Introduction
Esophageal carcinoma is the eighth most common type
of cancer globally (1,2). Squamous cell carcinoma (SCC) is the
predominant histological type of esophageal carcinoma (3), but its incidence varies widely by
region (4). High morbidity of
esophageal SCC has been reported in northern and central China,
Turkey, Kazakhstan and northeastern Iran (4,5).
Although radiotherapy is key to the treatment of esophageal cancer,
sustained remission and long-term survival remain moderate.
Autophagy is a natural homeostatic mechanism that
enables cells to maintain energy homeostasis by recycling cellular
components in response to nutrient deprivation and stress (6,7). The
association between radioresistance and autophagy has been
previously investigated (8).
Radiotherapy is crucial for the management of unresectable
esophageal carcinoma, but radioresistance of the tumor is a
hindrance to achieving sustained remission or long-term survival.
Autophagy has been reported to enhance the radioresistance of
non-small-cell lung cancer (9–11).
Preliminary studies indicated that autophagy inhibition improved
the radiosensitivity of breast cancer and esophageal SCC,
consequently enhancing the cytotoxicity of radiotherapy (12,13).
The aim of the present study was to elucidate the potential
therapeutic value of autophagy and its underlying mechanism in
esophageal SCC.
As the most common mechanism of deregulated cell
death in cancer, the mitochondrial pathway acts as 'an enemy
within' for cancer treatment (14). Cytochrome c, as one of
mitochondrial intermembrane space proteins released from damaged
mitochondria, is crucial for apoptosis (15). In cervical cancer cells,
mitochondria were reported to participate in ionizing radiation
(IR)-induced autophagic cell death (16), but their role in esophageal SCC has
not been fully elucidated.
Ample evidence suggests that interference with
autophagy of cancer cells may increase their sensitivity to
treatment, indicating that the promotion of protective autophagy
may be a potential mechanism of radiotherapy resistance in cancer
(17–21). However, existing studies are not
sufficient to confirm the role of autophagy and the detailed
underlying mechanisms in esophageal SCC following treatment with
IR. Therefore, further investigation into the role of autophagy in
esophageal SCC may determine whether antagonizing strategies can
help improve the outcome of radiotherapy.
Materials and methods
Reagents and antibodies
RPMI-1640 medium was purchased from Gibco; Thermo
Fisher Scientific (Grand Island, NY, USA). The autophagy inhibitors
3-methyladenine (3-MA) and LY294002 were obtained from
Sigma-Aldrich; Merck KGaA (St. Louis, MO, USA). Rabbit
anti-human/mouse/rat polyclonal antibody LC3 (E18-5402, 1:500),
GAPDH (E1C604, 1:2,000), cytochrome oxidase (COX) IV (E12-327,
1:1,000), cleaved caspase-3 (E11-0104L, 1:500), cleaved caspase-8
(E18-5267, 1:1,000), cleaved caspase-9 (E18-5240, 1:1,000), mouse
anti-human/mouse monoclonal antibody Bcl-2 (E10-30077, 1:1,000),
rabbit anti-human/mouse polyclonal antibody Bax (E11-0773B, 1:500),
Beclin-1 (E90562, 1:500), mouse anti-human/mouse/rat monoclonal
antibody cytochrome c (E12-378, 1:2000), goat anti-rabbit
(E0L3012), goat anti-mouse IgG secondary antibodies (E0L3032), Cell
Counting Kit-8 (CCK-8) and cell apoptosis analysis kit were all
purchased from EnoGene (New York, NY, USA). Mouse anti-human p53
polyclonal antibody was obtained from BD Pharmingen (554294; San
Jose, CA, USA). TRIzol reagent was obtained from Life Technologies;
Thermo Fisher Scientific (Carlsbad, CA, USA). RevertAid First
Strand cDNA Synthesis kit and QuantiFast SYBR Green PCR kit were
purchased from Thermo Fisher Scientific (Waltham, MA, USA).
Mitochondrial membrane potential assay kit with JC-1 was obtained
from Beyotime Biotechology (Shanghai, China).
Cell culture
The human esophageal carcinoma cell line Eca-109 was
acquired from the American Type Culture Collection (Manassas, VA,
USA). The cells were cultured in RPMI-1640 medium supplement with
10% fetal bovine serum (ScienCell, San Diego, CA, USA), 2 mmol/l
L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin.
All cells were maintained at 37°C under a humidified, 5%
CO2 atmosphere.
Animals
Athymic nude mice (BALB/c-nu, female), aged 4–5
weeks, were purchased from Nanjing Model Animal Research Center
(animal permission no., SYXK2012-0049). All experiments involving
mice were performed in conformity with the guidelines on animal
care and experiments on laboratory animals of the Center of
Experimental Animals, Nanjing University of Technology (Nanjing,
China), and were approved by the Ethics Committee for animal
experimentation.
Autophagy and IR
Eca-109 cells were pretreated with the Earle's
balanced salt solution (EBSS; NaCl 116.36 mM, KCl 5.37 mM,
CaCl2 1.80 mM, MgSO4·7H2O 0.81 mM,
Na2HPO4·12H2O 6.40 mM,
Na2HCO3 26.19 mM, and glucose 5.55 mM), in
order to create poor nutrient conditions and induce autophagosome
formation. For in vitro radiation experiments, following
EBSS treatment, Eca-109 cells were exposed to room temperature and
irradiated with a Cobalt-60 radiotherapy apparatus (Theratron 780c;
Best Theratronics Ltd., Ottawa, ON, Canada) at the indicated doses.
Following irradiation, cell cultures were placed in the cell
culture incubator and maintained at 37°C under 5% CO2.
Control cells were removed from the cell incubator and placed under
the IR source without radiation exposure for the same period. In
the combined treatment studies, indicated concentrations of 3-MA or
LY294002 were added into the medium prior to irradiation. Cells
were further collected for apoptosis analysis and measurement of
the relative protein and mRNA expression.
Electron microscopy
Following EBSS treatment, Eca-109 cells were
harvested by trypsinization and fixed with 2.5% glutaraldehyde for
at least 24 h. The cells were stained with
osmium-thiocarbohydrazide-osmium. Subsequently, the cells were
dehydrated in a series of graded ethanol concentrations (70–100%)
and were immersed serially in 1:1 hexamethyldisilazane followed by
absolute ethanol. Thin sections (1-μm) were cut, and the
gels were coated with 500 Å of gold in a JEOL vacuum sputter coater
and viewed under a JEOL T300 electron microscope with a scanning
attachment (JEOL, Tokyo, Japan).
Cell viability and apoptosis assay
For the cell viability assay, 10,000 cells per well
were seeded into a 96-well plate and cultured overnight. Following
treatment with 3-MA or LY294002, 10 μl CCK-8 solution was
added to each well and the optical density 450 absorbance was
measured by a multifunctional microplate reader (Thermo Fisher
Scientific) after a 2-h incubation at 37°C. For the cell apoptosis
assay, the cells were stained with Annexin V/propidium iodide (PI),
and cell apoptosis was analyzed using flow cytometry. In accordance
with the instruction of cell apoptosis analysis kit (EnoGene),
cells (5×105) were harvested and centrifuged at 1,000 ×
g for 5 min. Cell samples were then resuspended in 500 μl of
binding buffer. Annexin V-enhanced green fluorescent protein (5
μl) and PI (5 μl) were added to the samples and
incubated in the dark for 15 min. A total of 1×104 cells
were collected per sample and detected on FACSCalibur (BD
Biosciences, San Jose, CA, USA). The data were finally analyzed
using FlowJo software (FlowJo LLC, Ashland, OR, USA).
