc.), anti-N-cadherin (1:2,000 dilution; cat. no.
ab18203), anti-Snail (1:1,000 dilution; cat. no. ab82846),
anti-zinc finger E-box-binding homeobox 1 (ZEB1; 1:1,000 dilution;
cat. no. ab155249), anti-matrix metalloproteinase 2 (MMP2; 1:2,000
dilution; cat. no. ab37150) (all from Abcam), anti-MMP9 (1:1,000
dilution; cat. no. 54980; Arigo Biolaboratories, Hsinchu, Taiwan),
anti-phosphoinositide-3-kinase regulatory subunit 1 (PIK3R1;
1:1,000 dilution; cat. no. 13666), anti-AKT (1:1,000 dilution; cat.
no. 9272), anti-phosphorylated (p-)AKT (ser-473; 1:1,000 dilution;
cat. no. 4060) (all from Cell Signaling Technology, Inc.),
anti-extracellular signal-regulated kinases (ERK; 1:2,000 dilution;
cat. no. ab17942) and anti-p-ERK (ser-T202 and ser-T185; 1;1,000
dilution; cat. no. ab201015) (both from Abcam).
MTT assay
At 24 h post-transfection, the MDA-MB-231 and
HCC-1937 cells were seeded in 200 μl growth medium at
5×102 cells per well in 96-well plates (BD Biosciences,
Franklin Lakes, NJ, USA) and incubated overnight at 37°C in 5%
CO2. Every 24 h until 72 h, 20 μl MTT
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) solution was added
to each well and incubated at 37°C for 4 h. Next, 150 μl
dimethyl sulfoxide (Sigma-Aldrich; Merck KGaA) was added to each
well and agitated gently for 10 min to dissolve the MTT formazan
crystals after removing the supernatant. Cell viability was
measured by the recording absorbance at 490 nm with a microplate
reader (BioTek Instruments, Inc., Winooski, VT, USA).
Colony formation assay
A total of 1×103 transfected MDA-MB-231
and HCC-1937 cells from each group were seeded in a 6-well plate in
DMEM with 10% FBS. The plates were agitated to disperse the cells
equally. After 7 to 10 days of culturing, or when the colonies were
visible, the cell culture was terminated and the plates were washed
twice with PBS. Next, the cells were fixed in 95% ethanol for 10
min, dried and stained with 0.1% crystal violet solution for 10 min
at room temperature. Finally, the staining solution was washed away
and the number of colonies with diameters of >1.5 mm was counted
by eye. The experiments were performed in triplicate.
Wound-healing assay
To assay the migratory response of breast cancer
cells to ASPP2 expression, the transfected MDA-MB-231 and HCC-1937
cells were seeded into 6-well plates and cultured until the cells
reached ~90% confluence. Next, a scratch was made in each well
using a sterile pipette tip. Cells were washed with PBS to remove
cellular debris and allowed to migrate for 48 h. The process of
wound healing was observed under a light microscope and
representative images were acquired at 0 and 48 h post-wounding
with a digital camera system. All experiments were performed in
triplicate.
Transwell invasion assay
The transfected MDA-MB-231 and HCC-1937 cells at a
density of 5×104 were suspended in serum-free DMEM (200
μl) and added into the upper chamber of the Transwell, with
a Matrigel-coated (2 mg/ml) membrane containing 8-μm
diameter pores, to observe invasion following transfection.
Complete DMEM (500 μl) was then added to the bottom chamber
of 24-well plates to serve as a chemoattractant. Subsequent to 20 h
of incubation at 37°C in 5% CO2, the non-invading cells
on the upper surface were carefully removed with a cotton swab. The
cells that had invaded the lower surface of the membrane were fixed
with 10% formalin for 30 min prior to staining with crystal violet
for 15 min at room temperature, and then counted under a light
microscope at ×200 magnification. The cells were counted in five
random fields on each membrane. The experiments were conducted in
triplicate.
Apoptosis assay
For the measurement of apoptosis, at 24 h
post-transfection, the MDA-MB-231 and HCC-1937 cells
(2×105) were treated with 1μmol/l docetaxel for
36 h. The cells were then collected in centrifuge tubes (1,000 × g,
at room temperature for 5 min), and washed in chilled PBS.
Subsequently, the cells were re-suspended in 250 μl binding
buffer, and Annexin V/fluorescein isothiocyanate solution and
propidium iodide (PI) solution were added to the cell suspension.
Following incubation for 30 min, the rate of apoptosis was detected
by flow cytometry (FACSCanto™ II; BD Biosciences).
Statistical analysis
Data are presented as the mean ± standard deviation.
Two-way analysis of variance or Student’s t-test was used for
comparisons between groups. P<0.05 was used to indicate a
statistically significant difference. GraphPad Prism version 6.0
(GraphPad Software, Inc., La Jolla, CA, USA) or the SPSS program
(IBM Corp., Armonk, NY, USA) was used to perform the statistical
analyses.