Reverse transcription-polymerase chain
reaction (RT-PCR) analysis
Cells and tumor tissues were used to investigate the
expression of LC3 II and p53 mRNA. In accordance with the
manufacturer's protocol, total RNA was isolated with TRIzol reagent
(Life Technologies; Thermo Fisher Scientific), and was
reverse-transcribed to cDNA using a RevertAid First Strand cDNA
Synthesis kit (Thermo Fisher Scientific). The cDNA obtained was
amplified using the QuantiFast SYBR Green PCR kit. The assays were
performed on an ABI 7500 Fast Real-Time PCR system (Applied
Biosystems, Waltham, MA, USA). Specific primers were synthesized by
Invitrogen and the sequences are listed as follows: GAPDH, forward
5′-CCTCTGACTTCAACAGCGACAC-3′ and reverse
5′-CTGTTGCTGTAGCCAAATTCGT-3′; LC3 II, forward
5′-CAGGTTCACAGAACCCGCC-3′ and reverse 5′-GGTTGCGCTTCACAACTCAG-3′;
and p53, forward 5′-TTCGACATAGTGTGGTGG TGC-3′ and reverse
5′-GCTGTTCCGTCCCAGTAGATT-3′. Relative gene expression levels were
calculated by the 2−ΔΔCq method and normalized to GAPDH
expression as internal control (22).
Subcellular fractionation
Cells or tumor tissues were homogenized using a
Dounce homogenizer in an isotonic buffer (250 mM sucrose, 1 mM
EDTA, 50 mM Tris-HCl, 1 mM DTT, 1 mM PMSF, 1 mM benzamidine, 0.28
μ/ml aprotinin, 50 μg/ml leupeptin, and 7
μg/ml pepstain), and then centrifuged at 1,000 × g for 10
min. The resulting supernatant was centrifuged at 10,000 × g for 20
min and the pellet was collected as the crude mitochondrial
fraction. The remaining supernatant was centrifuged at 100,000 × g
for 1 h and the supernatant was collected as the cytosolic
fraction. The preparation was used for the following immunoblotting
assay.
Western blotting
Western blot analysis was performed according to
standard methods. Total cellular and tissue extracts or
mitochondrial fraction were prepared by RIPA buffer (Beyotime
Institute of Biotechnology, Shanghai, China) according to the
manufacturer's instructions. Protein concentrations were determined
using the Bradford assay. Equal amounts of protein (30–50
μg, depending on the protein) were separated by SDS-PAGE and
transferred onto PVDF membranes. The membranes were incubated with
the indicated primary antibodies and then horseradish
peroxidase-conjugated secondary antibodies. Protein bands were
developed using an ECL detection system. The grey density of target
bands was analyzed by ImageJ software (National Institutes of
Health, Bethesda, MD, USA) and normalized to GAPDH or the
mitochondrial marker COX IV.
Measurement of mitochondrial membrane
potential (MMP)
The MMP of Eca-109 cells was measured using the
cationic dye JC-1 (Beyotime Institute of Biotechnology). After
treatment, Eca-109 cells were incubated with JC-1 at 37°C and 5%
CO2 for 20 min. JC-1 monomers emit green fluorescence
spontaneously, whereas they emit red fluorescence upon entering the
mitochondria of normal cells. Thus, the ratio of red to green
reflects the value of MMP.-
Animal xenograft analysis
Eca-109 cells (5×106), in exponential
growth phase were injected subcutaneously into the right axilla of
nude mice to grow as tumor xenografts. When the tumor volume
reached 100 mm3, tumor-bearing mice were randomly
assigned to four treatment groups (n=6 per group) as follows:
Control, IR alone (8 Gy), 3-MA alone (10 mg/kg intraperitoneally
every other day for 2 weeks), and IR combined with 3-MA. Mice from
the IR group received localized tumor radiation with 8 Gy using a
Cobalt 60 radiotherapy apparatus (Theratron 780c; Best Theratronics
Ltd.). 3-MA was administrated intraperitoneally every other day for
2 weeks until the end of the experiment. The control group received
no IR. Tumor size was determined every other day by measuring the
tumor diameter with calipers. Tumor volume was calculated using the
following formula: Volume = width2 × length/2. The mice
were sacrificed when the tumors exceeded 20–25% of the body mass or
when the tumor volume reached 1,000–1,500 mm3. The
tumors were excised and weighed for analysis.
Statistical analysis
Data are expressed as means ± standard deviation.
Multiple groups were compared with one-way analysis of variance and
two groups with Dunnett's test, using GraphPad Prism 5 (GraphPad
Software, San Diego, CA, USA). Statistical significance was set at
P<0.05.
Results
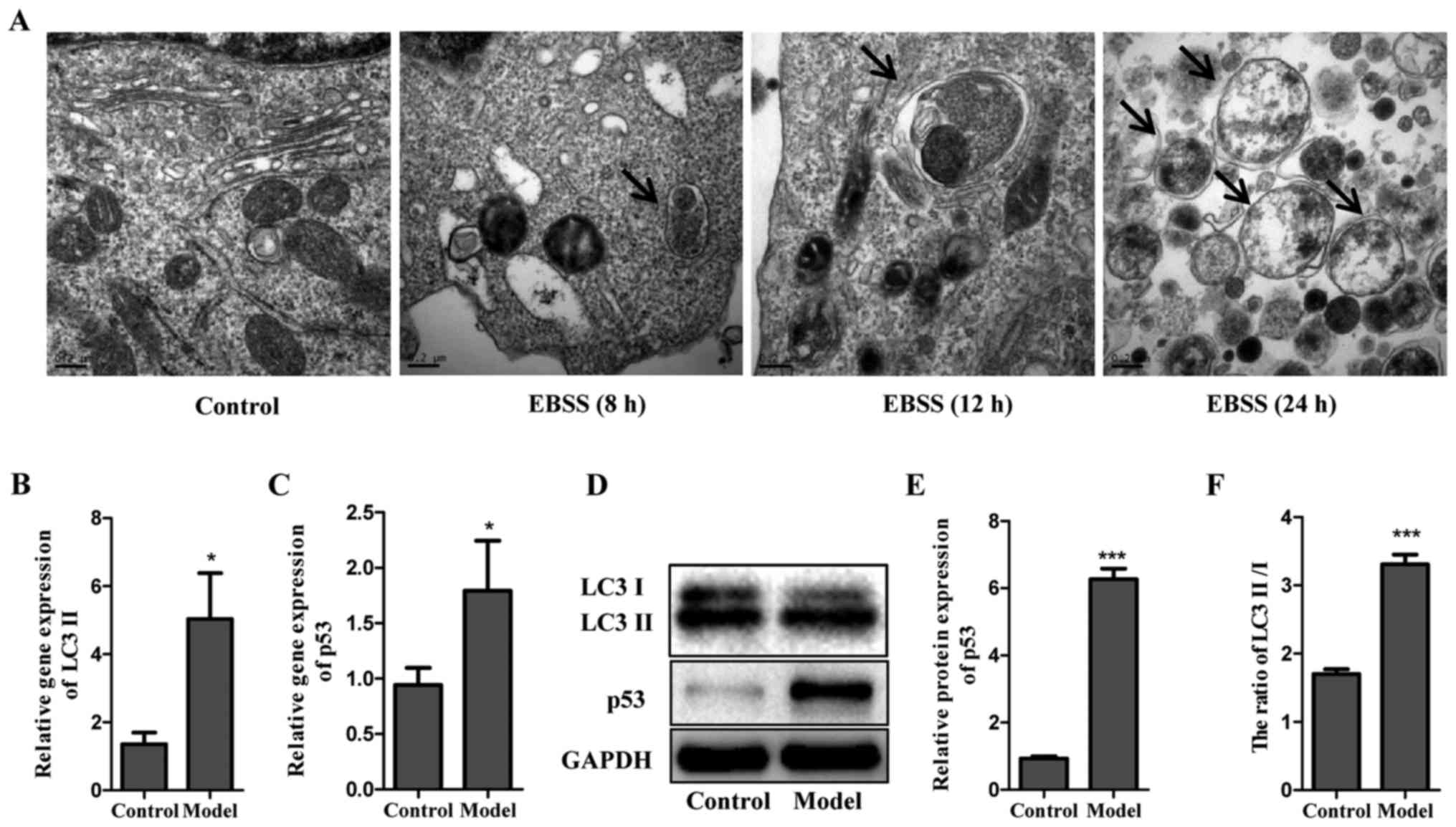
Induction of autophagy by EBSS
To evaluate the potential of EBSS on autophagy
induction, Eca-109 cells were examined at different time points
after being cultured in EBSS (8, 12 and 24 h). Electron microscopy
was used to determine the subcellular changes following exposure to
EBSS. As shown in Fig. 1A, no
autophagic vesicles were observed in untreated Eca-109 cells, in
contrast with those cultured in EBSS for 8, 12 and 24 h. Compared
with 8 h, the vesicles formed after 12 h of treatment with EBSS
were more remarkable, with lamellar structures and residual
digested material. However, only several empty vacuoles were
observed in the cells at 24 h. In addition, the protein and gene
levels of LC3 II were detected in cells cultured in EBSS for 12 h,
in which LC3 was used as a specific autophagy marker. The data
demonstrated that poor nutrient conditions provided by EBSS
markedly decreased LC3 I, but increased LC3 II, as demonstrated by
the increased ratio of LC3 II/LC3 I. Of note, a similar trend was
observed for p53 (Fig. 1B–F). The
observation of the subcellular structure and analysis of LC3 and
p53 formation suggested that EBSS-induced autophagy led to no
significant cell apoptosis until 12 h. Therefore, the process of
autophagy was examined post-EBSS treatment for 12 h in subsequent
experiments.