Results
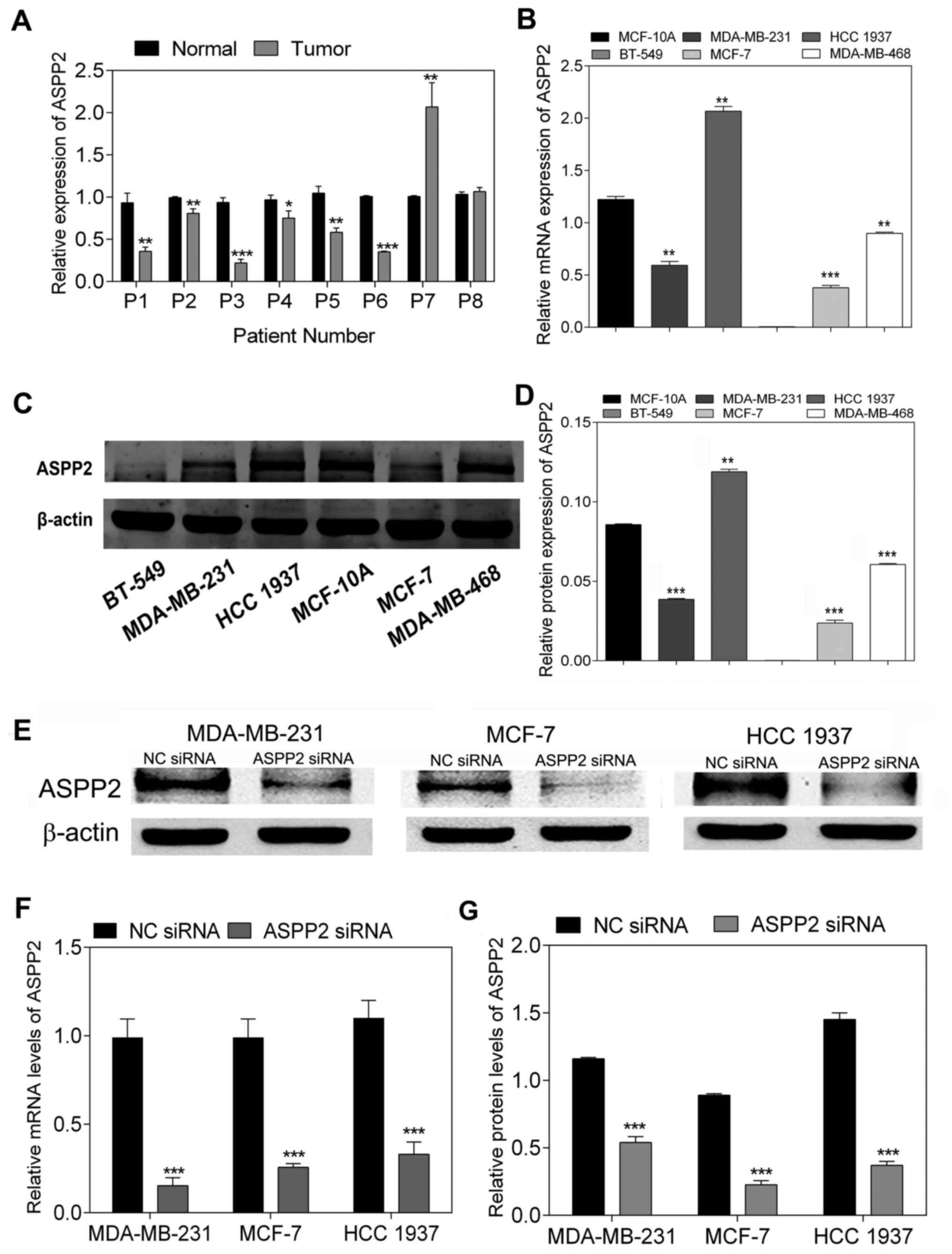
ASPP2 expression is downregulated in the
majority of breast cancer tissues and cell lines, and is inhibited
following siRNA transfection
To analyze the mRNA levels of ASPP2 expression in
breast cancer tissues compared with those in para-cancerous normal
tissues, the level of ASPP2 mRNA was determined by RT-qPCR. The
results showed that ASPP2 mRNA expression was suppressed in a
number of the breast cancer tissues compared with that in the
matched normal tissues (P<0.05; Fig. 1A). In addition, the mRNA and
protein expression of ASPP2 was examined in a panel of breast
cancer cell lines (BT-549, MDA-MB-231, HCC-1937, MDA-MB-468 and
MCF-7) compared with the expression in the breast epithelial cell
line (MCF-10A). Notably, with the exception of HCC-1937 cells, a
reduction in ASPP2 mRNA expression was found in all the remaining
cancer cell lines compared with that in MCF-10A, as measured by
RT-qPCR (P<0.001; Fig. 1B).
Meanwhile, western blot analysis showed that the majority of cancer
cell lines (with the exception of HCC-1937 cells) expressed lower
levels of ASPP2 protein compared with MCF-10A, which was consistent
with the tendency of the RT-qPCR results (P<0.001; Fig. 1C and D). For further investigation,
ASPP2 siRNA was transfected into three different cell lines
(MDA-MB-231, HCC-1937 and MCF-7 cells), and the interference effect
on endogenous ASPP2 expression was validated by RT-qPCR and western
blotting. Following transfection with ASPP2 siRNA, the expression
of ASPP2 decreased at the mRNA and protein levels in all three
different breast cancer cell lines (P<0.001; Fig. 1E–G). Accordingly, siRNA
transfection was considered to be effective for ASPP2 silencing in
breast cancer cells.
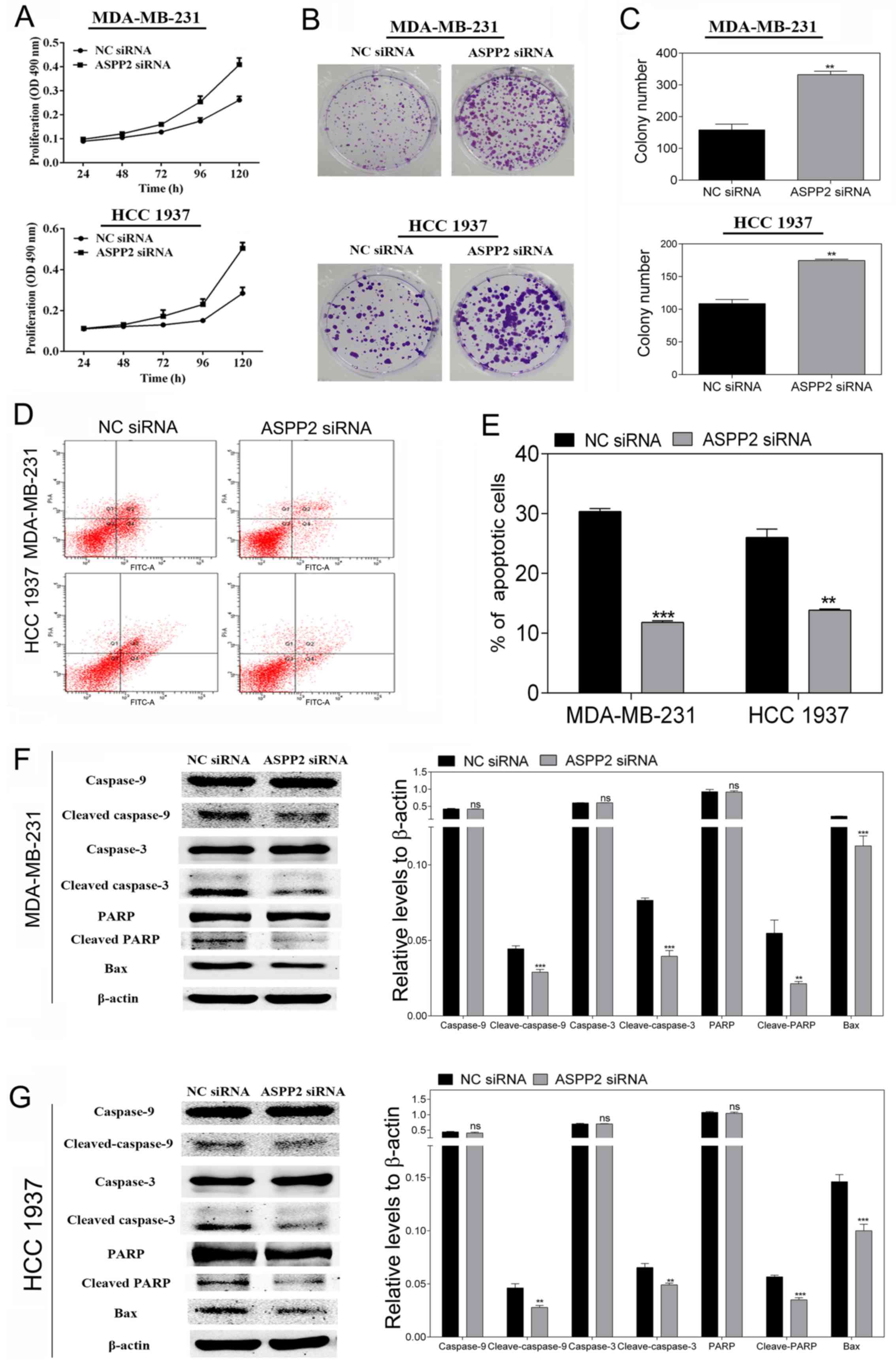
ASPP2 downregulation contributes to TNBC
cell proliferation and decreases cell apoptosis
For the determination of the impact of ASPP2 on the
cell viability and cell proliferation of TNBC cells, MDA-MB-231 and
HCC-1937 cells were transfected with ASPP2 siRNA and NC siRNA.
Subsequently, MTT and colony formation assays were performed. As
shown in Fig. 2A, as determined by
MTT assay, silencing ASPP2 clearly enhanced the cell proliferation
in a time-dependent manner in the MDA-MB-231 and HCC-1937 cells.
Likewise, the downregulation of ASPP2 caused a significant increase
in the colony formation number compared with the NC siRNA
transfected cells in the two cell lines (P<0.01; Fig. 2B and C). All the results indicated
that ASPP2 could promote MDA-MB-231 and HCC-1937 cellular
growth.
To investigate whether ASPP2 silencing could
increase cell viability by reducing cell apoptosis, docetaxel (1
μmol/l) was added to the transfected cells to induce
apoptosis following 24 h of transfection. Next, 36 h later, flow
cytometric analysis was performed to analyze the apoptosis in the
MDA-MB-231 and HCC-1937 cells. The total apoptosis rate of the
cells was reflected by the number of early and late apoptotic cells
in the Annexin V+/PI− and Annexin
V+/PI+ domains. As shown in Fig. 2D and E, in comparison with the NC
groups, the siRNA transfection group significantly decreased
apoptosis. Furthermore, the western blotting results showed that
the levels of apoptosis-related proteins, including cleaved
caspase-9, cleaved caspase-3, cleaved PARP and Bax, were
significantly decreased compared with those of the NC groups
(Fig. 2F and G). Caspase-9 is at
the top of the caspase cascade activation response, as the most
important promoter and key protease of mitochondrial apoptotic
pathway, the activation of which can further activate the
downstream Caspase family and then promote cell apoptosis (21). Bax can enhance the permeability of
the mitochondrial membrane for entry of cytochrome c into
the cytoplasm and then promote cell apoptosis (22). All the data indicated that ASPP2
silencing promoted cell proliferation and reduced cell apoptosis,
perhaps through the mitochondrial death pathway.