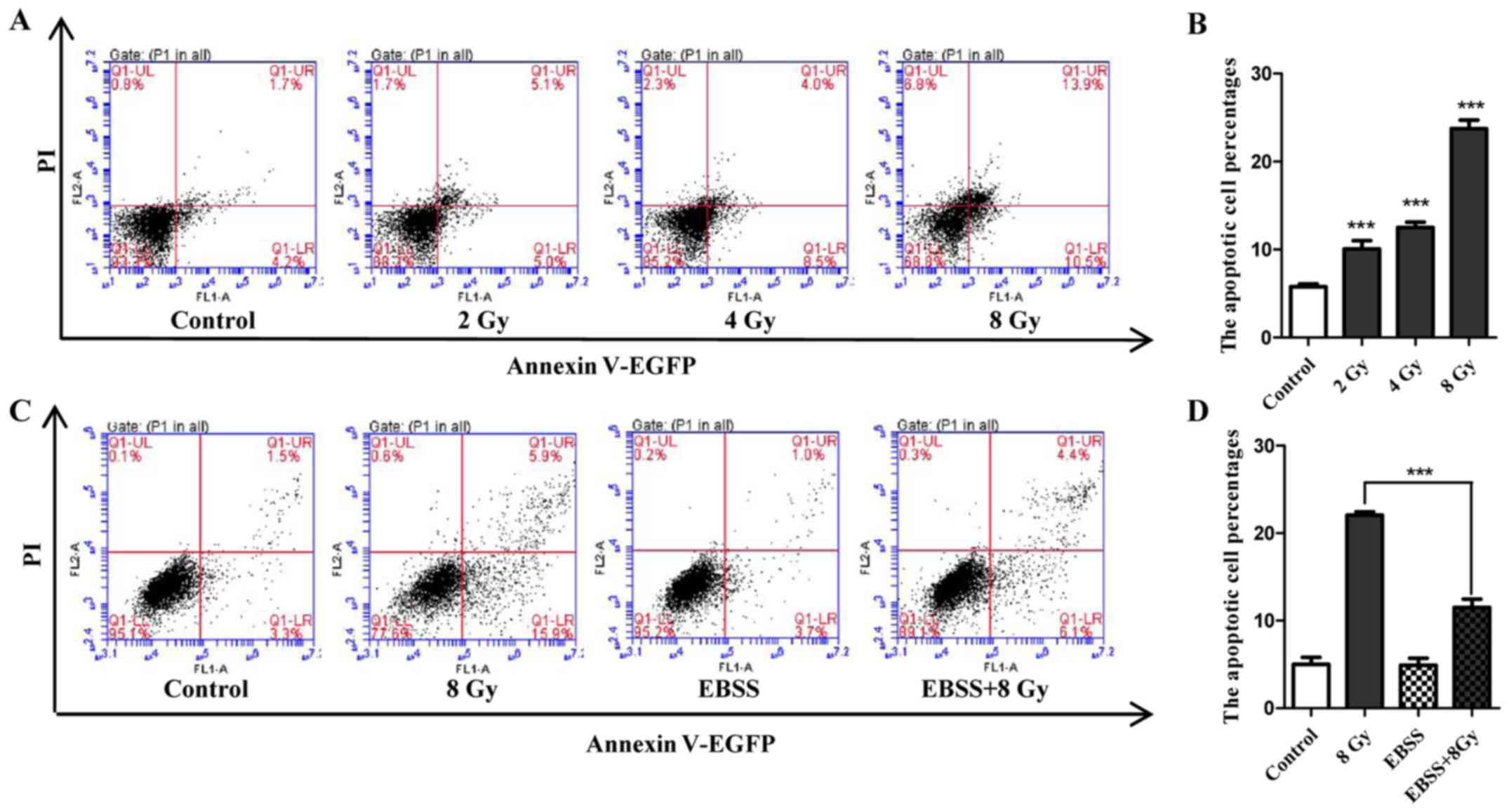
Radiosensitivity of Eca-109 cells and
attenuation effect of autophagy
Cell apoptosis induced by IR was determined in
Eca-109 cells. Following exposure to IR at the indicated dose, cell
apoptosis was assayed by flow cytometry. The results indicated that
the number of apoptotic cells increased in a dose-dependent manner
(Fig. 2A and B). To determine
whether autophagy could attenuate radiosensitivity, the apoptosis
rate of cells that were treated with IR under starvation conditions
was compared with that of cells that were only irradiated or
starved. As shown in Fig. 2C and
D, 12 h of EBSS treatment did not noticeably increase cell
apoptosis compared with untreated cells, but significantly weakened
the potential of apoptosis induction when combined with IR (8 Gy).
Collectively, these findings demonstrated that the radiosensitivity
of Eca-109 cells may be attenuated by starvation-induced
autophagy.