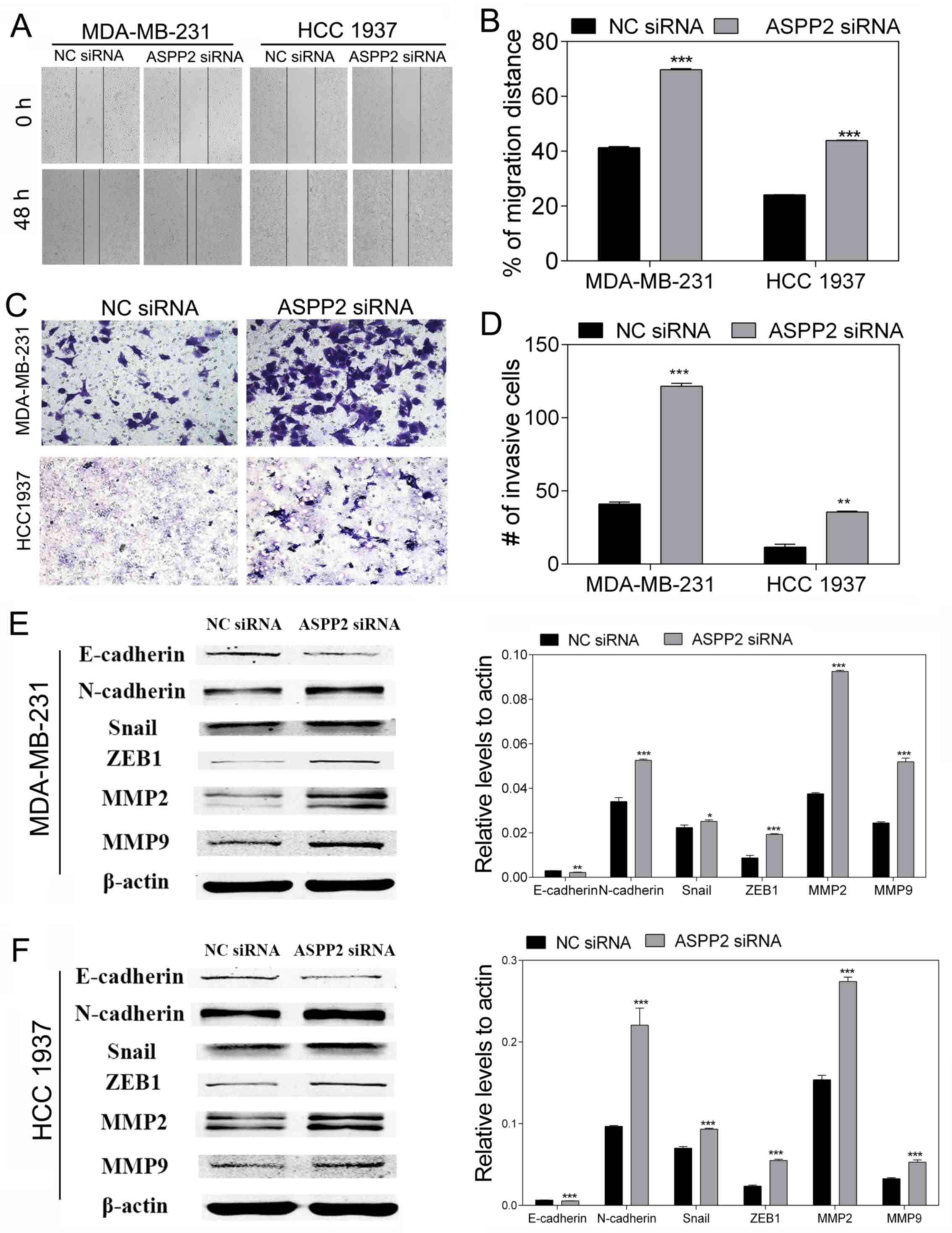
ASPP2 downregulation accelerates cell
migration, invasion and EMT in TNBC cells
To investigate the role of ASPP2 in cell migration
and invasion, wounding-healing and Transwell invasion assays were
performed. The wound-healing assay showed that ASPP2 silencing
significantly promoted the migration ability of the MDA-MB-231 and
HCC-1937 cells compared with the NC (both P<0.001; Fig. 3A and B). The Transwell invasion
assay showed that ASPP2 silencing increased the invasion ability of
the MDA-MB-231 and HCC-1937 cells (P<0.01 and P<0.001;
Fig. 3C and D). These results
indicated a direct association between ASPP2 and the motility of
TNBC cells. On the basis of the cell functional study, research on
the EMT-related proteins was performed using western blot to
further investigate the effect of ASPP2 on the migration and
invasion mechanism. Following depletion of ASPP2, the expression of
representative epithelial marker E-cadherin significantly
decreased, whereas the expression of mesenchymal marker N-cadherin
and other key markers, including Snail and ZEB1, all increased.
Furthermore, the levels of MMP2 and MMP9, which are involved in
EMT, also increased (Fig. 3E and
F). Taken together, these results suggest that ASPP2 silencing
may be responsible for EMT development, and this serves a vital
role in the progression of breast cancer.
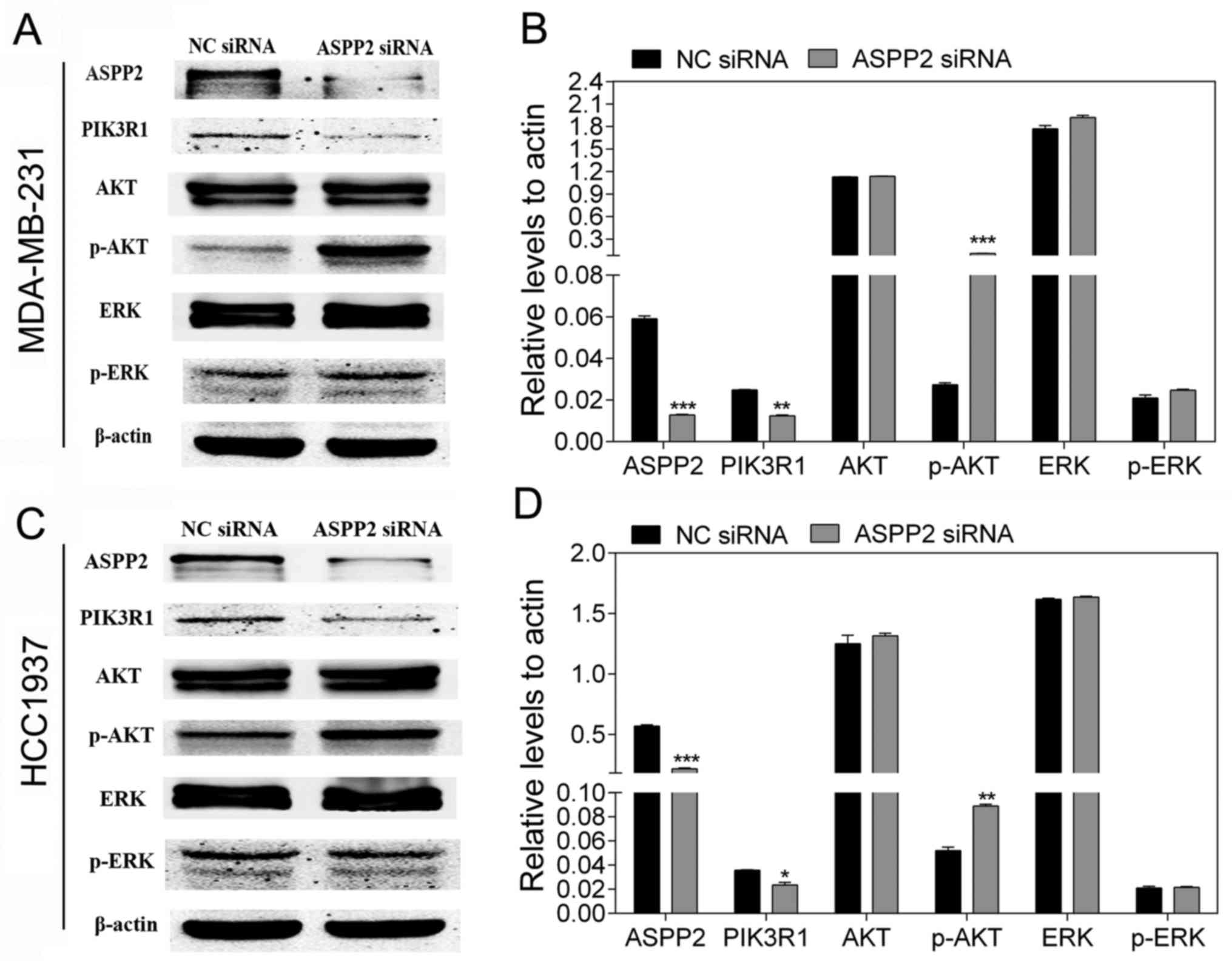
ASPP2 influences the PI3K/AKT signaling
pathway
Activation of the PI3K/AKT signaling pathway is
regarded as a crucial emblem in breast cancer that is associated
with its development, progress and metastatic spread (23). To further validate whether ASPP2 is
involved in the p53-independent pathway in TNBC, the effect of
ASPP2 on the PI3K/AKT pathway was investigated. The important
molecular markers associated with the PI3K/AKT pathway were then
detected. As shown in Fig. 4, the
downregulation of ASPP2 resulted in the decreased expression of
PIK3R1 (p85α) and the increased expression of p-AKT, whereas it had
no influence on the expression of p-ERK.
PIK3R1 (p85α) is the regulatory subunit of PI3K and
negatively regulates the PI3K pathway (24). The present results suggested that
the downregulation of ASPP2 was able to activate the PI3K/AKT
pathway in TNBC cells.
Discussion
TNBC is considered to be a cancer with one of the
worst prognoses of all the breast cancer subtypes. A growing body
of evidence has shown that the aberrant expression of certain genes
may result in tumor progression and metastasis. Previous studies
showed that ASPP2 was suppressed in breast cancer tissues, and that
the low expression of ASPP2 predicted a poor prognosis in
pancreatic cancer (15,25). Consistent with these results, the
present study found decreased ASPP2 mRNA levels in breast cancer
tissues compared with those in normal para-cancerous tissues. In
addition, ASPP2 expression was diminished in the majority of the
breast cancer cell lines, with the exception of the HCC-1937 cell
line, at the mRNA and protein levels. This finding suggests that
loss of ASPP2 may profoundly affect the pathogenesis of breast
cancer. The abnormally high expression in HCC-1937 cells attracted
was notable with regard to whether it had an unexpected role in
such TNBC cells.
Through knockdown of ASPP2 by specific siRNAs, cell
function findings demonstrated that the downregulation of ASPP2
promoted cell proliferation and increased the migration and
invasion abilities in HCC-1937 cells, in accordance with the
results in MDA-MB-231 cells; this affirmed the inhibitory role of
ASPP2 in TNBC and refuted the possibility of ASPP2 acting as an
oncogene and causing the high expression. The increased expression
of ASPP2 may arise from the failure to compensate for the abnormal
expression of certain genes. The BRCA1 DNA repair-associated gene
primarily promotes DNA repair in response to DNA damage, and
HCC-1937 cells are deficient in it (26). By binding to p53, ASPP2 enables p53
to selectively upregulate the expression of pro-apoptotic genes in
response to DNA damage (27), and
its expression can be upregulated in response to DNA damage
(28). Whether the expression of
the two genes are linked remains to be further investigated. In
contrast to previous results, one previous study showed that ASPP2
expression was downregulated in wild-type p53 tumor cells due to
promoter hypermethylation (29).
It was found that ASPP2 expression depends on the methylation
status, which remains to be assessed in breast cancer cell lines
for a better understanding of the mechanism of ASPP2
expression.