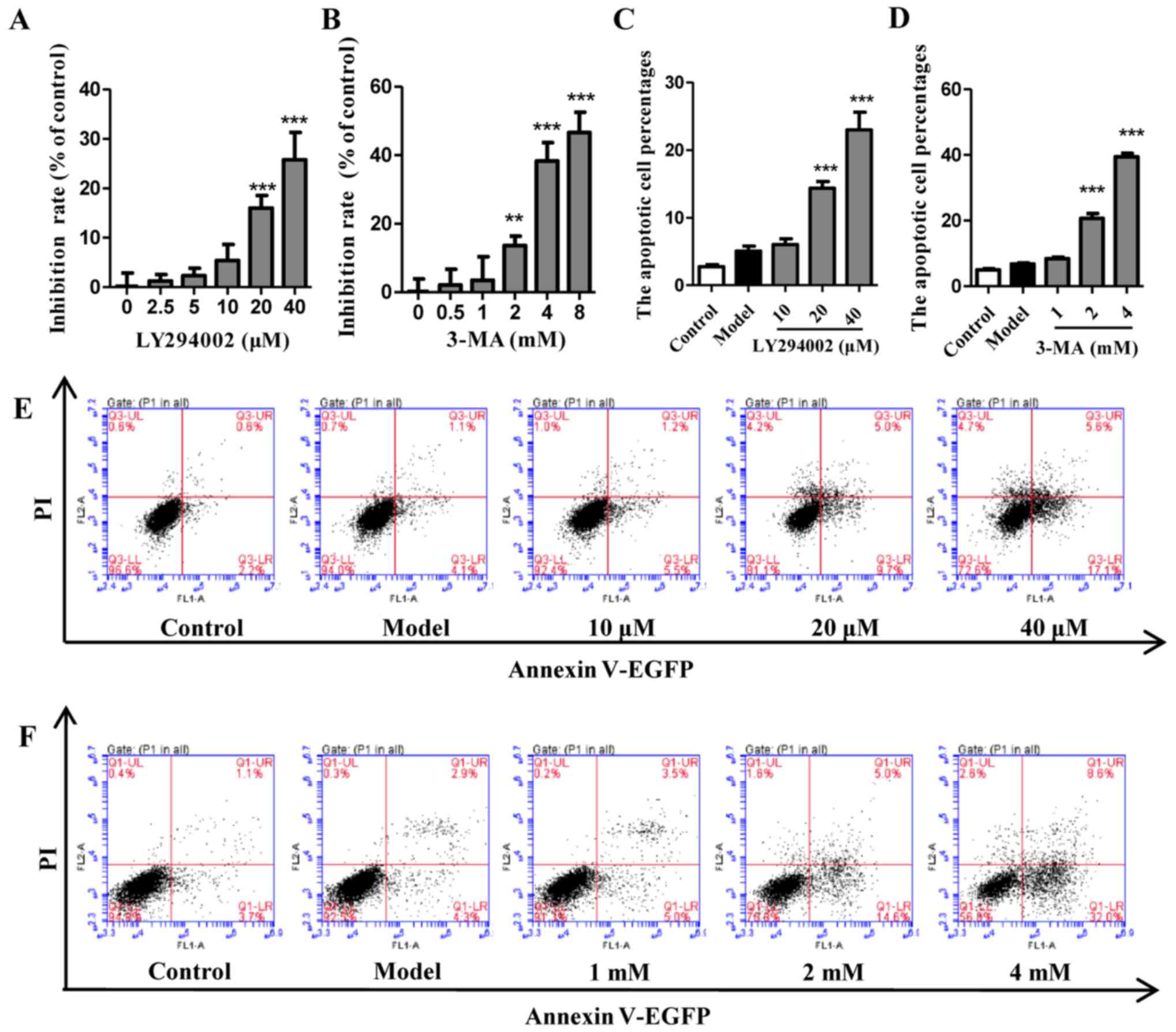
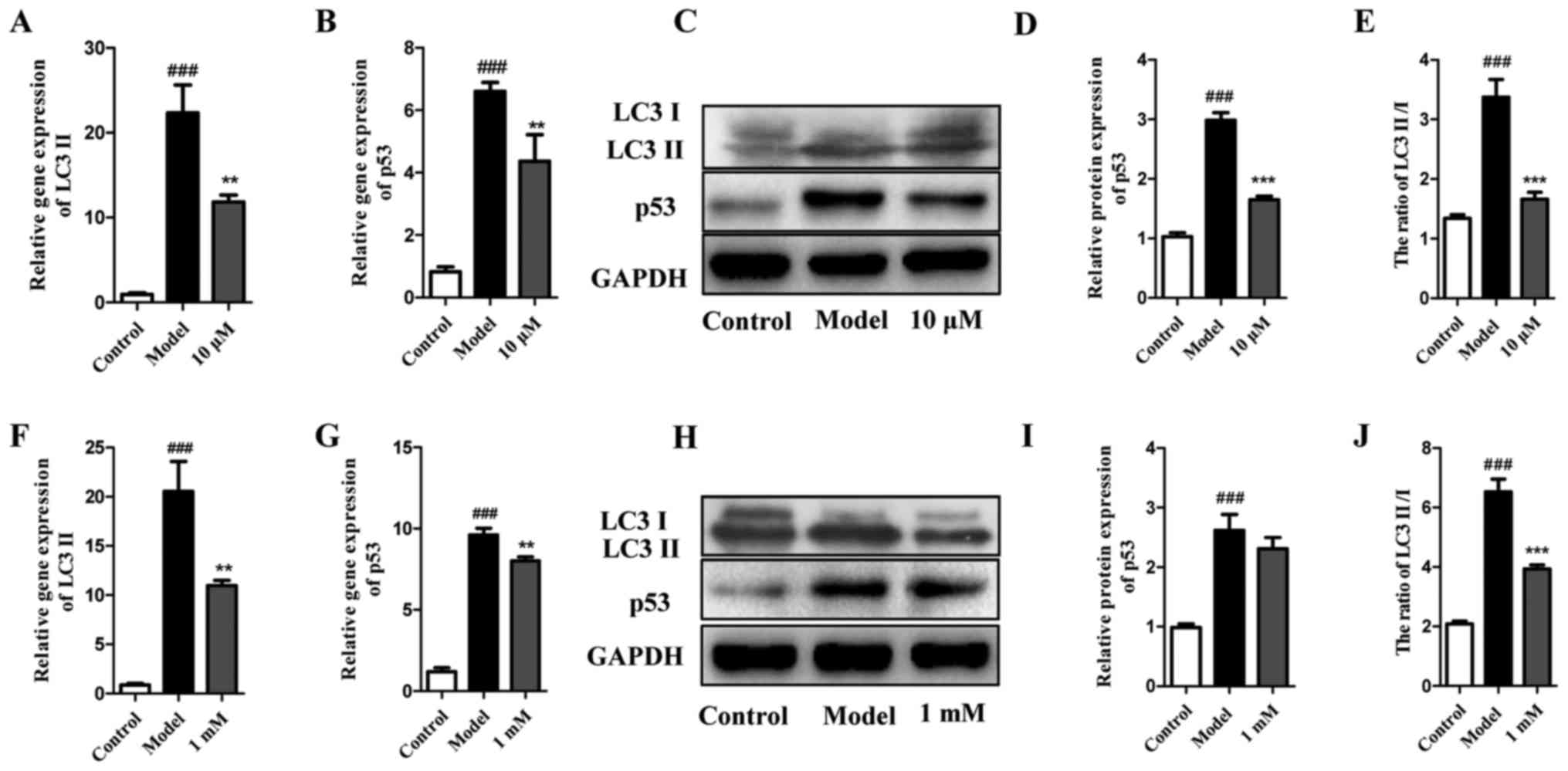
LY294002 or 3-MA alone inhibit autophagy
without induction of cell apoptosis within a certain concentration
range
The viability and apoptosis of cells exposed to EBSS
with different concentrations of LY294002 (Fig. 3A, C and E) or 3-MA (Fig. 3B, D and F) pretreatment were
analyzed. The results indicated that autophagy inhibitors LY294002
and 3-MA did not significantly inhibit cell viability or induce
cell apoptosis until the concentrations were increased to 20
μM for LY294002 and 2 mM for 3-MA. The autophagy of cells
that were pretreated with LY294002 (10 μM) and 3-MA (1 mM)
under EBSS starvation conditions was also evaluated by analyzing
LC3 and p53. As shown in Fig.
4A–E, LY294002 at 10 μM not only decreased the gene
expression of LC3 II and p53, but also significantly reduced the
protein expression ratio of LC3 II and LC3. Furthermore, despite
being increased in EBSS-treated cells, p53 was also decreased by
LY294002 at 10 μM. A similar trend was detected for 3-MA at
1 mM (Fig. 4F–J). Collectively,
these results indicate that autophagy inhibitors LY294002 and 3-MA
at a suitable concentration may significantly inhibit autophagy
without causing extensive cell death.
Inhibition of autophagy increases the
radiosensitivity of Eca-109 cells in vitro
To investigate the role of autophagy inhibitors in
the radiosensitivity of Eca-109 cells, the apoptosis rate of cells
irradiated at 8 Gy was detected with or without autophagy
inhibitors. As a result, treatment with 8 Gy alone led to a reduced
percentage of surviving cells compared with that in the sham group.
Furthermore, the apoptosis-induced potential of IR was markedly
enhanced following pretreatment with LY294002 and 3-MA (Fig. 5A and B). In addition, detection of
beclin-1, LC3 and p53 suggested that LY294002 and 3-MA accelerated
cell death and inhibited cell autophagy concurrently (Fig. 5C–F). In summary, these results
demonstrated that autophagy inhibition exhibited a radiosensitivity
potential in vitro.