ASPP2, as an anti-oncogene, functions primarily in
stimulating apoptosis and enhancing the expression of proapoptotic
genes. In previous studies, ASPP2 downregulation was found to be a
vital component of microRNA-548-3p, inducing cell proliferation and
reducing cell apoptosis (30). In
addition, ASPP2 suppressed cell autophagy and facilitated
oxaliplatin-induced colorectal cancer cell apoptosis (31). In the same way, reduced apoptosis
by silencing ASPP2 in the TNBC cells was observed in the present
study. Notably, it was also found that the decreased apoptosis was
accompanied by the deactivation of the caspase family and Bax in
the TNBC cells. The apoptosis pathway is traditionally divided into
two types: The death-receptor (extrinsic) pathway represented by
caspase-8 and cellular FADD-like IL-1β-converting enzyme-inhibitory
protein, and the mitochondrial (intrinsic) pathway represented by
caspase-9 and Bax (32). The
present study confirmed that ASPP2 induced apoptosis via the
mitochondrial pathway, supporting earlier findings (33). Nevertheless, whether the
death-receptor pathway or other mechanisms influenced ASPP2-induced
apoptosis remains to be evaluated.
EMT serves as a key promoter of the aggression,
invasion and metastasis of cancer, characterized by the loss or
reduction of epithelial markers (E-cadherin and cytokeratins),
together with the overexpression of mesenchymal markers (N-cadherin
and Vimentin) (34). ASPP2 can
suppress EMT by preventing β-catenin from entering the nucleus to
accelerate ZEB1 expression in accordance with its limiting ability
on oncogenic RAS, and the low level of ASPP2 is indicative of poor
patient survival and positive lymph node status in numerous cancer
types (12). Protein phosphatase
Mg2+/Mn2+-dependent 1D was found to promote
cell migration and invasion in pancreatic cancer via the
Wnt/β-catenin pathway on the basis of ASPP2 reduction (35). The present results showed that the
depletion of ASPP2 decreased the expression of E-cadherin in TNBC
cells, whereas the expression of other markers, including
N-cadherin, ZEB1, Snail, MMP2 and MMP9, was increased, all of which
predicted the EMT process occurring followed by a gain in the
ability of migration and invasion. These findings provide the
possible mechanism by which ASPP2 affects growth and metastasis in
TNBC.
The PI3K/AKT pathway is one of the most frequently
(~50%) dysregulated pathways in TNBC that is caused by key gene
mutants, leading to the overactivation of AKT or the functional
loss of regulation factors, including phosphatase and tensin
homolog protein (36). PI3K
inhibitors were reported to provide novel insights into TNBC
therapy, which has been confirmed to have more notable efficacy on
the tumor volume and mitotic activity in TNBC xenografts compared
with that in the luminal-like xenografts (37). To the best of our knowledge, ASPP2
serves key roles not only in a p53-dependent manner to regulate
apoptosis, but also with involvement in the other p53-independent
pathways, including the nuclear factor-кB (38), Hippo (11,39,40)
and Wnt/β-catenin (35) pathways.
As ASPP2 has almost no influence on mutant-p53, whereas p53
mutation is a common occurrence in TNBC, the p53-independent role
of ASPP2 deserves more attention. In consideration of the
importance of the PI3K/AKT pathway in TNBC, we hypothesized that
ASPP2 was associated with the PI3K/AKT pathway, acting in a
p53-independent manner in TNBC. The downregulation of ASPP2 was
demonstrated to result in the abatement of PIK3R1 (p85α). p85α has
been reported to have an inhibitory effect on the PI3K pathway, the
downregulation of which increased the p-AKT levels accordingly,
promoting breast cancer cell growth, migration and invasion
(41). Consistent with this
previous study, the p-AKT level in the present study was affirmed
to increase when ASPP2 was silenced, causing a reduction in p85α,
whereas the p-ERK level exhibited no significant change. In one
sense, this finding confirmed the aforementioned hypothesis.
However, another previous study showed that ASPP2 potentiated p-ERK
activation other than p-AKT activation through stimulating Ras
signaling in primary human fibroblasts (9). The two opposite results can possibly
be explained by the fact that the PI3K/AKT pathway is more involved
than Ras signaling in TNBC, resulting in the selective impact of
ASPP2 on the PI3K/AKT pathway. However, the detailed mechanism
between ASPP2 and the PI3K/AKT pathway in TNBC requires further
investigation.
Taken together, the present study results have
provided evidence that ASPP2 has an inhibitory influence on TNBC
growth and metastasis, and that this may rely on using the PI3K/AKT
pathway in a p53-independent manner. These findings may assist in
the development of more valuable strategies for the treatment of
TNBC.
Acknowledgments
We sincerely thank all the teachers at the Central
Laboratory of the Shanghai Tenth People’s Hospital for their help
and support.
Funding
This study was supported by grant no. 201640097 from
the Shanghai Municipal Health Bureau of Shanghai, China, and grant
no. 82172240 from the National Natural Science Foundation of
China.
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors’ contributions
TW, LF and HS conceived and designed the study. TW,
DX and BZ performed the experiments. TW, KH and JH wrote the paper.
HX, CW, YD and CJ reviewed and edited the manuscript. YD, JH and CJ
made contributions to the aquisition of patients’ data. HX, KH and
CW analyzed and interpreted the data. All authors read and approved
the manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
All experimental protocols were approved by the
Institutional Ethics Committees of Shanghai Tenth People’s Hospital
Affiliated to Tongji University (Shanghai, China).
Consent for publication
Not applicable.
Competing interests
The authors state that they have no competing
interests.
References
|
1
|
Miller KD, Siegel RL, Lin CC, Mariotto AB,
Kramer JL, Rowland JH, Stein KD, Alteri R and Jemal A: Cancer
treatment and survivorship statistics, 2016. CA Cancer J Clin.
66:271–289. 2016. View Article : Google Scholar
|
|
2
|
Pal SK, Childs BH and Pegram M: Triple
negative breast cancer: Unmet medical needs. Breast Cancer Res
Treat. 125:627–636. 2011. View Article : Google Scholar
|
|
3
|
Mayer IA, Abramson VG, Lehmann BD and
Pietenpol JA: New strategies for triple-negative breast
cancer–deciphering the heterogeneity. Clin Cancer Res. 20:782–790.