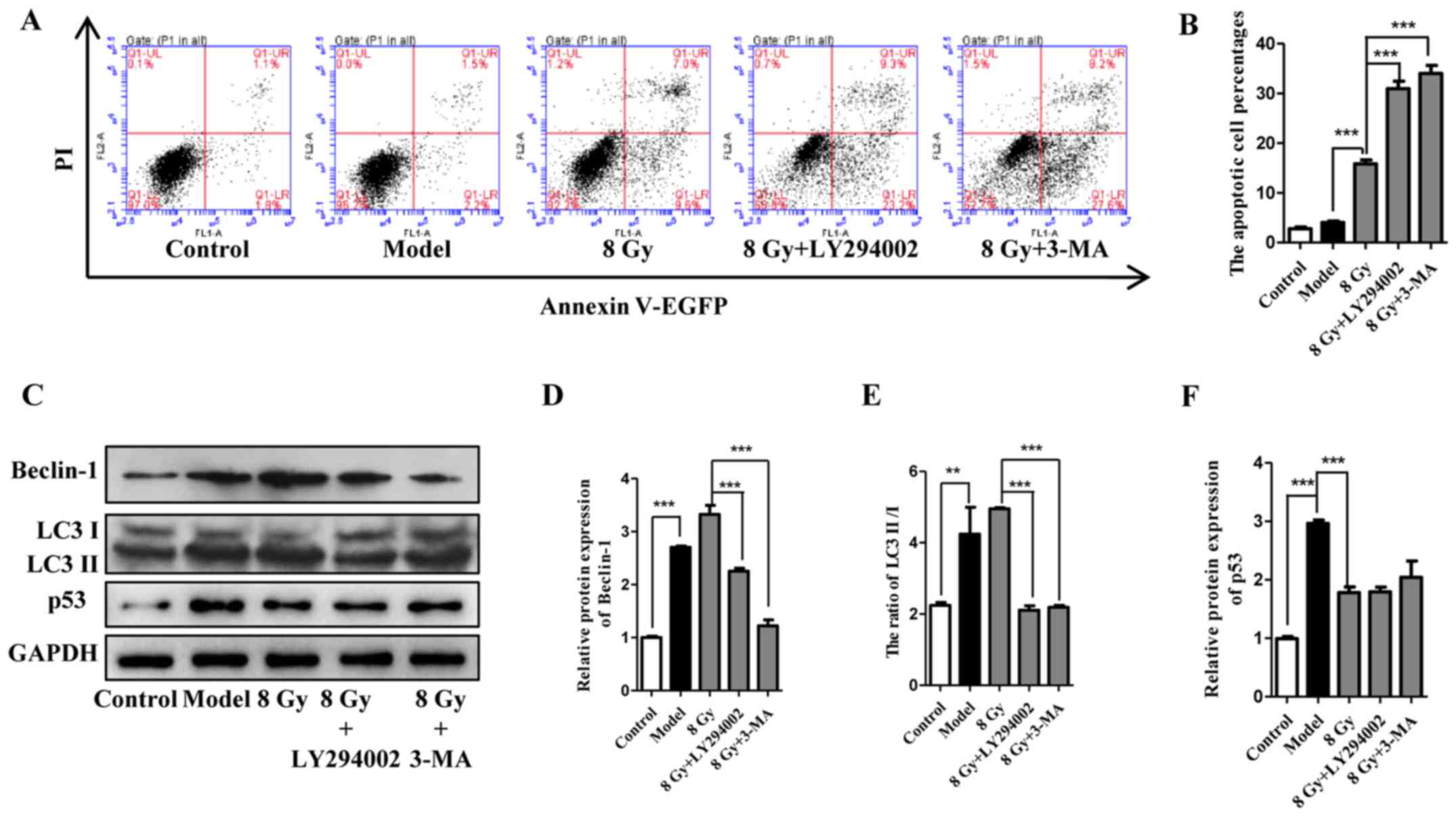 | Figure 5Effect of LY294002 and 3-MA on the
radiosensitivity of Eca-109 cells. (A and B) Cell apoptosis was
analyzed 12 h after treatment with IR (8 Gy) with or without
pretreatment with autophagy inhibitors (10 μM for LY294002
and 1 mM for 3-MA). (mean ± standard deviation, n=3,
***P<0.001). After treatment as indicated, Eca-109
cells were analyzed for the expression of Beclin-1, LC3 and p53
(mean ± standard deviation, n=3, **P<0.01 and
***P<0.001). 3-MA, 3-methyladenine; IR, ionizing
radiation; PI, propidium iodide; EGFP, enhanced green fluorescent
protein. |
Mitochondria are involved in enhancement
of radiosensitivity in Eca-109 cells induced by autophagy
inhibition
Mitochondria are closely associated with cell death.
To assess the changes and the role of mitochondria following
radiation and/or autophagy inhibition, apoptosis-related proteins,
cytochrome c and MMP were examined. As shown in Fig. 6C, following IR together with EBSS
treatment, increased expression of activated caspase-3, caspase-8
and caspase-9 and decreased levels of Bcl-2 were observed in
Eca-109 cells. We also observed increased accumulation of Bax in
mitochondria and a large release of cytochrome c into the
cytosolic fraction (Fig. 6A and
B). Compared with IR alone, the changes in cytochrome c,
Bcl-2 and Bax became more prominent following combination with
autophagy inhibitors (Fig. 6A–C).
As regards MMP, it was observed that radiation (8 Gy) prominently
reduced red fluorescence but enhanced green fluorescence. This
trend was further enhanced by autophagy inhibitors (Fig. 6D and E), suggesting that MMP was
reduced in Eca-109 cells. These results revealed that autophagy
inhibition increased the radiosensitivity of Eca-109 cells,
potentially through the mitochondrial apoptotic pathway.
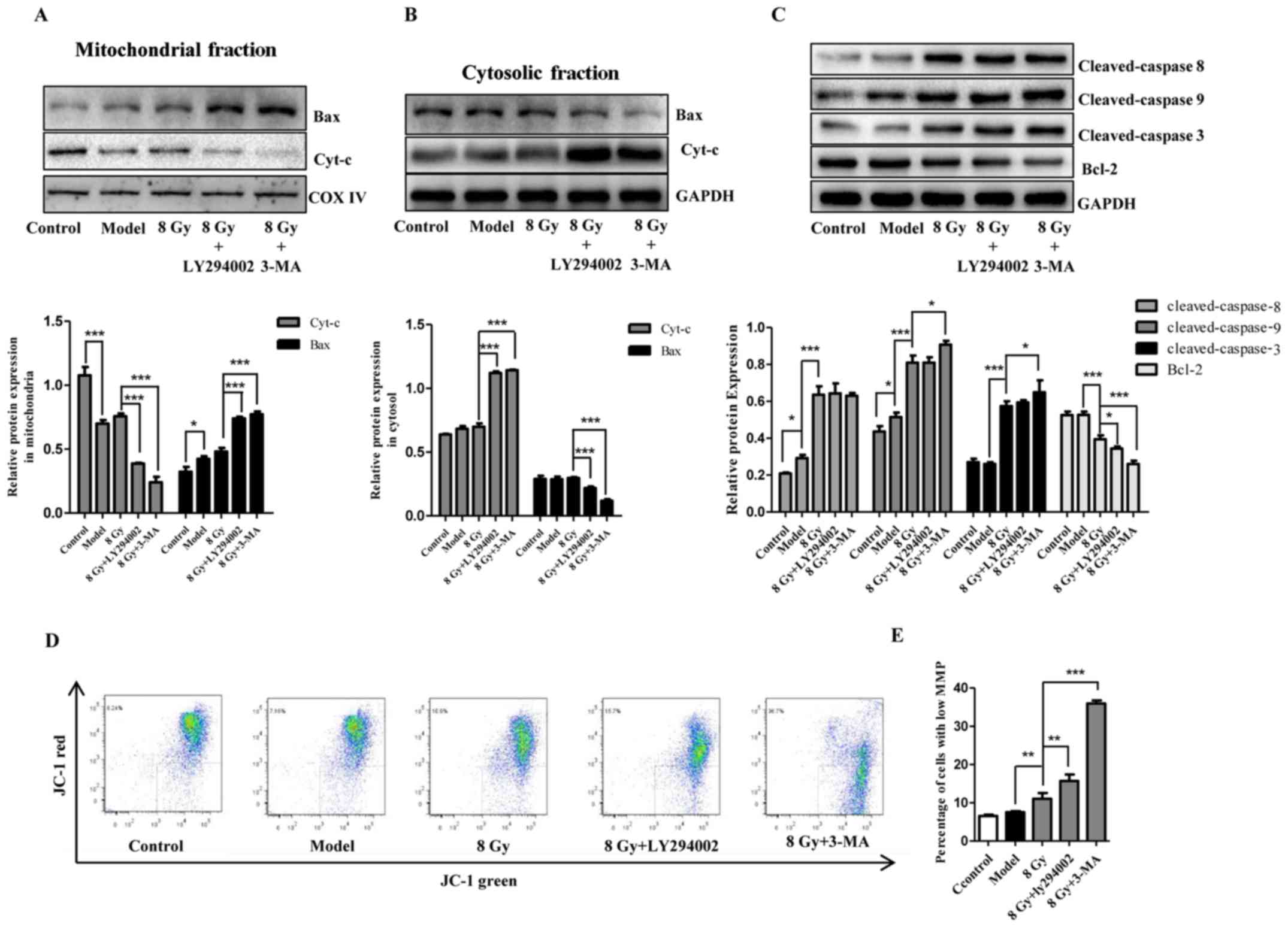 | Figure 6Involvement of mitochondrial
apoptotic pathway in radiosensitivity after autophagy inhibition in
Eca-109 cells. At 12 h after treatment with IR (8 Gy) with or
without pretreatment with autophagy inhibitors (10 μM for
LY294002 and 1 mM for 3-MA), cytochrome c release (A) and
Bax translocation to mitochondria (B), and the cellular apoptosis
initiators caspase-8 and caspase-9, the effector caspase-3, and the
apoptotic protein Bcl-2 (C) in Eca-109 cells were analyzed by
western blotting. GAPDH and COX IV were used as internal protein
loading controls for the cytosolic and mitochondrial fractions,
respectively. (D and E) After treatment as in A, representative
flow cytometric analysis of JC-1 assay was conducted, and the
depolarized cells exhibited decreased red fluorescence and enhanced
green fluorescence. The histogram presents the change of green
fluorescence intensity in Eca-109 cells after various treatments
(mean ± standard deviation, n=3, *P<0.05,
**P<0.01 and ***P<0.001). COX,
cytochrome oxidase; IR, ionizing radiation; 3-MA,
3-methyladenine. |
Combination of autophagy inhibitor and IR
suppresses the tumorigenesis of Eca-109 cells in a nude mouse
xenograft model
Since the additive effects of autophagy inhibition,
particularly by 3-MA, on the radiosensitity of Eca-109 cells has
been established, a nude mouse xenograft model was utilized to
validate the biological effects and underlying mechanisms by in
vivo 3-MA administration. Cell suspensions were injected
subcutaneously into the right axilla of athymic nude mice. The mice
were then randomly divided into the four indicated groups, which
were treated with DMSO (vehicle of 3-MA), radiation (8 Gy), 3-MA
(10 mg/kg), and radiation together with 3-MA. As shown in Fig. 7A–C, tumor volume and weight were
measured and the data suggested that the tumor size in the model
animals markedly enlarged compared with baseline. IR treatment
slightly delayed tumor growth. Furthermore, tumor growth was
significantly delayed when the animals were treated with IR and
3-MA, whereas treatment with 3-MA alone did not exert a noticeable
effect on tumor growth compared with the model group. The
immunoblotting analysis revealed that IR treatment promoted the
release of cytochrome c and Bax activation, along with
decreased levels of Bcl-2. Consistent with the autophagy inhibition
effect in vitro, this effect of IR treatment was markedly
potentiated by abolishing autophagy (Fig. 7D–F). In summary, these results
suggested that combination treatment with 3-MA and IR enhanced the
response of esophageal carcinoma cells to radiotherapy in
vivo.
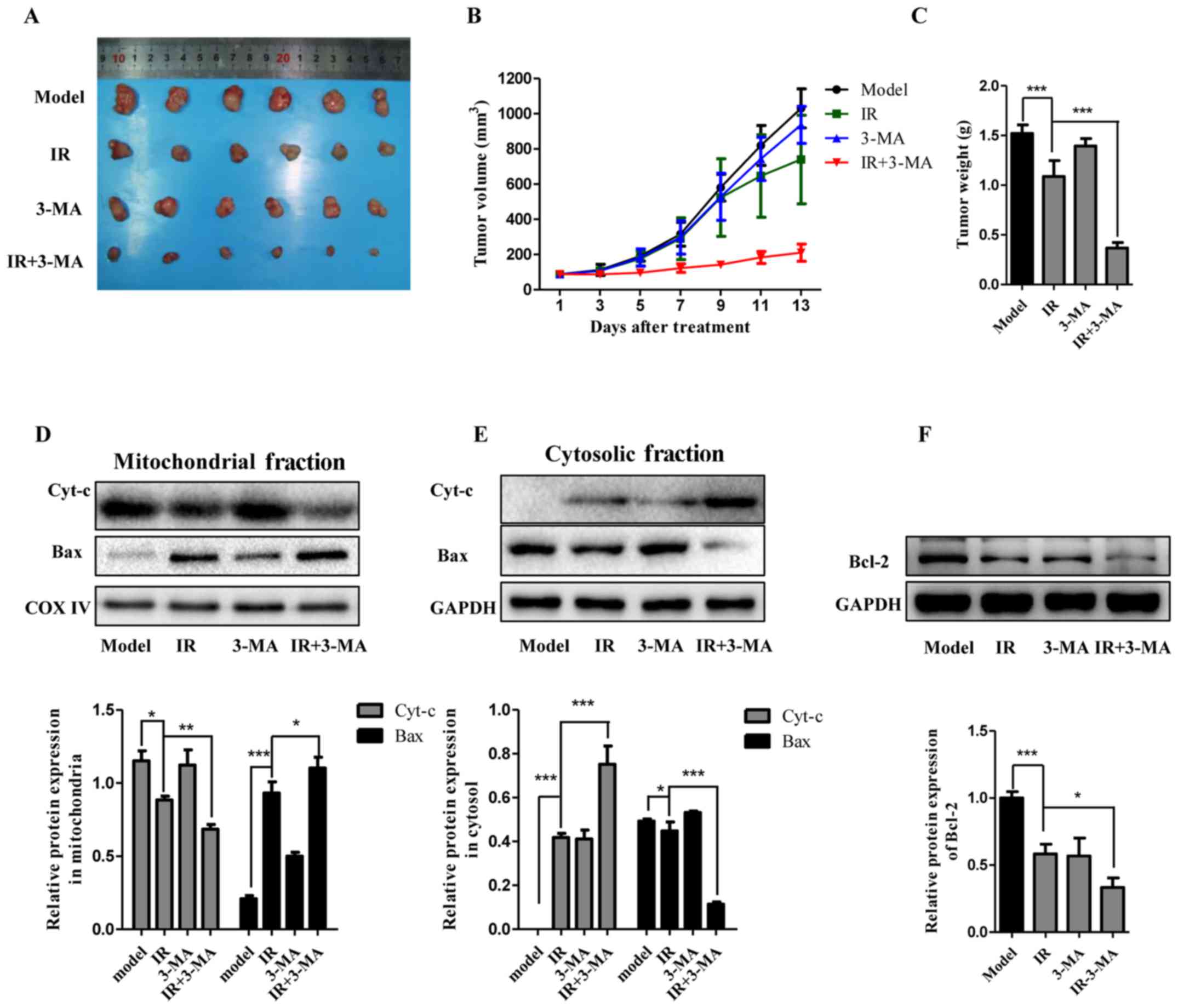 | Figure 7Autophagy inhibition markedly
increased the sensitivity of the tumor to IR in vivo. (A–C)
Representative images of tumors and tumor growth curve after cell
inoculation and treatment with a combination of IR and 3-MA in an
Eca-109 nude mouse xenograft model (mean ± standard deviation, n=6,
*P<0.05, **P<0.01 and
***P<0.001). (D–F) The apoptotic proteins Bax, Bcl-2,
and cytochrome c in xenografts were analyzed by western
blotting and the results of densitometric quantification are shown
using GAPDH or COX IV for normalization (mean ± standard deviation,
n=3, *P<0.05, **P<0.01 and
***P<0.001). COX, cytochrome oxidase; IR, ionizing
radiation; 3-MA, 3-methyladenine. |
Discussion
Autophagy enables cells to degrade intracellular
molecules to support cell survival, and it has historically been
observed in cells under nutrient deprivation conditions (6). As a characteristic of autophagy, the
formation of autophagic vacuoles encompasses cellular components to
be digested via fusion with lysosomes (23). In autophagy, organelles and
proteins to be degraded are sequestered into autophagosomes. During
its formation, the cytosolic isoform LC3 I is conjugated to
phosphatidylamine to form LC3 II. LC3 II is indispensable for the
expansion of autophagosomal membrane. Furthermore, as it is
correlated with the number of autophagosomes, increased LC3 II
levels indicate that autophagic activity in the cell is increased
(24,25). Even if the role of autophagy in
carcinogenesis and tumor progression remains controversial, it has
attracted significant attention as a potential anticancer target
(26,27).