2014. View Article : Google Scholar
|
|
4
|
Perou CM: Molecular stratification of
triple-negative breast cancers. Oncologist. 16(Suppl 1): 61–70.
2011. View Article : Google Scholar
|
|
5
|
Takahashi N, Kobayashi S, Jiang X,
Kitagori K, Imai K, Hibi Y and Okamoto T: Expression of 53BP2 and
ASPP2 proteins from TP53BP2 gene by alternative splicing. Biochem
Biophys Res Commun. 315:434–438. 2004. View Article : Google Scholar
|
|
6
|
Samuels-Lev Y, O’Connor DJ, Bergamaschi D,
Trigiante G, Hsieh JK, Zhong S, Campargue I, Naumovski L, Crook T
and Lu X: ASPP proteins specifically stimulate the apoptotic
function of p53. Mol Cell. 8:781–794. 2001. View Article : Google Scholar
|
|
7
|
Ahn J, Byeon IJ, Byeon CH and Gronenborn
AM: Insight into the structural basis of pro- and antiapoptotic p53
modulation by ASPP proteins. J Biol Chem. 284:13812–13822. 2009.
View Article : Google Scholar
|
|
8
|
Trigiante G and Lu X: ASPP [corrected] and
cancer. Nat Rev Cancer. 6:217–226. 2006. View Article : Google Scholar
|
|
9
|
Wang Z, Liu Y, Takahashi M, Van Hook K,
Kampa-Schittenhelm KM, Sheppard BC, Sears RC, Stork PJ and Lopez
CD: N terminus of ASPP2 binds to Ras and enhances Ras/Raf/MEK/ERK
activation to promote oncogene-induced senescence. Proc Natl Acad
Sci USA. 110:312–317. 2013. View Article : Google Scholar
|
|
10
|
Liu Z, Qiao L, Zhang Y, Zang Y, Shi Y, Liu
K, Zhang X, Lu X, Yuan L, Su B, et al: ASPP2 plays a dual role in
gp120-induced autophagy and apoptosis of neuroblastoma cells. Front
Neurosci. 11:1502017. View Article : Google Scholar
|
|
11
|
Royer C, Koch S, Qin X, Zak J, Buti L,
Dudziec E, Zhong S, Ratnayaka I, Srinivas S and Lu X: ASPP2 links
the apical lateral polarity complex to the regulation of YAP
activity in epithelial cells. PLoS One. 9:e1113842014. View Article : Google Scholar
|
|
12
|
Wang Y, Bu F, Royer C, Serres S, Larkin
JR, Soto MS, Sibson NR, Salter V, Fritzsche F, Turnquist C, et al:
ASPP2 controls epithelial plasticity and inhibits metastasis
through β-catenin-dependent regulation of ZEB1. Nat Cell Biol.
16:1092–1104. 2014. View
Article : Google Scholar
|
|
13
|
Zhao J, Wu G, Bu F, Lu B, Liang A, Cao L,
Tong X, Lu X, Wu M and Guo Y: Epigenetic silence of
ankyrin-repeat-containing, SH3-domain-containing, and
proline-rich-region- containing protein 1 (ASPP1) and ASPP2 genes
promotes tumor growth in hepatitis B virus-positive hepatocellular
carcinoma. Hepatology. 51:142–153. 2010. View Article : Google Scholar
|
|
14
|
Wang X, Yu M, Zhao K, He M, Ge W, Sun Y,
Wang Y, Sun H and Hu Y: Upregulation of MiR-205 under hypoxia
promotes epithelial-mesenchymal transition by targeting ASPP2. Cell
Death Dis. 7:e25172016. View Article : Google Scholar
|
|
15
|
Song B, Bian Q, Zhang YJ, Shao CH, Li G,
Liu AA, Jing W, Liu R, Zhou YQ, Jin G, et al: Downregulation of
ASPP2 in pancreatic cancer cells contributes to increased
resistance to gemcitabine through autophagy activation. Mol Cancer.
14:1772015. View Article : Google Scholar
|
|
16
|
Jabbour-Leung NA, Chen X, Bui T, Jiang Y,
Yang D, Vijayaraghavan S, McArthur MJ, Hunt KK and Keyomarsi K:
Sequential combination therapy of CDK inhibition and doxo-rubicin
is synthetically lethal in p53-mutant triple-negative breast
cancer. Mol Cancer Ther. 15:593–607. 2016. View Article : Google Scholar
|
|
17
|
Slee EA and Lu X: The ASPP family:
deciding between life and death after DNA damage. Toxicol Lett.
139:81–87. 2003. View Article : Google Scholar
|
|
18
|
Joerger AC, Ang HC, Veprintsev DB, Blair
CM and Fersht AR: Structures of p53 cancer mutants and mechanism of
rescue by second-site suppressor mutations. J Biol Chem.
280:16030–16037. 2005. View Article : Google Scholar
|
|
19
|
Barnes L, Eveson JW, Reichart P and
Sidransky D: WHO Classification of Tumours. 2005
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−ΔΔC(T)) method. Methods. 25:402–408. 2001. View Article : Google Scholar
|
|
21
|
Bratton SB and Salvesen GS: Regulation of
the Apaf-1-caspase-9 apoptosome. J Cell Sci. 123:3209–3214. 2010.
View Article : Google Scholar
|
|
22
|
Gross A, McDonnell JM and Korsmeyer SJ:
BCL-2 family members and the mitochondria in apoptosis. Genes Dev.