In the present study, Eca-109 cells were cultured in
EBSS medium to induce autophagosome formation and accumulation.
Herein, LC3 II/LC3 I was used as a surrogate marker for autophagy
levels in cells (28). We observed
that EBSS led to the formation of autophagic vacuoles and a
prominent increase in the LC3 II/LC3 I ratio in Eca-109 cells at 12
h post-EBSS treatment, which were markedly attenuated by
pre-treatment with LY294002 and 3-MA. In addition, the
apoptosis-inducing effect of IR and the implication of EBSS-induced
autophagy were investigated. The results suggested that cell
apoptosis is induced by IR in a dose-dependent manner; however, it
is prominently attenuated by EBSS-induced autophagy, indicating
that EBSS-induced autophagy in Eca-109 cells may play a role as a
cytoprotective mechanism, in consistence with previously reported
results (11).
Cancer cells escape death using a variety of means,
with prevention of apoptosis through loss of p53 in some cell types
being a classic example (29,30).
p53 was recently found to be involved in autophagy regulation
(31–33). In the development of pancreatic
cancer, p53 may play a key role in blocking tumor progression and
promoting cell death and senescence caused by inhibition of
autophagy (34). In the present
study, increased expression of p53 was observed with EBSS treatment
and was markedly attenuated by autophagy inhibition. In view of the
following results indicating that cell apoptosis was promoted by
the combinatorial use of IR and autophagy inhibitors, suppressing
the tumor suppressor p53 may provide a possible explanation as to
why autophagy inhibition improves the radiosensitivity of Eca-109
cells.
Autophagy inhibition in the present study was
performed by administration of LY294002 and 3-MA, and the effect of
these autophagy inhibitors on apoptosis and autophagy of starved
Eca-109 cells was investigated. The results demonstrated that
LY294002 and 3-MA effectively inhibited autophagy without inducing
extensive apoptosis at certain concentrations. To distinguish
between the apoptosis evoked by IR and that induced by autophagy
inhibitors and produce more reliable results, the optimal
concentrations of LY294002 and 3-MA were used in the following
experiments. Autophagy inhibition combined with IR exhibited
possible anticancer value, which has been reported in previous
studies (8,13,35).
In the present study, we also observed that the radiosensitivity of
Eca-109 cells was significantly enhanced following treatment with
LY294002 and 3-MA, which was indicated by increased apoptotic cell
numbers and Beclin-1 expression, together with an increased LC3
II/LC3 I ratio. Of note, tumor progression was markedly inhibited
in the xenograft nude mouse model following combined treatment with
autophagy inhibition and IR. Chen et al used the esophageal
carcinoma cell line EC9706 to investigate whether combining
radiation with autophagy inhibition enhances the suppression of
tumor growth and angiogenesis in esophageal cancer, and the results
support our findings (12),
suggesting that this may be a common phenomenon in esophageal
carcinoma.
To summarize, our results further validated the
vital role of autophagy inhibition in radiotherapy for esophageal
cancer. Mitochondria are vulnerable to various stimuli and are
crucial for regulating cell survival or death. Since a number of
studies support that mitochondria are closely associated with
autophagy and cancer cell death≈(16,36–38),
it is reasonable to hypothesize that the mitochondrial pathway may
contribute to enhanced radiosensitivity by autophagy inhibition in
esophageal SCC cells. The function of the mitochondrial pathway was
investigated in vitro as well as in vivo. It is well
known that mitochondrial apoptosis is controlled by proteins of the
Bcl-2 family, categorized as antiapoptotic proteins, BH3-only
proteins and effectors (39,40).
In the mitochondrial pathway, mitochondrial outer membrane
permeabilization (MOMP) is the defining event. MOMP may be
initiated directly by activating Bax or Bak. Subsequently,
cytochrome c and other factors are released and interact
with several cytosolic proteins, such as APAF-1, to evoke the
activation of caspase. Pro-caspase-9 is recruited and activated by
apoptosome formation and, in turn, the executioners caspase-3 and
-7 are cleaved and activated. Subsequently, the activity of
caspase-3 and -7 kills cells within minutes by cleaving abundant
substrates. Even in the absence of caspase activation, post-MOMP
mitochondria can hardly maintain cell survival due to their
inability to generate ATP (15,41,42).
The mechanisms that cancer cells utilize to inhibit mitochondrial
apoptosis may be classified as those inhibiting mitochondrial
permeabilization and those blocking caspase function (14). After IR, elevated expression of
activated caspases and Bax and decreased levels of Bcl-2 were
observed in starved Eca-109 cells, along with stimulated release of
cytochrome c and decreased MOMP. In addition, application of
autophagy inhibitors amplified the changes in mitochondrial
pathway-related proteins and MOMP. The role of the mitochondria
pathway was further studied in an Eca-109 xenograft nude mouse
model, the findings of which were consistent with those of the
in vitro experiments.
In conclusion, the results of the present study
demonstrated that autophagy inhibition markedly enhanced the
cytotoxicity of IR and suppressed tumor growth through the
mitochondrial pathway. Our results further supported the value of
autophagy inhibitors in the radiotherapy of esophageal SCC, and the
significance of mitochondria in cancer radiotherapy.
Abbreviations:
|
EBSS
|
Earle's balanced salt solution
|
|
IR
|
ionizing radiation
|
|
MMP
|
mitochondrial membrane potential
|
|
SCC
|
squamous cell carcinoma
|
Acknowledgments
The authors would like to thank Yuan Liu, Xi Yu, Lei
Ao and Lei Cao for providing experimental advice and technical
help.
Funding
The present study was supported by a grant from the
Jiangsu Cancer Hospital (no. ZM201207).
Ethics approval and consent to
participate
All experiments involving mice were performed in
conformity with the guidelines on animal care and experiments on
laboratory animals of the Center of Experimental Animals, Nanjing
University of Technology (Nanjing, China), and were approved by the
Ethics Committee for Animal Experimentation (no.
OGKQSPF/SQ-03).
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
HT wrote the manuscript. HT, PDQ and FJW designed
the experiments, HT, PDQ, JCL, YSG and HFZ performed experiments
and analysed the data. The final version of the manuscript has been
read and approved by all authors.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar
|
|
2
|
Jemal A, Siegel R, Ward E, Hao Y, Xu J and
Thun MJ: Cancer statistics, 2009. CA Cancer J Clin. 59:225–249.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Enzinger PC and Mayer RJ: Esophageal
cancer. N Engl J Med. 349:2241–2252. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kamangar F, Dores GM and Anderson WF:
Patterns of cancer incidence, mortality, and prevalence across five
continents: Defining priorities to reduce cancer disparities in
different geographic regions of the world. J Clin Oncol.
24:2137–2150. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Eslick GD: Epidemiology of esophageal
cancer. Gastroenterol Clin North Am. 38:17–25. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Murrow L and Debnath J: Autophagy as a
stress-response and quality-control mechanism: Implications for
cell injury and human disease. Annu Rev Pathol. 8:105–137. 2013.
View Article : Google Scholar
|
|
7
|
Martinet W, De Meyer GR, Andries L, Herman
AG and Kockx MM: In situ detection of starvation-induced autophagy.