13:1899–1911. 1999. View Article : Google Scholar
|
|
23
|
Azim HA, Kassem L, Treilleux I, Wang Q, El
Enein MA, Anis SE and Bachelot T: Analysis of PI3K/mTOR pathway
biomarkers and their prognostic value in women with hormone
receptor-positive, HER2-Negative Early Breast Cancer. Transl Oncol.
9:114–123. 2016. View Article : Google Scholar
|
|
24
|
Luo J and Cantley LC: The negative
regulation of phos-phoinositide 3-kinase signaling by p85 and it’s
implication in cancer. Cell Cycle. 4:1309–1312. 2005. View Article : Google Scholar
|
|
25
|
Van Hook K, Wang Z, Chen D, Nold C, Zhu Z,
Anur P, Lee HJ, Yu Z, Sheppard B, Dai MS, et al: ΔN-ASPP2, a novel
isoform of the ASPP2 tumor suppressor, promotes cellular survival.
Biochem Biophys Res Commun. 482:1271–1277. 2017. View Article : Google Scholar
|
|
26
|
Yoshida K and Miki Y: Role of BRCA1 and
BRCA2 as regulators of DNA repair, transcription, and cell cycle in
response to DNA damage. Cancer Sci. 95:866–871. 2004. View Article : Google Scholar
|
|
27
|
Slee EA and Lu X: The ASPP family:
Deciding between life and death after DNA damage. Toxicol Lett.
139:81–87. 2003. View Article : Google Scholar
|
|
28
|
Bergamaschi D, Samuels Y, Jin B,
Duraisingham S, Crook T and Lu X: ASPP1 and ASPP2: Common
activators of p53 family members. Mol Cell Biol. 24:1341–1350.
2004. View Article : Google Scholar
|
|
29
|
Liu ZJ, Lu X, Zhang Y, Zhong S, Gu SZ,
Zhang XB, Yang X and Xin HM: Downregulated mRNA expression of ASPP
and the hypermethylation of the 5’-untranslated region in cancer
cell lines retaining wild-type p53. FEBS Lett. 579:1587–1590. 2005.
View Article : Google Scholar
|
|
30
|
Song Q, Song J, Wang Q, Ma Y, Sun N, Ma J,
Chen Q, Xia G, Huo Y, Yang L, et al: miR-548d-3p/TP53BP2 axis
regulates the proliferation and apoptosis of breast cancer cells.
Cancer Med. 5:315–324. 2016. View
Article : Google Scholar
|
|
31
|
Shi Y, Han Y, Xie F, Wang A, Feng X, Li N,
Guo H and Chen D: ASPP2 enhances oxaliplatin (L-OHP)-induced
colorectal cancer cell apoptosis in a p53-independent manner by
inhibiting cell autophagy. J Cell Mol Med. 19:535–543. 2015.
View Article : Google Scholar
|
|
32
|
Hengartner MO: The biochemistry of
apoptosis. Nature. 407:770–776. 2000. View
Article : Google Scholar
|
|
33
|
Liu K, Jiang T, Ouyang Y, Shi Y, Zang Y,
Li N, Lu S and Chen D: Nuclear EGFR impairs ASPP2-p53
complex-induced apoptosis by inducing SOS1 expression in
hepatocellular carcinoma. Oncotarget. 6:16507–16516. 2015.
|
|
34
|
Sarrió D, Rodriguez-Pinilla SM, Hardisson
D, Cano A, Moreno-Bueno G and Palacios J: Epithelial-mesenchymal
transition in breast cancer relates to the basal-like phenotype.
Cancer Res. 68:989–997. 2008. View Article : Google Scholar
|
|
35
|
Wu B, Guo BM, Kang J, Deng XZ, Fan YB,
Zhang XP and Ai KX: PPM1D exerts its oncogenic properties in human
pancreatic cancer through multiple mechanisms. Apoptosis.
21:365–378. 2016. View Article : Google Scholar
|
|
36
|
Yuan TL and Cantley LC: PI3K pathway
alterations in cancer: Variations on a theme. Oncogene.
27:5497–5510. 2008. View Article : Google Scholar
|
|
37
|
Moestue SA, Dam CG, Gorad SS, Kristian A,
Bofin A, Mælandsmo GM, Engebråten O, Gribbestad IS and Bjørkøy G:
Metabolic biomarkers for response to PI3K inhibition in basal-like
breast cancer. Breast Cancer Res. 15:R162013. View Article : Google Scholar
|
|
38
|
Song X, Du J, Zhu W, Jin P and Ma F:
Identification and characterization of an apoptosis-stimulating
protein of p53 (ASPP) gene from Branchiostoma belcheri: Insights
into evolution of ASPP gene family. Fish Shellfish Immunol.
49:268–274. 2016. View Article : Google Scholar
|
|
39
|
Gao K, An J, Zhang Y, Jin X, Ma J, Peng J,
Tang Y, Yu L, Zhang P and Wang C: The E3 ubiquitin ligase Itch and
Yap1 have antagonistic roles in the regulation of ASPP2 protein
stability. FEBS Lett. 589:94–101. 2015. View Article : Google Scholar
|
|
40
|
Liu CY, Lv X, Li T, Xu Y, Zhou X, Zhao S,
Xiong Y, Lei QY and Guan KL: PP1 cooperates with ASPP2 to
dephosphorylate and activate TAZ. J Biol Chem. 286:5558–5566. 2011.
View Article : Google Scholar
|
|
41
|
Yan LX, Liu YH, Xiang JW, Wu QN, Xu LB,
Luo XL, Zhu XL, Liu C, Xu FP, Luo DL, et al: PIK3R1 targeting by
miR-21 suppresses tumor cell migration and invasion by reducing
PI3K/AKT signaling and reversing EMT, and predicts clinical outcome
of breast cancer. Int J Oncol. 48:471–484. 2016. View Article : Google Scholar
|