J Histochem Cytochem. 54:85–96. 2006. View Article : Google Scholar
|
|
8
|
Yang Y, Yang Y, Yang X, Zhu H, Guo Q, Chen
X, Zhang H, Cheng H and Sun X: Autophagy and its function in
radiosensitivity. Tumour Biol. 36:4079–4087. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen X, Wang P, Guo F, Wang X, Wang J, Xu
J, Yuan D, Zhang J and Shao C: Autophagy enhanced the
radioresistance of non-small cell lung cancer by regulating ROS
level under hypoxia condition. Int J Radiat Biol. 93:764–770. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lomonaco SL, Finniss S, Xiang C,
Decarvalho A, Umansky F, Kalkanis SN, Mikkelsen T and Brodie C: The
induction of autophagy by gamma-radiation contributes to the
radioresistance of glioma stem cells. Int J Cancer. 125:717–722.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chaachouay H, Ohneseit P, Toulany M,
Kehlbach R, Multhoff G and Rodemann HP: Autophagy contributes to
resistance of tumor cells to ionizing radiation. Radiother Oncol.
99:287–292. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen Y, Li X, Guo L, Wu X, He C, Zhang S,
Xiao Y, Yang Y and Hao D: Combining radiation with autophagy
inhibition enhances suppression of tumor growth and angiogenesis in
esophageal cancer. Mol Med Rep. 12:1645–1652. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liang DH, El-Zein R and Dave B: Autophagy
inhibition to increase radiosensitization in breast cancer. J Nucl
Med Radiat Ther. 6:62015. View Article : Google Scholar
|
|
14
|
Lopez J and Tait SW: Mitochondrial
apoptosis: Killing cancer using the enemy within. Br J Cancer.
112:957–962. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tait SW and Green DR: Mitochondria and
cell death: Outer membrane permeabilization and beyond. Nat Rev Mol
Cell Biol. 11:621–632. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen Z, Wang B, Yu F, Chen Q, Tian Y, Ma S
and Liu X: The roles of mitochondria in radiation-induced
autophagic cell death in cervical cancer cells. Tumour Biol.
37:4083–4091. 2016. View Article : Google Scholar
|
|
17
|
Bristol ML, Di X, Beckman MJ, Wilson EN,
Henderson SC, Maiti A, Fan Z and Gewirtz DA: Dual functions of
autophagy in the response of breast tumor cells to radiation:
Cytoprotective autophagy with radiation alone and cytotoxic
autophagy in radiosensitization by vitamin D 3. Autophagy.
8:739–753. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wilson EN, Bristol ML, Di X, Maltese WA,
Koterba K, Beckman MJ and Gewirtz DA: A switch between
cytoprotective and cytotoxic autophagy in the radiosensitization of
breast tumor cells by chloroquine and vitamin D. Horm Cancer.
2:272–285. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Apel A, Herr I, Schwarz H, Rodemann HP and
Mayer A: Blocked autophagy sensitizes resistant carcinoma cells to
radiation therapy. Cancer Res. 68:1485–1494. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kondo Y, Kanzawa T, Sawaya R and Kondo S:
The role of autophagy in cancer development and response to
therapy. Nat Rev Cancer. 5:726–734. 2005. View Article : Google Scholar
|
|
21
|
Sotelo J, Briceño E and López-González MA:
Adding chloroquine to conventional treatment for glioblastoma
multiforme: A randomized, double-blind, placebo-controlled trial.
Ann Intern Med. 144:337–343. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
23
|
Feng Y, He D, Yao Z and Klionsky DJ: The
machinery of macroautophagy. Cell Res. 24:24–41. 2014. View Article : Google Scholar :
|
|
24
|
Mizushima N and Yoshimori T: How to
interpret LC3 immunoblotting. Autophagy. 3:542–545. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T: LC3, a
mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Amaravadi RK, Lippincott-Schwartz J, Yin
XM, Weiss WA, Takebe N, Timmer W, DiPaola RS, Lotze MT and White E:
Principles and current strategies for targeting autophagy for
cancer treatment. Clin Cancer Res. 17:654–666. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Grandér D and Panaretakis T: Autophagy:
Cancer therapy's friend or foe? Future Med Chem. 2:285–297. 2010.
View Article : Google Scholar
|
|
28
|
He Y, Zhao X, Subahan NR, Fan L, Gao J and
Chen H: The prognostic value of autophagy-related markers beclin-1
and microtubule-associated protein light chain 3B in cancers: A
systematic review and meta-analysis. Tumour Biol. 35:7317–7326.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Clarke AR, Purdie CA, Harrison DJ, Morris
RG, Bird CC, Hooper ML and Wyllie AH: Thymocyte apoptosis induced
by p53-dependent and independent pathways. Nature. 362:849–852.
1993. View Article : Google Scholar
|
|
30
|
Lowe SW, Schmitt EM, Smith SW, Osborne BA
and Jacks T: p53 is required for radiation-induced apoptosis in
mouse thymocytes. Nature. 362:847–849. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Eisenberg-Lerner A, Bialik S, Simon HU and
Kimchi A: Life and death partners: Apoptosis, autophagy and the
cross-talk between them. Cell Death Differ. 16:966–975. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Crighton D, Wilkinson S, O'Prey J, Syed N,
Smith P, Harrison PR, Gasco M, Garrone O, Crook T and Ryan KM:
DRAM, a p53-induced modulator of autophagy, is critical for
apoptosis. Cell. 126:121–134. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Maiuri MC, Malik SA, Morselli E, Kepp O,
Criollo A, Mouchel PL, Carnuccio R and Kroemer G: Stimulation of
autophagy by the p53 target gene Sestrin2. Cell Cycle. 8:1571–1576.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rosenfeldt MT, O'Prey J, Morton JP, Nixon
C, MacKay G, Mrowinska A, Au A, Rai TS, Zheng L, Ridgway R, et al:
p53 status determines the role of autophagy in pancreatic tumour
development. Nature. 504:296–300. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Han MW, Lee JC, Choi JY, Kim GC, Chang HW,
Nam HY, Kim SW and Kim SY: Autophagy inhibition can overcome
radioresistance in breast cancer cells through suppression of TAK1
activation. Anticancer Res. 34:1449–1455. 2014.PubMed/NCBI
|
|
36
|
Chen Z, Liu X and Ma S: The roles of
mitochondria in autophagic cell death. Cancer Biother Radiopharm.
31:269–276. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Qiao ZY, Lai WJ, Lin YX, Li D, Nan XH,
Wang Y, Wang H and Fang QJ: Polymer-KLAK peptide conjugates induce
cancer cell death through synergistic effects of mitochondria
damage and autophagy blockage. Bioconjug Chem. 28:1709–1721. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Danese A, Patergnani S, Bonora M,
Wieckowski MR, Previati M, Giorgi C and Pinton P: Calcium regulates
cell death in cancer: Roles of the mitochondria and
mitochondria-associated membranes (MAMs). Biochim Biophys Acta.
1858:615–627. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kim H, Rafiuddin-Shah M, Tu HC, Jeffers
JR, Zambetti GP, Hsieh JJ and Cheng EH: Hierarchical regulation of
mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell
Biol. 8:1348–1358. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kim H, Tu HC, Ren D, Takeuchi O, Jeffers
JR, Zambetti GP, Hsieh JJ and Cheng EH: Stepwise activation of BAX
and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis.
Mol Cell. 36:487–499. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Colell A, Ricci JE, Tait S, Milasta S,
Maurer U, Bouchier-Hayes L, Fitzgerald P, Guio-Carrion A,
Waterhouse NJ, Li CW, et al: GAPDH and autophagy preserve survival
after apoptotic cytochrome c release in the absence of caspase
activation. Cell. 129:983–997. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lartigue L, Kushnareva Y, Seong Y, Lin H,
Faustin B and Newmeyer DD: Caspase-independent mitochondrial cell
death results from loss of respiration, not cytotoxic protein
release. Mol Biol Cell. 20:4871–4884. 2009. View Article : Google Scholar : PubMed/NCBI
|